Chapter 10 Inflammation
Inflammation is the local physiological response to tissue injury. It is not, in itself, a disease, but is usually a manifestation of disease. Inflammation may have beneficial effects, such as the destruction of invading micro-organisms and the walling-off of an abscess cavity, thus preventing spread of infection. Equally, it may produce disease; for example, an abscess in the brain would act as a space-occupying lesion compressing vital surrounding structures, or fibrosis resulting from chronic inflammation may distort the tissues and permanently alter their function.
Inflammation is usually classified according to its time course as:
The two main types of inflammation are also characterised by differences in the cell types taking part in the inflammatory response.
ACUTE INFLAMMATION

Acute inflammation is the initial tissue reaction to a wide range of injurious agents; it may last from a few hours to a few days. The process is usually described by the suffix ‘-itis’, preceded by the name of the organ or tissues involved. Thus, acute inflammation of the meninges is called meningitis. The acute inflammatory response is similar whatever the causative agent.
Causes of acute inflammation
The principal causes of acute inflammation are:
Microbial infections
One of the commonest causes of inflammation is microbial infection. Viruses lead to death of individual cells by intracellular multiplication. Bacteria release specific exotoxins—chemicals synthesised by them that specifically initiateinflammation—or endotoxins, which are associated with their cell walls. Additionally, some organisms cause immunologically mediated inflammation through hyper-sensitivity reactions (Ch. 9). Parasitic infections and tuberculous inflammation are instances where hypersensitivity is important.
Hypersensitivity reactions
A hypersensitivity reaction occurs when an altered state of immunological responsiveness causes an inappropriate or excessive immune reaction that damages the tissues. The types of reaction are classified in Chapter 9 but all have cellular or chemical mediators similar to those involved in inflammation.
Physical agents
Tissue damage leading to inflammation may occur through physical trauma, ultraviolet or other ionising radiation, burns or excessive cooling (‘frostbite’).
Essential macroscopic appearances of acute inflammation
The essential physical characteristics of acute inflammation were formulated by Celsus (30bc–ad38) using the Latin words rubor, calor, tumor and dolor. Loss of function is also characteristic.
Redness (rubor)
An acutely inflamed tissue appears red, for example skin affected by sunburn, cellulitis due to bacterial infection or acute conjunctivitis. This is due to dilatation of small blood vessels within the damaged area (Fig. 10.1).
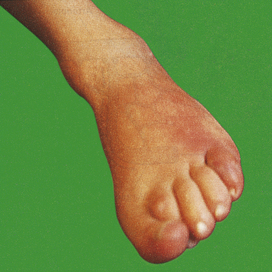
Heat (calor)
Increase in temperature is seen only in peripheral parts of the body, such as the skin. It is due to increased blood flow (hyperaemia) through the region, resulting in vascular dilatation and the delivery of warm blood to the area. Systemic fever, which results from some of the chemical mediators of inflammation, also contributes to the local temperature.
Swelling (tumor)
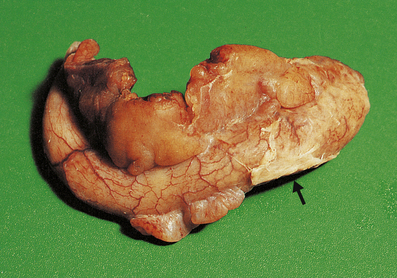
Swelling results from oedema—the accumulation of fluid in the extravascular space as part of the fluid exudate—and, to a much lesser extent, from the physical mass of the inflammatory cells migrating into the area (Fig. 10.2). As the inflammation response progresses, formation of new connective tissue contributes to the swelling.
Pain (dolor)
For the patient, pain is one of the best-known features of acute inflammation. It results partly from the stretching and distortion of tissues due to inflammatory oedema and, in particular, from pus under pressure in an abscess cavity. Some of the chemical mediators of acute inflammation, including bradykinin, the prostaglandins and serotonin, are known to induce pain.
Early stages of acute inflammation
In the early stages, oedema fluid, fibrin and neutrophil polymorphs accumulate in the extracellular spaces of the damaged tissue. The presence of the cellular component, the neutrophil polymorph, is essential for a histological diagnosis of acute inflammation. The acute inflammatory response involves three processes:
Changes in vessel calibre
The microcirculation consists of the network of small capillaries lying between arterioles, which have a thick muscular wall, and thin-walled venules. Capillaries have no smooth muscle in their walls to control their calibre, and are so narrow that red blood cells must past through them in single file. The smooth muscle of arteriolar walls forms precapillary sphincters which regulate blood flow through the capillary bed. Flow through the capillaries is intermittent, and some form preferential channels for flow while others are usually shut down (Fig. 10.3).
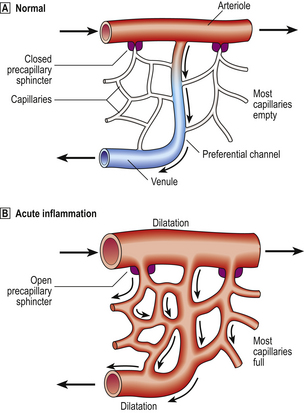
Fig. 10.3 Vascular dilatation in acute inflammation.  Normally, most of the capillary bed is closed down by precapillary sphincters.
Normally, most of the capillary bed is closed down by precapillary sphincters.  In acute inflammation, the sphincters open, causing blood to flow through all capillaries.
In acute inflammation, the sphincters open, causing blood to flow through all capillaries.
In blood vessels larger than capillaries, blood cells flow mainly in the centre of the lumen (axial flow), while the area near the vessel wall carries only plasma (plasmatic zone). This feature of normal blood flow keeps blood cells away from the vessel wall.
Changes in the microcirculation occur as a physiological response; for example, there is hyperaemia in exercising muscle and active endocrine glands. The changes following injury that make up the vascular component of the acute inflammatory reaction were described by Lewis in 1927 as ‘the triple response to injury’: a flush, a flare and a wheal. If a blunt instrument is drawn firmly across the skin, the following sequential changes take place:
The initial phase of arteriolar constriction is transient, and probably of little importance in acute inflammation. The subsequent phase of vasodilatation (active hyperaemia, in contrast to passive hyperaemia due to vascular distension from abnormally high venous pressure) may last from 15 minutes to several hours, depending upon the severity of the injury. There is experimental evidence that blood flow to the injured area may increase up to 10-fold.
As blood flow begins to slow again, blood cells begin to flow nearer to the vessel wall, in the plasmatic zone rather than the axial stream. This allows ‘pavementing’ of leukocytes (their adhesion to the vascular epithelium) to occur, which is the first step in leukocyte emigration into the extravascular space.
The slowing of blood flow that follows the phase of hyperaemia is due to increased vascular permeability, allowing plasma to escape into the tissues while blood cells are retained within the vessels. The blood viscosity is therefore increased.
Increased vascular permeability
Small blood vessels are lined by a single layer of endothelial cells. In some tissues, these form a complete layer of uniform thickness around the vessel wall, while in other tissues there are areas of endothelial cell thinning, known as fenestrations. The walls of small blood vessels act as a microfilter, allowing the passage of water and solutes but blocking that of large molecules and cells. Oxygen, carbon dioxide and some nutrients transfer across the wall by diffusion, but the main transfer of fluid and solutes is by ultrafiltration, as described by Starling. The high colloid osmotic pressure inside the vessel, due to plasma proteins, favours fluid return to the vascular compartment. Under normal circumstances, high hydrostatic pressure at the arteriolar end of capillaries forces fluid out into the extravascular space, but this fluid returns into the capillaries at their venous end, where hydrostatic pressure is low (Fig. 10.4). In acute inflammation, however, not only is capillary hydrostatic pressure increased, but there is also escape of plasma proteins into the extravascular space, increasing the colloid osmotic pressure there. Consequently, much more fluid leaves the vessels than is returned to them. The net escape of protein-rich fluid is called exudation; hence, the fluid is called the fluid exudate.
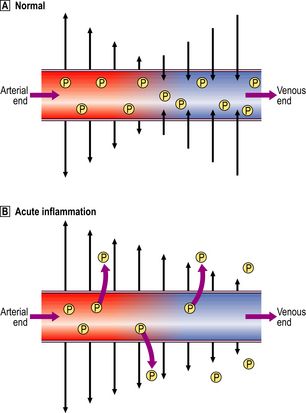
Fig. 10.4 Ultrafiltration of fluid across the small blood vessel wall. Normally, fluid leaving and entering the vessel is in equilibrium. In acute inflammation, there is a net loss of fluid together with plasma protein molecules (P) into the extracellular space, resulting in oedema.
Features of the fluid exudate
The increased vascular permeability means that large molecules, such as proteins, can escape from vessels. Hence, the exudate fluid has a high protein content of up to 50 g/l. The proteins present include immunoglobulins, which may be important in the destruction of invading micro-organisms, and coagulation factors, including fibrinogen, which result in fibrin deposition on contact with the extravascular tissues. Hence, acutely inflamed organ surfaces are commonly covered by fibrin: the fibrinous exudate. There is a considerable turnover of the inflammatory exudate; it is constantly drained away by local lymphatic channels to be replaced by new exudate.
Ultrastructural basis of increased vascular permeability
The ultrastructural basis of increased vascular permeability was originally determined using an experimental model in which histamine, one of the chemical mediators of increased vascular permeability, was injected under the skin. This caused transient leakage of plasma proteins into the extravascular space. Electron microscopic examination of venules and small veins during this period showed that gaps of 0.1–0.4 μm in diameter had appeared between endothelial cells. These gaps allowed the leakage of injected particles, such as carbon, into the tissues. The endothelial cells are not damaged during this process. They contain contractile proteins such as actin, which, when stimulated by the chemical mediators of acute inflammation, cause contraction of the endothelial cells, pulling open the transient pores. The leakage induced by chemical mediators, such as histamine, is confined to venules and small veins. Although fluid is lost by ultrafiltration from capillaries, there is no evidence that they too become more permeable in acute inflammation.
Other causes of increased vascular permeability
In addition to the transient vascular leakage caused by some inflammatory stimuli, certain other stimuli, e.g. heat, cold, ultraviolet light and X-rays, bacterial toxins and corrosive chemicals, cause delayed prolonged leakage. In these circum-stances, there is direct injury to endothelial cells in several types of vessel within the damaged area (Table 10.1).
Table 10.1 Causes of increased vascular permeability
| Time course | Mechanisms |
|---|---|
| Immediate transient | Chemical mediators, e.g. histamine, bradykinin, nitric oxide, C5a, leukotriene B4, platelet activating factor |
| Immediate sustained | Severe direct vascular injury, e.g. trauma |
| Delayed prolonged | Endothelial cell injury, e.g. X-rays, bacterial toxins |
Tissue sensitivity to chemical mediators
The relative importance of chemical mediators and of direct vascular injury in causing increased vascular permeability varies according to the type of tissue. For example, vessels in the central nervous system are relatively insensitive to the chemical mediators, while those in the skin, conjunctiva and bronchial mucosa are exquisitely sensitive to agents such as histamine.
Formation of the cellular exudate
The accumulation of neutrophil polymorphs within the extracellular space is the diagnostic histological feature of acute inflammation. The stages whereby leukocytes reach the tissues are shown in Figure 10.5.
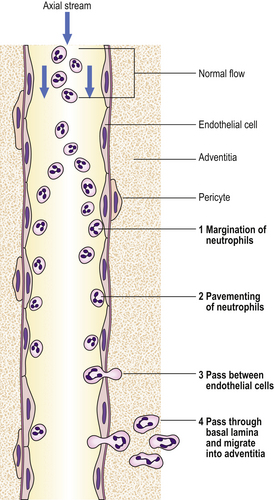
Fig. 10.5 Steps in neutrophil polymorph emigration. Neutrophils (1) marginate into the plasmatic zone; (2) adhere to endothelial cells; (3) pass between endothelial cells; and (4) pass through the basal lamina and migrate into the adventitia.
Margination of neutrophils
In the normal circulation, cells are confined to the central (axial) stream in blood vessels, and do not flow in the periph-eral (plasmatic) zone near to the endothelium. However, loss of intravascular fluid and increase in plasma viscosity with slowing of flow at the site of acute inflammation allow neutrophils to flow in this plasmatic zone.
Adhesion of neutrophils
The adhesion of neutrophils to the vascular endothelium that occurs at sites of acute inflammation is termed ‘pavementing’ of neutrophils. Neutrophils randomly contact the endothelium in normal tissues, but do not adhere to it. However, at sites of injury, pavementing occurs early in the acute inflammatory response and appears to be a specific process occurring independently of the eventual slowing of blood flow. The phenomenon is seen only in venules.
Increased leukocyte adhesion results from interaction between paired adhesion molecules on leukocyte and endothelial surfaces. There are several classes of such adhesion molecules: some of them act as lectins which bind to carbohydrates on the partner cell. Leukocyte surface adhesion molecule expression is increased by:
Endothelial cell expression of selectins, such as endothelial– leukocyte adhesion molecule-1 (ELAM-1), which establishes the first loose contact between leukocytes and endothelium (resulting in ‘rolling’), integrins, and intercellular adhesion molecule-1 (ICAM-1), to which the leukocytes’ surface adhesion molecules bond, is increased by:
In this way, a variety of chemical inflammatory mediators promote leukocyte–endothelial adhesion as a prelude to leukocyte emigration.
Neutrophil emigration
Leukocytes migrate by active amoeboid movement through the walls of venules and small veins, but do not commonly exit from capillaries. Electron microscopy shows that neutrophil and eosinophil polymorphs and macrophages can insert pseudopodia between endothelial cells, migrate through the gap so created between the endothelial cells, and then on through the basal lamina into the vessel wall. The defect appears to be self-sealing, and the endothelial cells are not damaged by this process.
Diapedesis
Red cells may also escape from vessels, but in this case the process is passive and depends on hydrostatic pressure forcing the red cells out. The process is called diapedesis, and the presence of large numbers of red cells in the extravascular space implies severe vascular injury, such as a tear in the vessel wall.
Later stages of acute inflammation
Chemotaxis of neutrophils
It has long been known from in vitro experiments that neutrophil polymorphs are attracted towards certain chemical substances in solution—a process called chemotaxis. Time-lapse cine photography shows apparently purposeful migration of neutrophils along a concentration gradient. Compounds that appear chemotactic for neutrophils in vitro include certain complement components, cytokines and products produced by neutrophils themselves. It is not known whether chemotaxis is important in vivo. Neutrophils may possibly arrive at sites of injury by random movement, and then be trapped there by immobilising factors (a process analogous to the trapping of macrophages at sites of delayed-type hypersensitivity by migration inhibitory factor; Ch. 9).
Chemical mediators of acute inflammation
The spread of the acute inflammatory response following injury to a small area of tissue suggests that chemical substances are released from injured tissues, spreading outwards into uninjured areas. Early in the response, histamine and thrombin released by the original inflammatory stimulus cause upregulation of P-selectin and platelet-activating factor (PAF) on the endothelial cells lining the venules. Adhesion molecules, stored in intracellular vesicles, appear rapidly on the cell surface. Neutrophil polymorphs begin to roll along the endothelial wall due to engagement of the lectin-like domain on the P-selectin molecule with sialyl Lewisx carbohydrate ligands on the neutrophil polymorph surface mucins. This also helps platelet-activating factor to dock with its corresponding receptor which, in turn, increases expression of the integrins’ lymphocyte function-associated molecule 1 (LFA-1) and membrane attack complex 1 (MAC-1). The overall effect of all these molecules is very firm neutrophil adhesion to the endothelial surface. These chemicals, called endogenous chemical mediators, cause:
Chemical mediators released from cells
Histamine.
This is the best-known chemical mediator in acute inflammation. It causes vascular dilatation and the immediate transient phase of increased vascular permeability. The immediate effect is assisted by its storage as preformed granules. In humans, mast cells are the most important source of histamine, but it is also present in basophil and eosinophil leukocytes, and platelets. Histamine release from these sites (for example, mast cell degranulation) is stimulated by complement components C3a and C5a, and by lysosomal proteins released from neutrophils.
Lysosomal compounds.
These are released from neutrophils and include cationic proteins, which may increase vascular permeability, and neutral proteases, which may activate complement.
Prostaglandins.
These are a group of long-chain fatty acids derived from arachidonic acid and synthesised by many cell types. Some prostaglandins potentiate the increase in vascular permeability caused by other compounds. Others include platelet aggregation (prostaglandin I2 is inhibitory while prostaglandin A2 is stimulatory). Part of the anti-inflammatory activity of drugs such as aspirin and the non-steroidal anti-inflammatory drugs is attributable to inhibition of one of the enzymes involved in prostaglandin synthesis.
Leukotrienes.
These are also synthesised from arachidonic acid, especially in neutrophils, and appear to have vasoactive properties. SRS-A (slow-reacting substance of anaphylaxis), involved in type I hypersensitivity (Ch. 9), is a mixture of leukotrienes.
5-Hydroxytryptamine (serotonin).
This is present in high concentration in platelets. It is a potent vasoconstrictor.
Chemokines.
This large family of 8–10-kD proteins selectively attracts various types of leukocytes to the site of inflammation. Some chemokines such as IL-8 are mainly specific for neutrophil polymorphs and to a lesser extent lymphocytes, whereas other types of chemokine are chemotactic for monocytes, natural killer (NK) cells, basophils and eosinophils. The various chemokines bind to extracellular matrix components such as heparin and heparan sulphate glycosaminoglycans, setting up a gradient of chemotactic molecules fixed to the extracellular matrix.
Plasma factors
The plasma contains four enzymatic cascade systems—complement, the kinins, the coagulation factors and the fibrinolytic system—which are inter-related and produce various inflammatory mediators.
Coagulation system.
The coagulation system (Ch. 23) is responsible for the conversion of soluble fibrinogen into fibrin, a major component of the acute inflammatory exudate.
Coagulation factor XII (the Hageman factor), once activated by contact with extracellular materials such as basal lamina, and various proteolytic enzymes of bacterial origin, can activate the coagulation, kinin and fibrinolytic systems. The inter-relationships of these systems are shown in Figure 10.6.
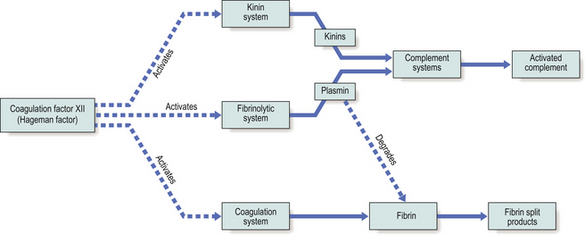
Fig. 10.6 Interactions between the systems of chemical mediators. Coagulation factor XII activates the kinin, fibrinolytic and coagulation systems. The complement system is in turn activated.
Kinin system.
The kinins are peptides of 9–11 amino acids; the most important is bradykinin. The kinin system is activated by coagulation factor XII (Fig. 10.7). Bradykinin is also a chemical mediator of the pain that is a cardinal feature of acute inflammation.
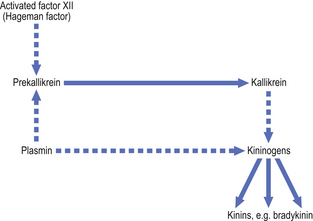
Fibrinolytic system.
Plasmin is responsible for the lysis of fibrin into fibrin split products, which may have local effects on vascular permeability.
Complement system.
The complement system is a cascade system of enzymatic proteins (Ch. 9). It can be activated during the acute inflammatory reaction in various ways:
The products of complement activation most important in acute inflammation include:
Table 10.2 summarises the chemical mediators involved in the three main stages of acute inflammation.
Table 10.2 Endogenous chemical mediators of the acute inflammatory response
| Status of acute inflammatory response | Chemical mediators |
|---|---|
| Vascular dilatation | HistamineProstaglandins PGE2/I2 VIP Nitric oxide PAF |
| Increased vascularpermeability | Transient phase—histamineProlonged phase—mediators such as bradykinin, nitric oxide, C5a, leukotriene B4 and PAF, potentiated by prostaglandins |
| Adhesion of leukocytes to endothelium | Up-regulation of adhesion molecules on endothelium, principally by IL-8, C5a, leukotriene B4, PAF, IL-1 and TNF-alpha |
| Neutrophil polymorph chemotaxis | Leukotriene B4, IL-8 and others |
Role of tissue macrophages
These secrete numerous chemical mediators when stimulated by local infection or injury. Most important are the cytokines interleukin-1 (IL-1) and tumour necrosis factor-alpha (TNF-alpha), whose stimulatory effect on endothelial cells occurs after that of histamine and thrombin. Other late products include E-selectin, an adhesion molecule that binds and activates neutrophils, and the chemokines IL-8 and epithelium-derived neutrophil attractant 78, which are potent chemotaxins for neutrophil polymorphs. Additionally, IL-1 and TNF-alpha cause endothelial cells, fibroblasts and epithelial cells to secrete MCP-1, another powerful chemotactic protein for neutrophil polymorphs.
Role of the lymphatics
Terminal lymphatics are blind-ended, endothelium-lined tubes present in most tissues in similar numbers to capillaries. The terminal lymphatics drain into collecting lymphatics, which have valves and so propel lymph passively, aided by contraction of neighbouring muscles, to the lymph nodes. The basal lamina of lymphatic endothelium is incomplete, and the junctions between the cells are simpler and less robust than those between capillary endothelial cells. Hence, gaps tend to open up passively between the lymphatic endothelial cells, allowing large protein molecules to enter.
In acute inflammation, the lymphatic channels become dilated as they drain away the oedema fluid of the inflammatory exudate. This drainage tends to limit the extent of oedema in the tissues. The ability of the lymphatics to carry large molecules and some particulate matter is important in the immune response to infecting agents; antigens are carried to the regional lymph nodes for recognition by lymphocytes (Ch. 9).
Role of the neutrophil polymorph
The neutrophil polymorph is the characteristic cell of the acute inflammatory infiltrate (Fig. 10.8). The actions of this cell will now be considered.
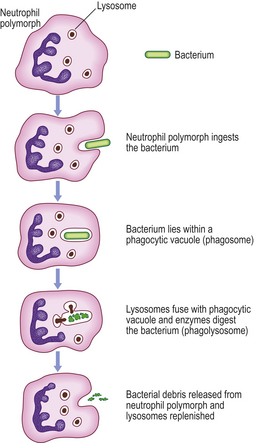
Fig. 10.8 Diagram of a neutrophil polymorph. The nucleus is polylobate and the cytoplasm shows dense granules which contain myeloperoxidase and other enzymes. Some of these enzymes are contained within lysosomes. These fuse with a phagocytic vacuole containing a phagocytosed bacterium, forming a phagolysosome in which the bacterium is digested by the enzymes.
Movement
Contraction of cytoplasmic microtubules and gel/sol changes in cytoplasmic fluidity bring about amoeboid movement. These active mechanisms are dependent upon calcium ions and are controlled by intracellular concentrations of cyclic nucleotides. The movement shows a directional response (chemotaxis) to the various chemicals of acute inflammation.
Adhesion to micro-organisms
Micro-organisms are opsonised (from the Greek word meaning ‘to prepare for the table’), or rendered more amenable to phagocytosis, either by immunoglobulins or by complement components. Bacterial lipopolysaccharides activate complement via the alternative pathway (Ch. 9), generating component C3b which has opsonising properties. In addition, if antibody binds to bacterial antigens, this can activate complement via the classical pathway, also generating C3b. In the immune individual, the binding of immunoglobulins to micro-organisms by their Fab components leaves the Fc component (Ch. 9) exposed. Neutrophils have surface receptors for the Fc fragment of immunoglobulins, and consequently bind to the micro-organisms prior to ingestion.
Phagocytosis
The process whereby cells (such as neutrophil polymorphs and macrophages) ingest solid particles is termed phagocytosis. The first step in phagocytosis is adhesion of the particle to be phagocytosed to the cell surface. This is facilitated by opsonisation. The phagocyte then ingests the attached particle by sending out pseudopodia around it. These meet and fuse so that the particle lies in a phagocytic vacuole (also called a phagosome) bounded by cell membrane. Lysosomes, membrane-bound packets containing the toxic compounds described below, then fuse with phagosomes to form phagolysosomes. It is within these that intracellular killing of micro-organisms occurs.
Intracellular killing of micro-organisms
Neutrophil polymorphs are highly specialised cells, containing noxious microbicidal agents, some of which are similar to household bleach. The microbicidal agents may be classified as:
Oxygen-dependent mechanisms.
The neutrophils produce hydrogen peroxide which reacts with myeloperoxidase in the cytoplasmic granules (Fig. 10.8) in the presence of halide, such as C1–, to produce a potent microbicidal agent. Other products of oxygen reduction also contribute to the killing, such as peroxide anions (O2–), hydroxyl radicals (•OH) and singlet oxygen (1O2).
Release of lysosomal products
Release of lysosomal products from the cell damages local tissues by proteolysis by enzymes such as elastase and collagenase, activates coagulation factor XII, and attracts other leukocytes into the area. Some of the compounds released increase vascular permeability, while others are pyrogens, producing systemic fever by acting on the hypothalamus.
The role of mast cells
Mast cells have an important role in acute inflammation. On stimulation by the C3a/C5a complement components they release preformed inflammatory mediators stored in their granules and metabolise arachidonic acid into newly synthesised inflammatory mediators such as leukotrienes, prostaglandins and thromboxanes.
Special macroscopic appearances of acute inflammation
The cardinal signs of acute inflammation are modified according to the tissue involved and the type of agent provoking the inflammation. Several descriptive terms are used for the appearances.
Serous inflammation
In serous inflammation, there is abundant protein-rich fluid exudate with a relatively low cellular content. Examples include inflammation of the serous cavities, such as peritonitis, and inflammation of a synovial joint, acute synovitis. Vascular dilatation may be apparent to the naked eye, the serous surfaces appearing injected (Fig. 10.2), i.e. having dilated, blood-laden vessels on the surface (like the appearance of the conjunctiva in ‘blood-shot’ eyes).
Catarrhal inflammation
When mucus hypersecretion accompanies acute inflammation of a mucous membrane, the appearance is described as catarrhal. The common cold is a good example.
Fibrinous inflammation
When the inflammatory exudate contains plentiful fibrinogen, this polymerises into a thick fibrin coating. This is often seen in acute pericarditis and gives the parietal and visceral pericardium a ‘bread and butter’ appearance.
Haemorrhagic inflammation
Haemorrhagic inflammation indicates severe vascular injury or depletion of coagulation factors. This occurs in acute pancreatitis due to proteolytic destruction of vascular walls, and in meningococcal septicaemia due to disseminated intravascular coagulation (see Ch. 23).
Suppurative (purulent) inflammation
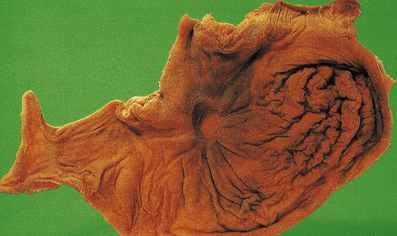
The terms ‘suppurative’ and ‘purulent’ denote the production of pus, which consists of dying and degenerate neutrophils, infecting organisms and liquefied tissues. The pus may become walled-off by granulation tissue or fibrous tissue to produce an abscess (a localised collection of pus in a tissue). If a hollow viscus fills with pus, this is called an empyema, for example empyema of the gallbladder (Fig. 10.9) or of the appendix (Fig. 10.10).
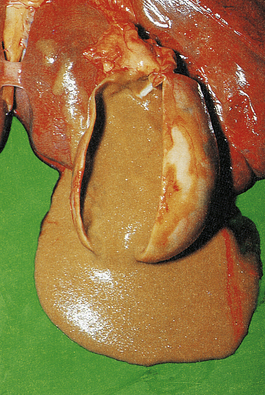
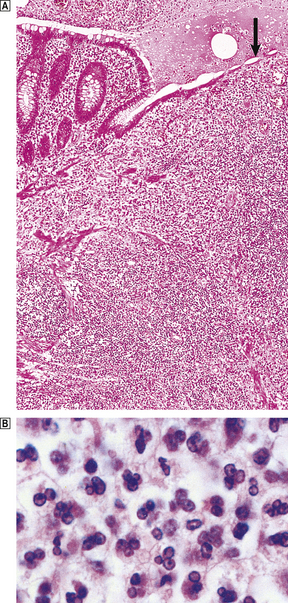
Fig. 10.10 Empyema of the appendix. The appendix lumen is filled with pus, there is focal mucosal ulceration (arrow) and the appendicular wall and mesoappendix (bottom) are thickened due to an acute inflammatory exudate. Pus in the lumen of the appendix. Pus consists of living and degenerate neutrophil polymorphs together with liquefied tissue debris.
Membranous inflammation
In acute membranous inflammation, an epithelium becomes coated by fibrin, desquamated epithelial cells and inflammatory cells. An example is the grey membrane seen in pharyngitis or laryngitis due to Corynebacterium diphtheriae.
Pseudomembranous inflammation
The term ‘pseudomembranous’ describes superficial mucosal ulceration with an overlying slough of disrupted mucosa, fibrin, mucus and inflammatory cells. This is seen in pseudomembranous colitis due to Clostridium difficile colonisation of the bowel, usually following broad-spectrum antibiotic treatment (Ch. 15).
Effects of acute inflammation
Acute inflammation has local and systemic effects, both of which may be harmful or beneficial. The local effects are usually clearly beneficial, for example the destruction of invading micro-organisms, but at other times they appear to serve no obvious function, or may even be positively harmful.
Beneficial effects
Both the fluid and cellular exudates may have useful effects. Beneficial effects of the fluid exudate are:
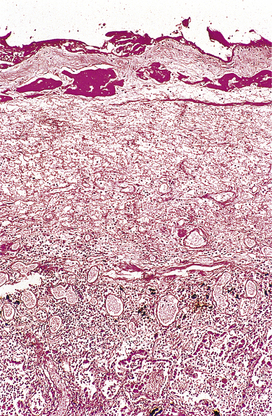
Fig. 10.11 Fibrinous exudate. Histology of the fibrinous exudate (dark-stained material) adherent to the pleura in acute lobar pneumonia.
The role of neutrophils in the cellular exudate has already been discussed. They have a life-span of only 1–3 days and must be constantly replaced. Most die locally, but some leave the site via the lymphatics. Some are actively removed by apoptosis. It is probable that apoptosis and its regulation play a major role in determining the outcome of episodes of inflammation. Blood monocytes also arrive at the site and, on leaving the blood vessels, transform into macrophages, becoming more metabolically active, motile and phagocytic. Phagocytosis of micro-organisms is enhanced by opsonisation by antibodies or by complement. In most acute inflammatory reactions, macrophages play a lesser role in phagocytosis compared with that of neutrophil polymorphs. Macrophages start to appear within a few hours of the commencement of inflammation, but do not predominate until the later stages when the neutrophils have diminished in number and the macrophage population has enlarged by local proliferation. They are responsible for clearing away tissue debris and damaged cells.
Both neutrophils and macrophages may discharge their lysosomal enzymes into the extracellular fluid by exocytosis, or the entire cell contents may be released when the cells die. Release of these enzymes assists in the digestion of the inflammatory exudate.
Harmful effects
The release of lysosomal enzymes by inflammatory cells may also have harmful effects:
Sequelae of acute inflammation
The sequelae of acute inflammation depend upon the type of tissue involved and the amount of tissue destruction, which depend in turn upon the nature of the injurious agent. The possible outcomes of acute inflammation are shown in Figure 10.12.
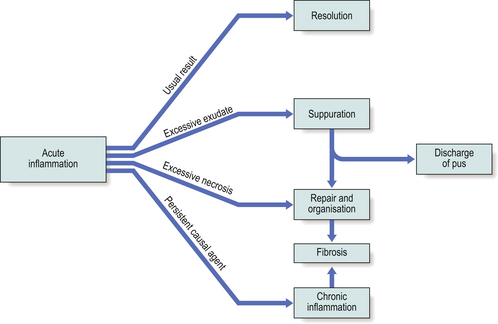
Fig. 10.12 The sequelae of acute inflammation. Resolution is the usual event, unless any of the adverse factors shown exist.
Resolution
The term resolution means the complete restoration of the tissues to normal after an episode of acute inflammation. The conditions that favour resolution are:
A good example of an acute inflammatory condition that usually resolves completely is acute lobar pneumonia (Ch. 14). The alveoli become filled with acute inflammatory exudate containing fibrin, bacteria and neutrophil polymorphs. The alveolar walls are thin and have many capillaries (for gas exchange) and lymphatic channels. The sequence of events leading to resolution is usually:
Following this, the lung parenchyma would appear histologically normal.
Suppuration
Suppuration is the formation of pus, a mixture of living, dying and dead neutrophils and bacteria, cellular debris and sometimes globules of lipid. The causative stimulus must be fairly persistent and is virtually always an infective agent, usually pyogenic bacteria (e.g. Staphylococcus aureus, Streptococcus pyogenes, Neisseria species or coliform organisms). Once pus begins to accumulate in a tissue, it becomes surrounded by a ‘pyogenic membrane’ consisting of sprouting capillaries, neutrophils and occasional fibroblasts; this is a manifestation of healing, eventually resulting in granulation tissue and scarring. Such a collection of pus is called an abscess, and bacteria within the abscess cavity are relatively inaccessible to antibodies and to antibiotic drugs (thus, for example, acute osteomyelitis, an abscess in the bone marrow cavity, is notoriously difficult to treat).
Abscess
An abscess (for example, a boil) usually ‘points’, then bursts; the abscess cavity collapses and is obliterated by organisation and fibrosis, leaving a small scar. Sometimes, surgical incision and drainage is necessary to eliminate the abscess.
If pus accumulates inside a hollow viscus (e.g. the gallbladder) the mucosal layers of the outflow tract of the viscus may become fused together by fibrin, resulting in an empyema (Fig. 10.9).
Such deep-seated abscesses sometimes discharge their pus along a sinus tract (an abnormal connection, lined by granulation tissue, between the abscess and the skin or a mucosal surface). If this results in an abnormal passage connecting two mucosal surfaces or one mucosal surface to the skin surface, it is referred to as a fistula. Sinuses occur particularly when foreign body materials are present, which are indigestible by macrophages and which favour continuing suppuration. The only treatment for this type of condition is surgical elimination of the foreign body material.
The fibrous walls of longstanding abscesses may become complicated by dystrophic calcification (Ch. 7).
Organisation
Organisation of tissues is their replacement by granulation tissue as part of the process of repair. The circumstances favouring this outcome are when:
During organisation, new capillaries grow into the inert material (inflammatory exudate), macrophages migrate into the zone and fibroblasts proliferate under the influence of TGF-beta, resulting in fibrosis and, possibly, scar formation. A good example of this is seen in the pleural space following acute lobar pneumonia. Resolution usually occurs in the lung parenchyma, but very extensive fibrinous exudate fills the pleural cavity (Fig. 10.11). The fibrin is not easily removed and consequently capillaries grow into the fibrin, accompanied by macrophages and fibroblasts (the exudate becomes ‘organised’). Eventually, fibrous adhesion occurs between the parietal and visceral pleura (Fig. 10.13). Fibrous adhesions also occur commonly in the peritoneal cavity after surgery or an episode of peritonitis; these can hamper further surgery and can also lead to intestinal obstruction.
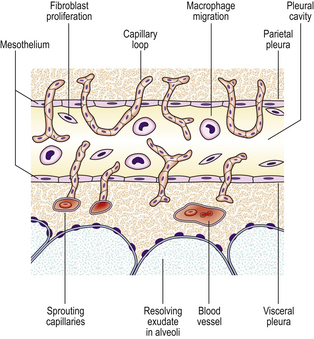
Progression to chronic inflammation
If the agent causing acute inflammation is not removed, the acute inflammation may progress to the chronic stage. In addition to organisation of the tissue just described, the character of the cellular exudate changes, with lymphocytes, plasma cells and macrophages (sometimes including multinucleate giant cells) replacing the neutrophil polymorphs (Fig. 10.14). Often, however, chronic inflammation occurs as a primary event, there being no preceding period of acute inflammation.
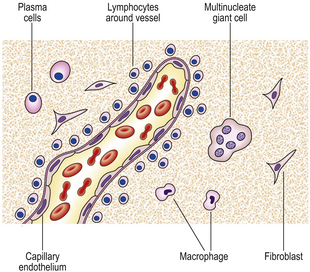
Fig. 10.14 The cells involved in chronic inflammation. Neutrophil polymorphs have disappeared from the site, and mononuclear cells such as lymphocytes and macrophages are prominent. Some specialised lymphocytes called plasma cells are present; these produce immunoglobulins. Some of the macrophages may become multinucleate giant cells. Fibroblasts migrate into the area and lay down collagen.
Systemic effects of inflammation
Apart from the local features of acute and chronic inflammation described above, an inflammatory focus produces systemic effects.
Pyrexia
Polymorphs and macrophages produce compounds known as endogenous pyrogens which act on the hypothalamus to set the thermoregulatory mechanisms at a higher temperature. Interleukin-2 probably has the greatest effect. Release of endogenous pyrogen is stimulated by phagocytosis, endotoxins and immune complexes.
Weight loss
Weight loss, due to negative nitrogen balance, is common when there is extensive chronic inflammation. For this reason, tuberculosis used to be called ‘consumption’.
Reactive hyperplasia of the reticulo-endothelial system
Local or systemic lymph node enlargement commonly accompanies inflammation, while splenomegaly is found in certain specific infections (e.g. malaria, infectious mononucleosis).
Haematological changes
Increased erythrocyte sedimentation rate
An increased erythrocyte sedimentation rate is a non-specific finding in many types of inflammation and is due to alterations in plasma proteins resulting in increased rouleaux formation of red cells.
Leukocytosis.
Neutrophilia occurs in pyogenic infections and tissue destruction; eosinophilia in allergic disorders and parasitic infection; lymphocytosis in chronic infection (e.g. tuberculosis), many viral infections and in whooping cough; and monocytosis occurs in infectious mononucleosis and certain bacterial infections (e.g. tuberculosis, typhoid).
CHRONIC INFLAMMATION
The word ‘chronic’ applied to any process implies that the process has extended over a long period of time. This is usually the case in chronic inflammation, but here the term ‘chronic’ takes on a much more specific meaning, in that the type of cellular reaction differs from that seen in acute inflammation. Chronic inflammation may be defined as an inflammatory process in which lymphocytes, plasma cells and macrophages predominate. As in acute inflammation, granulation and scar tissue are also formed, but in chronic inflammation they are usually more abundant. Chronic inflammation is usually primary, sometimes called chronic inflammation ab initio, but does occasionally follow acute inflammation.
Causes of chronic inflammation
Primary chronic inflammation
In most cases of chronic inflammation, the inflammatory response has all the histological features of chronic inflammation from the onset, and there is no initial phase of acute inflammation. Some examples of primary chronic inflammation are listed in Table 10.3.
Table 10.3 Some examples of primary chronic inflammation
| Cause of inflammation | Examples |
|---|---|
| Resistance of infective agent to phagocytosis and intracellular killing | Tuberculosis, leprosy, brucellosis, viral infections |
| Endogenous materials | Necrotic adipose tissue, bone, uric acid crystals |
| Exogenous materials | Silica, asbestos fibres, suture materials, implanted prostheses |
| Some autoimmune diseases | |
| Specific diseases of unknown aetiology | Chronic inflammatory bowel disease, e.g. ulcerative colitis |
| Primary granulomatous diseases | Crohn’s disease, sarcoidosis |
Progression from acute inflammation
Most cases of acute inflammation do not develop into the chronic form, but resolve completely. The commonest variety of acute inflammation to progress to chronic inflammation is the suppurative type. If the pus forms an abscess cavity that is deep-seated, and drainage is delayed or inadequate, then by the time that drainage occurs the abscess will have developed thick walls composed of granulation and fibrous tissues. The rigid walls of the abscess cavity therefore fail to come together after drainage, and the stagnating pus within the cavity becomes organised by the ingrowth of granulation tissue, eventually to be replaced by a fibrous scar.
Good examples of such chronic abscesses include: an abscess in the bone marrow cavity (osteomyelitis), which is notoriously difficult to eradicate; and empyema thoracis that has been inadequately drained.
Another feature that favours progression to chronic inflammation is the presence of indigestible material. This may be keratin from a ruptured epidermal cyst, or fragments of necrotic bone as in the sequestrum of chronic osteomyelitis (Ch. 25). These materials are relatively inert, and are resistant to the action of lysosomal enzymes. The most indigestible forms of material are inert foreign body materials, for example some types of surgical suture, wood, metal or glass implanted into a wound, or deliberately implanted prostheses such as artificial joints. It is not known why the presence of foreign body materials gives rise to chronic suppuration, but it is a well-established fact that suppuration will not cease without surgical removal of the material.
Foreign bodies have in common the tendency to provoke a special type of chronic inflammation called ‘granulomatous inflammation’ (p. 216), and to cause macrophages to form multinucleate giant cells called ‘foreign body giant cells’.
Recurrent episodes of acute inflammation
Recurring cycles of acute inflammation and healing eventually result in the clinicopathological entity of chronic inflammation. The best example of this is chronic cholecystitis, normally due to the presence of gallstones (Ch. 16); multiple recurrent episodes of acute inflammation lead to replacement of the gallbladder wall muscle by fibrous tissue and the predominant cell type becomes the lymphocyte rather than the neutrophil polymorph.
Macroscopic appearances of chronic inflammation
The commonest appearances of chronic inflammation are:

Microscopic features of chronic inflammation
The cellular infiltrate consists characteristically of lymphocytes, plasma cells and macrophages. A few eosinophil polymorphs may be present, but neutrophil polymorphs are scarce. Some of the macrophages may form multinucleate giant cells. Exudation of fluid is not a prominent feature, but there may be production of new fibrous tissue from granulation tissue (Figs 10.15-10.17). There may be evidence of continuing destruction of tissue at the same time as tissue regeneration and repair. Tissue necrosis may be a prominent feature, especially in granulomatous conditions such as tuberculosis. It is not usually possible to predict the causative factor from the histological appearances in chronic inflammation.
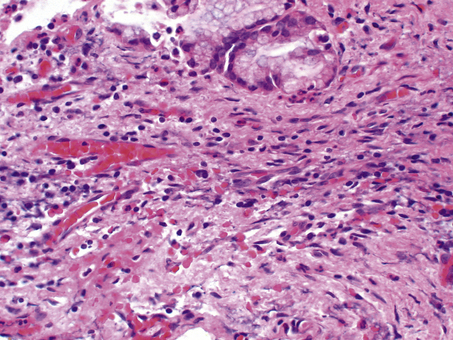
Fig. 10.17 Chronic inflammation in the wall of a gallbladder that has experienced previous episodes of acute cholecystitis. Aggregates of lymphocytes are appearing and there are ingrowing fibroblasts.
Paracrine stimulation of connective tissue proliferation
Healing involves regeneration and migration of specialised cells, while the predominant features in repair are angiogenesis followed by fibroblast proliferation and collagen synthesis resulting in granulation tissue. These processes are regulated by low molecular weight proteins called growth factors which bind to specific receptors on cell membranes and trigger a series of events culminating in cell proliferation (Table 10.4).
Table 10.4 Growth factors involved in healing and repair associated with inflammation
| Growth factor | Abbreviation | Main function |
|---|---|---|
| Epidermal growth factor | EGF | Regeneration of epithelial cells |
| Transforming growth factor-alpha | TGF-alpha | Regeneration of epithelial cells |
| Transforming growth factor-beta | TGF-beta | |
| Platelet-derived growth factor | PDGF | Mitogenic and chemotactic for fibroblasts and smooth muscle cells |
| Fibroblast growth factor | FGF | Stimulates fibroblast proliferation, angiogenesis and epithelial cell regeneration |
| Insulin-like growth factor-1 | IGF-1 | Synergistic effect with other growth factors |
| Tumour necrosis factor | TNF | Stimulates angiogenesis |
Cellular co-operation in chronic inflammation
The lymphocytic tissue infiltrate contains two main types of lymphocyte (described more fully in Ch. 9). B-lymphocytes, on contact with antigen, become progressively transformed into plasma cells, which are cells specially adapted for the production of antibodies. The other main type of lymphocyte, the T-lymphocyte, is responsible for cell-mediated immunity. On contact with antigen, T-lymphocytes produce a range of soluble factors called cytokines, which have a number of important activities.
These pathways of cellular co-operation are summarised in Figure 10.18.
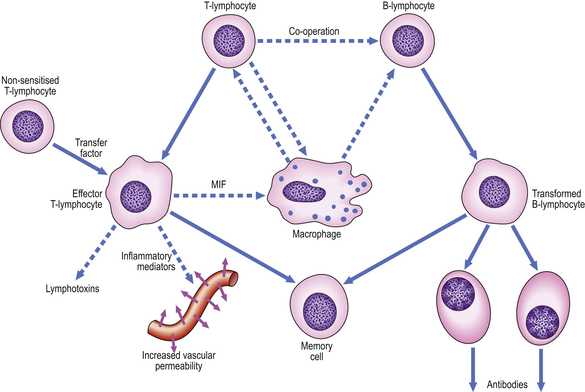
Macrophages in chronic inflammation
Macrophages are relatively large cells, up to 30 μm in diameter, that move by amoeboid motion through the tissues. They respond to certain chemotactic stimuli (possibly cytokines and antigen–antibody complexes) and have considerable phagocytic capabilities for the ingestion of micro-organisms and cell debris. When neutrophil polymorphs ingest micro-organisms, they usually bring about their own destruction and thus have a limited life-span of up to about 3 days. Macrophages can ingest a wider range of materials than can polymorphs and, being long-lived, they can harbour viable organisms if they are not able to kill them by their lysosomal enzymes. Examples of organisms that can survive inside macrophages include mycobacteria, such as Mycobacterium tuberculosis and M. leprae, and organisms such as Histoplasma capsulatum. When macrophages participate in the delayed-type hypersensitivity response (Ch. 9) to these types of organism, they often die in the process, contributing to the large areas of necrosis by release of their lysosomal enzymes.
Macrophages in inflamed tissues are derived from blood monocytes that have migrated out of vessels and have become transformed in the tissues. They are thus part of the mononuclear phagocyte system (Fig. 10.19), also known as the reticulo-endothelial system.
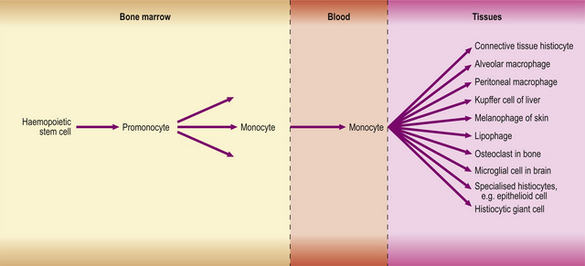
Fig. 10.19 The mononuclear phagocyte system. All of the differentiated cell types on the right are derived from blood monocytes.
The mononuclear phagocyte system, shown in Figure 10.19, is now known to include macrophages, fixed tissue histiocytes in many organs and, probably, the osteoclasts of bone. All are derived from monocytes, which in turn are derived from a haemopoietic stem cell in the bone marrow.
The ‘activation’ of macrophages as they migrate into an area of inflammation involves an increase in size, protein synthesis, mobility, phagocytic activity and content of lysosomal enzymes. Electron microscopy reveals that the cells have a roughened cell membrane bearing lamellipodia, while the cytoplasm contains numerous dense bodies—phagolysosomes (formed by the fusion of lysosomes with phagocytic vacuoles).
Macrophages produce a range of important cytokines, including interferon-alpha and -beta, interleukin-1, -6 and -8, and tumour necrosis factor-alpha (TNF) (see Ch. 9).
Specialised forms of macrophages and granulomatous inflammation
A granuloma is an aggregate of epithelioid histiocytes (Fig. 10.20). It may also contain other cell types such as lymphocytes and histiocytic giant cells. Granulomatous diseases comprise some of the most widespread and serious diseases in the world, such as tuberculosis and leprosy.
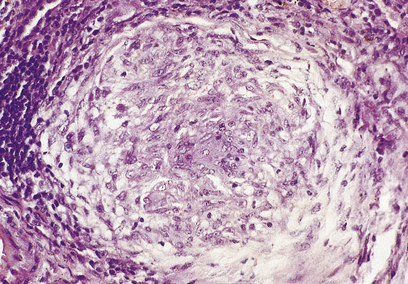
Fig. 10.20 A granuloma: a collection of epithelioid histiocytes. This example is from a case of sarcoidosis involving the liver.
Epithelioid histiocytes
Named for their vague histological resemblance to epithelial cells, epithelioid histiocytes have large vesicular nuclei, plentiful eosinophilic cytoplasm and are often rather elongated. They tend to be arranged in clusters. They have little phagocytic activity, but appear to be adapted to a secretory function. The full range, or purpose, of their secretory products is not known, although one product is angiotensin converting enzyme. Measurement of the activity of this enzyme in the blood can act as a marker for systemic granulomatous disease, such as sarcoidosis.
The appearance of granulomas may be augmented by the presence of caseous necrosis (as in tuberculosis) or by the conversion of some of the histiocytes into multinucleate giant cells. The association of granulomas with eosinophils often indicates a parasitic infection (e.g. worms). A common feature of many of the stimuli that induce granulomatous inflammation is indigestibility of particulate matter by macrophages. In other conditions, such as the systemic granulomatous disease sarcoidosis, there appear to be far-reaching derangements in immune responsiveness favouring granulomatous inflammation. In other instances, small traces of elements such as beryllium induce granuloma formation, but the way in which they induce the inflammation is unknown. Some of the commoner granulomatous conditions are shown in Table 10.5.
Table 10.5 Causes of granulomatous disease
| Cause | Example |
|---|---|
| Specific infections | |
| Materials that resist digestion | |
| Specific chemicals | Beryllium |
| Drugs | Hepatic granulomas due to allopurinol, phenylbutazone, sulphonamides |
| Unknown |
Histiocytic giant cells
Histiocytic giant cells tend to form where particulate matter that is indigestible by macrophages accumulates, for example inert minerals such as silica, or bacteria such as tubercle bacilli, which have cell walls containing mycolic acids and waxes that resist enzymatic digestion. Histiocytic giant cells form particularly when foreign particles are too large to be ingested by just one macrophage. The multinucleate giant cells, which may contain over 100 nuclei, are thought to develop ‘by accident’ when two or more macrophages attempt simultaneously to engulf the same particle; their cell membranes fuse and the cells unite. The multinucleate giant cells resulting have little phagocytic activity and no known function. They are given specific names according to their microscopic appearance.
Langhans’ giant cells
Langhans’ giant cells have a horseshoe arrangement of peripheral nuclei at one pole of the cell and are characteristically seen in tuberculosis, although they may be seen in other granulomatous conditions. (They must not be confused with Langerhans’ cells, the dendritic antigen-presenting cells of the epidermis; Ch. 9.)
Foreign body giant cells
So-called ‘foreign body giant cells’ are large cells with nuclei randomly scattered throughout their cytoplasm. They are characteristically seen in relation to particulate foreign body material.
Touton giant cells
Touton giant cells have a central ring of nuclei; the peripheral cytoplasm is clear due to accumulated lipid. They are seen at sites of adipose tissue breakdown and in xanthomas (tumour-like aggregates of lipid-laden macrophages).
Although giant cells are commonly seen in granulomas, they do not constitute a defining feature. Solitary giant cells in the absence of epithelioid histiocytes do not constitute a granuloma.
Role of inflammation in systemic and organ-specific diseases
Acute inflammation is involved in the cardiovascular system in the response to acute myocardial infarction (Ch. 13) and the generation of some complications of myocardial infarction such as cardiac rupture. It is also involved in infective endocarditis, pericarditis and myocarditis, and in some vasculitic syndromes. One mechanism of vasculitis is that immune complexes deposit in the vessel wall, activate complement, and thus excite an inflammatory response.
Commonly confused conditions and entities relating to inflammation
| Commonly confused | Distinction and explanation |
|---|---|
| Acute and chronic | In inflammation, acute and chronic denote both the dynamics and character of the process. Acute inflammation has a relatively rapid onset and, usually, resolution, and neutrophil polymorphs are the most abundant cells. Chronic inflammation has a relatively insidious onset, prolonged course and slow resolution, and lymphocytes, plasma cells and macrophages (sometimes with granuloma formation) are the most abundant cells. |
| Exudate and transudate | Exudates have a high protein content because they result from increased vascular permeability. Transudates have a low protein content because the vessels have normal permeability characteristics. |
| Granuloma and granulation tissue | A granuloma is an aggregate of epithelioid histiocytes and a feature of some specific chronic inflammatory disorders. Granulation tissue is an important component of healing and comprises small blood vessels in a connective tissue matrix with myofibroblasts. |
| Monocytes, macrophages and histiocytes | Monocytes are the newly formed cells of the mononuclear phagocyte system. After a few hours in the blood, they enter tissues and undergo further differentiation into macrophages. Some macrophages in tissues have specific features and names (e.g. Kupffer cells); others are referred to as histiocytes. |
| Fibrin and fibrous | Fibrin is deposited in blood vessels and tissues or on surfaces (e.g. in acute inflammation) as a result of the action of thrombin on fibrinogen. Fibrous describes the texture of a non-mineralised tissue of which the principal component is collagen (e.g. scar tissue). |
Chronic inflammation is involved in myocardial fibrosis after myocardial infarction.
Inflammation makes an important contribution to development of atheroma (Ch. 13). Macrophages adhere to endothelium, migrate into the arterial intima and, with T-lymphocytes, express cell adhesion molecules which recruit other cells into the area. The macrophages are involved in processing the lipids that accumulate in atheromatous plaques.
Inflammation also features in the tissue injury associated with neurodegenerative disorders of the central nervous system. Multiple sclerosis is a relatively common chronic demyelinating neurodegenerative disorder in which chronic inflammation plays an important role. Perivascular cuffing by plasma cells and T lymphocytes is seen in zones of white matter where macrophages break down myelin.
Badolato R., Oppenheim J.J.. Role of cytokines, acute-phase proteins, and chemokines in the progression of rheumatoid arthritis. Seminars in Arthritis and Rheumatism. 1996;26:526-538.
Balkwill F.. Cytokine amplification and inhibition of immune and inflammatory responses. Journal of Viral Hepatitis. 1997;4(Suppl 2):6-15.
Ballou S.P., Kushner I.. Chronic inflammation in older people: recognition, consequences, and potential intervention. Clinics in Geriatric Medicine. 1997;13:653-669.
Baumert P.W.Jr. Acute inflammation after injury. Quick control speeds rehabilitation. Postgraduate Medicine. 1995;97:35-36.
Borregard N., Sørensen O.E., Theilgaard-Mönch K.. Neutrophil granules: a library of innate immunity proteins. Trends in Immunology. 2007;28:340-345.
Brewer D.P.. Activities of the neutrophil polymorph. British Medical Journal. 1972;5810:396-400.
Burger D., Dayer J.M.. Inhibitory cytokines and cytokine inhibitors. Neurology. 1995;45(Suppl 6):S39-S43.
Cohen M.S.. Molecular events in the activation of human neutrophils for microbial killing. Clinical Infectious Diseases. 1994;18(Suppl 2):170-179.
Collins T.. Leukocyte recruitment, endothelial cell adhesion molecules, and transcriptional control. Insights for drug discovery. London: Kluwer; 2001.
Di Perry G., Bonora S., Allegranzi B., Concia E.. Granulomatous inflammation and transmission of infectious disease. Immunology Today. 20, 1999. 337–338
Edwards S.W.. Biochemistry and physiology of the neutrophil. Cambridge: Cambridge University Press; 1994.
Epstein W.L., Fukuyama K.. Mechanisms of granulomatous inflammation. Immunology Series. 1989;46:687-721.
Faurschou M., Borregaard N.. Neutrophil granules and secretory vesicles in inflammation. Microbes and Infection. 2003;5:1317-1327.
Formela L.J., Galloway S.W., Kingsnorth A.N.. Inflammatory mediators in acute pancreatitis. British Journal of Surgery. 1995;82:6-13.
Furie M.B., Randolph G.J.. Chemokines and tissue injury. American Journal of Pathology. 1995;146:1287-1301.
Galli S.J.. The Paul Kallos Memorial Lecture. The mast cell: a versatile effector cell for a challenging world. International Archives of Allergy and Immunology. 1997;113:14-22.
Goldblatt D., Thrasher A.J.. Chronic granulomatous disease. Clinical and Experimental Immunology. 1998;122:1-9.
Gordon S.. The role of the macrophage in immune regulation. Research in Immunology. 1998;149:685-688.
Kasama T., Miwa Y., Isozaki T., Odai T., Adachi M., Kunkel S.L.. Neutrophil-derived cytokines: potential therapeutic targets in inflammation. Current Drug Targets. 2005;4:273-279.
Kobayashi S.D., Voyich J.M., DeLeo T.. Regulation of neutrophil-mediated inflammatory response to infection. Microbes and Infection. 2003;5:1337-1344.
Kornfeld H., Mancino G., Colizzi V.. The role of macrophage in cell death and tuberculosis. Cell Death and Differentiation. 1999;6:71-78.
Lakhani S.R., Dilly S.A., Finlayson C.J., Dogan A.. Basic pathology, 3rd edn. London: Hodder Arnold; 2003.
Lawrence T., Gilroy D.W.. Chronic inflammation: a failure of resolution. International Journal of Experimental Pathology. 2007;88:85-94.
Lee W.Y., Chin A.C., Voss S., Parkos C.A.. In vitro neutrophil transepithelial migration. Methods in Molecular Biology. 341, 2006. 205–215
Leung D.Y.. Immunologic basis of chronic allergic diseases: clinical messages from the laboratory bench. Pediatric Research. 1997;42:559-568.
Levin N.W., Ronco C.. Chronic inflammation: an overview. Contributions to Nephrology. 137, 2002. 364–370
Levy J.H.. The human inflammatory response. Journal of Cardiovascular Pharmacology. 1996;27(Suppl 1):S31-S37.
Ley K.. Physiology of inflammation. Oxford: Oxford University Press; 2001.
Lue H., Kleemann R., Calandra T., Roger T., Bernhagen J.. Macrophage migration inhibitory factor (MIF): mechanisms of action and role in disease. Microbes and Infection. 2002;4:449-460.
Mahmoudi M., Curzen N., Gallagher P.J.. Atherogenesis: the role of inflammation and infection. Histopathology. 2007;50:535-546.
Majno G.. Chronic inflammation: links with angiogenesis and wound healing. American Journal of Surgical Pathology. 153, 1998. 1045–1049
Mannaioni P.F., Di Bello M.G., Masini E.. Platelets and inflammation: role of platelet-derived growth factor, adhesion molecules and histamine. Inflammation Research. 1997;46:4-18.
Sartor R.B.. Pathogenesis and immune mechanisms of chronic inflammatory bowel diseases. American Journal of Gastroenterology. 1997;92(Suppl):5S-11S.
Savill J.. Apoptosis in resolution of inflammation. Journal of Leukocyte Biology. 1997;61:375-380.
Schultz D.R., Tozman E.C.. Antineutrophil cytoplasmic antibodies: major autoantigens, pathophysiology, and disease associations. Seminars in Arthritis and Rheumatism. 1995;25:143-159.
Serhan C.N., Brain S.D., Buckley C.D., et al. Resolution of inflammation: state of the art, definitions and terms. FASEB Journal. 2007;21:325-332.
Thomson A.W., Lotze M.T.. The cytokine handbook, 4th edn. London: Academic Press; 2003.
Tremblay G.M., Janelle M.F., Bourbonnais Y.. Anti-inflammatory activity of neutrophil elastase inhibitors. Current Opinion in Investigational Drugs. 4, 2000. 556–565
Vallance P., Collier J., Bhagat K.. Infection, inflammation, and infarction: does acute endothelial dysfunction provide a link? Lancet. 1997;349:1391-1392.
Walker A., Ward C., Taylor E.L., et al. Regulation of neutrophil apoptosis and removal of apoptotic cells. Current Drug Targets. 2005;4:447-454.