Chapter 13 Scaffolds
Small globular proteins as antibody substitutes
Introduction
An emerging field in pharmaceutical biotechnology is represented by the generation of novel binding molecules based on small globular domains serving as protein frameworks (’scaffolds’). In this approach, certain amino acid residues of the surface of a single domain are combinatorially mutated to produce a protein library, which can then be screened for binding specificities of interest. Thus, the concept of a universal binding site from the antibody structure is transferred to alternative protein frameworks with suitable biophysical properties. On one hand these new proteins provide further insights into the processes of molecular recognition, on the other hand they also have commercial applications, as therapeutic agents, diagnostic reagents or affinity ligands.
Scaffolds
Monoclonal antibodies (mAbs) are an increasingly important class of therapeutic agents as discussed in preceding chapters. Over 25 recombinant antibodies are currently on the market, the top 10 best-selling of these products earned over $32 billion in global sales during 2008, and five mAb products (rituximab, infliximab, bevacizumab, trastuzumab and adalimumab) were included in the top 15 best-selling prescription drugs for that year (Reichert, 2010). The future prospects for therapeutic antibodies are clearly bright, the annual growth rate estimated to be 12% during 2010–2012. However, even antibody blockbusters suffer from certain drawbacks: human immunoglobulin G (IgG) antibody is a complex molecule composed of four protein chains with attached carbohydrates. A full-size IgG antibody is a Y-shaped complex of two identical light chains, each with a variable and constant domain, and two heavy chains, each with one variable and three constant domains (Fig. 13.1H). Therefore, full-size IgGs are always bivalent molecules and elicit effector functions through their Fc part, which may not be ideal for certain applications. With regards to expression, there is the requirement for a rather expensive mammalian cell production system and, in addition, antibodies depend on disulfide bonds for stability. Through the versatile techniques of genetic engineering, smaller antibody fragments have been produced to overcome some of the limitations of full-size IgGs (Holliger and Hudson, 2005), but some antibody fragments tend to aggregate and display limited solubility. Finally, the success, and consequently the extensive use, of antibodies has led to a complicated patent situation for antibody technologies and applications. Therefore, several research groups and small and medium-sized companies have recently focused on the development of small globular proteins as scaffolds for the generation of a novel class of versatile binding proteins.
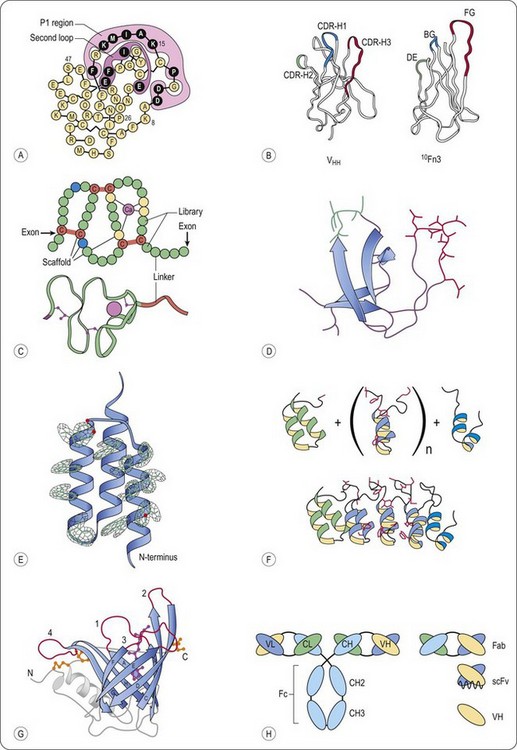
Fig. 13.1 Schematic representation of representative scaffolds. A, Kunitz type domain (LAC–D1) (Ley et al., 1996). Varied positions are depicted in black, the P1 and second loop positions are enclosed. B, Structural comparison of a llama VHH domain and the wild-type human 10Fn3 domain (Xu et al., 2002). Despite the lack of significant sequence identity, both domains fold into similar beta sheet sandwiches. The CDRs of the VHH domain and the residues randomized in the 10Fn3 domain are shown in color. C, Fixed and variable positions of the A-domain library, as well as the disulfide topology, are indicated (Silverman et al., 2005). Each circle represents an amino acid position. Calcium-coordinating scaffold residues are yellow, structural scaffold residues are blue, cysteine residues are red and variable positions are green. A ribbon diagram of a prototypical A-domain structure is included (Protein Data Bank entry 1AJJ). D, Fyn SH3 wt protein structure (Protein Data Bank entry 1M27) The RT-Src loop is in red, and the n-Src loop is in green. E, Electron density for all 13 mutated residues in the affibody (Hogbom et al., 2003). For clarity, only the electron density around the side chains is displayed. F, Schematic representation of the library generation of designed ankyrin repeat proteins (Binz et al., 2004). (upper part) Combinatorial libraries of ankyrin repeat proteins were designed by assembling an N-terminal capping ankyrin (green), varying numbers of the designed ankyrin repeat module (blue) and a C-terminal capping ankyrin (cyan); side chains of the randomized residues are shown in red. (lower part) Ribbon representation of the selected MBP binding ankyrin repeat protein (colours as above) This binder was isolated from a library of N-terminal capping ankyrin repeat, three designed ankyrin repeat modules and a C-terminal capping ankyrin repeat. G, General structure of human retinol-binding protein, a prototypic lipocalin (Schlehuber and Skerra, 2005). Ribbon diagram of the crystal structure of RBP with the bound ligand retinol (magenta, ball and stick representation). The eight antiparallel strands of the conserved β-barrel structure are shown in blue with labels A to H, and the four loops, which are highly variable among the lipocalin family, are colored red and numbered. The typical α-helix that is attached to the central β-barrel in all lipocalins, the loops at the closed end the N- and C-terminal peptide segments are shown in grey. The three disulfide bonds of RBP are depicted in yellow. H, Schematic representation of full-sized antibodies, multidomain and single-domain antigen-binding fragments (Saerens et al., 2008). Classical IgG consists of two light chains and two heavy chains. The light chain is composed of one variable (VL) and one constant (CL) domain, whereas the heavy chain has one variable (VH) and three constant domains (CH1, CH2, and CH3). The smaller antigen-binding antibody fragments are shown, that is, Fab, scFv, and the single-domain antibody (sdAb) fragment, VH.
As therapeutics, globular proteins with engineered binding properties might be particularly interesting, (i) if the neutralization of a target protein is the desired pharmacological effect (in contrast to a full length antibody, where the Fc portion may stimulate immune processes), (ii) as fusion proteins, for the targeted delivery of bioactive molecules to sites of disease, (iii) as receptor-binding drugs, thus interfering with the cell-signalling and (iv) as enzyme inhibitors.
Common to all approaches of finding a suitable scaffold are the following steps (Nygren and Uhlen, 1997; Smith 1998; Binz and Pluckthun, 2005):
• Choosing a small protein (domain) that is well expressed in bacteria or yeast and has good biophysical properties (stability, solubility)
• Creating a library of protein mutants by introducing diversity at a contiguous patch of the surface of the protein (e.g. loops)
• Using a genotype-phenotype display system (e.g. phage display) to select a range of binders to a therapeutic target of interest
• Screening the binders obtained by the selection procedure for the desired biological activity, e.g. by enzyme-linked immunosorbent assay (ELISA).
Mutations introduced in the protein scaffold to produce diversity may compromise the three-dimensional structure, stability and solubility of the protein scaffold, thus making the isolation of protein binders based on folding frameworks other than the immunoglobulin fold difficult. Nevertheless, more than 50 scaffolds have been described for the generation of new protein binders (reviewed in Binz et al., 2005; Gebauer and Skerra, 2009). Being mainly a topic of academic interest at first, nowadays the development and use of non-antibody classes of proteins is being pursued by small- and medium-sized biotechnology companies (Hey et al., 2005). The common basis for the successful development of well-designed protein drugs lies in the availability of both a well-behaving protein scaffold and an efficient selection or screening technology in order to find suitable candidate proteins in the large repertoire created by mutagenesis. The next sections will introduce selected classes of protein scaffolds (see also Figure 13.1) that have been successfully used and commercialized by small- and medium-sized companies. The first scaffold derived drug has been recently approved by the FDA for treatment of hereditary angioedema (see kunitz type domains), others are being investigated in clinical trials. Therefore, we will learn more in the near future about this interesting field of antibody alternatives.
Kunitz type domains
Proteases have important biological functions, they have been identified as key regulators of cellular processes such as ovulation, fertilization, wound healing, angiogenesis, apoptosis, peptide hormone release, coagulation and complement activation (Nixon and Wood, 2006). Because unregulated proteolysis in any of these processes will have undesirable effects, effective control of protease activity is critical. Indeed, unregulated activity has been implicated in the pathogenesis of many diseases (e.g. cancer, inflammatory diseases, chronic obstructive pulmonary disease (COPD), chronic pancreatitis, muscular dystrophy and Alzheimer’s disease (Naruse et al., 1999; Egeblad and Werb, 2002; Nunan and Small, 2002; Shapiro, 2002; Laval and Bushby, 2004; Parks et al., 2004), making proteases important target proteins. It is notable, that there are only a few marketed drugs (such as HIV protease inhibitors, angiotensin-converting enzyme inhibitors, the proteasome inhibitor bortezomib and the renin inhibitor aliskiren), since a key challenge in the discovery of new inhibitors is the ability to identify drugs which are both potent and specific. Structural similarities within the active site of most proteolytic enzyme families often result in a simultaneous inhibition of several family members, which can lead to an unacceptable toxicity profile. One solution to this problem represents the use of endogenous, engineered protease inhibitors that make more contacts with the target protease and therefore allow for tighter control over specificity. One type of endogenous inhibitors include the kunitz domain inhibitors, e.g. bovine pancreatic trypsin inhibitor (BPTI) (Ascenzi et al., 2003).
In 1992, Ladner and co-workers produced a small library of mutants of kunitz domain BPTI, displayed on phage and screened for the ability to bind to elastase (Roberts et al., 1992), a serine protease that is believed to play a causative role in a variety of lung diseases, including COPD and cystic fibrosis (Hautamaki et al., 1997). Isolates that were able to bind to elastase were expressed, purified and tested for inhibition of elastase. The P-loop (see Figure 13.1A), the region that was varied in the library, of the best inhibitor (Ki ∼1 pM) was transferred to a human kunitz domain without subsequent loss of potency (Ley et al., 1997). The resulting engineered molecule, depelestat (Dyax/Debiopharm SA), inhibited human neutrophile elastase (HNE) activity in the sputum of cystic fibrosis patients, was demonstrated to be safe in an aerolized format in monkeys and no adverse effects were reported in Phase I clinical trials (Delacourt et al., 2002; Grimbert et al., 2003; Nixon and Wood, 2006). Pharmacodynamic reports of a Phase IIa clinical trial have been shown to inhibit completely sputum HNE in 52.6% of the patients and to decrease interleukin-8 levels, a biomarker of inflammation, in the sputum of treated patients (Saudubray et al., 2003).
In 1996, another human kunitz domain, the lipoprotein-associated coagulation inhibitor (LACI-D1), was used as a scaffold for the successful isolation of a very specific and potent inhibitor of human kallikrein (Markland et al., 1996). Kallikrein is believed to be an important mediator of hereditary angioedema (HAE), a rare disorder with attacks of oedema in the hands, face, feet, abdomen and/or throat. This condition is caused by a genetic deficiency of C1 esterase inhibitor (C1-Inh), which is an endogenous inhibitor of plasma kallikrein. In Europe, HAE is treated with plasma-derived C1-Inh which attenuates attacks and may prove life-saving, but C1-Inh is expensive and requires the use of pooled blood product (Nixon and Wood, 2006). After an iterative selection and screening approach chosen by the authors (Markland et al., 1996), the plasma kallikrein inhibitor DX-88 (Dyax Corp/Genzyme Corp) was isolated and proved to be a potent inhibitor (Ki = 40 pM) with high selectivity (Table 1 in Williams and Baird, 2003). The use of DX-88 in a C1-Inh deficient mouse model demonstrated that DX-88 is effective in preventing changes in vascular permeability (Han et al., 2002). In December 2009, DX-88 (ecallantide) was approved by FDA for the treatment of HAE.
With the first approved drug kunitz type domains represent the most advanced scaffold in this field. The strategy of taking a human protease inhibitor as a scaffold and improving it by protein engineering works well for certain therapeutic applications. At the same time, kunitz type domains have the limitation of binding only to proteases, and thus, at present, they do not represent a universal source for the generation of binding proteins to a variety of targets.
Tenth type III domain of fibronectin (Adnectins)
The tenth type III domain of human fibronectin (10Fn3) is a 94 aminoacid residue structural protein with an immunoglobulin-like fold that is used as an antibody-mimic scaffold. High thermal stability (TM = 90°C) and solubility (>15 mg/mL), high expression yields in Escherichia coli and the lack of cysteines in its structure are attractive properties that make the 10Fn3 a good scaffold candidate (Xu et al., 2002). Three of the solvent exposed loops in 10Fn3, named BC, DE and FG are structurally analogous to the VH complementary-determining regions (CDR) (Figure 13.1B).
The first report on the use of the 10Fn3 domain as a protein with artificial binding sites was published in 1998 (Koide and Koide, 1998). In that study, a library of 108 distinct 10Fn3 mutants was made by deleting three residues in the FG loop and by randomizing five residues in the BC loop and four residues in the FG loop. The library, displayed on phage, was used in affinity selection experiments with immobilised ubiquitin as model target protein. After five rounds of panning, clones were randomly picked for sequencing. A clone designated as Ubi4-Fn3 dominated the population of selected protein mutants and was subjected to further analysis. Ubi4-Fn3 was demonstrated to bind in phage enzyme-linked immunosorbent assay (ELISA) experiments. However, when expressed as a single domain protein in E. coli, Ubi4-Fn3 exhibited low solubility at neutral pH, unspecific binding to the dextran matrices of the size-exclusion chromatography column and the dextran coated biosensor chips used for the subsequent characterization of Ubi-Fn3. More importantly, the affinity was relatively low; the IC50 determined by competition ELISA was 5 µM.
In order to select 10Fn3 domains binding to a target of interest with improved properties than Ubi4-Fn3, an alternative library design in combination with a fully in vitro selection system (mRNA display (Wilson et al., 2001)) was used for the isolation of 10Fn3 variants binding to tumour necrosis factor-α (TNF-α) (Xu et al., 2002). Clones from the ninth and tenth round of selection were cloned in E. coli, sequenced and expressed. Affinity constants were determined by incubating the in vitro translated 35S-methionine labelled proteins with biotinylated TNF-α at different concentrations. The solution containing the 10Fn3 mutants bound to TNF-α were aspirated by vacuum onto a membrane coated with streptavidin, therefore capturing protein complexes on the membrane. Binding was analysed by measuring the radioactivity on the membrane, the KD for selected mutants were found in the range of 1-24 nM. Further affinity maturation procedures revealed KD values around 100 pM, the best being 20 pM, and analytical gel filtration showed that the apparent molecular weight of purified, soluble wild-type (wt) and mutant domains was consistent with the variants being monomeric. The reported affinities, especially for TNF-α, are very high. However, these values should be evaluated critically because first, the capture step of the biotinylated TNF-α-Fn3 protein complexes was performed in a solid phase format, and second, TNF-α is homotrimeric protein, so avidity effects may contribute to the high apparent affinity observed.
Other 10Fn3 variants were shown to recognize ανβ3-integrin expressed on cell surface and inhibiting ανβ- dependent cell adhesion (Richards et al., 2003) and to bind to vascular endothelial growth factor receptor 2 (VEGF-R2) inhibiting the activation by VEGF-A, -C and –D (Parker et al., 2005; Getmanova et al., 2006). Indeed, the test case of 10Fn3 for the in vivo behaviour in human patients is the VEGF-R2 binding CT-322 (KD = 11 nM), currently in clinical development by Bristol Myers Squibb in cancer patients (Choy et al., 2002; Molckovsky and Siu, 2008). The CT-322 10Fn3 domain is coupled via a C-terminal cysteine to a 40-kDa polyethylene glycol (PEG) to increase the size of the molecule above the threshold for kidney-mediated clearance. In a Phase I study in patients with solid tumours, CT-322 administered intravenously at 1 mg/kg showed a terminal half-life of 68.7 hours, based on the first dose of a weekly dosing regimen (Molckovsky and Siu, 2008). The half-life in humans is substantially shorter than that of IgG (7–26 days), as expected for a molecule lacking FcRn binding, and also shorter than the 14-day half-life reported for Cimzia, a pegylated Fab that binds to TNF-α (Choy et al., 2002). Still, the pharmacokinetics of CT-322 supported weekly intravenous dosing, and a biomarker for VEGF-R2 blockade showed sustained elevation over several repeated weekly doses. Immunogenicity was also assessed in this Phase I trial showing that 31 out of 39 patients developed antibodies to CT-322 (Beyond Antibodies Conference, 2009, San Diego) but these did not lead to a clinical significant immunogenicity (Bloom and Calabro, 2009). As for other biologics, immunogenicity should be monitored carefully for this scaffold, especially for the generation of cross-reactive antibodies binding not only to the injected mutant, but also to the endogenous fibronectin domain. In the near future we will learn more about this scaffold, since patients are currently being enrolled in a Phase II trial in glioblastoma multiforme, in which CT-322 will be tested alone or in combination with irinotecan (Campto, Pfizer), a cytostatic agent inhibiting DNA-topoisomerase I. The technology of using the 10Fn3 domain was commercialized by Adnexus, which was acquired by Bristol Myers Squibb for $430 million in 2007, proving the interest and significant potential of antibody alternatives in the field of pharmaceutical biotechnology.
A-domains (Avimers)
A family of A-domains (Huang et al., 1999; North and Blacklow, 1999) has been described to be suitable as a scaffold for the generation of new binding proteins (Silverman et al., 2005). A-domains occur as strings of multiple domains in several cell-surface receptors (Figure 13.1C). Domains of this family bind over 100 different known targets, including small molecules, proteins and viruses (Krieger and Herz, 1994; Gliemann, 1998). Such a target is typically contacted by multiple A-domains with each domain binding independently to a unique epitope, thereby binding with high avidity. Each of the 217 human A-domains comprises approximately 35 amino acids and the domains are separated by linkers that average five amino acids in length. Native A-domains fold quickly and efficiently to a uniform, stable structure mediated by calcium binding and disulfide formation. A conserved scaffold motif of only 12 amino acids is required for this common structure (Koduri and Blacklow, 2001). Stemmer and co-workers have designed a phage display library of A-domains, consisting of a conserved consensus sequence in the scaffold motif and variable amino acids at different positions (Silverman et al., 2005). Like other directed evolution technologies, the process the authors developed to isolate binding proteins, included multiple recursive cycles, each consisting of library generation and screening in functional assays. However, rather than mutagenizing the protein between cycles (e.g. for affinity maturation purposes), they added a new domain library adjacent to the domain(s) selected in previous rounds.
The result of this process is a single protein chain containing multiple domains, each of which having a separate binding function. The A-domains are therefore called ‘avimers’, from avidity multimers. A heterotetramer consisting of three IL-6 binding A-domains and an IgG binding domain (named C326) showed a remarkable affinity (picomolar range) to its target and exhibited sub-picomolar IC50s in cell-proliferation assays (Silverman et al., 2005). Moreover, C326 completely abrogated acute-phase protein induction by human IL-6 in mice in a dose-dependent manner, suggesting that C326 inhibited IL-6 functions in vivo. The technology of using the modular platform of A-domains was commercialized by Avidia Inc. that was acquired by Amgen for $290 million in 2006. A placebo-controlled Phase I study of C326 was registered in the United States in September 2006, with patients suffering from Crohn’s disease. Since then, to our knowledge, new results have not been published in the public domain. Recent data (Beyond Antibodies Conference, 2009, San Diego) indicate that Amgen is further engineering avimers for the neutralization of IL-6 mediated effects.
SH3 domain of Fyn kinase (Fynomers)
Fyn kinase is a 59 kDa member of the Src family of tyrosine kinases (Cooke and Perlmutter, 1989; Resh, 1998). As a result of alternative splicing, the Fyn protein is expressed as two isoforms, differing in approximately 50 amino acids in a region between their SH2 and kinase domain. One form is found in thymocytes, splenocytes and some hematolymphoid cell lines (FynT), while the second form accumulates principally in the brain (Cooke and Perlmutter, 1989). Its biological functions are diverse and related to Fyn’s ability to associate and to phosphorylate a variety of intracellular signaling molecules. One of the best known functions of Fyn kinase is the phosphorylation of SLAM (signalling lymphocyte activation molecule) in T cells, inducing a signalling complex modulating interferon-γ expression (Li, 2005). The interaction between Fyn and SLAM occurs via the SH3 domain of Fyn and the adaptor protein SAP (SLAM associated protein), forming a ternary complex. Interestingly, Fyn SH3 associates with SAP through a surface–surface interaction that does not involve the canonical PXXP recognition motif of SH3 domains (Chan et al., 2003). The Fyn SH3 domain comprises 63 residues (amino acids 83–145 of the sequence reported by Kawakami et al., 1986; Semba et al., 1986) and shows the typical SH3 topology of two perpendicular β-sheets and a single turn of 310 helix (Figure 13.1D). Interacting regions of the Fyn SH3 domain include the RT-loop, n-src loop and single residues of the β3- and β4 strands (Chan et al., 2003). With its biophysical properties and its primary structure, Fyn SH3 perfectly matches important criteria for a scaffold to be used as alternative to antibodies: (i) it is expressed in bacteria at high level in soluble, monomeric form (Morton et al., 1996), (ii) it is stable (TM: 70.5° C) (Filimonov et al., 1999), (iii) it does not contain any cysteine residues, (iv) it is from human origin and (v) its amino acid sequence is conserved in mouse, rat, monkey (gibbon) and man. As mentioned above, its binding mode to SAP has been resolved and indicates that this particular domain does not necessarily need a PXXP core binding motif (Chan et al., 2003), which is an important prerequisite to be well suited for the isolation of binders against a variety of different target epitopes. Recently, we presented the design, construction, and characterization of a human Fyn SH3 phage library containing more than 1 billion individual clones, where both loops of the Fyn SH3 domain (the RT- and n-Src-loop) were randomized (Grabulovski et al., 2007). Using phage display technology, the isolation and in vitro characterization of Fyn SH3 proteins (termed Fynomers) binding to the extra-domain B (EDB) of fibronectin (Castellani et al., 1994; Kaspar et al., 2006), a marker of angiogenesis, was described. Fibronectin is a large glycoprotein that is present in large amounts in plasma and body tissues. EDB is a 91-amino acid type III homology domain that becomes inserted into the fibronectin molecule by a mechanism of alternative splicing at the level of the primary transcript (Zardi et al., 1987). The Fynomer clone termed D3 exhibited low nM affinities to EDB (Grabulovski et al., 2007). Furthermore, genetically fused homo-dimers of D3 were cloned, expressed and analysed for their capability to target tumoural neo-vasculature in vivo. In addition, we could demonstrate that neither Fyn SH3 WT nor the D3 mutant was immunogenic in mice after repeated intravenous injections (Grabulovski et al., 2007).
In another study, Bertschinger et al. successfully used Covalent DNA Display technology (Bertschinger and Neri, 2004) to isolate Fynomers binding to mouse serum albumin (Bertschinger et al., 2007). More recently, we have shown the construction of a novel, large Fyn SH3 library comprising more than 8 × 1010 individual clone members (Advancing Protein Therapeutics Conference, 2010, Frankfurt, Germany). In addition, a large collection of formats (homo-dimers, homo-trimers, bispecific dimers, bivalent and tetravalent Fc fusions) and sub-nanomolar Fynomer derivatives binding to human IL-17A have been presented. Overall, the single-pot Fyn SH3 library may provide useful reagents for many biochemical and biomedical applications as an alternative to more conventional IgG-based immunochemical technologies.
Affibodies: Z-domain of staphylococcal protein A
The immunoglobulin binding domain of staphylococcal protein A (SPA) is widely used as an immunochemical ligand for the purification or detection of antibodies and serves as affinity tag in fusion proteins (Uhlen et al., 1992). The IgG binding domain of SPA consists of five highly homologous three-helix bundle domains of approximately 58 amino acid residues each. Because SPA is known as a non-cysteine containing, highly soluble, proteolytically and thermally stable protein, an engineered version of one of the SPA domains, the so-called Z-domain (Nilsson et al., 1987), was chosen as a scaffold for the development of novel affinity proteins designated as ‘affibodies’ (Figure 13.1E). Utilizing structural data available for the complex between a native SPA domain and the Fc fragment of human IgG1, 13 positions distributed across two helices, located at the surface of the domain and involved in the Fc interaction, were chosen for random mutagenesis, and subsequently, for the creation of phage libraries (Nord et al., 1995). In 1997, from two medium-sized libraries comprising about 4 × 107 individual clones, binding proteins against three model target proteins (Taq DNA polymerase, human insulin, and a human apolipoprotein A-1 variant) were isolated, with binding affinities in the micromolar range (Nord et al., 1997). In the meantime, affibodies have been isolated against a variety of protein targets (e.g. human factor VIII (Nord et al., 2001), human immunoglobulin A (Ronnmark et al., 2002) and CD28 (Sandstrom et al., 2003)). In a more recent paper (Wikman et al., 2004), an affibody ligand binding to human epidermal growth factor receptor 2 (Her2) was isolated with a dissociation constant (KD) of 50 nmol/L. Dimerization of this affibody molecule resulted in improved target binding affinity (KD = 3 nmol/L), and radioiodination of the dimer allowed selective targeting and imaging of Her2-expressing xenografts in vivo (Steffen et al., 2006). Further affinity maturation strategies led to an affibody molecule with a dissociation constant in the range of 20 pmol/L and to better tumour targeting and imaging results in the same tumour mouse model (Orlova et al., 2006). A fully synthetic affibody molecules has also been described, site-specifically and homogenously conjugated with a DOTA (1,4,7,10-tetra-azacyclododecane-N,N’,N’’,N’’’-tetraacetic acid) chelator, produced in a single chemical process by peptide synthesis (Orlova et al., 2007).
Importantly, the first clinical investigation of 68Ga- and 111In-labelled anti Her2 affibody molecule (ABY-002) in three patients with metastatic breast cancer showed that Her2-specific affibody molecules have the potential to visualize Her2-expressing metastatic lesions (Löfblom et al., 2010). High contrast SPECT or PET images were obtained already 2–3 h after injection. Over-expression of Her2 in two metastases was confirmed on biopsy tissue samples using the HercepTest™ indicating that ABY-002 specifically targets Her2 also in humans. Further clinical studies are warranted to assess the sensitivity and specificity of radiolabelled Her2-targeting affibody molecules (Löfblom et al., 2010). For therapeutic applications, concerns about the immunogenic potential of this class of proteins should be seriously considered, owing to its bacterial origin.
Ankyrin repeat proteins (DARPins)
Ankyrin repeat (AR) proteins, first isolated in mammalian erythrocytes, are involved in the targeting, mechanical stabilization and orientation of membrane proteins to specialized compartments within the plasma membrane and endoplasmic reticulum. Natural ankyrin repeat proteins consist of many 33-amino-acid modules, each comprising a β-turn and two anti-parallel α-helices (Sedgwick and Smerdon, 1999). In most known complexes, the β-turn and the first α-helix mediate the interactions with the target, and different numbers of adjacent repeats are involved in binding. The reported target binding affinities of natural AR proteins are in the low nanomolar range (Suzuki et al., 1998). Ankyrin repeat proteins were built and diversified to create a library from which ankyrin variants were selected binding to maltose-binding-protein and two eukaryotic kinases (Binz et al., 2004; Amstutz et al., 2005; Zahnd et al., 2006). In the approach chosen by the authors, a consensus ankyrin repeat module consisting of six diversified potential interaction residues and 27 framework residues was designed based on sequence alignments and structural analyses (Figure 13.1F). Varying numbers of this repeat module were cloned between capping repeats, which are special terminal repeats of ankyrin domains shielding the hydrophobic core. Two libraries were created with two and three, respectively, randomized ankyrin repeats in between an N-terminal and a C-terminal cap.
Using a library with more than 1010 individual members in combination with the ribosome display selection methodology, binding variants (termed DARPins) were selected by performing four or five rounds of selection. Dissociation constants were determined by surface plasmon resonance and found to be in the range of 2–20 nM against these model target proteins. More recently, a so-called SRP phage display methodology was employed for efficient filamentous phage display of DARPins. Using a phage DARPin library with more than 1010 individual members resulted in isolation of well-behaved and highly specific DARPins against a broad range of target proteins (such as the Fc domain of human IgG, TNFalpha, ErbB1 (EGFR), ErbB2 (Her2) and ErbB4 (Her4)) having affinities as low as 100 pM directly from this library, without affinity maturation (Steiner et al., 2008).
In April 2010, the first cohorts of patients were enrolled in two separate Phase I trials with the first DARPin (MP0112) neutralizing VEGF for a single intravitreal injection in wet age-related macular degeneration (wet AMD) and diabetic macular edema (DME) (www.molecularpartners.ch). The trials are investigating the safety and tolerability of MP0112, but the studies will also allow a preliminary assessment of efficacy and the duration of action of MP0112. The DARPin was engineered to have a long ocular half-life, but fast systemic clearance. In general, as the DARPins were newly designed and are not found in nature in this format, their immunogenic potential should be investigated in detail, especially after repeated systemic administration.
Lipocalins
The lipocalins represent a family of functionally diverse, small proteins that comprise 160–180 amino acid residues and have weak sequence homology, but high similarity at the tertiary structural level (Skerra, 2000). Members of this family have important biological functions in a variety of organisms, from bacteria to humans. The majority of lipocalins are responsible for the storage and transport of compounds that have low solubility or are chemically sensitive, such as vitamins, steroids and metabolic products (Flower, 1995). An example of human lipocalin is the retinol binding protein (RBP), of which the three-dimensional-structure has been elucidated by X-ray cristallography (Cowan et al., 1990). RBP transports the poorly soluble and oxidation-prone vitamin A from the liver, where it is stored as a fatty acid ester, to several target tissues. Despite their extremely poor sequence homology, the lipocalins share a structurally conserved β-barrel as their central folding motif, which consists of eight antiparallel β-strands that are arranged in a cylindrical manner (Figure 13.1G). The binding specificity for low-molecular-weight compounds is well-characterized for many lipocalins: some bind their ligands with high specificity, whereas others form complexes with a considerable range of lipophilic molecules (Schlehuber and Skerra, 2005). Typically, the ligand affinities of lipocalins are moderate with dissociation constants of approximately 1 µM, which is consistent with their presumed function as a physiological buffer for the bound substance; however, there are notable exceptions, e.g. the tick histamine-binding protein (HBP)-2 with a KD of 1.7 nM (Paesen et al., 1999). The insect bilin binding protein (BBP) from Pieris brassicae served as a model lipocalin in initial studies to create artificial binding sites for several ligands. Sixteen amino acid positions located within the four loops at the open end of the β-barrel and adjoining regions of the β-strands were subjected to targeted random mutagenesis (Beste et al., 1999). The resulting molecular random library with about 4 × 108 members was subjected to selection towards several low-molecular-weight compounds using phage display. BBP variants, so-called ‘anticalins’, that specifically recognize fluorescein, digoxigenin, phthalic acid esters and doxorubicin were obtained, with affinities in the nM range (Beste et al., 1999; Schlehuber et al., 2000; Mercader and Skerra, 2002). In order to extend the concept of anticalins, several human lipocalins were subjected to random mutagenesis generating libraries that enable recognition of macromolecular protein targets. Because proteins have larger molecular dimensions than small compounds, they cannot penetrate into the ligand-binding pocket of lipocalins. Consequently, side chains of lipocalins at more exposed positions, close to the tips of the four loops at the open end of the β-barrel, were subjected to random mutagenesis. Following this strategy, a panel of anticalins based on human lipocalins with specificities for several therapeutic targets such as cytotoxic T-lymphocyte-associated antigen (CTLA-4), c-met, IL-4Ra, VEGF and other undisclosed targets were isolated (www.pieris-ag.com). The VEGF binding anticalin (PRS-050) is about to enter clinical trials and we will learn more about this scaffold in the near future.
Single domain antibodies
Although not belonging to the strict definition of antibody alternative protein ‘frameworks’, single domain antibody fragments are included as well in this chapter since they share a lot of common properties with the ‘classical’ scaffolds (e.g. small domains with high expression yields in bacteria). Moreover, they represent a valuable source for the generation of new binding proteins.
In a seminal early publication (Ward et al., 1989), mouse single variable domains (VH) were shown to be functional, and it was proposed that they could potentially target cryptic epitopes normally hidden for whole antibodies and even for smaller fragments thereof. However, they were described to be poorly soluble and often prone to aggregation, because of their exposed hydrophobic surface normally ‘capped’ by the VL domain. Interest was recently revived when it was discovered that at least two types of organisms, the camelids and cartilaginous fish have evolved high-affinity V-like single domains, mounted on an Fc-equivalent constant domain framework as an integral and crucial component of their immune system (De Genst et al., 2004; Dooley and Flajnik, 2005).
Unlike mouse VH domains (Ward et al., 1989), camelid VhH (termed nanobodies) and shark V-NAR domains are in general soluble and can be produced as stable in vitro reagents. However, for in vivo administration, humanization (or deimmunization) may be crucial to reduce immunogenicity. Human single domain antibodies (dAbs) (Figure 13.1H) would be even more preferable, and the problems of poor stability and solubility have been solved for some human V domains by the identification and design of mutations that minimize the hydrophobic VH/VL interface (Dottorini et al., 2004; Jespers et al., 2004a). Moreover, in a publication of Jespers and co-workers (2004b), aggregation-resistant domain antibodies could be directly selected on phage by heat denaturation. Starting from a DP47d domain antibody (a typical human VH dAb), which unfolds irreversibly and forms aggregates if heated above 55°C, a repertoire containing approximately 1 billion different mutants was cloned by diversification of the CDR loops. The library was multivalently displayed on phage and after three rounds of heat denaturation followed by selection on protein A (a ligand common to folded dAbs), 179 out of 200 colonies secreted dAb phage that retained more than 80% of protein A-binding activity after heating (in contrast to the starting protein DP47d, which loses binding by a factor of 560 after heating). Twenty clones were sequenced and revealed many unique dAb sequences with a large variability in the CDR length and sequences, which shows that mutations located only in the CDR loops of the dAbs are sufficient to confer resistance to aggregation. Interestingly, the TM of the mutants was not higher than the one of the parental clone DP47d, which shows that the selected dAbs are not heat stable, but can fold reversibly.
Most data in the public domain describe serum albumin-binding domain antibodies (AlbudAbs) fusions, which extend the serum half-life of otherwise short-lived proteins, such as the interleukin-1 receptor antagonist IL-1ra (Holt et al., 2008) and interferon (IFN)-α2b (Walker et al., 2010). In the latter study, IFN-α2b was fused to both human serum albumin (termed HSA-IFN-α2b) and to an AlbuDab (termed IFN-α2b-DOM7h-14). The fusion proteins were subjected to a pharmacokinetic (PK) analysis in rats following intravenous dosing. The in vivo half-life of both fusion proteins was significantly increased in comparison with the IFN-α standard (22.6 and 14.2 h for IFN-α2b-DOM7h-14 and HSA-IFN-α2b, respectively compared with 1.2 h for IFN-α standard). The t1/2 of IFN-α2b-DOM7h-14, interestingly, is approximately 1.5 times longer than that of HSA-IFN-α2b. Moreover, similar AUC values were obtained for IFN-α2b-DOM7h-14 and HSA-IFN-α2b, 737.5 and 689.2 h mg/mL, respectively, which in both cases represents a significant increase over the value of 18.8 h mg/mL observed with the IFN-α standard. A further study was carried out to determine PKs following subcutaneous administration in rats. As with the i.v. study the in vivo half-life of both fusion proteins was increased in comparison with IFN-α standard. Interestingly, the antiviral efficacy of the AlbuDab fusion IFN-α2b-DOM7h-14 was 5.8 fold greater in comparison with albumin fusion HSA-IFN-α2b, as determined by an in vitro antiviral assay with A549 human lung carcinoma cells, indicating that in this particular case fusion of IFN-α to the Albudab represents an attractive avenue to prolong its half-life.
Today, the single domain antibody technology is commercialized by Glaxo Smith Kline, which acquired Domantis Ltd. in December 2006 for £230 million, and certainly reflects the interest of big pharmaceutical companies in single domain protein scaffolds.
Other domains
The preceding sections introduced important domain scaffolds that have been commercialized and for which published data are available. A recent review by Gebauer and Skerra (2009) listed more than 50 scaffolds that have been described for the generation of new binding proteins. The majority of them have been used for research purposes only (e.g. probing the specificity determinants of WW-domains to their ligands (Dalby et al., 2000), elucidating target recognition rules of SH3 domains (Hiipakka et al., 1999; Panni et al., 2002) or identifying signal transduction pathways with the staphylococcal nuclease as scaffold (Norman et al., 1999)). Other domains have been commercialized by companies for in vitro applications (e.g. PDZ domains for the generation of high-affinity detection reagents (Biotech Studio; Ferrer et al., 2005) or for intracellular signalling interference applications (thioredoxin, Aptanomics; Kunz et al., 2006)). Antibody fragments of the constant domain have also been used as scaffolds, e.g. the CH2 domain of human IgG1 has been used as a library scaffold, and isolated CH2 domains with specific binding affinity to a HIV-1 gp120-CD4 complex have been described (Xiao et al., 2009). There, the new binding site is formed by engineered sequences in the BC and FG loops of the CH2 domain located at the N-terminal tip of the domain. Another approach was chosen by Wozniak-Knopp and co-workers (2010), where the whole Fc fragment of IgG1 was used as an alternative small-size antibody format. With the exception of the antigen binding site, the full Fc of an IgG1 has all the properties of a complete antibody, i.e. the ability to elicit effector functions via binding to Fc-gamma receptors and to the complement activator C1q, as well as the long half-life of antibodies mediated through binding to FcRn. Using their CH3 library and yeast display selection technologies these authors described recently the isolation of a Her2 binding Fc mutant with low nM affinities (Wozniak-Knopp et al., 2010).
A few other scaffolds have also been commercialized by small- and medium-sized companies; however, no refereed publications are available (ubiquitin by Scil Proteins, C-type lectin domain by Anaphore, and a not disclosed scaffold by Amunix; information obtained from the corresponding websites).
Concluding remarks
Overall, the engineering and practical uses of binding proteins derived from non-Ig scaffolds are established and these will push biological chemistry toward both basic research and applied science. Although some rational justification may be given for the choice of almost any scaffold described so far, the true potential of the various protein scaffolds for human therapy, including in vivo diagnostics, and considering the different approaches to the implementation of effector functions, will only become clear once a number of additional Phase I/II trials have been completed in the near future.
Amstutz P, Binz HK, Parizek P, et al. Intracellular kinase inhibitors selected from combinatorial libraries of designed ankyrin repeat proteins. Journal of Biological Chemistry. 2005;280:24715–24722.
Ascenzi P, Bocedi A, Bolognesi M, et al. The bovine basic pancreatic trypsin inhibitor (Kunitz inhibitor): a milestone protein. Current Protein & Peptide Science. 2003;4:231–251.
Bertschinger J, Neri D. Covalent DNA display as a novel tool for directed evolution of proteins in vitro. Protein Engineering, Design & Selection. 2004;17:699–707.
Bertschinger J, Grabulovski D, Neri D. Selection of single domain binding proteins by covalent DNA display. Protein Engineering, Design & Selection. 2007;20:57–68.
Beste G, Schmidt FS, Stibora T, et al. Small antibody-like proteins with prescribed ligand specificities derived from the lipocalin fold. Proceeding of the National Academy of Sciences of USA. 1999;96:1898–1903.
Binz K, Pluckthun A. Engineered proteins as specific binding reagents. Current Opinion in Biotechnology. 2005;16:459–469.
Binz HK, Amstutz P, Kohl A, et al. High-affinity binders selected from designed ankyrin repeat protein libraries. Nature Biotechnology. 2004;22:575–582.
Binz HK, Amstutz P, Plückthun A. Engineering novel binding proteins from nonimmunoglobulin domains. Nature Biotechnology. 2005;23:1257–1268.
Bloom L, Calabro V. FN3: a new protein scaffold reaches the clinic. Drug Discovery Today. 2009;14:949–955.
Castellani P, Viale G, Dorcaratto A, et al. The fibronectin isoform containing the ED-B oncofetal domain: a marker of angiogenesis. International Journal of Cancer. 1994;59:612–618.
Chan B, Lanyi A, Song HK, et al. SAP couples Fyn to SLAM immune receptors. Nature Cell Biology. 2003;5:155–160.
Choy EH, Hazleman B, Smith M, et al. Efficacy of a novel PEGylated humanized anti-TNF fragment (CDP870) in patients with rheumatoid arthritis: a phase II double-blinded, randomized, dose-escalating trial. Rheumatology (Oxford). 2002;41:1133–1137.
Cooke MP, Perlmutter RM. Expression of a novel form of the fyn proto-oncogene in hematopoietic cells. New Biology. 1989;1:66–74.
Cowan SW, Newcomer ME, Jones TA. Crystallographic refinement of human serum retinol binding protein at 2A resolution. Proteins. 1990;8:44–61.
Dalby PA, Hoess RH, DeGrado WF. Evolution of binding affinity in a WW domain probed by phage display. Protein Science. 2000;9:2366–2376.
De Genst E, Handelberg F, Van Meirhaeghe A, et al. Chemical basis for the affinity maturation of a camel single domain antibody. Journal of Biological Chemistry. 2004;279:53593–53601.
Delacourt C, Hérigault S, Delclaux C, et al. Protection against acute lung injury by intravenous or intratracheal pretreatment with EPI-HNE-4, a new potent neutrophil elastase inhibitor. American Journal of Respiratory Cell Molecular Biology. 2002;26:290–297.
Dooley H, Flajnik MF. Shark immunity bites back: affinity maturation and memory response in the nurse shark, Ginglymostoma cirratum. European Journal of Immunology. 2005;35:936–945.
Dottorini T, Vaughan CK, Walsh MA, et al. Crystal structure of a human VH: requirements for maintaining a monomeric fragment. Biochemistry. 2004;43:622–628.
Dyax Corp. (Ley AC, L. R, Guterman SK, Roberts Bruce L, Markland W, Ken RB) Engineered human-derived kunitz domains that inhibit human neutrophil elastase. US056633143 patent. 1997.
Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Review. Cancer. 2002;2:161–174.
Ferrer M, Maiolo J, Kratz P, et al. Directed evolution of PDZ variants to generate high-affinity detection reagents. Protein Engineering, Design & Selection. 2005;18:165–173.
Filimonov VV, Azuaga AI, Viguera AR, et al. A thermodynamic analysis of a family of small globular proteins: SH3 domains. Biophysical Chemistry. 1999;77:195–208.
Flower DR. Multiple molecular recognition properties of the lipocalin protein family. Journal of Molecular Recognition. 1995;8:185–195.
Gebauer M, Skerra A. Engineered protein scaffolds as next-generation antibody therapeutics. Current Opinion in Chemical Biology. 2009;13:245–255.
Getmanova EV, Chen Y, Bloom L, et al. Antagonists to human and mouse vascular endothelial growth factor receptor 2 generated by directed protein evolution in vitro. Chemical Biology. 2006;13:549–556.
Gliemann J. Receptors of the low density lipoprotein (LDL) receptor family in man. Multiple functions of the large family members via interaction with complex ligands. Biological Chemistry. 1998;379:951–964.
Grabulovski D, Kaspar M, Neri D. A novel, non-immunogenic Fyn SH3-derived binding protein with tumor vascular targeting properties. Journal of Biological Chemistry. 2007;282:3196–3204.
Grimbert D, Vecellio L, Delépine P, et al. Characteristics of EPI-hNE4 aerosol: a new elastase inhibitor for treatment of cystic fibrosis. Journal of Aerosol Medicine. 2003;16:121–129.
Han ED, MacFarlane RC, Mulligan AN, et al. Increased vascular permeability in C1 inhibitor-deficient mice mediated by the bradykinin type 2 receptor. Journal of Clinical Investment. 2002;109:1057–1063.
Hautamaki RD, Kobayashi DK, Senior RM, et al. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277:2002–2004.
Hey T, Fiedler E, Rudolph R, et al. Artificial, non-antibody binding proteins for pharmaceutical and industrial applications. Trends in Biotechnology. 2005;23:514–522.
Hiipakka M, Poikonen K, Saksela K. SH3 domains with high affinity and engineered ligand specificities targeted to HIV-1 Nef. Journal of Molecular Biology. 1999;293:1097–1106.
Hogbom M, Eklund M, Nygren PA, et al. Structural basis for recognition by an in vitro evolved affibody. Proceeding of the National Academy of Sciences of USA. 2003;100:3191–3196.
Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nature Biotechnology. 2005;23:1126–1136.
Holt LJ, Basran A, Jones K, et al. Anti-serum albumin domain antibodies for extending the half-lives of short lived drugs. Protein Engineering, Design & Selection. 2008;21:283–288.
Huang W, Dolmer K, Gettins PG. NMR solution structure of complement-like repeat CR8 from the low density lipoprotein receptor-related protein. Journal of Biological Chemistry. 1999;274:14130–14136.
Jespers L, Schon O, Famm K, et al. Aggregation-resistant domain antibodies selected on phage by heat denaturation. Nature Biotechnology. 2004;22:1161–1165.
Jespers L, Schon O, James LC, et al. Crystal structure of HEL4, a soluble, refoldable human V(H) single domain with a germ-line scaffold. Journal of Molecular Biology. 2004;337:893–903.
Kaspar M, Zardi L, Neri D. Fibronectin as target for tumor therapy. International Journal of Cancer. 2006;118:1331–1339.
Kawakami T, Pennington CY, Robbins KC. Isolation and oncogenic potential of a novel human src-like gene. Molecular Cell Biology. 1986;6:4195–4201.
Koduri V, Blacklow SC. Folding determinants of LDL receptor type A modules. Biochemistry. 2001;40:12801–12807.
Koide A, Koide S. The fibronectin type III domain as a scaffold for novel binding proteins. Journal of Molecular Biology. 1998;284:1141–1151.
Krieger M, Herz J. Structures and functions of multiligand lipoprotein receptors: macrophage scavenger receptors and LDL receptor-related protein (LRP). Annual Reviews in Biochemstry. 1994;63:601–637.
Kunz C, Borghouts C, Buerger C, et al. Peptide aptamers with binding specificity for the intracellular domain of the ErbB2 receptor interfere with AKT signaling and sensitize breast cancer cells to Taxol. Molecular Cancer Researchearch. 2006;4:983–998.
Laval SH, Bushby KM. Limb-girdle muscular dystrophies – from genetics to molecular pathology. Neuropathology and Applied Neurobiology. 2004;30:91–105.
Ley AC, LR, Guterman SK, Roberts Bruce L, et al. US056633143 patent 1997.
Ley AC, Markland W, Ladner RC. Obtaining a family of high-affinity, high-specificity protein inhibitors of plasmin and plasma kallikrein. Molecular Divers. 1996;2:119–124.
Li SS. Specificity and versatility of SH3 and other proline-recognition domains: structural basis and implications for cellular signal transduction. Biochemical Journal. 2005;390:641–653.
Löfblom J, Feldwisch J, Tolmachev V, et al. Affibody molecules: engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Letters. Epub ahead of print 11 April 2010.
Markland W, Ley AC, Ladner RC. Iterative optimization of high-affinity protease inhibitors using phage display. 2. Plasma kallikrein and thrombin. Biochemistry. 1996;35:8058–8067.
Mercader JV, Skerra A. Generation of anticalins with specificity for a nonsymmetric phthalic acid ester. Annals of Biochemstry. 2002;308:269–277.
Molckovsky A, Siu LL. First-in-class, first-in-human phase I results of targeted agents: highlights of the 2008 American Society of Clinical Oncology meeting. Journal of Hematology and Oncology. 2008;1:20.
Morton CJ, Pugh DJ, Brown EL, et al. Solution structure and peptide binding of the SH3 domain from human Fyn. Structure. 1996;4:705–714.
Naruse S, Kitagawa M, Ishiguro H. Molecular understanding of chronic pancreatitis: a perspective on the future. Molecular Medicine Today. 1999;5:493–499.
Nilsson B, Moks T, Jansson B, et al. A synthetic IgG-binding domain based on staphylococcal protein A. Protein Engineering. 1987;1:107–113.
Nixon AE, Wood CR. Engineered protein inhibitors of proteases. Current Opinion in Drug Discovery and Development. 2006;9:261–268.
Nord K, Nilsson J, Nilsson B, et al. A combinatorial library of an alpha-helical bacterial receptor domain. Protein Engineering. 1995;8:601–608.
Nord K, Gunneriusson E, Ringdahl J, et al. Binding proteins selected from combinatorial libraries of an alpha-helical bacterial receptor domain. Nature Biotechnology. 1997;15:772–777.
Nord K, Nord O, Uhlén M, et al. Recombinant human factor VIII-specific affinity ligands selected from phage-displayed combinatorial libraries of protein A. European Journal of Biochemistry. 2001;268:4269–4277.
Norman TC, Smith DL, Sorger PK, et al. Genetic selection of peptide inhibitors of biological pathways. Science. 1999;285:591–595.
North CL, Blacklow SC. Structural independence of ligand-binding modules five and six of the LDL receptor. Biochemistry. 1999;38:3926–3935.
Nunan J, Small DH. Proteolytic processing of the amyloid-beta protein precursor of Alzheimer’s disease. Essays in Biochemistry. 2002;38:37–49.
Nygren PA, Uhlen M. Scaffolds for engineering novel binding sites in proteins. Current Opinion in Structural Biology. 1997;7:463–469.
Orlova A, Magnusson M, Eriksson TL, et al. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Research. 2006;66:4339–4348.
Orlova A, Tolmachev V, Pehrson R, et al. Synthetic affibody molecules: a novel class of affinity ligands for molecular imaging of HER2-expressing malignant tumors. Cancer Research. 2007;67:2178–2186.
Paesen GC, Adams PL, Harlos K, et al. Tick histamine-binding proteins: isolation, cloning, and three-dimensional structure. Molecular Cell. 1999;3:661–671.
Panni S, Dente L, Cesareni G. In vitro evolution of recognition specificity mediated by SH3 domains reveals target recognition rules. Journal of. Biological Chemistry. 2002;277:21666–21674.
Parker MH, Chen Y, Danehy F, et al. Antibody mimics based on human fibronectin type three domain engineered for thermostability and high-affinity binding to vascular endothelial growth factor receptor two. Protein Engineering, Design & Selection. 2005;18:435–444.
Parks WC, Wilson CL, López-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nature Review. Immunology. 2004;4:617–629.
Reichert JM. Editorial for PEDS special issue on antibodies. Protein Engineering, Design & Selection. 2010;23:153–154.
Resh MD. Fyn, a Src family tyrosine kinase. International Journal of Biochemistry and Cell Biology. 1998;30:1159–1162.
Richards J, Miller M, Abend J, et al. Engineered fibronectin type III domain with a RGDWXE sequence binds with enhanced affinity and specificity to human alphavbeta3 integrin. Journal of Molecular Biology. 2003;326:1475–1488.
Roberts BL, Markland W, Ley AC, et al. Directed evolution of a protein: selection of potent neutrophil elastase inhibitors displayed on M13 fusion phage. Proceeding of the National Academy of Sciences of USA. 1992;89:2429–2433.
Ronnmark J, Grönlund H, Uhlén M, et al. Human immunoglobulin A (IgA)-specific ligands from combinatorial engineering of protein A. European Journal of Biochemistry. 2002;269:2647–2655.
Saerens D, Ghassabeh GH, Muyldermans S. Single-domain antibodies as building blocks for novel therapeutics. Current Opinion in Pharmacology. 2008;8:600–608.
Sandstrom K, Xu Z, Forsberg G, et al. Inhibition of the CD28–CD80 co-stimulation signal by a CD28-binding affibody ligand developed by combinatorial protein engineering. Protein Engineering. 2003;16:691–697.
Saudubray F, Labbé A, Durieu I. Phase IIa clinical study of a new human neutrophil elastase inhibitor (HNE), EPI-HNE4 (DX-890), with repeated administration by inhalation in adult cystic fibrosis patients. Journal of Cystic Fibrosis. 2003;2:A85.
Schlehuber S, Beste G, Skerra A. A novel type of receptor protein, based on the lipocalin scaffold, with specificity for digoxigenin. Journal of Molecular Biology. 2000;297:1105–1120.
Schlehuber S, Skerra A. Lipocalins in drug discovery: from natural ligand-binding proteins to anticalins. Drug Discovery Today. 2005;10:23–33.
Sedgwick SG, Smerdon SJ. The ankyrin repeat: a diversity of interactions on a common structural framework. Trends in Biochemical Science. 1999;24:311–316.
Semba K, Nishizawa M, Miyajima N, et al. yes-related protooncogene, syn, belongs to the protein-tyrosine kinase family. Proceeding of the National Academy of Sciences of USA. 1986;83:5459–5463.
Shapiro SD. Proteinases in chronic obstructive pulmonary disease. Biochemical Society Transactions. 2002;30:98–102.
Silverman J, Liu Q, Bakker A, et al. Multivalent avimer proteins evolved by exon shuffling of a family of human receptor domains. Nature Biotechnology. 2005;23:1556–1561.
Skerra A. Lipocalins as a scaffold. Biochimica et Biophysica Acta. 2000;1482:337–350.
Smith G. Patch engineering: a general approach for creating proteins that have new binding activities. Trends in Biochemical Science. 1998;23:457–460.
Steffen AC, Orlova A, Wikman M, et al. Affibody-mediated tumour targeting of HER-2 expressing xenografts in mice. European Journal of Nuclear Medicine and Molecular Imaging. 2006;33:631–638.
Steiner D, Forrer P, Plückthun A. Efficient selection of DARPins with sub-nanomolar affinities using SRP phage display. Journal of Molecular Biology. 2008;382:1211–1227.
Suzuki F, Goto M, Sawa C, et al. Functional interactions of transcription factor human GA-binding protein subunits. Journal of. Biological Chemistry. 1998;273:29302–29308.
Tomlinson IM. Next-generation protein drugs. Nature Biotechnology. 2004;22:521–522.
Uhlen M, Forsberg G, Moks T, et al. Fusion proteins in biotechnology. Current Opinion in Biotechnology. 1992;3:363–369.
Walker A, Dunlevy G, Rycroft D, et al. Anti-serum albumin domain antibodies in the development of highly potent, efficacious and long-acting interferon. Protein Engineering, Design & Selection. 2010;23:271–278.
Ward ES, Güssow D, Griffiths AD, et al. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli. Nature. 1989;341:544–546.
Wikman M, Steffen AC, Gunneriusson E, et al. Selection and characterization of HER2/neu-binding affibody ligands. Protein Engineering, Design & Selection. 2004;17:455–462.
Williams A, Baird LG. DX-88 and HAE: a developmental perspective. Transfusion and Apheresis Science. 2003;29:255–258.
Wilson DS, Keefe AD, Szostak JW. The use of mRNA display to select high-affinity protein-binding peptides. Proceedings of the National Academy of Sciences of USA. 2001;98:3750–3755.
Wozniak-Knopp G, Bartl S, Bauer A, et al. Introducing antigen-binding sites in structural loops of immunoglobulin constant domains: Fc fragments with engineered HER2/neu-binding sites and antibody properties. Protein Engineering, Design & Selection. 2010;23:289–297.
Xiao X, Feng Y, Vu BK, et al. A large library based on a novel (CH2) scaffold: identification of HIV-1 inhibitors. Biochemical and Biophysical Research Communications. 2009;387:387–392.
Xu L, Aha P, Gu K, et al. Directed evolution of high-affinity antibody mimics using mRNA display. Chemical Biology. 2002;9:933–942.
Zahnd C, Pecorari F, Straumann N, et al. Selection and characterization of Her2 binding-designed ankyrin repeat proteins. Journal of Biological Chemistry. 2006;281:35167–35175.
Zardi L, Carnemolla B, Siri A, et al. Transformed human cells produce a new fibronectin isoform by preferential alternative splicing of a previously unobserved exon. The EMBO Journal. 1987;6:2337–2342.