17 Antibacterial Agents
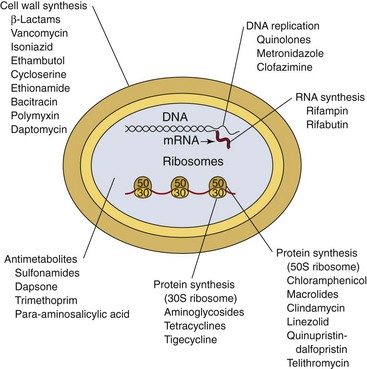
This chapter provides an overview of the mechanisms of action and spectrum of the most commonly used antibacterial antibiotics, as well as a description of the common mechanisms of bacterial resistance. The terminology appropriate for this discussion is summarized in Box 17-1, and the basic mechanisms and sites of antibiotic activity are summarized in Table 17-1 and Figure 17-1, respectively.
Box 17-1
Terminology
Antibacterial spectrum: Range of activity of an antimicrobial against bacteria. A broad-spectrum antibacterial drug can inhibit a variety of gram-positive and gram-negative bacteria, whereas a narrow-spectrum drug is active against a limited variety of bacteria.
Bacteriostatic activity: Level of antimicrobial activity that inhibits the growth of an organism. This is determined in vitro by testing a standardized concentration of organisms against a series of antimicrobial dilutions. The lowest concentration that inhibits the growth of the organism is referred to as the minimum inhibitory concentration (MIC).
Bactericidal activity: Level of antimicrobial activity that kills the test organism. This is determined in vitro by exposing a standardized concentration of organisms to a series of antimicrobial dilutions. The lowest concentration that kills 99.9% of the population is referred to as the minimum bactericidal concentration (MBC).
Antibiotic combinations: Combinations of antibiotics that may be used to (1) broaden the antibacterial spectrum for empirical therapy or the treatment of polymicrobial infections, (2) prevent the emergence of resistant organisms during therapy, and (3) achieve a synergistic killing effect.
Antibiotic synergism: Combinations of two antibiotics that have enhanced bactericidal activity when tested together compared with the activity of each antibiotic.
Antibiotic antagonism: Combination of antibiotics in which the activity of one antibiotic interferes with the activity of the other (e.g., the sum of the activity is less than the activity of the most active individual drug).
β-Lactamase: An enzyme that hydrolyzes the β-lactam ring in the β-lactam class of antibiotics, thus inactivating the antibiotic. The enzymes specific for penicillins, cephalosporins, and carbapenems are the penicillinases, cephalosporinases, and carbapenemases, respectively.
Table 17-1 Basic Mechanisms of Antibiotic Action
| Antibiotic | Action |
|---|---|
| Disruption of Cell Wall | |
| Binds PBPs and enzymes responsible for peptidoglycan synthesis | |
| β-Lactam/β-lactamase inhibitor | Binds β-lactamases and prevents enzymatic inactivation of β-lactam |
| Vancomycin | Inhibits cross-linkage of peptidoglycan layers |
| Daptomycin | Causes depolarization of cytoplasmic membrane, resulting in disruption of ionic concentration gradients |
| Bacitracin | Inhibits bacterial cytoplasmic membrane and movement of peptidoglycan precursors |
| Polymyxins | Inhibits bacterial membranes |
| Isoniazid Ethionamide |
Inhibits mycolic acid synthesis |
| Ethambutol | Inhibits arabinogalactan synthesis |
| Cycloserine | Inhibits cross-linkage of peptidoglycan layers |
| Inhibition of Protein Synthesis | |
| Aminoglycosides | Produces premature release of aberrant peptide chains from 30S ribosome |
| Tetracyclines | Prevents polypeptide elongation at 30S ribosome |
| Glycylcyclines | Binds to 30S ribosome and prevents initiation of protein synthesis |
| Oxazolidinone | Prevents initiation of protein synthesis at 50S ribosome |
| Prevents polypeptide elongation at 50S ribosome | |
| Inhibition of Nucleic Acid Synthesis | |
| Quinolones | Binds α subunit of DNA gyrase |
| Rifampin | Prevents transcription by binding DNA-dependent RNA polymerase |
| Rifabutin | |
| Metronidazole | Disrupts bacteria DNA (is cytotoxic compound) |
| Antimetabolite | |
| Sulfonamides | Inhibits dihydropteroate synthase and disrupts folic acid synthesis |
| Dapsone | Inhibits dihydropteroate synthase |
| Trimethoprim | Inhibits dihydrofolate reductase and disrupts folic acid synthesis |
DNA, Deoxyribonucleic acid; PBPs, penicillin-binding proteins; RNA, ribonucleic acid.
The year 1935 was an important one for the chemotherapy of systemic bacterial infections. Although antiseptics had been applied topically to prevent the growth of microorganisms, the existing antiseptics were ineffective against systemic bacterial infections. In 1935, the dye protosil was shown to protect mice against systemic streptococcal infection and to be curative in patients suffering from such infections. It was soon found that protosil was cleaved in the body to release p-aminobenzene sulfonamide (sulfanilamide), which was shown to have antibacterial activity. This first “sulfa” drug ushered in a new era in medicine. Compounds produced by microorganisms (antibiotics) were eventually discovered to inhibit the growth of other microorganisms. For example, Alexander Fleming was the first to realize the mold Penicillium prevented the multiplication of staphylococci. A concentrate from a culture of this mold was prepared, and the remarkable antibacterial activity and lack of toxicity of the first antibiotic, penicillin, were demonstrated. Streptomycin and the tetracyclines were developed in the 1940s and 1950s, followed rapidly by the development of additional aminoglycosides, semisynthetic penicillins, cephalosporins, quinolones, and other antimicrobials. All these antibacterial agents greatly increased the range of infectious diseases that could be prevented or treated. Although the development of new antibacterial antibiotics has lagged in recent years, some new classes of agents have been introduced, including the ketolides (e.g., telithromycin), glycylcyclines (tigecycline), lipopeptides (daptomycin), streptogramins (quinupristin-dalfopristin), and oxazolidinones (linezolid).
Unfortunately, with the introduction of new chemotherapeutic agents, bacteria have shown a remarkable ability to develop resistance. Thus antibiotic therapy will not be the magical cure for all infections, as predicted; rather, it is only one weapon, albeit an important one, against infectious diseases. It is also important to recognize that because resistance to antibiotics is often not predictable, physicians must rely on their clinical experience for the initial selection of empirical therapy and then refine their treatment by selecting antibiotics demonstrated to be active by in vitro susceptibility tests. Guidelines for the management of infections caused by specific organisms are discussed in the relevant chapters of this text.
Inhibition of Cell Wall Synthesis
The most common mechanism of antibiotic activity is interference with bacterial cell wall synthesis. Most of the cell wall–active antibiotics are classified as β-lactam antibiotics (e.g., penicillins, cephalosporins, cephamycins, carbapenems, monobactams, β-lactamase inhibitors), so named because they share a common β-lactam ring structure. Other antibiotics that interfere with construction of the bacterial cell wall include vancomycin, daptomycin, bacitracin, and the following antimycobacterial agents: isoniazid, ethambutol, cycloserine, and ethionamide.
β-Lactam Antibiotics
The major structural component of most bacterial cell walls is the peptidoglycan layer. The basic structure is a chain of 10 to 65 disaccharide residues consisting of alternating molecules of N-acetylglucosamine and N-acetylmuramic acid. These chains are then cross-linked with peptide bridges that create a rigid mesh coating for the bacteria. The building of the chains and cross-links is catalyzed by specific enzymes (e.g., transpeptidases, transglycosylases, carboxypeptidases) that are members of a large family of serine proteases. These regulatory enzymes are also called penicillin-binding proteins (PBPs), because they are the targets of β-lactam antibiotics. When growing bacteria are exposed to these antibiotics, the antibiotic binds to specific PBPs in the bacterial cell wall and inhibits assembly of the peptidoglycan chains. This, in turn, activates autolysins that degrade the cell wall, resulting in bacterial cell death. Thus the β-lactam antibiotics generally act as bactericidal agents.
Bacteria can become resistant to β-lactam antibiotics by three general mechanisms: (1) prevention of the interaction between the antibiotic and the target PBP, (2) modification of the binding of the antibiotic to the PBP, and (3) hydrolysis of the antibiotic by bacterial enzymes, β-lactamases. The first mechanism of resistance is seen in gram-negative bacteria (particularly Pseudomonas species). Gram-negative bacteria have an outer membrane that overlies the peptidoglycan layer. Penetration of β-lactam antibiotics into gram-negative rods requires transit through pores in this outer membrane. Changes in the proteins (porins) that form the walls of the pores can alter the size of the pore opening or charge of these channels and result in the exclusion of the antibiotic.
Resistance can also be acquired by modification of the β-lactam antibiotic binding to the PBP. This can be mediated by (1) an overproduction of PBP (a rare occurrence), (2) acquisition of a new PBP (e.g., methicillin resistance in Staphylococcus aureus), or (3) modification of an existing PBP through recombination (e.g., penicillin resistance in Streptococcus pneumoniae) or a point mutation (penicillin resistance in Enterococcus faecium).
Finally, bacteria can produce β-lactamases that inactivate the β-lactam antibiotics. Interestingly, the β-lactamases are in the same family of serine proteases as the PBPs. More than 200 different β-lactamases have been described. Some are specific for penicillins (i.e., penicillinases), cephalosporins (i.e., cephalosporinases), or carbapenems (i.e., carbapenemases), whereas others have a broad range of activity, including some that are capable of inactivating most β-lactam antibiotics. An exhaustive discussion of β-lactamases is beyond the scope of this chapter; however, a brief discussion is germane for understanding the limitations of β-lactam antibiotics. By one classification scheme, β-lactamases have been separated into four classes (A to D). The most common class A β-lactamases are SHV-1 and TEM-1, penicillinases found in common gram-negative rods (e.g., Escherichia, Klebsiella), with minimal activity against cephalosporins. Unfortunately, simple point mutations in the genes encoding these enzymes have created β-lactamases with activity against all penicillins and cephalosporins. These β-lactamases are referred to as extended-spectrum β-lactamases (ESBLs) and are particularly troublesome, because most are encoded on plasmids that can be transferred from organism to organism. The class B β-lactamases are zinc-dependent metalloenzymes that have a broad spectrum of activity against all β-lactam antibiotics, including the cephamycins and carbapenems. The class C β-lactamases are primarily cephalosporinases that are encoded on the bacterial chromosome. Expression of these enzymes is generally repressed, although this can be altered by exposure to certain “inducing” β-lactam antibiotics or by mutations in the genes controlling expression of the enzymes. Expression of this class of β-lactamases is particularly troublesome, because they are active against the most potent expanded-spectrum cephalosporins. The class D β-lactamases are penicillinases found primarily in gram-negative rods.
Penicillins
Penicillin antibiotics (Table 17-2) are highly effective antibiotics with an extremely low toxicity. The basic compound is an organic acid with a β-lactam ring obtained from culture of the mold Penicillium chrysogenum. If the mold is grown by a fermentation process, large amounts of 6-aminopenicillanic acid (the β-lactam ring is fused with a thiazolidine ring) are produced. Biochemical modification of this intermediate yields antibiotics that have increased resistance to stomach acids, increased absorption in the gastrointestinal tract, resistance to destruction by penicillinase, or a broader spectrum of activity that includes gram-negative bacteria.
| Antibiotics | Spectrum of Activity |
|---|---|
| Natural penicillins: benzylpenicillin (penicillin G), phenoxymethyl penicillin (penicillin V) | Active against all β-hemolytic streptococci and most other species; limited activity against staphylococci; active against meningococci and most gram-positive anaerobes; poor activity against aerobic and anaerobic gram-negative rods |
| Penicillinase-resistant penicillins: methicillin, nafcillin, oxacillin, cloxacillin, dicloxacillin | Similar to the natural penicillins, except enhanced activity against staphylococci |
| Broad-spectrum penicillins: aminopenicillins (ampicillin, amoxicillin); carboxypenicillins (carbenicillin, ticarcillin); ureidopenicillins (piperacillin) | Activity against gram-positive cocci equivalent to the natural penicillins; active against some gram-negative rods, with piperacillin the most active |
| β-Lactam with β-lactamase inhibitor (ampicillin-sulbactam, amoxicillin-clavulanate, ticarcillin-clavulanate, piperacillin-tazobactam) | Activity similar to natural β-lactams, plus improved activity against β-lactamase producing staphylococci and selected gram-negative rods; not all β-lactamases are inhibited; piperacillin/tazobactam is the most active |
Penicillin G is inactivated by gastric acid; thus it is used mainly as an intravenous drug for the treatment of infections caused by the limited number of susceptible organisms. Penicillin V is more resistant to acid and is the preferred oral form for the treatment of susceptible bacteria. Penicillinase-resistant penicillins such as methicillin and oxacillin, are used to treat infections caused by susceptible staphylococci. Unfortunately, resistance to this group of antibiotics has become commonplace in both hospital-acquired and community-acquired staphylococcus infections. Ampicillin was the first broad-spectrum penicillin, although the spectrum of activity against gram-negative rods was limited primarily to Escherichia, Proteus, and Haemophilus species. Other penicillins (e.g., carbenicillin, ticarcillin, piperacillin) are effective against a broader range of gram-negative bacteria, including Klebsiella, Enterobacter, and Pseudomonas species.
Selected penicillins have been combined with β-lactamase inhibitors. The β-lactamase inhibitors (e.g., clavulanic acid, sulbactam, tazobactam) are relatively inactive by themselves but, when combined with some penicillins (i.e., ampicillin, amoxicillin, ticarcillin, piperacillin), are effective in treating some infections caused by β-lactamase-producing bacteria. The inhibitors irreversibly bind and inactivate susceptible bacterial β-lactamases (although not all are bound by these inhibitors), permitting the companion drug to disrupt bacterial cell wall synthesis.
Cephalosporins and Cephamycins
The cephalosporins (Table 17-3) are β-lactam antibiotics derived from 7-aminocephalosporanic acid (the β-lactam ring is fused with a dihydrothiazine ring) that was originally isolated from the mold Cephalosporium. The cephamycins are closely related to the cephalosporins, except that they contain oxygen in place of sulfur in the dihydrothiazine ring, rendering them more stable to β-lactamase hydrolysis. The cephalosporins and cephamycins have the same mechanism of action as the penicillins; however, they have a wider antibacterial spectrum, are resistant to many β-lactamases, and have improved pharmacokinetic properties (e.g., longer half-life).
Table 17-3 Selected Examples of Cephalosporins and Cephamycins
| Antibiotics | Spectrum of Activity |
|---|---|
| Narrow spectrum (cephalexin, cephalothin, cefazolin, cephapirin, cephradine) | Activity equivalent to oxacillin against gram-positive bacteria; some gram-negative activity (e.g., Escherichia coli, Klebsiella, Proteus mirabilis) |
| Expanded-spectrum cephalosporins (cefaclor, cefuroxime) | Activity equivalent to oxacillin against gram-positive bacteria; improved gram-negative activity to include Enterobacter, Citrobacter, and additional Proteus species |
| Expanded-spectrum cephamycins (cefotetan, cefoxitin) | Activity similar to expanded-spectrum cephalosporins but less susceptible to β-lactamases |
| Broad spectrum (cefixime, cefotaxime, ceftriaxone, ceftazidime) | Activity equivalent to oxacillin against gram-positive bacteria; improved gram-negative activity to include Pseudomonas |
| Extended spectrum (cefepime, cefpirome) | Activity equivalent to oxacillin against gram-positive bacteria; marginally improved gram-negative activity |
Biochemical modifications in the basic antibiotic molecule resulted in the development of antibiotics with improved activity and pharmacokinetic properties. The cephalosporins have enhanced activity against gram-negative bacteria compared with the penicillins. This activity, in turn, varies among the different “generations” of cephalosporins. The activity of narrow-spectrum, first-generation antibiotics is primarily restricted to Escherichia coli, Klebsiella species, Proteus mirabilis, and oxacillin-susceptible gram-positive cocci. Many of the expanded-spectrum, second-generation antibiotics have additional activity against Haemophilus influenzae, Enterobacter, Citrobacter, and Serratia species, and some anaerobes, such as Bacteroides fragilis. The broad-spectrum, third-generation antibiotics and extended-spectrum, fourth-generation antibiotics are active against most Enterobacteriaceae and Pseudomonas aeruginosa. Extended-spectrum antibiotics offer the advantage of increased stability to β-lactamases. Unfortunately, gram-negative bacteria have rapidly developed resistance to most cephalosporins and cephamycins (primarily as the result of β-lactamase production), which has significantly compromised the use of all these agents.
Carbapenems and Monobactams
Other classes of β-lactam antibiotics (Table 17-4) are the carbapenems (e.g., imipenem, meropenem, ertapenem, doripenem) and monobactams (e.g., aztreonam). The carbapenems are important, widely prescribed broad-spectrum antibiotics that are active against many groups of organisms. In contrast, the monobactams are narrow-spectrum antibiotics that are active only against aerobic, gram-negative bacteria. Anaerobic bacteria and gram-positive bacteria are resistant. The advantage of narrow-spectrum antibiotics is that they can be used to treat susceptible organisms without disruption of the patient’s normal, protective bacterial population. Despite this advantage, monobactams are not widely used.
Table 17-4 Other β-Lactam Antibiotics
| Antibiotics | Spectrum of Activity |
|---|---|
| Carbapenems (imipenem, meropenem, ertapenem, doripenem) | Broad-spectrum antibiotics active against most aerobic and anaerobic gram-positive and gram-negative bacteria except oxacillin-resistant staphylococci, most Enterococcus faecium, and selected gram-negative rods (e.g., some Burkholderia, Stenotrophomonas, some Pseudomonas) |
| Monobactam (aztreonam) | Active against selected aerobic gram-negative rods but inactive against anaerobes or gram-positive cocci |
In recent years, resistance to carbapenems mediated by production of carbapenemases has become problematic, in part because the in vitro susceptibility tests may not reliably detect these resistance bacteria. As mentioned earlier, the β-lactamases are separated into four classes, A to D. Class A carbapenemases have been found in a broad range of bacteria, including Pseudomonas and Enterobacteriaceae (the most common one is the Klebsiella pneumoniae carbapenemase or KPC), renders organisms producing this carbapenemase resistant to all β-lactams, and is only reliably detected with special in vitro susceptibility tests (modified Hodge test). The class B carbapenemase is a metallo-β-lactamase (requires zinc for activity), is widely distributed in gram-negative bacteria, and also cannot be detected reliably by conventional susceptibility tests (demonstrated by showing the organism is susceptible to the carbapenemase when zinc is removed from the susceptibility test medium). Organisms producing class B carbapenemases (most common is New Delhi metallo-β-lactamase [NDM], named for its origin) are resistant to all β-lactam antibiotics, with the possible exception of aztreonam. Finally, the class D carbapenemases are primarily found in Acinetobacter, are detected by conventional susceptibility tests, and encode resistance to all β-lactam antibiotics. This group of carbapenemases is important, because Acinetobacter strains producing this carbapenemase are generally resistant to all antibiotics with few exceptions.
Glycopeptides
Vancomycin, originally obtained from Streptomyces orientalis, is a complex glycopeptide that disrupts cell wall peptidoglycan synthesis in growing gram-positive bacteria. Vancomycin interacts with the D-alanine-D-alanine termini of the pentapeptide side chains, which interferes sterically with the formation of the bridges between the peptidoglycan chains. Vancomycin is used for the management of infections caused by oxacillin-resistant staphylococci and other gram-positive bacteria resistant to β-lactam antibiotics. Vancomycin is inactive against gram-negative bacteria, because the molecule is too large to pass through the outer membrane pores and reach the peptidoglycan target site. In addition, some organisms are intrinsically resistant to vancomycin (e.g., Leuconostoc, Lactobacillus, Pediococcus, and Erysipelothrix) because the pentapeptide terminates in D-alanine-D-lactate, which does not bind vancomycin. Intrinsic resistance is also found in some species of enterococci that contain a D-alanine-D-serine terminus (i.e., Enterococcus gallinarum, Enterococcus casseliflavus). Finally, some species of enterococci (particularly E. faecium and Enterococcus faecalis) have acquired resistance to vancomycin. The genes for this resistance (primarily vanA and vanB), which also mediate changes in the pentapeptide terminus, can be carried on plasmids and have seriously compromised the usefulness of vancomycin for the treatment of enterococcal infections. More importantly, the gene for vancomycin resistance contained within a transposon on a multiresistance conjugative plasmid has been transferred in vivo from E. faecalis to a multiresistant S. aureus. The transposon then moved from the E. faecalis plasmid and recombined and integrated into the S. aureus resistance plasmid. This resulted in an S. aureus plasmid that encoded resistance to β-lactams, vancomycin, aminoglycosides, and other antibiotics—a plasmid that could be transferred to other staphylococci by conjugation. Interestingly, these resistant strains of Staphylococcus have primarily been restricted to Michigan; however, if this resistance becomes widespread, the medical implications are profound.
Lipopeptides
Daptomycin, a naturally occurring cyclic lipopeptide produced by Streptomyces roseosporus, binds irreversibly to the cytoplasmic membrane, resulting in membrane depolarization and disruption of the ionic gradients, leading to cell death. It has potent activity against gram-positive bacteria, but gram-negative bacteria are resistant to daptomycin, because the drug cannot penetrate through the cell wall to the cytoplasmic membrane. Daptomycin has good activity against multidrug resistant staphylococci, streptococci, and enterococci (including vancomycin-resistant strains.
Polypeptides
Bacitracin, which was isolated from Bacillus licheniformis, is a mixture of polypeptides used in topically applied products (e.g., creams, ointments, sprays) for the treatment of skin infections caused by gram-positive bacteria (particularly those caused by Staphylococcus and group A Streptococcus). Gram-negative bacteria are resistant to this agent. Bacitracin inhibits cell wall synthesis by interfering with dephosphorylation and the recycling of the lipid carrier responsible for moving the peptidoglycan precursors through the cytoplasmic membrane to the cell wall. It may also damage the bacterial cytoplasmic membrane and inhibit ribonucleic acid (RNA) transcription. Resistance to the antibiotic is most likely caused by failure of the antibiotic to penetrate into the bacterial cell.
The polymyxins are a group of cyclic polypeptides derived from Bacillus polymyxa. These antibiotics insert into bacterial membranes like detergents, by interacting with lipopolysaccharides and the phospholipids in the outer membrane, producing increased cell permeability and eventual cell death. Polymyxins B and E (colistin) are capable of causing serious nephrotoxicity. Thus their use has been limited historically to the external treatment of localized infections such as external otitis, eye infections, and skin infections caused by sensitive organisms. However, because some organisms such as Acinetobacter and Pseudomonas are only susceptible to colistin, this antibiotic is used to treat some systemic infections. These antibiotics are most active against gram-negative rods, because gram-positive bacteria do not have an outer membrane.
Isoniazid, Ethionamide, Ethambutol, and Cycloserine
Isoniazid, ethionamide, ethambutol, and cycloserine are cell wall–active antibiotics used for the treatment of mycobacterial infections. Isoniazid (isonicotinic acid hydrazide [INH]) is bactericidal against actively replicating mycobacteria. Although the exact mechanism of action is unknown, the synthesis of mycolic acid is affected (the desaturation of the long-chain fatty acids and the elongation of fatty acids and hydroxy lipids are disrupted). Ethionamide, a derivative of INH, also blocks mycolic acid synthesis. Ethambutol interferes with the synthesis of arabinogalactan in the cell wall, and cycloserine inhibits two enzymes, D-alanine-D-alanine synthetase and alanine racemase, which catalyze cell wall synthesis. Resistance to these four antibiotics results primarily from reduced drug uptake into the bacterial cell or alteration of the target sites.
Inhibition of Protein Synthesis
The primary action of the agents in the second largest class of antibiotics is the inhibition of protein synthesis (see Table 17-1).
Aminoglycosides
The aminoglycoside antibiotics (Table 17-5) consist of amino sugars linked through glycosidic bonds to an aminocyclitol ring. Streptomycin, neomycin, kanamycin, and tobramycin were originally isolated from Streptomyces species, and gentamicin and sisomicin were isolated from Micromonospora species. Amikacin and netilmicin are synthetic derivatives of kanamycin and sisomicin, respectively. These antibiotics exert their effort by passing through the bacterial outer membrane (in gram-negative bacteria), cell wall, and cytoplasmic membrane to the cytoplasm, where they inhibit bacterial protein synthesis by irreversibly binding to the 30S ribosomal proteins. This attachment to the ribosomes has two effects: production of aberrant proteins as the result of misreading of the messenger RNA (mRNA), and interruption of protein synthesis by causing the premature release of the ribosome from mRNA.
Table 17-5 Inhibitors of Protein Synthesis
| Antibiotics | Spectrum of Activity |
|---|---|
| Aminoglycosides (streptomycin, kanamycin, gentamicin, tobramycin, amikacin) | Primarily used to treat infections with gram-negative rods; kanamycin with limited activity; tobramycin slightly more active than gentamicin versus Pseudomonas; amikacin most active; streptomycin and gentamicin combined with cell wall–active antibiotic to treat enterococcal infections; streptomycin active versus mycobacteria and selected gram-negative rods |
| Aminocyclitol (spectinomycin) | Active versus Neisseria gonorrhoeae |
| Tetracyclines (tetracycline, doxycycline, minocycline) | Broad-spectrum antibiotics active against gram-positive and some gram-negative bacteria (Neisseria, some Enterobacteriaceae), mycoplasmas, chlamydiae, and rickettsiae |
| Glycylcyclines (tigecycline) | Spectrum similar to tetracyclines but more active against gram-negative bacteria and rapidly growing mycobacteria |
| Oxazolidinone (linezolid) | Active against Staphylococcus (including methicillin-resistant and vancomycin-intermediate strains), Enterococcus, Streptococcus, gram-positive rods, and Clostridium and anaerobic cocci; not active against gram-negative bacteria |
| Macrolides (erythromycin, azithromycin, clarithromycin, roxithromycin) | Broad-spectrum antibiotics active against gram-positive and some gram-negative bacteria, Neisseria, Legionella, Mycoplasma, Chlamydia, Chlamydophila, Treponema, and Rickettsia; clarithromycin and azithromycin active against some mycobacteria |
| Ketolides (telithromycin) | Broad-spectrum antibiotic with activity similar to macrolides; active against some macrolides-resistant staphylococci and enterococci |
| Lincosamide (clindamycin) | Broad-spectrum activity against aerobic gram-positive cocci and anaerobes |
| Streptogramins (quinupristin-dalfopristin) | Primarily active against gram-positive bacteria; good activity against methicillin-susceptible and -resistant staphylococci, streptococci, vancomycin-susceptible and -resistant Enterococcus faecium (no activity against E. faecalis), Haemophilus, Moraxella, and anaerobes (including Bacteroides fragilis); not active against Enterobacteriaceae or other gram-negative rods |
The aminoglycosides are bactericidal because of their ability to bind irreversibly to ribosomes and are commonly used to treat serious infections caused by many gram-negative rods (e.g., Enterobacteriaceae, Pseudomonas, Acinetobacter) and some gram-positive organisms. Penetration through the cytoplasmic membrane is an aerobic, energy-dependent process; so, anaerobes are resistant to aminoglycosides, and susceptible organisms in an anaerobic environment (e.g., abscess) do not respond to treatment. Streptococci and enterococci are resistant to aminoglycosides, because the aminoglycosides fail to penetrate through the cell wall of these bacteria. Treatment of these organisms requires co-administration of an aminoglycoside with an inhibitor of cell wall synthesis (e.g., penicillin, ampicillin, vancomycin) that facilitates uptake of the aminoglycoside.
The most commonly used antibiotics in this class are amikacin, gentamicin, and tobramycin. All three aminoglycosides are used to treat systemic infections caused by susceptible gram-negative bacteria. Amikacin has the best activity and is frequently reserved for treatment of infections caused by gram-negative bacteria that are resistant to gentamicin and tobramycin. Streptomycin is not readily available but has been used for the treatment of tuberculosis, tularemia, and gentamicin-resistant streptococcal or enterococcal infections (in combination with a penicillin).
Resistance to the antibacterial action of aminoglycosides can develop in one of four ways: (1) mutation of the ribosomal binding site, (2) decreased uptake of the antibiotic into the bacterial cell, (3) increased expulsion of the antibiotic from the cell, or (4) enzymatic modification of the antibiotic. The most common mechanism of resistance is enzymatic modification of aminoglycosides. This is accomplished by the action of phosphotransferases (aminoglycoside phosphotransferase [APHs]; seven described), adenyltransferases (adenine nucleotide translocases [ANTs]; four described), and acetyltransferases (acetyl-CoA carboxylases [AACs]; four described) on the amino and hydroxyl groups of the antibiotic. The differences in the antibacterial activity among the aminoglycosides are determined by their relative susceptibility to these enzymes. The other mechanisms by which bacteria develop resistance to aminoglycosides are relatively uncommon. Resistance caused by alteration of the bacterial ribosome requires systematic mutation of the multiple copies of the ribosomal genes that exist in the bacterial cell. Resistance caused by inhibited transport of the antibiotic into the bacterial cell is occasionally observed with Pseudomonas but is more commonly seen with anaerobic bacteria. This mechanism produces low-level cross-resistance to all aminoglycosides. Active efflux of aminoglycosides occurs only in gram-negative bacteria and is rarely observed.
Tetracyclines
The tetracylines (see Table 17-5) are broad-spectrum, bacteriostatic antibiotics that inhibit protein synthesis in bacteria by binding reversibly to the 30S ribosomal subunits, thus blocking the binding of aminoacyl-transfer RNA (tRNA) to the 30S ribosome–mRNA complex. Tetracyclines (i.e., tetracycline, doxycycline, minocycline) are effective in the treatment of infections caused by Chlamydia, Mycoplasma, and Rickettsia species and other selected gram-positive and gram-negative bacteria. All tetracyclines have a similar spectrum of activity, with the primary difference among the antibiotics being in their pharmacokinetic properties (doxycycline and minocycline are easily absorbed and have a long half-life). Resistance to the tetracyclines can stem from decreased penetration of the antibiotic into the bacterial cell, active efflux of the antibiotic out of the cell, alteration of the ribosomal target site, or enzymatic modification of the antibiotic. Mutations in the chromosomal gene encoding the outer membrane porin protein, OmpF, can lead to low-level resistance to the tetracyclines, as well as to other antibiotics (e.g., β-lactams, quinolones, chloramphenicol).
Researchers have identified a variety of genes in different bacteria that control the active efflux of the tetracyclines from the cell. This is the most common cause of resistance. Resistance to the tetracyclines can also result from the production of proteins similar to elongation factors that protect the 30S ribosome. When this happens, the antibiotic can still bind to the ribosome, but protein synthesis is not disrupted.
Glycylclines
Tigecycline, the first representative of this new class of antibiotics, is a semisynthetic derivative of minocycline. It inhibits protein synthesis in the same manner as the tetracyclines. Tigecycline has a higher binding affinity for the ribosome and is less affected by efflux or enzymatic modification. It has a broad spectrum of activity against gram-positive, gram-negative, and anaerobic bacteria, although Proteus, Morganella, Providencia, and P. aeruginosa are generally resistant.
Oxazolidinones
The oxazolidinones are a narrow-spectrum class of antibiotics, with linezolid being the agent currently used. Linezolid blocks initiation of protein synthesis by interfering with the formation of the initiation complex consisting of tRNA, mRNA, and the ribosome. The drug binds to the 50S ribosomal subunit, which distorts the binding site for tRNA, thus inhibiting formation of the 70S initiation complex. Because of this unique mechanism, cross-resistance with other protein inhibitors does not occur. Linezolid has activity against all staphylococci, streptococci, and enterococci (including those strains resistant to penicillins, vancomycin, and the aminoglycosides). Because the multidrug-resistant enterococci are difficult to treat, use of linezolid is generally reserved for these infections.
Chloramphenicol
Chloramphenicol has a broad antibacterial spectrum similar to that of tetracycline but is not commonly used in the United States. The reason for its limited use is that besides interfering with bacterial protein synthesis, it disrupts protein synthesis in human bone marrow cells and can produce blood dyscrasias, such as aplastic anemia (1 per 24,000 treated patients). Chloramphenicol exerts its bacteriostatic effect by binding reversibly to the peptidyl transferase component of the 50S ribosomal subunit, thus blocking peptide elongation. Resistance to chloramphenicol is observed in bacteria producing plasmid-encoded chloramphenicol acetyltransferase, which catalyzes the acetylation of the 3-hydroxy group of chloramphenicol. The product is incapable of binding to the 50S subunit. Less commonly, chromosomal mutations alter the outer membrane porin proteins, causing the gram-negative rods to be less permeable.
Macrolides
Erythromycin, derived from Streptomyces erythreus, is the model macrolide antibiotic (see Table 17-5). The basic structure of this class of antibiotics is a macrocyclic lactone ring bound to two sugars, desosamine and cladinose. Modification of the macrolide structure led to the development of azithromycin, clarithromycin, and roxithromycin. Macrolides exert their effect by their reversible binding to the 23S ribosomal RNA (rRNA) of the 50S ribosomal subunit, which blocks polypeptide elongation. Resistance to macrolides most commonly stems from the methylation of the 23S rRNA, preventing binding by the antibiotic. Other mechanisms of resistance include inactivation of the macrolides by enzymes (e.g., esterases, phosphorylases, glycosidase) or mutations in the 23S rRNA and ribosomal proteins. Macrolides are bacteriostatic antibiotics with a broad spectrum of activity. They have been used to treat pulmonary infections caused by Mycoplasma, Legionella, and Chlamydia species, as well as to treat infections caused by Campylobacter species and gram-positive bacteria in patients allergic to penicillin. Most gram-negative bacteria are resistant to the macrolides. Azithromycin and clarithromycin have also been used to treat infections caused by mycobacteria (e.g., Mycobacterium avium complex).
Ketolides
Ketolides are semisynthetic derivative of erythromycin, modified to increase stability in acid. Telithromycin is currently the only ketolide available for use in the United States. As with the macrolides, telithromycin binds to the 50S ribosomal subunit and blocks protein synthesis. Mutations in 23S rRNA or the ribosomal proteins can lead to resistance. Telithromycin has good activity against staphylococci (except strains that have consititutive resistance to erythromycin), S. pneumoniae, other respiratory pathogens (e.g., H. influenzae, Moraxella catarrhalis), gram-positive rods, and some anaerobes. It is not active against B. fragilis and most aerobic gram-negative rods (e.g., Enterobacteriaceae, Pseudomonas, Acinetobacter, Stenotrophomonas). Telithromycin also has good activity against intracellular pathogens (e.g., Legionella, Mycoplasma, Chlamydia, Chlamydiophila), Rickettsia, Bartonella, Coxiella, Francisella, and M. avium.
Clindamycin
Clindamycin (in the family of lincosamide antibiotics) is a derivative of lincomycin, which was originally isolated from Streptomyces lincolnensis. Like chloramphenicol and the macrolides, clindamycin blocks protein elongation by binding to the 50S ribosome. It inhibits peptidyl transferase by interfering with the binding of the amino acid–acyl-tRNA complex. Clindamycin is active against staphylococci and anaerobic gram-negative rods but is generally inactive against aerobic gram-negative bacteria. Methylation of the 23S rRNA is the source of bacterial resistance. Because both erythromycin and clindamycin can induce this enzymatic resistance (also plasmid mediated), cross-resistance between these two classes of antibiotics is observed.
Streptogramins
The streptogramins are a class of cyclic peptides produced by Streptomyces species. These antibiotics are administered as a combination of two components, group A and group B streptogramins, which act synergistically to inhibit protein synthesis. The antibiotic currently available in this class is quinupristin-dalfopristin. Dalfopristin binds to the 50S ribosomal subunit and induces a conformational change that facilitates binding of quinupristin. Dalfopristin prevents peptide chain elongation, and quinupristin initiates premature release of peptide chains from the ribosome. This combination drug is active against staphylococci, streptococci, and E. faecium (but not E. faecalis). Use of the antibiotic has been restricted primarily to treating vancomycin-resistant E. faecium infections.
Inhibition of Nucleic Acid Synthesis
Quinolones
The quinolones (Table 17-6) are one of the most widely used classes of antibiotics. These are synthetic chemotherapeutic agents that inhibit bacterial DNA topoisomerase type II (gyrase) or topoisomerase type IV, which are required for DNA replication, recombination, and repair. The DNA gyrase-A subunit is the primary quinolone target in gram-negative bacteria, whereas topoisomerase type IV is the primary target in gram-positive bacteria. The first quinolone used in clinical practice was nalidixic acid. This drug was used to treat urinary tract infections caused by a variety of gram-negative bacteria, but resistance to the drug developed rapidly, causing it to fall out of use. This drug has now been replaced by newer, more active quinolones, such as ciprofloxacin, levofloxacin, and moxifloxacin. Modifying the two-ring quinolone nucleus made these newer quinolones (referred to as fluoroquinolones). These antibiotics have excellent activity against gram-positive and gram-negative bacteria, although resistance can develop rapidly in Pseudomonas, oxacillin-resistant staphylococci, and enterococci. In particular, the newer extended-spectrum quinolones have significant activity against gram-positive bacteria.
| Antibiotics | Spectrum of Activity |
|---|---|
| Narrow spectrum (nalidixic acid) | Active against selected gram-negative rods; no useful gram-positive activity |
| Broad spectrum (ciprofloxacin, levofloxacin) | Broad-spectrum antibiotics with activity against gram-positive and gram-negative bacteria |
| Extended spectrum (gatifloxacin, moxifloxacin) | Broad-spectrum antibiotics with enhanced activity against gram-positive bacteria (particularly streptococci and enterococci) compared with early quinolones; activity against gram-negative rods similar to that of ciprofloxacin and related quinolones |
Resistance to the quinolones is mediated by chromosomal mutations in the structural genes for DNA gyrase and topoisomerase type IV. Other mechanisms include decreased drug uptake caused by mutations in the membrane permeability regulatory genes, and overexpression of efflux pumps that actively eliminate the drug. Each of these mechanisms is primarily chromosomally mediated.
Rifampin and Rifabutin
Rifampin, a semisynthetic derivative of rifamycin B produced by Streptomyces mediterranei, binds to DNA-dependent RNA polymerase and inhibits the initiation of RNA synthesis. Rifampin is bactericidal for Mycobacterium tuberculosis and is very active against aerobic gram-positive cocci, including staphylococci and streptococci.
Because resistance can develop rapidly, rifampin is usually combined with one or more other effective antibiotics. Rifampin resistance in gram-positive bacteria results from a mutation in the chromosomal gene that codes for the beta subunit of RNA polymerase. Gram-negative bacteria are resistant intrinsically to rifampin, because of decreased uptake of the hydrophobic antibiotic. Rifabutin, a derivative of rifamycin, has a similar mode and spectrum of activity. It is particularly active against M. avium.
Metronidazole
Metronidazole was originally introduced as an oral agent for the treatment of Trichomonas vaginitis. However, it was also found to be effective in the treatment of amebiasis, giardiasis, and serious anaerobic bacterial infections (including those caused by B. fragilis). Metronidazole has no significant activity against aerobic or facultatively anaerobic bacteria. The antimicrobial properties of metronidazole stem from the reduction of its nitro group by bacterial nitroreductase, thereby producing cytotoxic compounds that disrupt the host DNA. Resistance results either from decreased uptake of the antibiotic or from elimination of the cytotoxic compounds before they can interact with host DNA.
Antimetabolites
The sulfonamides are antimetabolites that compete with p-aminobenzoic acid, thereby preventing the synthesis of the folic acid required by certain microorganisms. Because mammalian organisms do not synthesize folic acid (required as a vitamin), sulfonamides do not interfere with mammalian cell metabolism. Trimethoprim is another antimetabolite that interferes with folic acid metabolism by inhibiting dihydrofolate reductase, thereby preventing the conversion of dihydrofolate to tetrahydrofolate. This inhibition blocks the formation of thymidine, some purines, methionine, and glycine. Trimethoprim is commonly combined with sulfamethoxazole to produce a synergistic combination active at two steps in the synthesis of folic acid. Dapsone and p-aminosalicylic acid are also antifolates that have proved to be useful for treating mycobacterial infections.
Sulfonamides are effective against a broad range of gram-positive and gram-negative organisms, such as Nocardia, Chlamydia, and some protozoa. Short-acting sulfonamides, such as sulfisoxazole, are among the drugs of choice for the treatment of acute urinary tract infections caused by susceptible bacteria, such as E. coli. Trimethoprim-sulfamethoxazole is effective against a large variety of gram-positive and gram-negative microorganisms and is the drug of choice for the treatment of acute and chronic urinary tract infections. The combination is also effective in the treatment of infections caused by Pneumocystis jirovecii, bacterial infections of the lower respiratory tract, otitis media, and uncomplicated gonorrhea.
Resistance to these antibiotics can stem from a variety of mechanisms. Bacteria such as Pseudomonas are resistant as the result of permeability barriers. A decreased affinity of dihydrofolate reductase can be the source of trimethoprim resistance. In addition, bacteria that use exogenous thymidine (e.g., enterococci) are also intrinsically resistant.
Other Antibiotics
Clofazimine is a lipophilic antibiotic that binds to mycobacterial DNA. It is highly active against M. tuberculosis, is a first-line drug for the treatment of Mycobacterium leprae infections, and has been recommended as a secondary antibiotic for the treatment of infections caused by other mycobacterial species.
Pyrazinamide (PZA) is active against M. tuberculosis at a low pH, such as that found in phagolysosomes. The active form of this antibiotic is pyrazinoic acid, produced when PZA is hydrolyzed in the liver. The mechanism by which PZA exerts its effect is unknown.
1. Describe the mode of action of the following antibiotics: penicillin, vancomycin, isoniazid, gentamicin, tetracycline, erythromycin, polymyxin, ciprofloxacin, and sulfamethoxazole.
2. Name the three mechanisms bacteria use to become resistant to β-lactam antibiotics. What is the mechanism responsible for oxacillin resistance in Staphylococcus? Imipenem resistance in Pseudomonas? Penicillin resistance in S. pneumoniae?
3. By what three mechanisms have organisms developed resistance to aminoglycosides?
4. What mechanism is responsible for resistance to the quinolones?
5. How do trimethoprim and the sulfonamides differ in their mode of action?
1. Penicillin interferes with cell wall synthesis by binding to specific penicillin-binding proteins (PBPs), the regulatory enzymes (e.g., transpeptidases, transglycosylases, carboxypeptidases) responsible for construction of the peptidoglycan layer of the cell wall. Vancomycin also disrupts cell wall peptidoglycan synthesis, in this case in gram-positive bacteria. This is accomplished by vancomycin interacting with the D-alanine-D-alanine termini of the pentapeptide side chains that form bridges between the peptidoglycan chains. Isoniazid disrupts the synthesis of mycolic acid, an important component of the cell wall in mycobacteria. Gentamicin, tetracycline, and erythromycin inhibit protein synthesis in bacteria. Gentamicin binds irreversibly to the 30S ribosomal proteins, leading to misreading of mRNA and premature release of the ribosome from mRNA. The tetracyclines bind reversibly to the 30 S ribosomal subunits and block the binding of aminoacyl-transfer RNA to the 30S ribosome–mRNA complex. Erythromycin, a macrolide antibiotic, binds reversibly to the 23S rRNA of the 50S ribosomal subunit and blocks polypeptide elongation. Polymyxin inserts into bacterial membranes similar to detergents by interacting with the lipopolysaccharides and the phospholipids in the outer membrane, producing increased cell permeability. Ciprofloxacin, a fluoroquinolone, inhibits bacterial DNA topoisomerase type II (gyrase), which is required for DNA replication, recombination, and repair. Sulfamethoxazole is an antimetabolite that prevents the synthesis of folic acid.
2. Bacteria can become resistant to β-lactam antibiotics by (1) degrading the antibiotic with β-lactamases; (2) modifying the target (i.e., PBP) so that either a new PBP is acquired by the organism or an existing PBP is altered, producing an enzymatically active PBP that is not recognized by the antibiotic; or (3) preventing access to the target by creating a permeability barrier (e.g., a change in porins in the gram-negative cell wall). Staphylococcus aureus organisms become resistant to oxacillin and related β-lactams by acquiring a new PBP that is enzymatically active (e.g., can be used to build the peptidoglycan layer in the cell wall) but is not bound and inactivated by the antibiotic. Streptococcus pneumoniae organisms become resistant to penicillin when they acquire an altered PBP (through recombination). Pseudomonas aeruginosa can become resistant to imipenem by one of two mechanisms: (1) acquisition of a β-lactamase that degrades the carbapenem antibiotic or (2) alteration in the outer membrane of the cell wall (i.e., porin mutation) that prevents entry of the antibiotic into the cell.
3. Organisms can become resistant to aminoglycosides by (1) enzymatic modification of the antibiotic (the most common method), (2) decreased uptake of the antibiotic into the bacterial cell, (3) increased expulsion of the antibiotic from the cell, and (4) mutation of the ribosomal binding site.
4. Bacteria become resistant to quinolones by chromosomal mutations in the structural genes of the targets: DNA gyrase and topoisomerase IV. Other less common methods include decreased drug uptake caused by mutations in the membrane permeability regulatory genes and overexpression of efflux pumps that actively eliminate the drug.
5. Trimethoprim interferes with folic acid metabolism by inhibiting dihydrofolate reductase, preventing the conversion of dihydrofolate to tetrahydrofolate. The sulfonamides inhibit dihydropteroic acid synthase, which functionally also inhibits folic acid synthesis but at a different step.