The Child with a Condition of the Blood, Blood-Forming Organs, or Lymphatic System
1 Define each key term listed.
2 Summarize the components of blood.
3 Recall normal blood values of infants and children.
4 List two laboratory procedures commonly performed on children with blood disorders.
5 Compare and contrast four manifestations of bleeding into the skin.
6 List the symptoms, prevention, and treatment of iron-deficiency anemia.
7 Recommend four food sources of iron for a child with iron-deficiency anemia.
8 Examine the pathophysiology and the signs and symptoms of sickle cell disease.
9 Describe four types of sickle cell crises.
10 Devise a nursing care plan for a child with sickle cell disease.
11 Recognize the effects on the bone marrow of increased red blood cell production caused by thalassemia.
12 Review the effects of severe anemia on the heart.
13 Recall the pathophysiology and the signs and symptoms of hemophilia A and hemophilia B.
14 Identify the nursing interventions necessary to prevent hemarthrosis in a child with hemophilia.
15 Plan the nursing care of a child with leukemia.
16 Review the nursing care of a child receiving a blood transfusion.
17 Discuss the effects of chronic illness on the growth and development of children.
18 Recall the stages of dying.
19 Contrast age-appropriate responses to a sibling’s death and the nursing interventions required.
20 Formulate techniques the nurse can use to facilitate the grieving process.
21 Discuss the nurse’s role in helping families to deal with the death of a child.
 , p. 636)
, p. 636) , p. 634)
, p. 634) , p. 625)
, p. 625) , p. 633)
, p. 633) , p. 634)
, p. 634) , p. 625)
, p. 625) , p. 631)
, p. 631) , p. 626)
, p. 626) , p. 626)
, p. 626) , p. 627)
, p. 627) , p. 641)
, p. 641) , p. 626)
, p. 626) http://evolve.elsevier.com/Leifer
http://evolve.elsevier.com/Leifer
The blood and blood-forming organs make up the hematological system. Blood is vital to all body functions. Blood dyscrasias or disorders occur when blood components fail to form correctly or when blood values exceed or fail to meet normal standards.
Plasma and blood cells are formed at about the second week of gestation, primarily in the yolk sac. Later, blood forms in the spleen, liver, thymus, lymph system, and bone marrow. In the fetus, blood is formed primarily in the liver until the last trimester of pregnancy. During childhood, the red blood cells (RBCs) are formed in the marrow of the long bones (such as the tibia and femur); by adolescence, hematopoiesis (blood formation) takes place in the marrow of the ribs, sternum, vertebrae, pelvis, skull, clavicle, and scapulae. The rate of RBC production is regulated by erythropoietin. This substance is produced by the liver of the fetus, but at birth the kidneys take over erythropoietin production.
The lymphatic system includes lymphocytes, lymphatic vessels, lymph nodes, the spleen, the tonsils, the adenoids, and the thymus gland. The lymphatic system drains regions of the body to lymph nodes, where infectious organisms are destroyed and antibody production is stimulated. Lymph nodes are not palpable in the newborn, but the cervical, axillary, and inguinal nodes may be palpable by childhood. Lymphadenopathy is an enlargement of lymph nodes that is indicative of infection or disease. Figure 27-1 summarizes some of the differences between the child’s and the adult’s lymphatic systems.
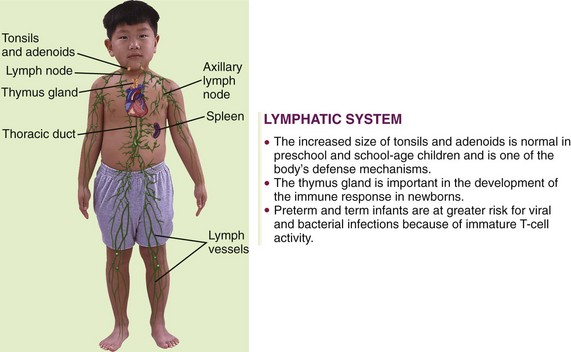
FIGURE 27-1 Summary of lymphatic system differences between the child and the adult. The lymphatic system is a subsystem of the circulatory system. It returns excess tissue fluid to the blood and defends the body against disease.
Figure 27-2 depicts the main types of blood cells in the circulating blood. Circulating blood consists of two portions: plasma and formed elements. The formed elements are erythrocytes (red blood cells), leukocytes (white blood cells [WBCs]), and thrombocytes (platelets). Erythrocytes primarily transport oxygen and carbon dioxide to and from the lungs and tissues. Leukocytes act as the body’s defense against infections. Thrombocytes, along with portions of blood plasma, are involved with blood coagulation. In the young child, every available space in the bone marrow is involved with blood formation.
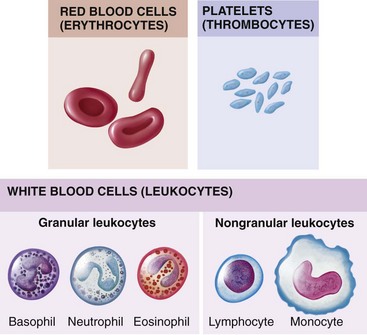
FIGURE 27-2 The formed elements of the blood. Red blood cells (RBCs; erythrocytes), white blood cells (WBCs; leukocytes), and platelets (thrombocytes) constitute the formed elements of the blood.
Lymphocytes, unlike other WBCs, are produced in the lymphoid tissues of the body. They travel in the circulation but are more commonly found in the lymph tissue. They are released into the body to fight infection and provide immunity. Their numbers greatly increase in chronic inflammatory conditions. The spleen is the largest organ of the lymphatic system. One of the main functions of the spleen is to bring blood into contact with lymphocytes. Aside from trauma and rupture, the most commonly seen pathological condition of the spleen is enlargement. This is termed splenomegaly. The spleen enlarges during infections, congenital and acquired hemolytic anemias, and liver malfunction.
Bone marrow aspiration is a procedure helpful in determining disorders of the blood. Numerous types of blood counts are used as well. Many are specific to a particular disease. The skin is sometimes an indicator of certain conditions of the blood. Petechiae (pinpoint hemorrhagic spots) and purpura (large petechiae) are often seen and should alert the nurse to the possibility of blood dyscrasias. The physician examines the liver and spleen by palpation and percussion to determine if they are enlarged.
Anemias
Anemia can result from many different underlying causes. A reduction in the amount of circulating hemoglobin reduces the oxygen-carrying ability of the blood. A hemoglobin level below 8 g/dL results in an increased cardiac output and a shunting of blood from the periphery of the body to the vital organs. Pallor, weakness, tachypnea, shortness of breath, and congestive heart failure can result.
Iron-Deficiency Anemia
Pathophysiology: The most common nutritional deficiency of children in the United States today is anemia caused by insufficient amounts of iron in the body. The incidence is highest during infancy and adolescence—two rapid growth periods. Anemia (an, “without,” and emia, “blood”) is a condition in which there is a reduction in the amount and size of the RBCs or in the amount of hemoglobin, or both. Iron-deficiency anemia may be caused by severe hemorrhage, the child’s inability to absorb the iron received, excessive growth requirements, or an inadequate diet. Researchers have also found that feeding whole cow’s milk to infants can precipitate gastrointestinal bleeding, resulting in anemia.
Prevention of iron-deficiency anemia begins with good prenatal care to ensure that the mother has a suitable intake of iron during pregnancy. During the first few months after birth, the newborn relies on iron that was stored in the system during fetal life. Preterm infants may be deprived of a sufficient supply, because iron is obtained late in the prenatal period. In addition, the iron stores of low-birth-weight infants and infants from multiple births are relatively small.
The highest incidence of iron-deficiency anemia occurs from the ninth to the twenty-fourth month. During this rapid growth period, the infant outgrows the limited iron reserve that was in the body; in addition, iron-fortified formula and infant cereals may have been eliminated from the diet. Poorly planned meals or feeding problems also contribute to this deficiency. The mother may sometimes rely too heavily on bottle feedings to avoid conflict at meals. Unfortunately, milk contains very little iron. Instead, the amounts of solid food should be increased and the amount of milk decreased. Boiled egg yolk, liver, leafy green vegetables, Cream of Wheat, dried fruits (apricots, peaches, prunes, raisins), dry beans, crushed nuts, and whole-grain bread are good sources of iron. Iron-fortified cereals eaten out of the box provide a nutritious snack. Unfortunately, not all the iron found in a food source is absorbed by the body. The bioavailability of iron in vegetables is less than that in meat.
Manifestations: The symptoms of iron-deficiency anemia are pallor, irritability, anorexia, and a decrease in activity. Many infants are overweight because of excessive consumption of milk (so-called “milk babies”). Blood tests for anemia may include RBC count, hemoglobin and hematocrit levels, and determination of morphological cell changes and iron concentration. The stool may be tested for occult blood. A dietary history is also obtained. Sometimes a slight heart murmur is heard. The spleen may be enlarged.
Untreated iron-deficiency anemias progress slowly, and in severe cases the heart muscle becomes too weak to function. If this happens, heart failure follows. Children with long-standing anemia may also show growth retardation and cognitive changes. Screening procedures are suggested at 9 and 24 months for full-term infants and earlier for low-birth-weight infants.
Treatment: Iron-deficiency anemia responds well to treatment. Iron, usually ferrous sulfate, is given orally two or three times a day between meals. Vitamin C aids in the absorption of iron; therefore giving juice when administering iron is suggested. Liquid preparations are taken through a straw to prevent temporary discoloration of the teeth. (Some iron preparations without this disadvantage are available.) The toddler needs solid foods that are rich sources of iron. An iron-dextran mixture (Imferon) given intramuscularly is also highly effective. It must be injected deep in a large muscle using the Z-track technique to minimize staining and tissue irritation.
Parent Education: Parents need explicit instructions on proper foods for the infant. The nurse stresses the importance of breastfeeding for the first 6 months and the use of iron-fortified formula throughout the first year of life (the absorption of iron from human milk is much better than from cow’s milk). The amount of milk consumed during the day and night is determined. Solid food intake is reviewed, and specific iron-enriched nutrients are suggested. The nurse considers financial, ethnic, and family preferences in teaching plans. Behavior concerns at mealtime may also have to be addressed.
The stools of infants who are taking oral iron supplements are tarry green. An absence of this finding may indicate poor parental compliance with therapy. Oral iron preparations are not to be given with milk, which interferes with absorption. To increase absorption, these preparations should be given between meals, when digestive acid concentration is highest. It is important to emphasize that both dietary changes and supplemental iron therapy are necessary to eradicate iron-deficiency anemia. Good dietary practices must be lifelong to maintain good health. Parents are encouraged to return for periodic evaluation of the child’s blood status. Nursing Care Plan 27-1 specifies interventions for the child with iron-deficiency anemia.
27-1  Nursing Care Plan
Nursing Care Plan
The Child with Iron-Deficiency Anemia
A parent brings a 10-month-old boy to the clinic. The child is pale and listless. Laboratory results show iron-deficiency anemia. The parent states that the child loves to drink milk.

Imbalanced nutrition: less than body requirements, related to iron-poor diet

1. The mother of a child diagnosed with anemia comes to the clinic and states she is worried about the pallor and lack of energy her child continues to show. She states she works outside the home but has her child in “good day care” that provides good meals. What further questions would be appropriate for the nurse to ask in this interview?
 Medication Safety Alert!
Medication Safety Alert!Sickle Cell Disease
Pathophysiology: Sickle cell disease is an inherited defect in the formation of hemoglobin. It occurs mainly in people of African descent, but it is also carried by some persons of Mediterranean descent, such as some Arabs, Greeks, Maltese, Sicilians, as well as those originating from other Mediterranean areas. Many researchers believe that the gene for sickle cell disease developed in these populations as protection against malaria. Sickling (clumping) caused by decreased blood oxygen levels may be triggered by dehydration, infection, physical or emotional stress, or exposure to cold. Laboratory examination of the affected child’s blood shows that the RBC has changed its shape to resemble that of a sickle blade, from which the name of the disorder is derived (Figure 27-3).
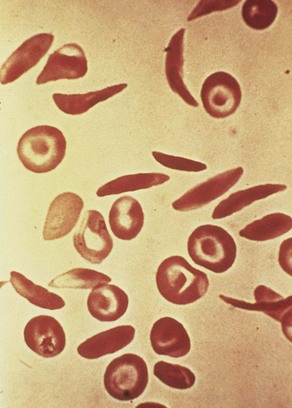
FIGURE 27-3 Scanning electron micrograph of erythrocytes. Note that the normal red blood cell is round. The sickle-shaped cell can clump as it flows through the circulation, causing a vasoocclusive sickle cell crisis.
Sickle cells contain an abnormal form of hemoglobin termed hemoglobin S (the sickling type). The membranes of these cells are fragile and easily destroyed. Their crescent shape makes it difficult for them to pass through the capillaries, causing a pileup of cells in the small vessels. This clumping may lead to a thrombosis (clot) and cause an obstruction. Infarcts, or areas of dead tissue, may result when the tissue is denied proper blood supply. These generally develop in the spleen but may also be seen in other areas of the body, such as the brain, heart, lungs, gastrointestinal tract, kidneys, and bones. The patient feels acute pain in the affected area.
There are two types of sickle cell disease: an asymptomatic (a, “without,” and symptoma, “symptom”) version (sickle cell trait) and much more severe forms that necessitate intermittent hospitalization (sickle cell anemia).
Sickle cell trait: This form of the disease occurs in about 10% of the African American population in the United States. The blood of the patient contains a mixture of normal (hemoglobin A) and sickle (hemoglobin S) hemoglobins. The proportions of hemoglobin S are low because the disease is inherited from only one parent. The physician can distinguish sickle cell trait from the more severe disease by electrophoresis study of the patient’s RBCs and hemoglobin. Sickling is more rapid and extreme with sickle cell disease. In sickle cell trait the hemoglobin and RBC counts are normal.
Sickle cell trait does not develop into sickle cell disease. Although there is no need to treat the patient with sickle cell trait, the patient is a carrier, and genetic counseling is important.
Sickle cell anemia: This severe form of sickle cell disease results when the abnormality is inherited from both parents (Figure 27-4). Each offspring has a one in four chance of inheriting the disease (which is not the same as one in four children inheriting it). In general, the clinical symptoms do not appear until the last part of the first year of life. There may be an unusual swelling of the fingers and toes. The symptoms of sickle cell disease are caused by enlarging bone marrow sites that impair circulation to the bone and the abnormal sickle cell shape that causes clumping, obstruction in the vessel, and ischemia to the organ that the vessel supplies.
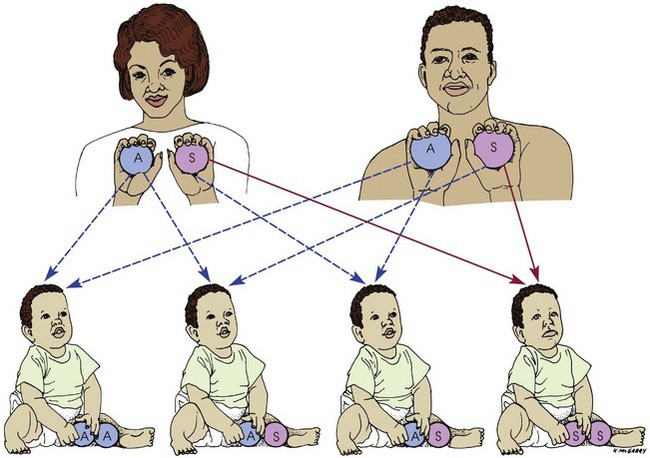
FIGURE 27-4 Transmission of sickle cell disease from parents to children. Parents who are carriers of the sickle cell trait do not show symptoms of the disease because hemoglobin A (the normal form of hemoglobin) in their red blood cells protects them from hemoglobin S (the sickling form). When two carriers become parents, however, the possibilities are as follows: One child in four will inherit all normal hemoglobin and thus be free of the disease (AA); two children in four will inherit both hemoglobin A and hemoglobin S and thus become carriers (AS) of the trait (like their parents); and one child in four will inherit all sickling hemoglobin and thus be affected by sickle cell disease (SS).
There is chronic anemia. The hemoglobin level ranges from 6 to 9 g/dL or lower. The child is pale, tires easily, and has little appetite. These manifestations of anemia are complicated by characteristic episodes called sickle cell crises, which are painful and can be fatal. Specific types of crises have been defined. They differ in their pathological causes and manifestations and may necessitate somewhat different treatments (Table 27-1). Unfortunately, in some cases the sickle cell crisis is the first obvious manifestation of the condition. The patient appears acutely ill. There is severe abdominal pain. Muscle spasms, leg pain, or painful swollen joints may be seen. Fever, vomiting, hematuria, convulsions, stiff neck, coma, or paralysis can result, depending on the organs involved. Children with sickle cell disease have a risk for stroke as a complication of a vasoocclusive sickle cell crisis.
Table 27-1
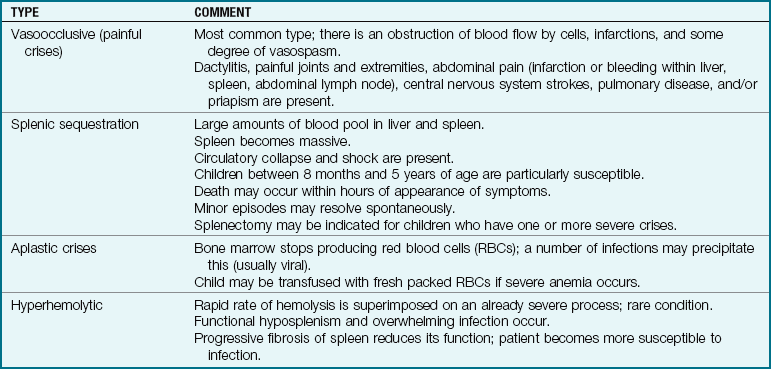
Data from Jackson, P., Vessey, J., & Schapiro, N (2009). Primary care of the child with a chronic condition (5th ed.). Philadelphia: Saunders; Kliegman, R., Behrman, R., Jenson, H., & Stanton, B. (2007). Nelson textbook of pediatrics (18th ed.). Philadelphia: Saunders.
The sickle cell crises recur periodically throughout childhood; however, they tend to decrease with age. Patients should be kept in good health between episodes. Immunizations of these children are particularly important, including those for Haemophilus influenzae, hepatitis A, and hepatitis B as well as the pneumococcal vaccine. Patients should refrain from becoming overly tired. They also should avoid situations such as flying in an unpressurized airplane or exercising at a high altitude, because oxygen concentrations in their blood are already reduced. Extra stress and exposure to cold may lower resistance, causing additional problems. Overheating, which can lead to dehydration, is also to be avoided. Oral intake of iron is of no value.
Diagnosis: Sickle cell disease can be detected before birth by chorionic villi sampling. Early diagnosis by mandatory screening of all newborns in the United States allows early detection before symptoms occur. The sickling test (Sickledex) is commonly used for screening. Hemoglobin electrophoresis (“fingerprinting”) is used when the result is positive. This procedure separates and records the various peptide patterns of the blood. It distinguishes between patients with the trait and those with the disease.
Treatment and Nursing Care: When the infant or child is hospitalized during a crisis, the treatment is supportive and symptomatic. The patient is confined to bed. Analgesics are given to relieve pain. Children in severe pain may need a continuous intravenous (IV) infusion containing a narcotic. Meperidine (Demerol) is not recommended for children with sickle cell disease because of the risk of normeperidine-induced seizures (National Institutes of Health [NIH], 2002). A patient-controlled analgesia (PCA) pump enables children older than 7 years to maintain control and participate in care. Every effort is made to combat dehydration and acidosis. Small blood transfusions may be administered to increase the hemoglobin count, but the results are only temporary. Packed RBCs are often given for this purpose. An accurate record of intake and output is kept. The patient’s body position is changed frequently but gently. Hemopoietic stem cell transplantation may provide a cure in the near future.
Sickle cell disease may take a wide variety of courses. As always, the individual patient’s progress is followed. Sometimes it is difficult to distinguish between abdominal pain caused by a sickle cell crisis and abdominal pain caused by appendicitis. The nurse must remember that pain experienced by a child with sickle cell disease may also be caused by an unrelated condition.
Prevention of infection and prevention of dehydration are important goals in the care of a child with sickle cell disease. Pneumococcal and meningococcal vaccinations and annual influenza vaccination are recommended. Oral penicillin prophylaxis before any invasive treatment, including dental care, is also advised (NIH, 2002). Multiple transfusions of packed cells may be required to maintain adequate hemoglobin levels. The nurse should observe the child for reactions to the blood transfusion. Hemosiderosis (the deposit of iron into organs and tissues in the body) is a complication of this type of hemolytic disease. An exchange transfusion may reduce the number of circulating sickle cells and prevent complications such as thrombus formation and stroke. High-dose IV methylprednisolone may be used to decrease severe pain (Dover & Platt, 2003).
The main goals of therapy are to prevent sickling, dehydration, hypoxia, and infection, which can cause a sickle cell crisis. Erythropoietin and some chemotherapy regimens can increase the production of fetal hemoglobin and reduce complications. Bone marrow transplantation and gene replacement therapy may hold a promising cure (Jackson et al., 2009).
Surgery: The use of splenectomy in children with sickle cell disease has been conservative. A recurrence of acute splenic sequestration becomes less likely after age 5 years. Routine splenectomy is not recommended because the spleen generally atrophies on its own because of fibrotic changes that take place in patients with sickle cell disease.
Thalassemia
Pathophysiology: The thalassemias are a group of hereditary blood disorders in which the patient’s body cannot produce sufficient adult hemoglobin. The RBCs are abnormal in size and shape and are rapidly destroyed. This abnormality results in chronic anemia. The body attempts to compensate by producing large amounts of fetal hemoglobin. Thalassemias are caused by a deficiency in the normal synthesis of hemoglobin polypeptide chains. They are categorized according to the Greek letters designating the polypeptide chain affected as α-, β-, γ-, or δ-thalassemia.
The most common variety of thalassemia involves impaired production of beta chains and is known as β-thalassemia. This variety consists of two forms: thalassemia minor and thalassemia major. Thalassemia major is also called Cooley’s anemia. Thalassemia occurs mainly in persons of Mediterranean origin, such as Greeks, Syrians, and Italians, and their descendants elsewhere. The term is derived from the Greek thalassa, which means “sea.” Thalassemia can also occur from spontaneous mutations.
Thalassemia minor: Thalassemia minor, which is also termed β-thalassemia trait, occurs when the child inherits a thalassemia gene from only one parent (heterozygous inheritance). It is associated with mild anemia. Hemoglobin concentration averages 2 to 3 g/dL, lower than normal age-related values. These patients are often misdiagnosed as having an iron-deficiency anemia. Symptoms are minimal. The patient is pale, and the spleen may be enlarged. The patient may lead a normal life, with the illness going undetected. This condition is of genetic importance, particularly if both parents are carriers of the trait. Prenatal blood samples can detect thalassemia major in such cases.
Thalassemia major (Cooley’s anemia): When two thalassemia genes are inherited (homozygous inheritance), the child is born with a more serious form of the disease. A progressive, severe anemia becomes evident within the second 6 months of life.
The child is pale and hypoxic, has a poor appetite, and may have a fever. Jaundice, which at first is mild, progresses to a muddy bronze color resulting from hemosiderosis, a deposit of iron (released by blood cell destruction) into the tissues. The liver enlarges, and the spleen grows enormously. Abdominal distention is great, which causes pressure on the organs of the chest. Cardiac failure caused by the profound anemia is a constant threat. Bone marrow space enlarges to compensate for an increased production of blood cells. Hematopoietic (hema, “blood,” and poiesis, “to make”) defects and a massive expansion of the bone marrow in the face and skull result in changes in the facial contour that give the child a characteristic appearance (Figure 27-5). The teeth protrude because of an overgrowth of the upper jawbone; the bone becomes thin and is subject to pathological fracture.
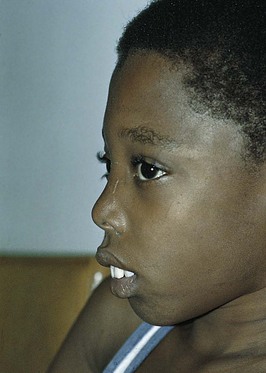
FIGURE 27-5 Appearance of child with thalassemia major (Cooley’s anemia). Note the overgrowth of the upper jawbone (maxillary hyperplasia).
Diagnosis is aided by a family history of thalassemia, radiographic bone growth studies, and blood tests. Hemoglobin electrophoresis is helpful in diagnosing the type and severity of the various thalassemias. Prenatal screening and diagnosis are available, and genetic counseling is advised.
Treatment and Nursing Care: The goals of care for children with thalassemia are to (1) maintain hemoglobin levels at 9.5 g/dL to prevent overgrowth of bone marrow and resultant deformities and (2) provide for growth and development and normal physical activity. Prevention or early treatment of infection is important. Some patients require splenectomy. Bone marrow transplantation holds promise for the future treatment of this condition (Jackson et al., 2009).
The mainstay of treatment for thalassemia major is frequent blood transfusions to maintain the hemoglobin level above 9.5 g/dL. As a result of repeated blood transfusions, excessive deposits of iron may be stored in the tissues. This is termed hemosiderosis and is seen especially in the spleen, liver, heart, pancreas, and lymph glands. Deferoxamine mesylate (Desferal), an iron-chelating agent, is given to counteract hemosiderosis. Severe splenomegaly may occur in some children. Splenectomy may make the patient more comfortable, increase the ability to move about, and allow for more normal growth. After surgery, these children are given prophylactic antibiotics to prevent infection.
Nursing measures adhere to the principles of long-term care. The observation of the patient during a blood transfusion is discussed on p. 637. Monitoring of vital signs is necessary to detect irregularities of the heart. Whenever possible, the same nurse cares for the patient during transfusions, blood tests, and other unpleasant procedures to provide security. Children are taught to regulate their activities according to their own tolerance.
The emotional health of the child and parents calls for special consideration by the nurse. Every attempt to ease the strain of this prolonged illness must be made. Home care arrangements can be provided through community agencies. The family can be referred to the Cooley’s Anemia Foundation for support and education. Older children need special support to accept changes in their body image caused by the disease. Suggestions applicable to the care of the chronically ill child are discussed throughout the text. Care of the dying child is discussed later in this chapter.
Bleeding Disorders
Pathophysiology: Hemophilia is one of the oldest hereditary diseases known to humanity. In this disorder the blood does not clot normally, and even the slightest injury can cause severe bleeding. It has been called the disease of kings because it has occurred in the children of several royal families in Russia and Europe. This congenital disorder is confined almost exclusively to males but is transmitted by symptom-free females.
Hemophilia is inherited as a sex-linked recessive trait. It is termed sex-linked because the defective gene is located on the X, or female, chromosome. Different combinations of genes account for the fact that some children inherit the disease, some become carriers, and others neither inherit nor carry the trait. New mutations do occur, and the reason for this is unclear. The sex of the fetus can be determined by amniocentesis. Fetal blood sampling detects hemophilia. Some carrier women can also be identified.
There are several types of hemophilia. More than 10 identified factors in blood are involved in the clotting mechanism. A deficiency in any one of the factors will interfere with normal blood clotting. The two most common types of hemophilia are hemophilia B, or Christmas disease (a factor IX deficiency), and hemophilia A (a deficiency in factor VIII). This discussion is limited to classic hemophilia, or hemophilia A, which accounts for approximately 84% of cases.
Hemophilia A is caused by a deficiency of coagulation factor VIII, or antihemophilic globulin (AHG). The severity of the disease depends on the level of factor VIII in the plasma of the patient’s blood. Some patients’ lives are endangered by minor injury, whereas a child with a mild case of hemophilia might just bruise a little more easily than the normal person. The degree of severity tends to remain constant within a given family. The aim of therapy is to increase the level of factor VIII high enough to ensure clotting. It is possible to determine the level of factor VIII in the blood by means of a test called the partial thromboplastin time (PTT), which can help to diagnose and assess the child’s condition. Prenatal diagnosis by amniocentesis is possible.
Manifestations: Hemophilia can be diagnosed at birth because maternal factor VIII cannot cross the placenta and be transferred to the fetus. However, it is usually not apparent in the newborn unless abnormal bleeding occurs at the umbilical cord or after circumcision. As the child grows older and becomes more subject to injury, the slightest bruise or cut can induce extensive bleeding. Normal blood clots in about 3 to 6 minutes. In a patient with severe hemophilia, however, the time required for clotting may be 1 hour or longer.
Anemia, leukocytosis, and a moderate increase in the number of platelets may be seen in the hemorrhaging child. There may also be signs of shock. Spontaneous hematuria is seen. Death can result from excessive bleeding anywhere in the body, but particularly when hemorrhage into the brain or neck occurs. Severe headache, vomiting, and disorientation may reflect cranial bleeding. Bleeding into the neck can cause airway obstruction. Bleeding into the ears and eyes can affect hearing and vision. Bleeding into the spinal column can lead to paralysis.
The circumstances leading to diagnosis may be the inability of a parent to stop a child’s bleeding from a cut around the mouth or gums. A deciduous tooth loss may precipitate problems in a child who has a bleeding disorder. Hematomas may develop after immunizations. An injured knee, elbow, or ankle presents particular problems. Hemorrhage into the joint cavity, or hemarthrosis (hema, “blood,” arthron, “joint,” and osis, “condition of”), is considered a classic symptom of hemophilia. The effusion (ex, “out,” and fundere, “to pour”) into the joint is very painful because of the pressure buildup. Repeated hemorrhages may cause permanent deformities that could incapacitate the child. This deformity is sometimes referred to as an ankylosis (ankyle, “stiff joint,” and osis, “condition of”).
Treatment and Nursing Care: In a newborn with a family history of hemophilia, circumcision, heel sticks, and intramuscular injections are delayed to prevent bleeding and tissue injury. The principal therapy for hemophilia is to prevent bleeding by replacing the missing factor. The development of recombinant antihemophilic factor, a synthetic product, has eliminated the need for repeated blood transfusions and its accompanying dangers (such as human immunodeficiency virus [HIV] and hepatitis infection). The missing factor is replaced by reconstituting the product with sterile water and administering it through an implanted IV port. Once the parents and child are taught how to administer the medication, treatment is carried out in the home. Diagnosed infants may receive prophylactic factor replacements at regular intervals to prevent hemarthrosis. Desmopressin acetate (DDAVP) is a nasal spray that increases factor VIII in the blood, which leads to decreased bleeding. It may be the treatment of choice for mild cases of hemophilia. Aminocaproic acid (Amicar) is an antifibrinolytic agent that can control bleeding that may occur because of dental care or other oral bleeding.
Prophylactic factor VIII concentrate replacement combined with specific treatment before planned invasive procedures, such as dental extraction or minor surgery, and also education concerning the prevention of injuries that can cause bleeding enable the hemophiliac child to live a normal life. Because young children often fall while playing, the joints of knees, hips, and elbows can be protected by padding their play outfits. Appropriate sports activities should be selected to prevent undue injury and the risk of bleeding into joints. When bleeding does occur, the traditional approach to care includes rest, ice, compression, and elevation (RICE). A medical alert identification band should be worn at all times.
Home care programs are the treatment of choice. These greatly reduce the cost of treatment and decrease the risk of psychological trauma. A multidisciplinary approach to care assists families to develop healthy coping strategies to deal with a child who has a chronic illness. The multidisciplinary approach to care is supported by the National Hemophilia Foundation.
It is difficult for parents not to be overly protective. The struggle to protect these children and still foster independence and a sense of autonomy may seem monumental to parents who work away from home. Allowing children to participate in decision making about their care and focusing on their strengths is helpful.
Parent groups and professional counseling may provide support to enable children and parents to develop a healthy attitude toward the child’s medical condition.
Platelet Disorders
The reduction or destruction of platelets in the body interferes with the clotting mechanism. Skin lesions that are common to these disorders include petechiae, a bluish, nonblanching, pinpoint-sized lesion; purpura, groups of adjoining petechiae; ecchymosis, an isolated bluish lesion larger than a petechia; and hematoma, a raised ecchymosis.
Idiopathic (Immunological) Thrombocytopenia Purpura
Pathophysiology: Idiopathic (immunological) thrombocytopenic purpura (ITP) is an acquired platelet disorder that occurs in childhood. It is the most common of the purpuras, a group of disorders affecting the numbers of platelets or their function. The cause is unknown, but it is thought to be an autoimmune system reaction to a virus. Platelets become coated with antiplatelet antibody, are “perceived” as foreign material, and are eventually destroyed by the spleen. ITP occurs in all age-groups, with the main incidence seen between 2 and 4 years of age.
Manifestations: The classic symptom of ITP is easy bruising, which results in petechiae (pinpoint hemorrhagic spots beneath the skin) and purpura (hemorrhage into the skin). Approximately 30% of the patients also have nosebleeds. There may have been a recent history of rubella, rubeola, or viral respiratory infection. The interval between exposure and onset is about 2 weeks. The platelet count is below 20,000/mm3 (normal range is between 150,000/mm3 and 400,000/mm3). Diagnosis is confirmed by bone marrow aspiration to rule out leukemia.
Treatment and Nursing Care: When platelet counts are low, the greatest danger is spontaneous intracranial bleeding. Neurological assessments are therefore a priority of care. Treatment is not indicated in most cases of ITP. Spontaneous remission occurs in about 6 weeks to 4 months. A few children progress to chronic ITP. Drugs that interfere with platelet function should be avoided to prevent bleeding. These include aspirin, phenylbutazone (Butazolidin), and phenacetin, acetylsalicylic acid, and caffeine (APC). Activity is limited during the acute stage to prevent bruises from falls and trauma. Nursing considerations for the more acutely ill child focus on observing the patient for signs of bleeding. The child should use soft toothbrushes for oral hygiene to minimize tissue trauma. Packed RBCs may be administered when there has been a large amount of blood loss. Platelets are usually not given because they are destroyed by the disease process. Corticosteroids such as prednisone may be prescribed. IV gamma globulin may be used to elevate platelet counts. In some patients, infusion with anti-D antibody may be an effective treatment when there is no active bleeding present. A splenectomy may be indicated for cases of chronic ITP. Complications of ITP include bleeding from the gastrointestinal tract, hemarthrosis, and intracranial hemorrhage. Mortality in childhood ITP is less than 1%. All children must keep recommended immunizations against the viral diseases of childhood current to prevent this complication from occurring.
Disorders of White Blood Cells
Leukemia refers to a group of malignant diseases of the bone marrow and lymphatic system. There are many types and classifications, each with its own therapy and prognosis. The two most common forms are acute lymphoid leukemia (ALL) and acute nonlymphoid (myelogenous) leukemia (AMLL or AML). Cytochemical markers, chromosome studies and immunological markers differentiate the types of leukemia. The discussion in this test refers to ALL, the most common type of childhood leukemia.
Pediatric oncologists (physicians who specialize in the treatment of tumors) are challenged with the treatment of cancer in children because irradiation, surgery, and chemotherapy often have adverse effects on growth and development. Most children with cancer are treated in large medical centers to maximize the availability of high technology and newer treatment methods.
Survival rates for children diagnosed with leukemia have greatly improved. Current long-term survival of acute lymphoid leukemia approaches 80%, and nonlymphoid leukemia survival has increased to 50% (Pizzo & Poplack, 2005). However, close monitoring of late side effects of leukemia therapy is essential, and long-term follow-up care should be monitored.
Pathophysiology: Leukemia (leuko, “white,” and emia, “blood”) is a malignant disease of the blood-forming organs of the body that results in an uncontrolled growth of immature WBCs. The immature cells are termed blasts, or stem cells. This term comes from the Greek blastos, meaning “germ” or “formative cell.” The nurse may see the terms lymphoblasts or myeloblasts referred to in descriptive histories. Leukemia is the most common form of childhood cancer. It was considered fatal in the past, but the prognosis has improved greatly with modern treatments and medication. Approximately 2000 new cases of childhood leukemia are diagnosed in the United States every year. About 4 children per 100,000 under 15 years of age are affected annually, with the typical onset occurring between 2 and 5 years of age. Chromosomal abnormalities can be identified in 80% of cases of acute lymphocytic leukemia (Alterkruse et al., 2009).
The leukemias involve a disruption of bone marrow function caused by the overproduction of immature WBCs in the marrow. Although the WBC count can be as high as 50,000/mm3 to 100,000/mm3, the cells are immature and do not function as healthy WBCs to fight infection. Increased susceptibility to infection results. The WBCs take over the centers that are designed to form RBCs, and anemia results. When the WBCs infiltrate and take over the marrow centers that form platelets, the reduced platelet counts cause bleeding tendencies. The invasion of the bone marrow causes weakening of the bone, and pathological fractures can occur.
Leukemia cells can infiltrate the spleen, liver, and lymph glands, resulting in fibrosis and diminished function. The cancerous cells invade the central nervous system and other organs, draining these organs of their nutrients and finally causing metabolic starvation of the body.
Manifestations: The most common symptoms during the initial phase of leukemia are low-grade fever, pallor, bruising tendency, leg and joint pain, listlessness, abdominal pain, and enlargement of lymph nodes. These symptoms may develop gradually or may be sudden in onset. As the disease progresses, the liver and spleen become enlarged. The skin may have an unusual lemon yellow color. Petechiae and purpura may be early objective symptoms. Anorexia, vomiting, weight loss, and dyspnea are also common. The kidneys and testicles may enlarge, and the patient may develop hematuria.
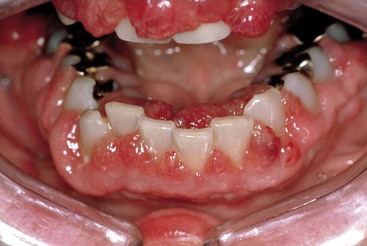
Because the WBCs are not functioning normally, bacteria easily invade the body. Ulcerations develop around the mucous membranes of the mouth and anal regions and have a tendency to bleed (Figure 27-6). Anemia becomes severe despite transfusions. The child may die as a direct result of the disease or from secondary infection. The symptoms are the same regardless of the type of WBC affected, but they vary widely with each patient depending on the parts of the body involved.
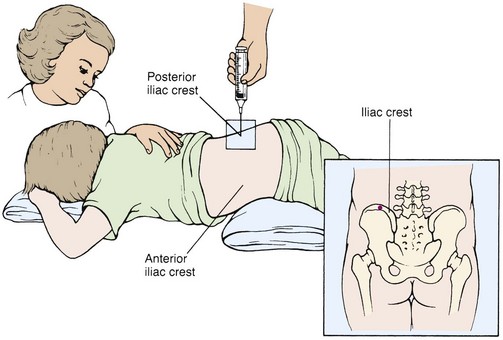
Diagnosis: The diagnosis of leukemia is based on the history and symptoms of the patient and the results of extensive blood tests that demonstrate the presence of leukemic blast cells in the blood, bone marrow, or other tissues. Because many WBCs and RBCs are formed in the bone marrow, a bone marrow aspiration is commonly performed. A piece of bone marrow is aspirated from the sternum or, more often in children, from the iliac crest. A special needle is used to obtain the sample, and the marrow is studied in the laboratory (Figure 27-7). X-ray films of the long bones show changes. After the diagnosis has been confirmed, a spinal tap determines central nervous system involvement. Kidney and liver function studies are also performed because normal functioning of these organs is absolutely necessary for chemotherapy to be safely used in treating the disease.
Treatment and Nursing Care: Long-term care is given on an outpatient basis whenever possible. The treatment of a child with cancer involves the multidisciplinary health care team (pediatrician, pathologist, oncologist, nurse, radiotherapist, nutritionist, psychologist, and school personnel). As with any diagnosis of cancer, the child with leukemia is usually referred to a specialized center where facilities for required care are available. Chemotherapy is carried out in specialized units with specially trained and certified personnel.
Although chemotherapy may be effective in reducing leukemic cells, the side effects as well as the long-term effects of treatments must be addressed. In chemotherapy, bone marrow suppression makes it essential for the family to be taught about infection prevention. Adequate hydration should be emphasized to minimize kidney damage. Active routine immunizations must be delayed while the child is receiving immunosuppressive drugs, because the body will not be able to manufacture antigens as expected. Parents should report any exposure to infections such as chickenpox so that immunoglobulin can be administered. Chickenpox can be life threatening to a child who is immunosuppressed. Nausea and vomiting are common complications of chemotherapy and result in decreased appetite, weight loss, and generalized weakness. Presenting the child’s favorite foods in an attractive manner may help stimulate the appetite. Total parenteral nutrition (TPN) may be indicated to support nutritional needs. An intake and output record is maintained, and meticulous oral hygiene is given.
The nurse should refer parents to available support groups. The Ronald McDonald house and hospice programs help parents and families cope with this illness. Because hair loss (alopecia) is a side effect of chemotherapy, the child can be offered a hat or a wig to help preserve a positive body image. School tutoring and counseling should be continuous in the hospital setting and in the home during home care to provide optimal growth and development.
 Nursing Tip
Nursing Tip
Four priority challenges in the care of leukemic children are (1) the complications of anemia from decreased red blood cell (RBC) infection, (2) infection from neutropenia, (3) bleeding from decreased platelets, and (4) fractures due to the involvement of the bone marrow.
Chemotherapy includes the following components:
• Central nervous system prophylaxis for high-risk patients
The list of medications for treatment of this disease is growing. A combination of drugs used to induce remissions includes prednisone, vincristine sulfate, and daunorubicin or l-asparaginase. They work within 4 to 6 weeks in about 95% of children with ALL. The therapeutic effects of these drugs are of short duration, and therefore it is necessary to use additional drugs that help to maintain the remissions. The steroid prednisone has the side effects of masking the symptoms of infection, increasing fluid retention, inducing personality changes, and causing the child’s face to take on a moon-shaped appearance. Methotrexate and 6-mercaptopurine are useful in maintaining remissions because they act against chemicals vital to the life of the WBC. These powerful medications produce side effects of varying degrees, such as nausea, diarrhea, rash, hair loss (alopecia), fever, anuria, anemia, and bone marrow depression. Peripheral neuropathy may be signaled by severe constipation caused by decreased nerve sensations to the bowel. The nurse should consult a pharmacology text for information about the particular drugs used for the patient to anticipate potential problems.
A bone marrow transplant may be useful. An autologous transplant uses the child’s own bone marrow that has been purged of malignant cells. An allogeneic bone marrow transplant is taken from a donor who matches the child. Transplanted marrow rejection is a risk. When the child is hospitalized, protective environment precautions and transmission-based precautions may prevent health care–associated infections (see Appendix A). Hemopoietic stem cell transplantation (HSCT) has been used successfully in children with AML and children with ALL who do not respond to chemotherapy. Prevention of infection is the challenge for the health care team after transplant. HSCT is a risky procedure because the immune system of the child must first be destroyed before the transplant. If the transplant fails the child is left immunologically defenseless and may die.
Children’s anxiety often centers on their symptoms. They fear that the treatments necessary to correct their problems may be painful, as indeed some are, such as venipunctures, bone marrow aspirations, and blood transfusions. Their trust in others is in a precarious balance. Nurses must inform children of what they are about to do and why it is necessary. The explanation is given in terms the child will understand.
The child may ask the nurse the inevitable question, “Am I going to die?” One suggestion is to reply with a question, such as “Why do you ask that? Do you feel sick today?” This may encourage the child to verbalize feelings. The pediatric nurse who gives patients permission to discuss their concerns will find opportunities to clear up misconceptions and to decrease children’s feelings of isolation.
The patient is frequently observed for signs of infection. Particular attention is given to potential sites of infection, such as the patient’s mucous membranes and puncture breaks in the skin from laboratory or therapeutic procedures. Pierced ears or other body parts are observed for inflammation. Vital signs are observed for subtle variances, because steroid therapy may mask these indicators. The patient is turned often and is observed for skin breakdown, particularly in the perianal area. Nutritious meals and supplemental feedings that are high in protein and calories are offered. Parents and children are taught what to look for and report.
Thrombocytopenic bleeding is a common complication of leukemia. The nurse observes the patient’s skin for petechiae and ecchymosis. Nosebleeds are common and are treated by the application of cold and pressure.
The mouth is inspected daily for ulcerations and bleeding from the gums. It may be rinsed with a prescribed solution of one part hydrogen peroxide to four parts saline solution. Commercial mouthwashes are used with caution because they may alter normal flora and may cause fungal overgrowth. A Waterpik is helpful in massaging and toughening the gums. If the child is comatose, mouth care supplies are kept at the bedside. A soft tooth sponge is helpful. The nurse may also clean food particles from the patient’s teeth with a piece of gauze wrapped around the finger. Lip balm is applied to dry, cracked lips.
Care of a child receiving a transfusion: Platelets and packed RBCs may be given to the child. Hemolytic reactions caused by mismatched blood are rare. Nevertheless, the registered nurse should positively identify donor and recipient blood types and groups on labels and the patient’s chart together with another licensed professional. Blood is infused through a blood filter to exclude impurities. Medications are never added to blood. Blood is administered slowly. The IV site is frequently checked for infiltration. The patient is observed for signs of transfusion reaction, which include chills, itching, rash, fever, headache, and pain in the back. If such a reaction occurs, the tubing should be clamped off immediately, the line kept open with normal saline solution, and the nurse in charge notified.
Transfusions with piggyback setups are common. Blood, normal saline, or other suitable IV solutions are connected by a stopcock. When a blood transfusion must be stopped, tube patency can be maintained by opening the saline line. Necessary emergency medications can thus be administered and the site preserved for future infusions.
Circulatory overload is always a danger with children. An infusion pump is routinely used to regulate blood flow. Dyspnea, precordial pain, rales, cyanosis, dry cough, and distended neck veins are indicative of circulatory overload. Apprehension can also be a warning signal of air emboli or electrolyte disturbance. The nurse must maintain a high level of alertness for such signs, particularly in children whose conditions warrant repeated transfusions. If a reaction occurs, the blood bag and tubing are saved and returned to the blood bank. Most transfusion reactions occur within the first 10 minutes of administration; nevertheless, the patient is carefully monitored throughout this treatment. Diphenhydramine (Benadryl) may be ordered for allergic reactions. Aminophylline may be ordered for wheezing. Oxygen may be necessary to relieve dyspnea and cyanosis. Blood transfusions administered through central lines must be warmed by a blood warmer to prevent cardiac dysrhythmias. Baseline data (temperature, pulse, respiration, and blood pressure) are established before transfusion, and the nurse monitors for changes. It is helpful if the parents remain with the child during this time. Suitable diversions minimize boredom.
Hodgkin’s Disease
Pathophysiology: Hodgkin’s disease is a malignancy of the lymph system that primarily involves the lymph nodes. It may metastasize to the spleen, liver, bone marrow, lungs, or other parts of the body. The presence of giant multinucleated cells called Reed-Sternberg cells is diagnostic of the disease. Hodgkin’s disease is rarely seen before 5 years of age, with the incidence increasing during adolescence and early adulthood. It is twice as common in boys as in girls.
Manifestations: The presenting symptom of Hodgkin’s disease is generally a painless lump along the neck. Characteristically, there are few other manifestations. In general the swelling is first noted by the patient or parents. In more advanced cases, there may be unexplained low-grade fever, anorexia, unexplained weight loss, night sweats, general malaise, rash, and itching. Diagnosis is confirmed by x-ray films, blood tests, body scan, and a biopsy of the node. The stages of Hodgkin’s disease are defined in Table 27-2.
Table 27-2
Criteria for Staging Hodgkin’s Disease
| STAGE | CRITERIA |
| I | Restricted to single site or localized in a group of lymph nodes; asymptomatic |
| II | Involves two or more lymph nodes in area or on same side of diaphragm |
| III | Involves lymph node regions on both sides of diaphragm; involves adjacent organ or spleen |
| IV | Is diffuse disease; least favorable prognosis |
Treatment: Well-established treatment regimens are now being used to combat this illness. Both radiation therapy and chemotherapy are used in accordance with the clinical stage of the disease. The combination of cyclophosphamide and vincristine (Oncovin), procarbazine hydrochloride, and prednisone is a common protocol. It is referred to as the COPP regimen. Long-term prognosis is excellent, but long-term effects of therapy have to be monitored.
Nursing Care: Nursing care is mainly directed toward symptomatic relief of the side effects of radiation therapy and chemotherapy. Education of the patient and family is paramount because most patients are cared for in the home. The nurse should explain the myriad diagnostic tests to be performed and prepare the child for the typical procedures and their aftereffects. After a lymphangiogram, for example, the skin and urine may take on a bluish color.
Children and parents should be prepared to deal with the impact on self-image. The school nurse should be contacted to implement a schedule that will promote growth and development while preventing overfatigue. A common side effect of irradiation is malaise. The adolescent tires easily and may be irritable and anorectic. The skin in the treated area may be sensitive and must be protected against exposure to sunlight and irritation. After treatment, a sun-blocking agent containing para-aminobenzoic acid (PABA) should be used to prevent burning. The attending physician may prescribe an ointment to relieve itching. Nothing should be applied to the treatment area without the recommendation of the physician. There may be diarrhea after abdominal irradiation. The patient does not become radioactive during or after therapy.
After splenectomy, the patient faces the long-term risk of serious infection. This risk is explained to the parents and adolescent. Temperature elevations must be monitored carefully. There may also be infection with little or no fever as a result of masking by certain medications. In such cases, cultures of blood, urine, sputum, or stool may have to be taken. Parents and the adolescent are instructed to feel free to call the clinic, particularly if there is a change in the condition or if there is apprehension or confusion about symptoms. Medication readjustments should not be attempted unless specifically advised by the physician.
Emotional support of the adolescent is age appropriate. Nurses must particularly be prepared for periods of anger, which may be directed at them. Suitable outlets, such as the use of a punching bag, allow for the safe direction of anger. Routine use helps to prevent a buildup of tension. Activity is generally regulated by the patient. The physician advises the patient if special precautions are necessary.
The appearance of secondary sexual characteristics and menstruation may be delayed in pubescent patients. Sterility is often a side effect of the treatment. This can be a source of anxiety. Adolescents may be interested in sperm banking before immunosuppressive therapy is initiated. The nurse respects the patient concerns and can be most effective by listening empathically (Nursing Care Plan 27-2).
27-2 Nursing Care Plan
The Adolescent Receiving Cancer Chemotherapy
An adolescent is admitted for initiation of chemotherapy. She appears thin, anxious, and fearful. She asks if she is going to die.



Nursing Care of the Chronically Ill Child
Chronic illness during childhood often affects growth and development (Table 27-3). Specific programs that foster feelings of security and independence within the limits of the situation are essential. Behavior problems are lessened when patients can verbalize specific concerns with persons sensitive to their problems. To be in school and to be considered one of the group is very important to children. If they feel rejected by and different from their peers, they may be prone to depression. Hospital school programs provide familiarity and enable patients to keep pace with their classmates. The recreational therapist may also be helpful in combating boredom and providing outlets for tension.
Table 27-3
The Effects of Chronic Illness on Growth and Development*

*Chronic illness can impede growth and development. The nurse should reinforce teaching concerning the developmental needs of chronically ill children at different age levels to promote self-acceptance and a positive self-esteem.
Nurses must help patients to accept their body with all its strengths and imperfections. They must develop an awareness of the adolescent’s particular fears of forced dependence, body invasion, mutilation, rejection, and loss of face, especially within peer groups. The nurse anticipates a certain amount of reluctance to adhere to hospital regulations, which reflects the adolescent’s need for self-determination. Recognizing this as an asset rather than a liability enables the nurse to respond creatively.
Developmental Disabilities
Children who have a developmental disability that affects their intellect or ability to cope face some unique difficulties. They may often be overprotected, unable to break away from supervision, and deprived of necessary peer relationships. The pubertal process with its emerging sexuality concerns parents and may precipitate a family crisis.
Home Care
Most children with acute and chronic conditions are cared for in the home. Home health care and other community agencies work together to provide holistic care. Respite care provides trained workers who come into the home for brief periods to relieve parents of the responsibility of caring for the child. This enables the parents to shop, take care of business transactions, or simply take a much-needed vacation. The school systems also share in the responsibility of care, which is crucial if a family is to be successful in home care. One mother, whose 13-year-old daughter has a severe developmental disability (cerebral palsy, blindness, scoliosis, mental retardation), offered the following suggestions for the health care worker assisting in the home:
• Observe how the parents interact with the child.
• Do not wait for the child to cry out for attention, because the youngster may be unable to communicate in this way.
• Watch for facial expressions and body language.
• Post signs above the bed denoting special considerations, such as “Never position on left side” and “Do not feed with plastic spoon.”
• Listen to the parents and observe how they attend to the physical needs of the youngster.
• Do not be afraid to ask questions or discuss apprehensions you may feel about your ability to care for the child.
• Be attuned to the needs of other children in the home.
• Be creative in exploring avenues for socialization, because these adolescents are seldom invited to birthday or slumber parties.
• Explore community facilities or support groups that might benefit the family.
The Chronically Ill Child as Family Member
The chronically ill child must be a contributing member to the family unit. Faced with having to choose, the child and parent will often discard the health care practice when health care activities inhibit development and prevent peer socialization opportunities. The child must be treated normally and overprotection and overrestriction avoided. Focusing on what the child can do and providing successful experiences are more effective than focusing on the disability.
Involvement of the entire family with the care of the chronically ill child aids in normal family interaction. Respite care opportunities are needed to provide parents with a normal spousal relationship. The child should be integrated into, rather than isolated from, the community and society. Nurses can assist the child and the family to develop strategies to cope with chronic illness and to promote optimal growth and development. The wellness of the child should be the center of the child’s life, rather than the disability.
Nursing Care of the Dying Child
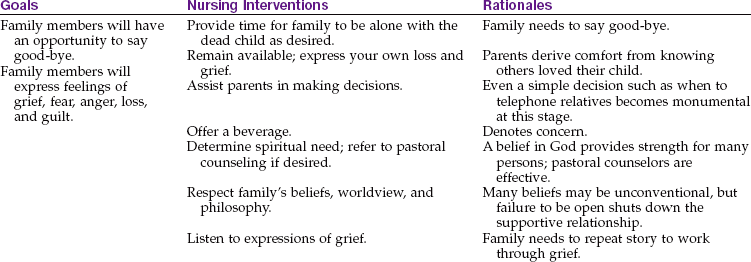
Facing death is often a difficult personal issue for the nurse. The nurse must understand the grieving process; personal and cultural views concerning that process; the views of a parent losing a child; and the perceptions of the child facing death. Integrating these understandings and helping all involved to cope successfully involves a multidisciplinary approach. The response to a child’s death is influenced by whether there was a long period of uncertainty before the death or whether it was a sudden, unexpected event. The nurse must show compassion but function in a clinically competent, professional manner. Demonstrating a nonjudgmental approach when the personal or cultural practices of the family conflict with the nurse’s own values presents a challenge. Sensitive, effective care can be provided only if the nurse is aware of these needs of the family. The nurse can facilitate the grief process by anticipating psychological and somatic responses and maintaining open communication. The family’s efforts to cope, adapt, and grieve must be supported.
The response of the family to the death of the child may initially be manifested by somatic distress such as weakness, anguish, or shortness of breath. A family member may feel detached from the world and have a sense of unreality or disbelief. A sense of guilt and blame may follow (“I should have” or “I could have”). Hostility is a normal response and may drive away those who do not understand its normalcy in the acute grieving process. A restlessness and general irritability or inability to function may follow. Assistance in the care of other children or household responsibilities may be necessary. Nursing priorities include being a patient and family advocate, providing support, and facilitating the grieving process.
Self-Exploration
One important, if not the most important, task to prepare for working with the dying patient is self-exploration. Our own attitudes about life and death affect our nursing practice. Emotions buried deep within can form barriers to effective communication unless they are recognized and released. How nurses have or have not dealt with their own losses affects present lives and the ability to relate to patients. Nurses must recognize that coping is an active and ongoing process. At times, nurses need compassionate detachment from patients and their families to become revitalized. We must find constructive outlets, such as exercise and music, to maintain equilibrium. An active support system consisting of nonjudgmental people who are not threatened by natural expressions of feelings is crucial. Proper channeling of these emotions can be a valuable part of the empathetic response to others. It is vital that nurses support one another in the work environment.
The Child’s Reaction to Death
Each child, like each adult, approaches death in an individual way, drawing on limited experience. Nurses must become well acquainted with patients and view them within the context of the family and social culture. Their anxiety often centers on symptoms. They fear that treatments may be painful. Nurses must be honest and inform patients about the upcoming procedures in terms the child will understand. Expressing feelings is encouraged: “You seem angry.” Sufficient time should be given for a response. Children should be allowed to have as much control as possible regarding what happens to them. This is fostered by including them in decisions that concern their welfare. However, the child should not be offered a choice when there is none. Children often communicate symbolically. The nurse listens to what they say to adults, to their toys, and to other children. Crayons and paper are provided for self-expression.
Although age is a factor, the child’s level of cognitive development, rather than chronological age, affects the response to death. Children younger than 5 years of age are mainly concerned with separation from their parents and abandonment. (Even adults are threatened by thoughts of dying alone.) Preschool children respond to questions about death by relying on their experience and by turning to fantasy. They may believe death is reversible or that they are in some way responsible. Children do not develop a realistic concept of death as a permanent biological process until 9 or 10 years of age.
 Health Promotion
Health Promotion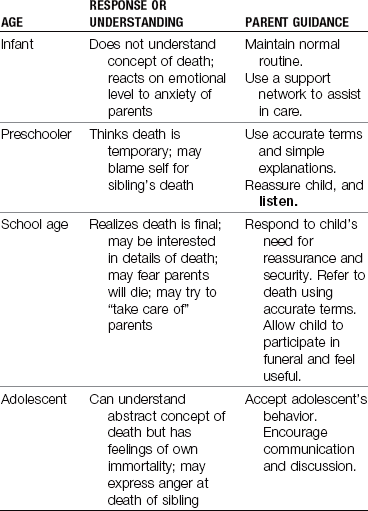
Dying adolescents face conflicts between their treatment regimens and their need to establish independence from their parents and conformity with their peers. This leads to anger and resentment, which are often displaced onto hospital staff members. An atmosphere of acceptance and nonjudgmental listening allows adolescents freedom to vent their hostility in a nonthreatening environment. Nursing Care Plan 27-3 specifies nursing interventions for the dying child.

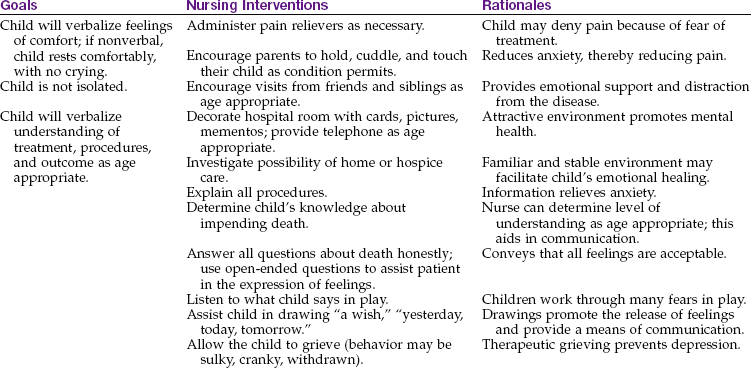
The Child’s Awareness of His or Her Condition
Surprising as it may seem, many investigators have shown that terminally ill children are generally aware of their condition, even when it is carefully concealed. This is reflected in their drawings and play and can be detected through psychological testing. Failure to be honest with children leaves them to suffer alone, unable to express their fears and sadness or even to say good-bye. The family should be referred as needed for support and social services.
Physical Changes of Impending Death
The physical changes that occur with impending death include cool, mottled, cyanotic skin and the slowing of all body processes. There may be a loss of consciousness, but hearing is intact. Rales in the chest may be heard, which result from increased secretions pooling in the lungs. Movement and neurological signs lessen. If thrashing or groaning occurs, the patient is assessed for pain, and pain relief should be provided.
Stages of Dying
The stages of dying as detailed by Kübler-Ross (1975)—denial, anger, bargaining, depression, acceptance, and reaching out to help others—can be applied to parents and siblings as well as to the sick child. (Nurses may also respond with similar feelings.) It is important to accept and support each participant at whatever stage has been reached and to refrain from directing progress. Nurses should be available and make their availability known (Box 27-1).
Parents are encouraged to assist in the care of their child. This is facilitated by hospice care and the movement toward supervised home care. It is therapeutic for children to be in their own surroundings whenever possible. Siblings involved in patient care feel less neglected, and the sacrifices they must make become more meaningful. Discussions before death allow them to make amends for their hostilities toward the sick child. The family’s religious and spiritual philosophy can be a source of strength and support, as can caring neighbors and friends.
Statistics show a high correlation between the death of a child and divorce. Nurses must observe signs of tension between parents so that suitable intervention may be implemented. Each parent grieves in an individual time and way, often making it difficult for spouses to be supportive of each other. The suppression of strong feelings of guilt, helplessness, and outrage can be devastating. Feelings left unexpressed can cause depression and/or physical illness.
Kübler-Ross (1969) reminds us that dying is the easy part. Helping patients to live until they die is the real challenge. She discusses this beautifully in A Letter to a Child With Cancer, which she wrote in response to a child’s question, “What is life, what is death, and why do little children have to die?” A library may be consulted to locate this classic book and others published especially to help children and parents with dying and grief. There are several hospices in the United States that limit their services to children. St. Mary’s Hospice in Bay Side, New York, is credited with being the first.
Get Ready for the NCLEX® Examination!
Key Points
• Circulating blood consists of two portions: plasma and formed elements.
• Bone marrow aspiration is one procedure that is helpful in determining disorders of the blood.
• The most common nutritional deficiency of children in the United States is iron-deficiency anemia.
• Sickle cell disease is an inherited defect in the formation of hemoglobin. The cells become crescent shaped and clump together.
• Pain control with meperidine (Demerol) should not be used for children with sickle cell anemia.
• Massive expansion of the bone marrow in thalassemia causes changes in the contour of the child’s skull and face.
• Hemophilia A results from a deficiency in coagulation factor VIII, and hemophilia B (Christmas disease) involves a deficiency of factor IX.
• Hemarthrosis (bleeding into the joints) is a characteristic sign of hemophilia A.
• Hemosiderosis (deposits of iron in the organs and tissues) is a complication of multiple transfusions in hemolytic blood disorders.
• Signs of transfusion reactions include chills, itching rash, fever, and headache.
• Petechiae are bluish pinpoint lesions on the skin. Purpura are groups of adjoining petechiae, ecchymosis is an isolated bluish lesion larger than a petechiae, and a hematoma is a raised ecchymosis.
• Leukemia is the most common form of childhood cancer.
• Four priority challenges in the care of a child with leukemia are anemia, bleeding, infection, and fractures.
• Diagnostic procedures for patients with blood disorders are often invasive or painful. The nurse prepares and supports the patient and family during these procedures.
• Maintenance of schooling, adequate hydration and nutrition, prevention of infection, promotion of a positive self-image, and meticulous oral hygiene are essential components of nursing care of a child with leukemia.
• Reed-Sternberg cells are diagnostic for Hodgkin’s disease.
• Children who are chronically ill must be aided in mastering developmental tasks.
• The stages of dying according to Kübler-Ross include denial, anger, bargaining, depression, acceptance, and reaching out to help others.
• The nurse can help the family of a dying child by listening and assessing their needs, reinforcing information, providing privacy, and using appropriate phrases and open-ended concrete questions and statements.
Additional Learning Resources
 Go to your Study Guide for additional learning activities to help you master this chapter content.
Go to your Study Guide for additional learning activities to help you master this chapter content.
Go to your Evolve website (http://evolve.elsevier.com/Leifer) for the following FREE learning resources:
• Answer Guidelines for Critical Thinking Questions
• Answers and Rationales for Review Questions for the NCLEX® Examination
• Glossary with English and Spanish pronunciations
• Interactive Review Questions for the NCLEX® Examination
• Patient Teaching Plans in English and Spanish
 Online Resources
Online Resources• Cooley’s Anemia Foundation: www.thalassemia.org
• National Hemophilia Foundation. www.hemophilia.org
• Non-Hodgkin’s lymphoma: www.oncologychannel.com/nonhodgkins
Review Questions for the NCLEX® Examination
1. When the patient experiences apprehension and urticaria while receiving a blood transfusion, the nurse:
1. slows the transfusion and takes the patient’s vital signs.
2. observes the child for further transfusion reactions.
3. stops the transfusion, allows normal saline solution to run slowly, and notifies the charge nurse.
4. stops what he or she is doing and obtains the patient’s history.
2. A child who is in a vasoocclusive crisis caused by sickle cell anemia is experiencing acute pain. The nurse understands that Demerol (meperidine) is not an appropriate pain medication to administer to this child because it:
3. Which principle should the nurse teach the parent concerning administering liquid iron preparations to the child with iron-deficiency anemia?
1. Allow the preparation to mix with saliva and bathe the teeth before swallowing.
4. Thalassemia major (Cooley’s anemia) is treated primarily with:
5. What is a characteristic manifestation of Hodgkin’s disease?