43 Herpesviruses
Herpes simplex; varicella and zoster; infectious mononucleosis; B cell lymphomas; cytomegalovirus disease; exanthem subitum; Kaposi’s sarcoma; herpes B
Key points
• All herpesviruses persist for the lifetime of the host and establish a latent (non-replicating) state from which they may be reactivated under certain conditions.
• Cell-mediated immunity, especially the action of cytotoxic T lymphocytes, is essential in the control of herpesvirus infections.
• Acute necrotizing sporadic viral encephalitis caused by HSV is a medical emergency requiring urgent treatment with high-dose intravenous aciclovir that must be instigated empirically without delay and prior to laboratory confirmation.
• Genital herpes simplex may be due to HSV 1 or 2, and the individuals are often unaware that they have this recurring infection.
• A live-attenuated VZV vaccine can protect against chickenpox (varicella); it may boost immunity in older age groups with a reduction in the symptoms of zoster (shingles), which arises as a result of VZV reactivation.
• Glandular fever is a clinical presentation of primary EBV infection, mainly in the 15–25 years age group; most individuals have an asymptomatic infection in childhood.
• EBV is a recognized human tumour virus associated with certain epithelial and lymphoid tumours (e.g. B cell lymphoma post transplant).
• CMV is commonly acquired without symptoms; however, in the immunocompromised host, CMV disease is serious and merits prophylaxis or pre-emptive therapy.
• Less well known herpesviruses (HHV 6 and 7) cause a common childhood rash illness (exanthem subitum), whereas HHV 8 is associated with Kaposi’s sarcoma.
Species-specific herpesviruses have been described for most animals and they share several features including their structure, mode of replication and the capacity to establish lifelong latent infections from which virus may be reactivated. Together, they form the herpesviridae family.
Latent infection
Latency reflects persistent infection (as manifest by the presence of the viral genome) during which no infectious virus is produced, except during intermittent episodes of reactivation.
Reactivation
Reactivation from the latent state may be restricted to asymptomatic virus shedding or manifest as clinical disease.
Recurrence or recrudescence
These terms are used when reactivated virus produces clinically apparent disease.
The herpesviridae family is subdivided into 3 subfamilies: alpha (α)-, beta (β)- and gamma (γ)- herpesvirinae (see Table 43.1). The human α- herpesvirinae contain the genera simplexvirus (herpes simplex virus (HSV) 1 and 2) and varicellovirus (varicella zoster virus, VZV); the β-herpesvirinae consist of cytomegalovirus (cytomegalovirus, CMV) and roseolovirus (human herpes virus, HHV 6 and 7) genera; and the γ-herpesvirinae contain the lymphocryptovirus (Epstein–Barr virus, EBV) and rhadinovirus (Kaposi sarcoma-associated herpesvirus, KSHV) genera. Individual viruses are denoted by the taxonomic unit (family/subfamily) followed by an Arabic number (e.g. human herpesvirus (HHV) 4). However, vernacular and approved names are often used interchangeably (e.g. HHV 4 and EBV). At present, eight human herpesviruses (HHV 1–8) are recognized, and infection with each of HHV 1–7 has been shown to be common in all populations; studies with HHV 8 suggest it is an uncommon infection in developed countries. The herpes B virus of monkeys can be transmitted to man accidentally.

Description
Herpesviruses have a characteristic morphology (Fig. 43.1). The icosahedral protein capsid (average diameter 100 nm) consists of 162 hollow hexagonal and pentagonal capsomeres with an electron-dense core containing the DNA genome (together forming the nucleocapsid). Outside the capsid (in mature virus particles) is an amorphous proteinaceous layer, the tegument, surrounded by a lipid envelope derived from host cell membranes. Projecting from the trilaminar lipid host-derived envelope are spikes of viral glycoproteins. Cryo-electron micrographs indicate that the capsid is organized into at least three layers, with viral DNA inserted in the innermost layer. The average enveloped particle is approximately 200 nm in diameter.
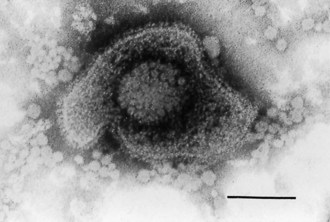
Fig. 43.1 Electron micrograph of HSV. Negative staining (2% phosphotungstic acid). Bar = 100 nm.
(Prepared by Dr BW McBride.)
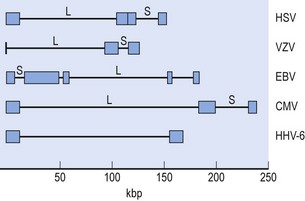
The herpesvirus genome is linear double-stranded (ds) DNA that varies in length from 125–248 kbp with a base content ranging from 42–69 G+C mol% for the human herpesviruses. The presence of unique long and short (UL, US) regions bounded by repeated and inverted short segments allows recombination and isomeric forms in some cases (Fig. 43.2). Genes coding for viral glycoproteins, major capsid proteins, enzymes involved in DNA replication, and some transcripts associated with latency, have been identified. Conserved sequences appear in certain regions, and some genes show homology with regions of human chromosomes. Restriction endonuclease and genome sequence analysis permits epidemiological comparison of strains (‘fingerprinting’) within herpesvirus species.
Biological classification
Broad characteristics of the three herpes subfamilies include:
1. Alphaherpesvirinae (e.g. HSV, VZV and B virus): rapid growth in cultured cells, latency in sensory ganglia.
2. Betaherpesvirinae (e.g. CMV): slow growth in cultured cells, restricted host range.
The viruses are relatively thermo-labile and readily inactivated by lipid solvents such as alcohols and detergents.
Replication
After initial attachment of viral receptor-binding proteins to cell surface proteoglycans followed by specific binding to target cell receptors, the envelope of herpesviruses fuses with the cell membrane. The nucleocapsids cross the cytoplasm to the nuclear membrane; replication of viral DNA and assembly of capsids takes place within the nucleus. With HSV, it is known that the tegument proteins partake in transactivation of the first set of genes. Between 65 and 100 viral proteins are synthesized in an orderly sequence or cascade. Briefly, using HSV as an example, circularization of the herpesvirus genome in the cell nucleus results in the orderly transcription (by host RNA polymerase II) of a cascade of immediate early (IE; α), early (E; β) and late (L; γ) genes. IE proteins activate expression of E genes that include viral-derived enzymes (including DNA polymerase and thymidine kinase (TK)) involved in replication which is thought to proceed in a ‘rolling circle’ manner yielding a concatomer that is cut to genome-length DNA units. Induction by E gene products of L genes results in production of viral structural proteins that package around progeny DNA to give rise to new nucleocapsids.
The viral capsid proteins migrate from the cytoplasm to the nucleus where capsid assembly occurs, and new viral DNA is inserted and located in the inner shell. Viral glycoproteins are processed in the Golgi complex and incorporated into host cell membranes, from which the viral envelope is acquired, usually from the inner layer of the nuclear membrane as the virus buds out from the nucleus. It then passes by way of membranous vacuoles to reach the cell surface. Productively infected cells generally do not survive (lytic infection).
Herpes simplex virus
HSV is ubiquitous, infecting the majority of the world’s population early in life and persisting in a latent form from which reactivation with shedding of infectious virus occurs, thus maintaining the transmission chain.
Description
In contrast to other members of the group, HSV can be grown relatively easily in cells from a wide variety of animals so that far more extensive studies have been undertaken with this virus.
There are two distinct types of HSV: type 1 (HSV 1) and type 2 (HSV 2). These two types are generally (but not exclusively) associated with different sites of infection in patients (see below); type 1 strains are associated primarily with the mouth, eye and central nervous system (CNS), whereas type 2 strains are found most often in the genital tract.
HSV glycoproteins
The envelope of HSV contains at least 11 glycoproteins. Three of the glycoproteins (g) are essential for production of infectious virus: gB and gD, which are involved in adsorption to and penetration into cells (alongside gC), and gH (together with gL), involved in fusion at entry and in the release of virus. Some of the glycoproteins have common antigenic determinants shared by HSV 1 and 2 (gB and gD) whereas others have specific determinants for one type only (gG).
Pathogenesis
Primary infection
The typical lesion produced by HSV is the vesicle, a ballooning degeneration of intra-epithelial cells, which contains infectious fluid. The basal epithelium is usually intact as vesicles penetrate the subepithelial layer only occasionally. The base of the vesicle contains multinucleate cells (Tzanck cells) and infected nuclei contain eosinophilic inclusion bodies. The roof of the vesicle breaks down and an ulcer forms. This happens rapidly on mucous membranes and non-keratinizing epithelia; on the skin, the ulcer crusts over, forming a scab, and then heals. A mononuclear cellular immune reaction is usual with the vesicle fluid becoming cloudy and cellular infiltration in the subepithelial tissue. After resorption, or loss, of the vesicle fluid, the damaged epithelium is regenerated. Natural killer (NK) cells play a significant role in early defence by recognizing and destroying HSV-infected cells. Herpesvirus glycoproteins synthesized during virus growth are inserted into host cell membranes, and some are secreted into extracellular fluid. The host’s adaptive immune system responds to all these foreign antigens producing both cytotoxic (CD4+ and CD8+) and helper (CD4+) T lymphocytes which activate primed B lymphocytes to produce specific antibodies, and are also involved in the induction of delayed hypersensitivity. The different glycoproteins have significant roles in generating these various cell responses; many induce neutralizing antibodies.
During the replication phase at the site of entry in the epithelium, virus particles enter the sensory nerve endings that penetrate to the parabasal layer of the epithelium and are transported, probably as nucleocapsids, along the axon to the nerve body (neurone) in the sensory (dorsal root) ganglion by retrograde axonal flow. Virus replication in a neurone ends in lytic infection and neuronal cell death; however, in some ganglion cells, a latent infection is established in which surviving neurones harbour the viral genome. Neurones other than those in sensory ganglia can be the site of herpesvirus latency. It is not clear whether true latency occurs at epithelial sites; there is some evidence of persistence of virus at peripheral sites, but this may be due to a reactivation with a low level of virus replication.
Antibody reduces the severity of infections although it does not prevent recurrences. Thus, neonates receiving maternal antibody transplacentally are protected against the worst effects of neonatal herpes. HSV 2 infection seems to protect against HSV 1, but previous HSV 1 infection only partly modifies HSV 2 disease.
Latent infection
Latent infection of sensory neurones is a feature of the neurotropic herpesviruses, HSV and VZV; HSV 1 latency is the best understood. Only a small proportion (about 1%) of cells in the affected ganglion carry the viral genome as free circular episomes – estimated about 20 copies per infected cell. Very few virus genes are expressed in the latent state; in HSV, some viral RNA transcripts (latency-associated transcripts, LATs) are found in the nuclei, but no virus-encoded proteins have been demonstrated in the cells, so these infected neurones are not recognized by the immune system. Latent HSV genomes have been detected in post mortem studies on excised ganglia and other neuronal tissues. HSV 1 is regularly detected in:
HSV 2 latency in the sacral ganglia has been demonstrated. Either type may become latent in other ganglia.
Reactivation and recrudescence
Reactivation processes are still not clearly understood. It is suggested that HSV DNA passes along the nerve axon back to the nerve ending where infection of epithelial cells may occur. Not all reactivation will result in a visible lesion; there may be asymptomatic shedding of virus only detectable by culture or DNA detection methods.
The factors influencing the development of recrudescent lesions are not yet clearly identified. An increase of CD8+ T suppressor lymphocyte activity is common at the time of recurrences. Some mediators (e.g. prostaglandins), and a temporary decrease in immune effector cell function, particularly delayed hypersensitivity, may enhance spread of HSV. Certainly, the known triggers for recurrences are accompanied by a local increase in prostaglandin levels, and depression of cell-mediated immunity predisposes to herpes recurrence. The mechanism of reactivation is not known, but the event can be predicted accurately in those known to harbour latent virus. Thus, after bone marrow transplant, or high-dose chemotherapy, herpes simplex recurs in 80% of cases at a median interval of 18 days in the absence of prophylaxis.
In whatever way reactivation is achieved, it is a feature of HSV infection. It occurs naturally, and can be induced by a variety of stimuli such as:
The interval between the stimulus and the appearance of a clinically obvious lesion is 2–5 days; this has been demonstrated regularly in patients undergoing neurological interference with their trigeminal ganglion, a common site of herpes latency.
Clinical features
Primary infection
Primary infection refers to the patient’s initial acquisition of virus and, thus, occurs in those with no antibody against HSV (Table 43.2). It usually involves the mucous membranes of the mouth, but may include the lips, skin of the face, nose or any other site, including the eye and genital tract.
Table 43.2 Types of herpes simplex virus (HSV) infection
| Type of infection | Antibody status of patient |
|---|---|
| Primary: first HSV infection (any type at any site) | Seronegative |
| Latent: no symptoms | Seropositive |
| Recurrent: recrudescence of the latent HSV type(s) | Seropositive |
| Initial (non-primary): first episode of the heterologous HSV in a seropositive patient | Seronegative for infecting type; seropositive for latent type |
| Re-infection (exogenous): with a strain that differs from the latent HSV type | Seropositive |
Recurrence or recrudescence
Symptomatic recurrence is heralded by a prodrome in two-thirds of people, who experience pain or paresthesiae (tingling, warmth, itch) at the site followed by erythema and a papule, usually within 24 h. Progression to a vesicle and ulcer, with subsequent crusting, takes 8–12 days before natural healing occurs. Because of their association with febrile illness, the lesions are popularly known as cold sores or fever blisters. The most common sites are at the mucocutaneous junction of the lip (seldom inside the mouth), on the chin, or inside the nose. However, recurrent lesions can manifest at any site innervated by the affected neurone, determined by the site of initial infection.
Severe pain, extensive mucosal ulceration and delayed healing are features of recurrent herpes in severely immunocompromised patients. The ulcers provide entry for other infections. HSV viraemia after reactivation is uncommon, even in the immunocompromised individual, but can lead to disseminated infection in internal organs. Some sufferers experience erythema multiforme following their recurrent herpes, associated with reaction to certain herpes antigens; this may take the form of the Stevens–Johnson syndrome.
Oral infection
Classically, primary HSV infection presents as an acute, febrile gingivostomatitis in pre-school children. Vesicular lesions ulcerate rapidly and are present in the front of the mouth and on the tongue (stomatitis). Gingivitis is usually present. Vesicles may also develop on the lips and skin around the mouth (herpetic dermatitis), and cervical lymphadenopathy can occur. The child is miserable for 7–10 days in an untreated case before the lesions heal. However, the majority of primary infections go unrecognized, the episode being attributed to teething or mistaken for ‘thrush’ (candida infection). In primary infection in older children and adults, there may also be an associated mononucleosis; pharyngitis is also notable. Viraemia with dissemination of HSV to internal organs is rare except in pregnancy (primary infection, with hepatitis), the neonate (see below), or the immunocompromised patient.
Skin infection
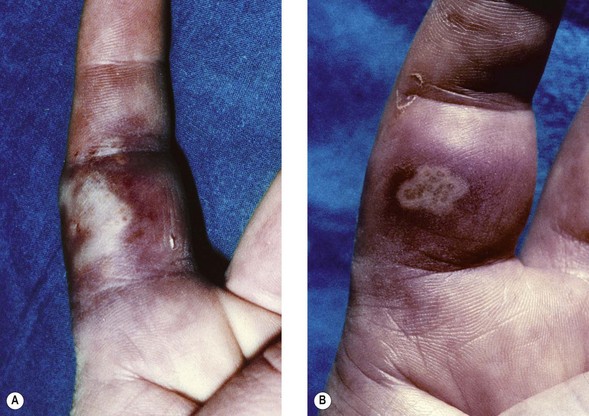
Herpetic whitlow
Hand infections with HSV are not uncommon, but other sites may be involved [e.g. as a result of sports such as herpes gladiatorum in wrestlers or rugby players (as a result of direct skin-to-skin contact)]. Three presentations of hand infections may be observed:
1. The classical primary lesion on the fingers or thumb of the toddler with herpetic stomatitis, due to autoinoculation.
2. Another classical and often primary infection is acquired by accidental inoculation in health-care workers (traumatic herpes). These infections may recur; the majority are HSV 1.
3. The most common hand lesions are recurrent, associated with HSV 2 and genital herpes, and are seen in young adults. Pain and swelling occur, and the vesicles become pustular, but, if in well keratinized areas, do not always ulcerate. Associated lymphangitis is common. Primary lesions take up to 21 days to heal, recurrent ones 10 days. Figure 43.3 shows recurrent lesions at an interval of 10 years.
Eczema herpeticum
A severe form of cutaneous herpes may occur in children with atopic eczema – eczema herpeticum or Kaposi’s varicelliform eruption. Vesicles resembling those of chickenpox may appear, mainly on already eczematous areas. Extensive ulceration results in protein loss and dehydration, and viraemia can lead to disseminated disease with severe, even fatal, consequences. A similar picture is seen occasionally in adults with pemphigus who develop herpes simplex. Patients with burns are also at risk. In each instance, early recognition and prompt antiviral therapy can be life-saving.
Eye infection
HSV infection of the eye may be initiated during a childhood primary infection, or occur from transfer of virus from a cold sore. There may be periorbital herpetic dermatitis together with conjunctivitis, or keratoconjunctivitis associated with corneal ulceration. Typically, branching (dendritic) corneal ulcers are found. If these recur untreated, the result is corneal scarring and impairment of vision. More extensive ulceration occurs if steroids have been used, but deeper infiltrates are common in long-standing cases and benefit from combined steroid and antiviral therapy. The presence of typical herpes vesicles on eyelid margins is a useful clinical guide but is not always seen. The majority of eye infections are with HSV 1, and most patients with recurring eye disease are aged over 50 years. More than half of the corneal grafting performed in the UK is for HSV corneal scarring, although the disease may recur in the graft. Acute retinal necrosis associated with HSV 1 or 2 is also recognized.
Central nervous system infection
HSV may reach the brain in several ways. Viraemia has been detected during primary herpetic stomatitis, and infection may be carried within cells into the brain and meninges. Direct infection from the nasal mucosa along the olfactory tract is another possibility, but the most likely route is central spread from the trigeminal ganglia.
HSV encephalitis
Encephalitis caused by HSV is a rare condition, but is the most common sporadic fatal encephalitis recognized in developed countries (1–2 cases per million population annually). The infection has a high mortality rate and significant morbidity in survivors of the acute necrotizing form. It presents:
Some 70% of cases are in people with serological evidence, and often a clinical history, of previous HSV infection. Recurrent lesions are seldom apparent at the same time as encephalitis. A prodrome of fever and malaise is followed by headache and behavioural change, sometimes associated with a sudden focal episode such as a seizure, or paralysis; coma usually precedes death. The temporal lobe is most frequently affected, and virus replication in neurones, followed by the oedema associated with the inflammatory response, accounts for the haemorrhagic necrosis and space-occupying nature of this form of the disease. More diffuse, milder disease has been recorded. Brainstem encephalitis is another serious manifestation.
Examination of cerebrospinal fluid (CSF) often demonstrates the presence of red cells and a lymphocytic response, but these findings are non-specific. Neuro-imaging can often suggest the diagnosis, the typical feature being the presence of focal lesions on CT or MRI (more sensitive) scans. Virus-specific diagnosis is considered below. Early clinical recognition, leading to prompt antiviral therapy, significantly reduces the 70% mortality rate and serious morbidity of untreated cases, but therapy must be started as soon as possible before confirmation of the diagnosis. HSV 1 is responsible for most cases of encephalitis where virus has been identified (outside the neonatal period; see below). HSV 2 encephalitis does occasionally occur in immunocompromised adults and may be seen more frequently in those infected with the human immunodeficiency virus (HIV).
HSV meningitis
Aseptic meningitis caused by HSV is much less serious than encephalitis. HSV 2 is the usual cause and it reaches the CSF following radiculitis during genital herpes (see below). A lymphocytic reaction is seen in CSF, and there may be recurrent bouts of this meningitis (sometimes known as Mollaret’s meningitis). HIV-infected patients may present with HSV meningitis.
Genital tract infection
Both types of HSV can infect the genital tract. Although the more common association has been with type 2 virus, type 1 infection is not infrequent, particularly in young women, where it may account for more than half of genital infections. Genital infection may be acquired by autoinoculation from lesions elsewhere on the body, but most often results from intimate sexual contact, including oro-genital contact. The lesions are vesicular at first, but rapidly ulcerate.
• In the male, the glans and shaft of the penis are the most frequent sites of infection.
• In the female, the labia and vagina, or cervix, may be involved.
• In both sexes, lesions may spread to surrounding skin sites.
The incubation period is 2–20 days with an average of 7 days. A primary infection is usually the most severe, especially in women. Fever and malaise are accompanied by regional lymphadenopathy and urethritis, and vaginal discharge may be present. The whole episode lasts for 3–4 weeks, and high titres of virus are shed. In some cases, a lymphocytic meningitis develops, and urinary retention can also be a problem; these are manifestations of sacral radiculopathy. Where the infection is an initial genital herpes (but not a primary HSV) infection (see Table 43.2), the attack is generally less severe, but may still last for about 2 weeks.
Recurrent genital herpes
This can be as frequent as six or more episodes a year. Although the attacks are milder and shorter than first episodes (around 7–10 days), the results are socially and psychologically distressing. Some patients experience prodromal symptoms in the distribution of the sacral nerves, but the patient is already infectious by this stage. Virus shedding from the genital tract is often asymptomatic. The patient or general practitioner may not recognize recurrent lesions as herpetic in origin, so that, in many instances, the risk of transmission to sexual partners is not appreciated. HSV 1 genital infection recurs less often than HSV 2, and, thus, carries a better prognosis. Either type is capable of transmission from mother to infant. Transplacental passage resulting in intra-uterine damage to the fetus has been recorded but is very rare, and probably limited to cases with substantial maternal viraemia. Ascending infection from the cervix may be more significant, especially when the membranes are ruptured for some time before delivery.
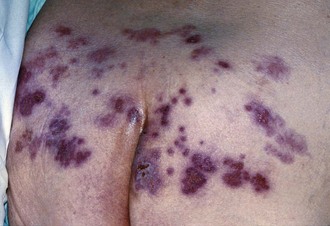
Genital herpes can be a significant problem in immunosuppressed patients, and may be seen as persistent, severe, peri-anal lesions (with or without proctitis) in many HIV-infected men that have sex with men (MSM). Genital herpetic ulcers are known to increase the risk of transmission of infection with HIV. Figure 43.4 shows recurrent HSV 2 over the sacrum in an elderly person; the latent virus in the sacral ganglion travels to the skin dermatome served as well as the internal mucosal site; the patient had been nursed on a rubber ring.
Neonatal herpes
A rare (1.65 per 100 000 live births in the UK) but very serious infection, untreated neonatal herpes has a case fatality rate exceeding 60% with half of the survivors severely damaged. Virus, commonly HSV 2 (but this will vary according to a region’s prevalence of HSV 1 infection of the genital tract), is acquired by passage through an infected genital tract. The greatest risk, with a 50% transmission rate (because there is more virus present and no transplacental antibody transfer has occurred), is when the mother has a primary HSV infection at the time of delivery. In contrast, with recurrent herpes at term, the transmission rate is only 5% or less.
Neonatal infection may present in 3 different ways:
1. Skin, eye and mucous membrane (SEM) disease at about 10–12 days post-partum. Vesicular lesions on the skin may be absent, or few in number, and are commonly located on the presenting part (e.g. the scalp; sites of trauma). Virus dissemination is the most serious complication and, without prompt antiviral treatment, disseminated disease (see below) will occur in 75% of cases with signs of general sepsis, including fever, poor feeding and irritability.
2. Disseminated disease presents during the first few days (often around day 6) of life with pneumonitis and hepatitis (manifest as hepatomegaly and jaundice) with or without signs of aseptic meningitis or encephalitis. Progressive liver failure with coagulopathy leads to death around day 16 in the most serious disseminated form.
3. Encephalitis (with or without dissemination to internal organs) manifests itself at around 2–3 weeks of age and, in the absence of prompt antiviral treatment, causes death or severe neurological morbidity.
Prompt high-dose antiviral therapy is the key to survival of the neonate with minimal morbidity, although local recurrent lesions can be expected, especially at skin sites, in the first year.
The prevention of neonatal herpes is difficult, the vast majority of infections occurring in babies born to women with no past history of genital herpes, and in whom the infection at term was either asymptomatic, or unrecognized clinically. Routine pre-term screening for virus shedding, particularly as applied to those with a past history, does not predict babies at risk. With the availability of type-specific antibody tests, susceptible women at risk of acquiring HSV 2 from partners can be identified. The discovery of lesions compatible with primary HSV infection during pregnancy necessitates consideration of antiviral therapy for the mother and (when occurring during the latter stages of pregnancy) caesarean section. If suspicious lesions are seen during labour, swabs (in virus transport medium, VTM) for virus culture and/or DNA amplification by PCR should be taken. Caesarean section may reduce the risk of infection if performed in the early stages of labour (before rupture of membranes, or shortly thereafter). However, if the mother is known to have moderate levels of antibody, the baby is unlikely to develop the disease.
Some cases of neonatal herpes are acquired just after birth from contact with sources of HSV other than the mother’s genital tract. The clinical presentation is similar, although the virus is likely to be HSV 1 from oral or skin lesions of attendants or relatives.
In a case of suspected neonatal herpes, nasopharyngeal secretions, swabs (in VTM) from the skin, mouth and conjunctiva alongside CSF, urine and blood samples should be cultured and/or analysed by PCR.
Laboratory diagnosis
Virus detection (by culture and/or PCR) and serological studies both have their place in the diagnosis of infection with HSV. In all instances of acute infection, be it primary or recurrent, virus detection (e.g. using direct vesicle swabs sent in VTM) is the method of choice, as antibody responses are much less informative. Indeed, in recurrent episodes, the antibody titre may not vary. However, sensitive assays for immunoglobulin (Ig) G antibody, including type-specific antibody, have an important place in prospective testing.
Herpesvirus detection
Direct diagnosis of HSV infection is available and should be sought in cases where there is any doubt as to the clinical diagnosis, or where rapid confirmation is required to guide the choice of therapy or other management.
Detection of viral DNA by PCR is the most sensitive and specific method of diagnosis. The two HSV types can be differentiated by using either type-specific primers in the PCR, or common primers followed by analysis with restriction enzymes or hybridization probes for each type.
Isolation of HSV is carried out in cultures of human diploid fibroblast cells. Growth is rapid, and within 24 h a cytopathic effect (CPE) may be visible, presenting as rounded, ballooned cells in foci that later expand and eventually involve the whole cell sheet. Virus is released from infected cells into the culture fluid – hence, the rapid spread of infection.
Herpesvirus particles may be demonstrated by electron microscope (EM) analysis of vesicle fluid or tissue preparations. Detection of viral antigens in cells by immunostaining (the use of labelled monoclonal antibodies directed against viral antigens) provides a rapid diagnosis on cells scraped from the base of lesions. In the most serious infections, easy access to the site of infection may not be possible.
Antibody tests for HSV
Complement fixation tests (CFTs) are useful in the diagnosis of primary infections, when a significant change in antibody titre can be expected, but the assays are not widely available any longer in the UK. In the case of herpes encephalitis, a serological diagnosis depends on the demonstration of intrathecal synthesis of antibody to HSV. CFTs can be used, testing serum and CSF in parallel against HSV antigens and another unrelated antigen. In health, there is no antibody detectable by such tests in the CSF. If the serum antibody to CSF antibody ratio is diminished from the normal 200 : 1 to 40 : 1 (or less), and the blood–CSF barrier integrity is confirmed by other antibody (or albumin) being excluded from the CSF, intrathecal antibody synthesis is demonstrated. It is also helpful to check for the possible transfer of antibody from blood to CSF by means of an IgG index performed on the same serum and CSF samples. Tests on serum alone cannot confirm herpes encephalitis.
Enzyme immuno-assays (EIAs) are much more sensitive and specific than CFTs. When type-specific antigen preparations based on gG are used, type-specific antibody can be detected. Therapy often results in delayed appearance of these type-specific antibodies, and late convalescent samples (at 6 weeks) should be included.
Treatment
Specific antiviral therapy has revolutionized the management of HSV infections over the past 30 years. Before the development of agents suitable for systemic use, topical application of the relatively non-selective idoxuridine was used successfully in the treatment of eye and skin infections. Aciclovir has a better therapeutic ratio and proven efficacy, when used early enough in appropriate dosage, for the whole range of acute HSV infections. Latency is not eradicated by this agent, which inhibits viral DNA synthesis. Aciclovir can also be used prophylactically to prevent reactivation in the immunocompromised individual, and long-term suppressive therapy has been particularly successful in the management of frequently recurring genital herpes and HSV-related erythema multiforme. Patient-initiated early treatment can also abort or modify recurrences.
Aciclovir is the most widely used treatment for HSV, and the drug has an excellent safety record; it is available in preparations for topical, oral and intravenous use. It can be used in pregnancy as there is no evidence of adverse effects in infants of treated women although such use is not specifically licensed in the UK and, thus, requires careful risk assessment. Topical cream or ointment is suitable only for mild epithelial lesions, such as recurrent cold sores. Oral or intravenous therapy with aciclovir should be given for:
• any lesions that are not simply mild and superficial ones
• HSV-associated disease in the immunocompromised host
• CNS and systemic infection (intravenous therapy is required)
Dosage varies considerably, depending on the site of infection and whether the aim is suppression of recurrence, or therapy of established disease. Because the level of aciclovir achieved in the CSF is only half that in plasma, the dosage for the treatment of encephalitis has to be twice that for other systemic disease. Therapy must be maintained until clinical signs indicate a favourable response. In serious systemic disease, or in the severely immunocompromised host, therapy is continued for 2–3 weeks, or longer. In the neonate, 3 weeks’ therapy at a higher (intravenous) dose should be followed by suppressive oral treatment for 6–12 months.
The poor bio-availability of oral aciclovir has led to the development of a pro-drug, valaciclovir, which is rapidly converted into aciclovir, producing significantly higher plasma levels after oral dosage. Another effective antiherpes agent, penciclovir, is given in the form of an oral pro-drug, famciclovir. Both of these agents are licensed for treatment of genital herpes, and may be administered less frequently than aciclovir.
Resistance to aciclovir can develop. The most common forms are HSV strains with deficient or altered virus-encoded thymidine kinase (TK); hence they cannot phosphorylate aciclovir to a monophosphate form which is key to further phosphorylation by cellular kinases into the triphospate active form. This has not been a significant clinical problem but it must be remembered, particularly in the severely immunocompromised host (e.g. acquired immune deficiency syndrome (AIDS), or following bone marrow transplant). A few resistant viruses with altered virus-encoded DNA polymerase have also been isolated and associated with clinical disease. With widespread use of aciclovir, more resistant strains may arise, and monitoring of the antiviral sensitivity of HSV isolates may be necessary. Strains resistant to aciclovir are generally also resistant to famciclovir, and foscarnet may be used as it does not require activation by viral TK.
Epidemiology
HSV is probably transferred by direct contact with vesicular lesions and/or infectious fluid. Many children, especially in over-crowded conditions, acquire oral HSV 1 infections in the first years of life. Spread may not occur so readily in better social conditions with the result that primary infection is often delayed into young adulthood in developed countries. This is the usual time of exposure to genital herpes and, as a result, primary infections may be HSV 2 or HSV 1.
Sensitive EIAs, and type-specific assays, have shown that:
• some 60–90% of adults have had an HSV 1 infection
• many more adults have had HSV 2 infection than give a history of genital herpes.
Neonatal herpes is a rare complication in the UK. The rate of cases has increased in some populations as genital herpes has become more common.
Control
Transmission of herpes simplex can be reduced by:
• simple hygiene measures (e.g. attention to adequate hand hygiene)
Reference has already been made to the prevention of neonatal infection as has the use of prophylactic antiviral regimens to control predictable recurrence. Progress in understanding latency and reactivation will provide approaches to preventing reactivation. Protection from ultraviolet light, and the use of inhibitors of prostaglandin synthesis, may be useful in this context.
Experimental vaccines are under investigation, but none is licensed for use. Research into subunit vaccines based on the viral glycoproteins, or other significant viral proteins, may lead to an appropriate preparation to elicit the immune responses important in control of HSV. Recent trials have shown limited protection against genital tract disease. A vaccine that stimulates T cell immune responses may be required.
Varicella–zoster virus (VZV)
Infection with VZV presents in two forms:
1. Primary infection: varicella (or chickenpox) is a generalized eruption.
2. Reactivated infection: zoster (or shingles) is localized to one (or a few) dermatomes.
Description
The viruses isolated from varicella and zoster are identical. The virus has the morphology of all herpesviruses. Seventy genes code for 67 different proteins, including five families of glycoprotein genes. The glycoproteins gE, gB and gH are abundant in infected cells, and are present in the viral envelope (Fig. 43.5).
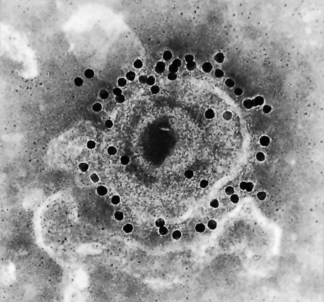
Fig. 43.5 VZV showing the virus envelope glycoprotein I (gE) labelled with monoclonal antibody and goat anti-mouse IgG conjugated with 15 nm colloidal gold (original magnification ×150 000).
(Micrograph taken by C. Graham, supplied by Dr E Dermott, Department of Microbiology and Immunobiology, Queen’s University, Belfast, UK.)
Following attachment (mediated primarily by gB) to cell surface glycosaminoglycans and fusion, the nucleocapsid enters the cell and viral DNA is released into the nucleus where virus replication takes place. Histological examination of infected epidermis reveals typical nuclear inclusions and multinucleate giant cells identical to those of HSV. Human fibroblast cell cultures are most often used for isolation. Enveloped virus released from the nucleus remains closely attached to microvilli along the cell surface; studies of this ‘cell-associated’ characteristic, with infection being passed from cell to cell, have been limited compared with studies of the lytic HSV. The typical CPE appears in cell cultures in 3 days to 2–3 weeks.
Recently, the presence of geographically distinct VZV genotypes has been described, but little is known about the clinical significance of these findings. Antibodies to the three main glycoproteins all neutralize virus infectivity. One of these glycoproteins, gB, shares 49% amino acid identity with the gB of HSV, and this may account for the cross-reactive, anamnestic, antibody response that may be detected during infections with either virus.
Pathogenesis
Varicella
This is a disease predominantly of children characterized by a vesicular skin eruption (chickenpox; the origin of the term is unclear: from chickpea (the bean; refers to the look of the lesion) or the Old English term gican (‘to itch’)). Virus enters through the upper respiratory tract or conjunctivae, and may multiply in local lymph tissue for a few days before entering the blood and being distributed throughout the body. After replication in reticulo-endothelial sites, a second viraemic stage precedes the appearance of the skin and mucosal lesions. An alternative model has been proposed, based on studies in a humanised severe combined immunodeficient (hu-SCID) mouse model (bearing human skin xenografts). The work showed that activated human tonsillar T lymphocytes could be infected and, if given to the mice intravenously, homed rapidly to the skin grafts where VZV could be detected by 7 days (although the typical epidermal vesicular lesion did not appear for 10–21 days).
The VZV vesicles lie in the middle of the epidermis, and the fluid contains numerous free virus particles. Within 3 days, the fluid becomes cloudy with the influx of leukocytes; fibrin and interferon are also present. The pustules then dry up, scabs form, and they desquamate. Lesions in all stages are present at any one time while new ones are appearing. The clearance of virus-infected cells is dependent on functional T lymphocyte-mediated immune mechanisms and antibody-dependent cell cytotoxicity (ADCC). Persons deficient in these responses, and in interferon production, have prolonged vesicular phases and great difficulty in controlling the infection.
Zoster
The pathogenesis of zoster (shingles) is not so well established as that of HSV recurrence. The latent virus is found in neurones and satellite cells in sensory ganglia, and more than one region of the genome is transcribed, although the state of the latent VZV is largely unknown. It seems likely that virus reaches the ganglion from the periphery by travelling up nerve axons, as HSV does, but there is also the possibility that, during viraemia, some virus enters ganglion cells. Another difference from HSV latency lies in the persistent VZV expression that has been detected in some mononuclear cells; this may have a role in VZV disease such as post-herpetic neuralgia (see below).
Reactivation of VZV as zoster can occur at any age in a person who has had a primary infection, which may or may not have been apparent clinically. The rate is greatest in persons aged ≥60 years and, as most primary infection takes place before the age of 20 years, there is usually a latent period of several decades. However, a much shorter latent period is seen in immunocompromised patients, and also in those who acquired primary infection in utero (see below) or in the first year of life. More than one episode of zoster is uncommon in any individual. The stimulus to reactivation is largely unknown, but virus travels from sensory ganglia to the peripheral site. Zoster is usually limited to one dermatome; in adults, this is most commonly in the thoracic or upper lumbar regions, or in the area supplied by the ophthalmic division of the trigeminal nerve. It is thought that this distribution is related to the density of the original varicella rash. There are associations with preceding trauma to the dermatome – injury or injections – with an interval of 2–3 weeks before the zoster appears. There is an associated suppression of specific T cell-mediated responses in acute zoster, but rapid secondary antibody responses are usually found. Reactivation occurs more commonly in T cell immunodeficiency states.
Viraemia may occur in the course of zoster but is unusual with pre-existing immunity normally being boosted rapidly. In the immunocompromised host, however, viraemia may lead to dissemination of zoster, either to internal organs or in a generalized manner reminiscent of varicella (‘disseminated zoster’).
Clinical features
Varicella
The incubation period averages 14–15 days but may range from 10 to 21 days. The patient is infectious for 2 days before, and for some days after, onset, while new vesicles are appearing and until all vesicles have crusted over.
The rash of varicella (chickenpox) is usually centripetal, being most dense on the trunk and head. Initially macular, the rash rapidly evolves through papules to the characteristic clear vesicles (‘dew drops’).
Presentations vary widely – from the clinically inapparent through to a severe febrile illness with a widespread itchy rash, especially in secondary cases in older members of a household. Usually a relatively mild infection in the young child, the complications of varicella, much more likely in adults, may cause significant morbidity and even mortality.
In the past, the main differential diagnosis was smallpox (Table 43.3). Another possibility is monkeypox if the travel or occupational history (or exotic pet handling) is suggestive of such exposure.
Table 43.3 Key differences between rashes of varicella and smallpox
| Feature | Varicella | Smallpox |
|---|---|---|
| Characteristic lesions | Blisters: domed small superficial single vesicles with clear fluid | Several layers of cells cover multifocal vesicles; early umbilication seen |
| Distribution | Centripetal: most on trunk, neck, face, proximal parts of limbs | Centrifugal: face, forearms and palms, soles of feet |
| Appearance at any one time | Pleomorphic: papules, vesicles and crusts may all be seen in one area | Lesions all at same stage in one affected area |
Secondary bacterial infection of skin lesions is the most common complication of varicella, mainly in the young child, and increases the amount of residual scarring. Thrombocytopaenic purpura occurs, especially in the immunocompromised host, and this may lead to haemorrhagic chickenpox which is life-threatening. A variety of organs may rarely be affected producing arthritis, myocarditis, hepatitis, glomerulonephritis and/or appendicitis. However, the two most frequent problems are related to the lungs and the CNS.
Pneumonia
VZV pneumonitis is a serious complication, even in immunocompetent individuals. It is more likely to occur in adults, smokers, pregnant women, and immunocompromised hosts. Severity may vary from subclinical (evident only on radiography) to life-threatening. Cough, dyspnoea, tachypnoea and chest pains begin a few days after the rash. Nodular infiltrates are seen in the lungs radiologically. Prompt antiviral therapy (see below) must be used in sufficient dosage and started at the first sign of pneumonia. Early investigations (imaging and gas exchange) are indicated in all smokers and immunocompromised patients with chickenpox.
Central nervous system
Neurological complications include the common but benign cerebellar ataxia syndrome, and aseptic meningitis. Acute encephalitis is rare but more serious, and occurs mostly in the immunocompromised host. It may be confused with post-infectious encephalopathy, which, with other post-infectious manifestations such as transverse myelitis or Guillain–Barré syndrome, is immunologically mediated and not related to viral cytopathogenicity.
Varicella in pregnancy
VZV can cross the placenta following maternal viraemia and infect the fetus. The infection may be more serious for the mother herself in pregnancy, with pneumonia being the major problem. Two types of intra-uterine infection are noted:
1. Congenital varicella syndrome (CVS) is a rare consequence of fetal infection in the first half of pregnancy (<1% of fetuses if maternal infection occurs during first 12 weeks of gestation; 2% if maternal infection occurs during 13–20 weeks of gestation). The characteristic features, usually unilateral, are scarring of the skin, damage to the musculoskeletal system (muscular atrophy, hypoplasia of the limbs, rudimentary or missing digits), as well as to the CNS (cortical atrophy, psychomotor retardation) and eyes (chorioretinitis, cataracts). Fetal infection is not inevitable. Silent intra-uterine infection can also occur: no damage is seen, but the baby is born with latent VZV infection, which may manifest as zoster in the first year of life.
2. Neonatal varicella is defined as varicella developing within the first 4 weeks of life, usually acquired from maternal varicella in late pregnancy (although, rarely, the exposure may occur post-natally, e.g. to an older sibling with chickenpox). The interval between maternal viraemia and delivery is important. If the maternal rash begins 7 days or more before delivery, her antibody response will have developed and been transferred across the placenta so that the baby does not develop disease. However, the infant is at serious risk if maternal varicella occurs 6 days or less before, or up to 2 days after, delivery; this allows viraemic spread across the placenta, before antibody is made and transferred to the baby. Because the usual respiratory entry route has been bypassed, the incubation period is reduced to (on average) 10 days. If infection arises, serious (and even fatal) disseminated disease may develop including pneumonitis and encephalitis. Such exposed neonates should be given passive immunization (see below) and consideration of prompt antiviral therapy, as should neonates of seronegative mothers exposed to any other source of VZV within the first 4 weeks of life.
Zoster
This is the manifestation of reactivated VZV infection. Zoster takes the form of a localized eruption, is unilateral, and typically confined to one dermatome. Prodromal paresthesiae and pain in the area supplied by the affected sensory nerve are common before the skin lesions develop; these are identical to those of varicella except in their distribution. The evolution of the rash is similar, with some new vesicles appearing whilst the earliest ones are crusting; however, the whole episode in the majority of cases is confined to the affected dermatome and heals in 1–3 weeks. Acute pain is not always a feature, but its presence should alert to the possibility of zoster, and a search for early lesions, perhaps internal, is indicated. Occasionally there are no skin lesions – ‘zoster sine herpete’.
Disseminated zoster is indicated by lesions appearing in the skin at distant sites (resembling the clinical picture of chickenpox) or, more seriously, by involvement of internal organs such as the lung and CNS (meningitis, encephalitis or myelitis). In the immunocompromised host, this results in severe disease with occasional fatalities. Patients with internal organ zoster may or may not present with a typical skin rash.
Post-herpetic neuralgia
This is the most common complication of zoster, a risk in 50% of patients aged over 60 years, and results in significant morbidity in around 20% of cases. It is defined as intractable pain persisting for 1 month or more after the skin rash. Constant pain at the site, or stabbing pains or paraesthesiae, may continue for 1 year, or much longer in a number of individuals. This is an exhausting and disabling condition for which no satisfactory cure has been found; adequate early antiviral therapy may reduce the incidence.
Ophthalmic zoster
Involvement of the ophthalmic division of the trigeminal nerve occurs in up to one-quarter of zoster episodes, with ocular complications in more than half of the patients. Corneal ulceration, stromal keratitis and anterior uveitis may result in permanent scarring, so this complication may threaten sight when the nasociliary branch is involved. Ocular complications are reduced in patients given oral aciclovir early in ophthalmic zoster. Occasionally, acute retinal necrosis is identified.
A contralateral hemiparesis due to granulomatous cerebral angiitis in the weeks following acute ophthalmic zoster is a recognized neurological complication. A more acute one, such as the Ramsay–Hunt syndrome (facial palsy with aural zoster vesicles), suggests that motor neurones can also be involved. Sympathetic ganglia may also be the site of latency, as indicated by cases in which the initial recrudescence has been in gastric mucosa with subsequent dissemination.
Recurrent and chronic VZV
Immunocompromised individuals, most particularly those with CD4+ T cell lymphopenia due to HIV infection, may develop recurrent and chronic VZV infection. New lesions continue to appear, or re-appear after aciclovir therapy, often presenting an atypical hyperkeratotic appearance. Aciclovir-resistant VZV has been isolated in this situation.
Laboratory diagnosis
Typical presentations of varicella or zoster seldom need laboratory confirmation; however, atypical presentations merit investigation, especially in the immunocompromised host. Vesicular rashes due to enterovirus are sometimes confused with varicella and, in immunosuppressed individuals, various vesicular lesions may be mistaken for zoster. Localized vesicular lesions other than those on the face or genitalia are commonly misdiagnosed as due to zoster (see Fig. 43.4); many are due to recurrence of HSV, and this is readily shown by antigen detection and virus isolation, or by PCR. Thus, the approach to testing of vesicular lesions usually involves simultaneous HSV and VZV assessment by PCR.
Virus detection
Early vesicular lesions provide the best diagnostic material. Vesicle fluid is collected in a capillary tube, aspirated with a fine needle and syringe, or lesions are swabbed directly (and sent in VTM). Direct examination by EM will reveal herpes particles; some of the fluid can be diluted in VTM and inoculated into tissue culture for virus isolation which takes between 5 days and 3 weeks. More rapid detection is possible with centrifugation-enhanced cultures (‘shell vials’). If cells swabbed from the base of lesions are available, or biopsy tissue, virus antigens may be sought by immunostaining assays. VZV DNA amplification by PCR is used for the detection of VZV in CSF or aqueous humour, and routinely now for all types of samples. The latter two methods (immunostaining and PCR) are the ones most used for rapid diagnosis.
Serological diagnosis
Antibody testing with VZV antigens can confirm a diagnosis of varicella by demonstration of seroconversion or rising titres of antibody between acute and convalescent serum samples; CFTs are still useful for this purpose but are not widely available any longer. CFTs are not sufficiently sensitive to determine past infection and, to assess immune status, assays need to be based on enzyme or radiolabelled methods, or immunostaining of infected cells. IgM to VZV is detectable by IgM capture systems in both varicella and zoster, appearing early in zoster. It is increasingly common to test for past infection (and therefore immunity) by measuring IgG antibody to VZV in those who are, or will become, immunocompromised, in women exposed antenatally to VZV, or in healthcare workers (or other adults) who are to be offered VZV immunization (see below).
Treatment
VZV is not as sensitive to aciclovir as HSV, with 50% inhibitory dose (ID50) values ranging from 4–17 µM aciclovir compared with 0.1–1.6 µM for HSV. This means that frequent high-dose oral or intravenous therapy is required against the virus. High-dose aciclovir given intravenously is effective in the treatment of varicella and zoster in the immunocompromised host. Oral aciclovir can be used to accelerate healing and reduce new lesion formation during zoster in immunocompetent patients if given early enough and may lower the rate of postherpetic neuralgia. Alternative preparations such as valaciclovir and famciclovir require less frequent dosing. Trials have shown that high-dose oral aciclovir shortens the course of varicella in immunocompetent children by 1 day if commenced within 24 h of the onset of the rash. The practical difficulties of achieving such an early start of treatment rule out routine use of aciclovir for all cases of varicella in immunocompetent children, but consideration should be given to treating all adults, and (where possible) adolescents and family contact cases who are known to develop more extensive disease. Treatment of VZV infection is given primarily to all ‘high risk of complication’ groups and should, thus, be considered for:
Epidemiology
Varicella is partly seasonal, being spread mainly by the respiratory route in winter and early spring. Some cases result from contact with zoster and occur sporadically in any season. Varicella is highly infectious to susceptible close contacts (as a result of respiratory spread), as in a household; a past history of varicella is a good indicator of immunity since the clinical picture is quite distinct. The majority of children contract VZV between the ages of 4 and 10 years in western countries with around 8% of young adults remaining susceptible (Fig. 43.6). However, a much higher proportion of young adults remain susceptible in sub-tropical countries (for reasons that are not entirely clear). The rate of pneumonitis as a complication of varicella is surprisingly high in otherwise healthy adults (1 in 200; in children, the figure is 1 in 200 000), particularly pregnant women and smokers, who develop pneumonia in up to 10% of cases. Adult VZV pneumonitis may prove fatal without prompt antiviral therapy. Zoster is associated with decreased T cell function, and occurs with increased incidence in:
• the pre-AIDS phase of HIV infection
• patients receiving chemotherapy or radiotherapy for lymphoid malignancies.
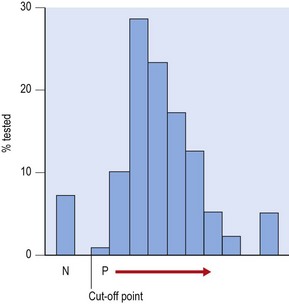
Fig. 43.6 Distribution of antibody (IgG) to VZV in a young adult population (southern England). The proportion confirmed as negative (N) was 7.8%. P →, increasingly positive result.
In developed countries, the increasing survival of people beyond 65 years of age means that the incidence of zoster will increase. This may be exacerbated by less natural boosting of immune responses as VZV decreases in countries offering childhood immunization for varicella.
Control
Passive immunization
Passive immunity is partly protective for varicella as seen in infants with maternal antibody or patients given varicella–zoster immunoglobulin (VZIG) within 72 h of exposure. VZIG (in the UK), or another similar high-titre antibody preparation, is available for neonates, non-immune pregnant contacts, or immunocompromised contacts, of VZV. As most pregnant contacts will be immune, testing for VZV IgG antibody after exposure may prevent unnecessary use of costly VZIG. Current preparations seldom prevent infection, but do ameliorate disease. Importantly, VZIG reduces the rate of transmission to the fetus.
Varicella vaccine
A live-attenuated varicella vaccine has been in use for some years in Japan and some European countries and was approved in the USA for routine childhood immunization in 1995. The vaccine strain (Oka), which can be distinguished from wild-type VZV by molecular analysis, is given by intramuscular injection. The vaccine is immunogenic in children with leukaemia in remission and in healthy children and adults; vaccinees have resisted infection on close exposure to varicella. Some symptoms are noted around 10 days post-vaccination, and vesicles appear at the site of injection in up to 5% of individuals. Immunization does not prevent latency developing; however, the incidence of zoster in vaccinees compared with that in the naturally infected is not increased. In the USA, surveillance has confirmed a significant reduction in the incidence of varicella, especially in the 1–4 years age group, and a substantial fall in the mortality and complication rates. There have been reports of ‘breakthrough’ epidemics locally, even in well vaccinated cohorts, and a booster dose may be necessary. A large trial of vaccination in the elderly (over 60 years old) showed a 60% reduction in the incidence of zoster and post-herpetic neuralgia in those immunized with a high-titre preparation of Oka virus-based vaccine. Currently, in the UK, the VZV vaccine is not offered as part of the childhood immunization programme but, instead, to susceptible individuals who are in regular contact with those at risk of developing serious VZV illness. Thus, the vaccine is offered to non-immune health-care workers and healthy contacts of immunocompromised patients. The primary course consists of two vaccine doses administered 4–8 weeks apart.
Epstein–Barr virus
In 1964, Epstein, Barr and Achong described herpesvirus particles in cells from a lymphoma in African children studied by Burkitt, who suspected an infectious aetiology of the tumour. The link between the new herpesvirus, denoted Epstein–Barr virus (EBV), and a variety of epithelial and lymphoid tumours is now clear. Primary infection with EBV
• is most often acquired in childhood when it is generally asymptomatic
• gives rise to infectious mononucleosis (also known as glandular fever) in up to a quarter of individuals when infection is delayed into adolescence.
Man is the only natural host, but EBV infection can be transmitted experimentally to some non-human (New World) primates (tamarin, marmoset; Old World primates are naturally immune as a result of infection with simian EBV homologues).
Description
The characteristic morphology of EBV seen on EM is that of a herpesvirus. EBV cannot be grown in human fibroblast or epithelial cell lines and there is no completely productive or permissive system for culture of EBV. This lymphotropic virus is classified in the gammaherpesvirinae subfamily, genus lymphocryptovirus.
Replication
The full replication (productive, or lytic) cycle of EBV can take place in certain differentiated epithelial cells although it is not clear whether this is part of the natural life cycle of EBV infection in the human host. The B lymphocyte is the principal and essential cell infected through attachment of the major viral envelope glycoprotein gp350/220 to EBV receptors (primarily, the CD21 molecule) expressed on mature resting B lymphocytes. There are other receptors on cells of stratified squamous epithelium (e.g. in the oropharynx and ectocervix) and these epithelial cells can be infected through their basal aspect. Viral production is restricted to the differentiated cells of the granular layer and above, and virus is shed from the superficial cells. It is difficult to grow differentiating epithelia in culture, and most work on the lytic cycle of EBV has been done in B lymphoblastoid cell lines (BLCLs) in which a small proportion of the EBV-infected cells can be induced to produce virus.
The organization of the EBV genome differs from that of HSV, and some genes are present in one and not in the other. Approximately 80 proteins are encoded; many glycoproteins (gp) are known, including the major gp350/220, which mediates attachment to CD21, as well as gp25, gp42 and gp85, which bind a co-receptor (MHC class 2 molecules) on the target cell and are involved in membrane fusion. The latent (non-productive) state of EBV infection is established in a subset of resting memory B lymphocytes in which the EBV genome is maintained in the nucleus as multiple full-length covalently closed circular episomes. Specific EBV-encoded small RNA species (EBERs) are found in all cells infected with the virus. A variable number of EBV genes are expressed in the different forms of the latent state. These are principally genes coding for one or more of six EBV nuclear antigens (EBNA leader protein (LP), 1, 2, 3a-c) and two latent membrane proteins (LMP 1, 2). EBNA 1 maintains the EBV episome as the B cells divide, and is the only latent protein expressed in all types of EBV-associated tumours (see below). EBNA 2 and LMP 1 are both viral oncogenes that promote B cell proliferation. Latency involves different expression patterns of these latent viral proteins: all are expressed by BLCLs in culture as well as at the outset of primary EBV infection (unrestricted latency) which is followed by down-regulation of some, or all, of them (restricted latency) in the course of infection. Whilst all the lytic and latent proteins are involved in recognition by EBV-specific T cells that mediate immune control of the virus, such down-regulation of latent genes is one mechanism that facilitates persistent infection and immune evasion. An unrestricted latent virus expression profile is observed during infectious mononucleosis and in EBV-associated post-transplant lymphoproliferative disease whilst more restricted patterns are seen in other malignancies associated with this virus (see below). Interestingly, EBV encodes a homologue of interleukin (IL) 10 (viral IL 10, vIL10) that may play a role in immune modulation.
Although there are two types of EBV, type 1 and 2, their disease association is similar and no unique clinical significance has been attributed to either type.
The full lytic cycle of EBV replication is accompanied by the production of a large number of lytic proteins that include virus structural antigens resulting in the assembly of progeny virus. Although EBV does encode a viral TK, aciclovir is phosphorylated by cellular kinases in cells producing EBV. The viral DNA polymerase is sensitive to the active aciclovir triphosphate, and treatment with aciclovir reduces EBV production (at least in vitro) but has no effect on latency or the B cell proliferation induced by the virus.
Pathogenesis
In primary infection, virus in saliva is thought to infect oropharyngeal B lymphocytes in tonsillar crypts, and the exact role of epithelial cells in the process is still unclear. This leads to activation of the infected cells, which progress to the local lymph nodes and on through the circulation with the potential to enter a productive (lytic) phase and release of progeny virus elsewhere in the body. Most shedding of virus takes place in the oral cavity (epithelial cells may have a role in this aspect of infection), and EBV can be detected regularly in the saliva of asymptomatic hosts, the amount increasing in immunosuppressed states.
Activated B lymphocytes secrete immunoglobulin, and EBV is a potent polyclonal activator of antibody production by B cells, independent of any accessory cells.
Recovery from primary EBV infection is associated with humoral and, primarily, cellular immune responses involving T cells; any delay in cellular control, or over-vigorous responses, will contribute to the severity of the infection. Thus, large initial infective doses may result in high numbers of circulating infected B lymphocytes followed by a marked T cell response. The polyclonal B cell activation results in the transient appearance of a variety of antibodies (predominantly IgM), both autophile and heterophile. The cellular response is detected as a mononucleosis with large numbers of atypical lymphocytes in blood and infiltrating tissues that are EBV-specific T cells.
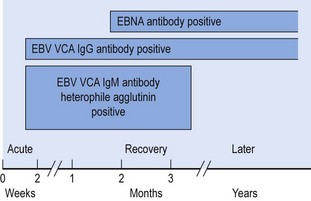
Antibody responses after EBV infection follow a characteristic pattern, with the initial IgM response to virus capsid antigens (VCA) persisting for some months. The latent EBNA complex elicits antibodies in late convalescence only, perhaps after release from B cells lysed by EBV-specific T cells (Fig. 43.7). Failure to produce antibody to the EBNAs is a feature of immunodeficiency states that may also be associated with increased levels of antibodies to EBV lytic cycle antigens [early antigen (EA) and VCA], reflecting a high virus replication rate. High IgA levels to VCA are found in those at risk of developing EBV-associated nasopharyngeal carcinoma (see below). Antibodies against the major viral envelope glycoprotein gp350/220 are neutralizing and may protect against re-infection.
Clinical features
Primary infection with EBV is usually mild and unrecognized in the vast majority who acquire it in the first years of life.
Infectious mononucleosis/glandular fever
Infectious mononucleosis (IM; or glandular fever, GF) is seen predominantly in the 15–25 years age group when primary EBV infection is delayed into adulthood. The incubation period is 30–50 days, and the onset is abrupt with a triad of sore throat, cervical lymphadenopathy and fever, often accompanied by malaise, headache, sweating (particularly at night) and gastrointestinal discomfort. A mononucleosis of atypical lymphocytes is observed in blood. Pharyngitis may be severe, accompanied by a greyish-white membrane and gross tonsillar enlargement. Lymphadenopathy becomes generalized, often with splenic enlargement and tenderness, mild hepatomegaly (and biochemical hepatitis) in some individuals and clinical jaundice in 5–10% of cases. Intermittent fevers with drenching sweats may occur daily over 2 weeks. A faint transient morbilliform rash may be seen; a maculopapular rash may follow ampicillin administration, due to immune complexes with antibody to ampicillin. The illness can last for several weeks, and prolonged and debilitating fatigue and lack of concentration are common in the aftermath.
Complications of infectious mononucleosis/glandular fever
Other EBV-associated disease, tumours and immunosuppression
EBV is associated with an increasing number of diseases, including malignant tumours (Table 43.4). The role played by EBV in these conditions is not clear in all cases. Cellular immunodeficiency, associated with impaired T cell control of EBV-induced B cell proliferation, may result in EBV-driven B cell lymphoproliferations and lymphomas. This may be inherited (e.g. X-linked lymphoproliferative (or Duncan’s) syndrome (X-LPS) due to a defective immunomodulatory SAP gene), acquired (e.g. in AIDS), or due to iatrogenic immunosuppression following transplantation surgery (post transplant lymphoproliferative disease, PTLD). In African Burkitt’s lymphoma (BL), characterized by translocation of the cellular proto-oncogene c-myc gene into chromosomal locations driven by the highly active immunoglobulin promoters, EBV infection at an early age combines with chronic immunosuppression due to holoendemic malaria to promote further the development of highly aggressive BL tumours (endemic BL, eBL). Sporadic BL (sBL), that arises outwith equatorial Africa, is also associated with EBV in up to 50% of cases. Hodgkin’s lymphoma (HL) is associated with EBV in approximately 50% of cases (particularly the mixed cellularity subset), and the anaplastic subset of nasopharyngeal carcinoma (NPC) is almost always associated with the virus.
Table 43.4 Diseases associated with EBV
| Disease | Cells infected | Link |
|---|---|---|
| Infectious mononucleosis (‘glandular fever’) | Naive B lymphocytes | Causal; acute primary infection |
| Oral hairy leukoplakia (seen in AIDS) | Differentiated epithelium along edge of tongue | Causal; productive recurrence in immunocompromised host |
| Nasopharyngeal carcinoma (especially in South-East Asia and China) | Anaplastic/undifferentiated nasopharyngeal epithelium (long latent period, >30 years) | All malignant cells contain EBV; co-factor(s) play a role and there is genetic risk |
| African (endemic) Burkitt’s lymphoma | Monoclonal B cell tumour (short latent period, >5 years) | All malignant cells contain EBV; holoendemic malaria plays a role |
| Immunoblastic lymphoma (post-transplant/AIDS; X-linked lymphoproliferative syndrome) | Activated B lymphocytes | Over 90% of malignant cells contain EBV; immunodeficiency states, genetic defect |
| Hodgkin’s lymphoma | Hodgkin-Reed-Sternberg cells (germinal-centre B lymphocytes) | 30–90% of malignant cells contain EBV (particular disease subsets) |
| Anaplastic gastric carcinoma | Epithelial cells | Malignant cells contain EBV (particular disease subsets) |
| T/NK cell lymphoma | T/NK lymphocytes | 30–100% of malignant cells contain EBV (particular disease subsets) |
Laboratory diagnosis
IM/GF is accompanied by the production of heterophile agglutinins that can be detected by a rapid slide agglutination test (the ‘Paul–Bunnell test’; related tests include the ‘heterophile antibody test’ and the ‘monospot test’). Agglutination of horse or sheep red blood cells by serum absorbed to exclude a natural antibody is the basis of this test. Atypical lymphocytes, accounting for 20% of the lymphocytosis common in this condition, are seen in blood films. Definitive diagnosis requires the demonstration of IgM antibody to EBV VCA, or seroconversion of VCA IgG antibody. These tests, using indirect immunostaining (immunofluorescence) or, more commonly, EIAs, are generally available. Other serological tests may be applicable to aid serological diagnosis in special situations (e.g. EBNA and EA antibody tests by EIA).
Culture of EBV, from saliva or throat washings, is a research technique. Tissue sections can be immunostained for EBNA or other EBV proteins (Fig. 43.8), or probed for EBERs using in-situ hybridization techniques; these approaches, as well as PCR for EBV DNA in blood, are important in the diagnosis of disease in the immunocompromised host. The role of assessing levels of EBV DNA by PCR following transplantation to predict, diagnose or monitor treatment responses to PTLD is still being evaluated. Such measurements are not straight-forward in the solid organ transplant (SOT) setting although greater success has been achieved using EBV PCR to monitor high risk haematopoietic stem cell transplant (HSCT) recipients.
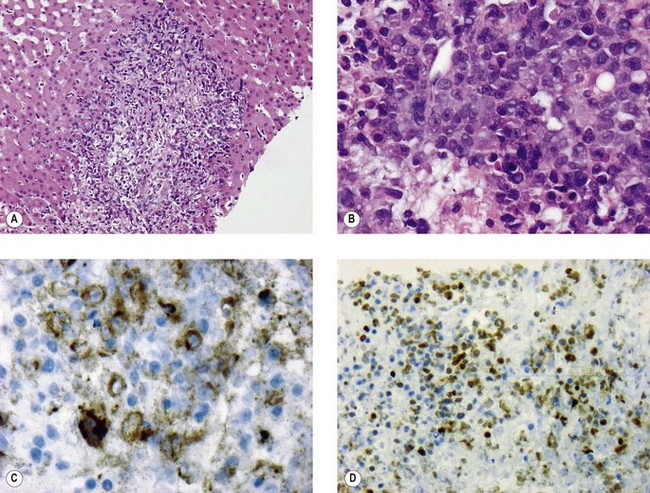
Fig. 43.8 Liver needle biopsy of PTLD in a liver transplant recipient. (A) Portal area infiltrated with high-grade lymphoma showing slight spill-over into adjacent and relatively normal-looking liver tissue. (B) High-power view of the neoplastic infiltrate showing large, atypical, lymphoid blast cells. (C) EBV LMP immunohistochemistry decorates many of the neoplastic lymphoid cells. (D) EBNA 2 immunohistochemistry also highlights many tumour cell nuclei (brown is positive).
(Courtesy of Dr COC Bellamy, Department of Laboratory Medicine, The Royal Infirmary of Edinburgh, UK.)
Treatment
Aciclovir is of little value against EBV disease although it is sometimes included in regimes against PTLD. Whilst reducing immunosuppression in transplant recipients suffering PTLD is an option, this may put the grafted organ(s) at risk. Adoptive humoral immunotherapy using the agent rituximab, a humanized murine monoclonal antibody against the pan-B cell surface marker CD20, is effective against EBV-associated B cell tumours (including PTLD). Similarly, adoptive cellular immunotherapy employing autologous, or MHC-matched allogeneic, EBV-specific T lymphocytes has been shown to be effective in individuals that have not responded to any other treatment for PTLD.
Epidemiology
EBV is transmitted by saliva, and a potential role for sexual transmission has also been proposed. Rarely, transmission has been reported following transfusion of blood products to seronegative recipients. Infection is widespread, with most of the population infected in early life, even in developed countries. Increasing numbers of severely immunocompromised hosts are at risk of developing EBV-driven malignant lymphomas, including SOT and HSCT recipients. This can occur when donor virus is transmitted in grafted tissues or following reactivation of the recipient’s own isolate. Primary EBV infection post transplant is a high-risk situation for PTLD development which explains why paediatric SOT recipients are at particular risk of the disease.
Control
Subunit vaccines based on the major membrane glycoprotein gp350/220 have been undergoing trials and shown to protect against tumour-inducing doses of EBV in a cottontop tamarin model and against IM/GF in seronegative healthy human populations (although not through sterile immunity as EBV infection is not prevented). Screening for IgA antibodies to EBV VCA is used in populations at risk of NPC to detect pre-clinical cases although EBV PCR analysis is rapidly replacing such an approach.
Cytomegalovirus
Human cytomegalovirus (CMV) infects man, but there are other cytomegaloviruses that are specific for other animal species (e.g. murine CMV). The name derives from the CPE of CMV infection that results in a swollen state of infected cells (cytomegaly) in culture and in tissues. Nuclei of productively infected cells contain a large inclusion body, giving a typical ‘owl’s eye’ appearance as can be seen by immunohistochemical staining under the light microscope. Whilst CMV is slowly proliferative in tissue culture, it replicates rapidly in the human body and can pose a serious threat to vulnerable individuals.
Description
CMV has the same general structure as other herpesviruses but its target cell surface receptor(s) is not yet known for certain. Human fibroblast cells are required for isolation of the virus in vitro. In contrast, CMV replicates in vivo in epithelial cells in:
CMV remains highly cell associated, and is sensitive to freezing and thawing. Virus shed in urine is stable at 4°C for many days.
Replication
The temporal regulation of viral protein synthesis in the growth cycle is more obvious in laboratory culture of the slower-growing CMV than with HSV. Non-structural IE protein (p72) appears in nuclei within 16 h of inoculation, whereas structural L proteins are produced after DNA synthesis; the typical CPE is often not recognizable for 5–21 days. Foci of swollen cells expand slowly as infection passes from cell to cell (Fig. 43.9). Passage and storage of virus are best achieved by trypsinization and passage as infected cells.
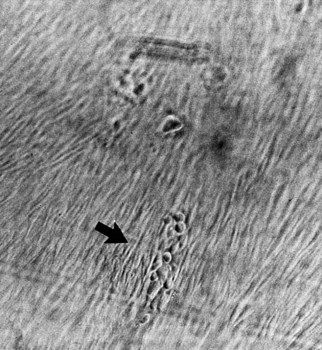
Fig. 43.9 Focus of CMV infection (arrowed) in a tissue culture monolayer of human embryo fibroblasts.
Human CMV does not produce a virus-specified TK; instead, the protein kinase product of CMV gene UL97 carries out initial phosphorylation of ganciclovir in CMV-infected cells, and cellular kinases produce the active triphosphate form of the drug which inhibits the CMV DNA polymerase.
There are several families of glycoproteins in CMV, and these are important antigenic targets. Most neutralizing antibody is directed against gB.
Pathogenesis
Primary infection with CMV may be acquired at any time, possibly from conception onwards and, similar to other herpesviruses, CMV persists in the host for life. Reactivation is common, and virus is shed in body secretions such as urine, saliva, semen, breast milk and cervical fluid. Mononuclear cells carry the latent virus genome and viral RNA transcripts of early genes have been detected in such cells. Bone marrow progenitor cells of the myeloid line may be the prime site of latency. Once their descendants have been activated to differentiate into tissue macrophages, the virus can enter the replication cycle. Recurrent infections may follow reactivation of latent (endogenous) virus, or re-infection with another (exogenous) strain. Isolates can be distinguished by restriction endonuclease analysis or PCR amplification followed by sequence analysis, and by variations in envelope glycoproteins (gB and gH). Endothelial giant cells (multinucleate cells) have been found in the circulation during disseminated CMV infection. These cells are fully permissive for CMV replication.
Intra-uterine infection
Maternal viraemia may result in fetal infection in approximately 1 in 3 cases of primary CMV infection during pregnancy, and may lead to disease in the fetus. Infection may also be acquired in utero when the mother suffers CMV reactivation, but this rarely results in disease. Transplacental infection is probably carried by infected cells, and transmission is associated with a high viraemic load. CMV causes damage to target cells once they have formed rather than acting as a teratogenic agent.
Perinatal infection
This is predominantly acquired from infected maternal genital tract secretions, or from breast-feeding (3–5% of pregnant women in Europe reactivate CMV). In the past, perinatal blood transfusion was rarely the source, but, as leukodepletion (removal of white blood cells) is now practised, this risk has been reduced.
Postnatal infection
This can be acquired in many ways. Saliva containing CMV is spread among young children, and, at older ages, by kissing. Semen can contain high titres of virus, and may be a source of sexual transmission or artificial insemination-associated infection. Whole blood transfusion used to be (and donated organs remain) an important source of CMV. It is not known which donors are most likely to transmit infection, so all CMV IgG antibody-positive (‘seropositive’) cell and organ donors are considered as potentially infectious as the presence of antibody reflects the presence of persistent virus.
Host responses
The host immune response to primary CMV includes humoral (IgM, IgG) and cellular (T lymphocyte) responses. Some of the T cell responses may contribute to immunopathology by reacting with MHC molecules induced by CMV. CMV early genes transactivate other viral and cellular genes, and there may be an important interaction with HIV, leading to the production of HIV from latently infected cells. Because CMV infects mononuclear cells, there is a degree of immunosuppression associated with the acute infection. Cell-mediated responses are crucial to the control of CMV as evident by the serious consequences of disseminated infection in the immunocompromised host. The incubation period for primary infection is 4–6 weeks; reactivation, after transplantation for instance, appears from 3 weeks onwards.
Clinical features
Congenital CMV infection
Congenital CMV infection is asymptomatic at birth in approximately 90% of infected babies, but approximately 10% of these may show sensorineural deafness and/or intellectual impairment in childhood as a result of progressive CMV infection. All congenitally infected infants excrete abundant virus in urine during the first year. Congenitally-infected babies that show symptoms at birth are said to have cytomegalic inclusion disease (CID) with:
Mononucleosis
Postnatal infection with CMV is seldom recognized clinically. Respiratory tract infection is common in infancy and a mononucleosis syndrome (reminiscent of symptomatic primary EBV infection) is seen occasionally, especially in young adults or when CMV is acquired from blood transfusion. Hepatitis, fever and atypical lymphocytosis are noted, but pharyngitis and lymphadenopathy are unusual and heterophile agglutinins are not found.
Infection in the immunocompromised patient
Immunocompromised patients may develop symptoms as a result of primary, or recurrent, CMV infection. Dissemination of the virus in the blood, as indicated by a hectic fever, is a bad prognostic sign. The complications of CMV infection in cellular immunodeficiency include:
• pneumonitis: a high mortality rate in recipients of bone marrow allografts
• retinitis: may occur on its own (10–40% of patients with AIDS)
CMV infection in transplant recipients is a significant cause of direct (caused by the virus) and indirect (caused by virus interactions with the immune system) morbidity that may culminate in loss of the grafted organ and even death. The mortality rate is high, particularly in allogeneic bone marrow recipients who develop an immunopathological pneumonitis associated with graft-versus-host disease. Transplant protocols all include prophylaxis or pre-emptive therapy for the prevention of CMV disease. The former approach entails routine administration of valganciclovir for a period of time post transplantation, and the latter involves regular measurements of CMV viral load in blood by PCR to detect virus with a view to instigating early treatment prior to symptomatic CMV disease.
Retinitis due to CMV recurrence is a feature of late-stage AIDS. Early recognition, antiviral treatment and maintenance therapy are important in slowing the progression towards blindness.
Laboratory diagnosis
Detection of CMV at the site of disease is the aim (if possible). Samples should include urine, saliva, broncho-alveolar lavage fluid, and/or biopsy tissue (if available), and peripheral blood collected in suitable (usually EDTA) anticoagulant.
Virus detection
Rapid diagnostic methods can detect either viral DNA using hybridization or PCR assays, or CMV early antigen in cell cultures 24 h post-inoculation (Fig. 43.10). Conventional culture is used to isolate virus but this approach has been largely replaced by genome detection assays. High titres of CMV produce a CPE very quickly in tissue culture, but most cultures require 2–3 weeks. CMV must be demonstrated in a urine (or saliva) sample taken within the first 3 weeks of life to demonstrate congenital infection as later samples may reflect virus acquired in the postnatal period. Confirmation that CMV is related to a disease process comes from showing the presence of virus (by immunohistochemical or genome detection methods) in the affected tissues, or, in some cases, that a primary infection has occurred. The demonstration of CMV viraemia is a significant prognostic finding, by detection of leukocytes containing CMV late phosphoprotein antigen (pp65) or (nowadays) CMV DNA (by PCR); above certain levels, these approaches are predictive of clinical disease. The quantitative PCR assay has become important in detecting and monitoring CMV following transplantation.
Serology for CMV
CFTs are adequate for showing sero-conversion after primary CMV infection in immunocompetent hosts but the assays are not widely available now. To screen for ‘seropositive’ status, a more sensitive assay, such as EIA for CMV IgG or total antibody (or latex agglutination assay), is appropriate. These tests can be done urgently for organ donor–recipient assessments. CMV IgM is found after primary or secondary infection, but it may not be possible to detect IgM in the neonate or immunocompromised patient.
Treatment
Antiviral agents for CMV infections are available but serious side effects limit their use to life- or sight-threatening complications. Ganciclovir is the agent most often used, given intravenously. Marrow toxicity results in neutropenia, and there can be long-term loss of spermatogenesis. Clinically, treatment has been successful in CMV hepatitis, colitis and encephalitis, and progression of CMV retinitis in AIDS can be controlled with prolonged maintenance therapy. Ganciclovir-resistant virus has been found; usually a mutation in the protein kinase gene (UL97) is responsible but a DNA polymerase (product of the UL54 gene) mutation may also occur. Foscarnet is an alternative agent that does not require phosphorylation but has serious side-effects (e.g. nephrotoxicity). Aciclovir is not effective therapy for CMV, but some benefit from high-dose prophylaxis against CMV in transplant recipients has been noted. Oral ganciclovir has been used for prophylaxis in transplant recipients for several years but has now been replaced by an oral pro-drug, valganciclovir, which provides systemic levels of ganciclovir equivalent to those achieved by intravenous therapy. Cidofovir is an alternative antiviral agent.
Epidemiology
Primary CMV infection is acquired by 40–60% of persons by mid-adult life, and by more than 90% of those with multiple intimate exposures. Fewer than 5% of units of whole blood from seropositive donors result in transmission to seronegative recipients, whereas 80% of kidneys transmit infection from seropositive donors. The full extent of congenital CMV disease is not known, but this infection occurs in approximately 3 of 1000 live births in the UK and is the most common viral cause of congenital infection.
Control
Screening of organ donors (D) and recipients (R) is carried out prior to transplantation to prevent, where possible, a seronegative recipient (R−) receiving a solid organ from a seropositive donor (D+), in order to avoid the high risk D+/R− SOT situation. In the bone marrow transplant setting, a similar high risk situation involves D−/R+ HSCT procedures. Such tissue matching (when possible) reduces morbidity and mortality significantly for all forms of allogeneic transplant. Blood donor screening to select CMV-seronegative units for support of seronegative patients in transplant programmes, and for premature babies, is less important now with the use of leukodepletion. Antiviral prophylaxis (starting from time of transplant surgery), or pre-emptive treatment (starting from time of virus detection in blood), are routine procedures now in SOT and HSCT programmes for those at risk of CMV disease.
No CMV vaccine is licensed for use but trials continue. Experimental live-attenuated vaccines have been tried, but hopes rest on subunit vaccines, or on a combined approach.
Human herpesviruses 6 and 7
Description
The existence of further herpesviruses infecting man was not suspected until one was isolated in 1986 from the blood of patients with lymphoproliferative disorders, some of whom had AIDS. EM revealed a virus with characteristic herpesvirus features. DNA sequence studies showed this virus, now officially named human herpesvirus 6 (HHV 6), to be distinct from the then five known human herpesviruses, but closer to human CMV, with which it has some homology. Two variants have been identified on sequence analysis: HHV 6A and 6B. Although first isolated from B cells, HHV 6 was shown to infect CD4+ T cells preferentially. HHV 6B is the cause of a common disease of infancy called exanthem subitum or roseola infantum (‘sixth disease’) and may cause febrile convulsions in childhood, whereas variant 6A has much less clear disease association. Both variants do seem to play a role in disease (bone marrow suppression, encephalitis) in the immunosuppressed state following transplantation. The cell receptor for HHV 6 is CD46, a complement regulatory glycoprotein present on all nucleated cells. Bone marrow progenitor cells contain truly latent HHV 6; low-level persistent infection is found in salivary glands and brain cells.
In 1990, another new human herpesvirus was isolated in similar circumstances, and has been shown to be sufficiently distinct from other HHV to be denoted HHV 7. Primary infection with HHV 7 has been identified in some cases of exanthem subitum and febrile convulsions in childhood but its role post transplantation is unclear. A new genus, roseolovirus, has been established for HHV 6 and 7, which are members of the subfamily betaherpesvirinae, distantly related to human CMV, but with much smaller genomes.
Pathogenesis and clinical features
Both HHV 6B and 7 are shed persistently in saliva, and both have been found in female genital secretions. Only HHV 7 is found in breast milk. It is known that HHV 6 may integrate into the human chromosome and can, thus, be inherited from either parent. Such congenital infection leads to a persistently high level of viral DNA within cells in blood. In most people, only low numbers of copies of HHV 6 or 7 are found in circulating cells.
Exanthem subitum (Roseola infantum)
Exanthem subitum (ES) was long considered to be an infection caused by a virus, and transmission by blood was confirmed experimentally years before HHV 6 was isolated. The infection is extremely common in the first years of life, presenting between 6 months and 3 years of age with a sudden onset of fever (up to 39°C). The acute febrile illness due to these viruses accounts for 20% of all fevers of early childhood. The child is not usually ill, remaining alert and playful, with some throat congestion and cervical lymphadenopathy. Sometimes more pronounced respiratory symptoms occur with febrile convulsions. Fever usually persists for 3 days, when the temperature falls suddenly; a widespread maculopapular rash appears in around 10% of cases. HHV 6B has been isolated from peripheral blood in the acute febrile phase, in patients who subsequently produced a rash and in many who did not. Seroconversion is noted in convalescence, confirming a primary infection with HHV 6B. Some cases are associated with HHV 7 infection, but no other firm disease association has yet been found for HHV 7.
Neurological disease
A British survey has confirmed the importance of roseoloviruses in neurological illness (febrile convulsions and encephalitis) in early childhood. Diagnosis is difficult as viral DNA is not always detected in CSF (and may then represent integrated HHV 6), but seroconversion to one of the viruses may confirm a primary infection. HHV 6 strains appear to remain thereafter in brain, and these viruses are now considered to be commensal in CNS. No clear association with demyelinating diseases, particularly multiple sclerosis, has been shown.
Other associations
In the rare adult case, hepatitis and lymphadenopathy have been found, and sometimes a heterophile agglutinin-negative mononucleosis. Reactivation in immunocompromised hosts may be diagnosed by viral load or on the basis of serology, but this is difficult to interpret. HHV 6 and 7 reactivate in the first weeks after transplant and appear to make CMV disease worse, but the full extent of disease in the transplant recipient has still to be established. HHV 6 may cause bone marrow suppression and encephalitis following transplantation (HHV 6B is more often implicated than 6A).
Both HHV 6 and 7 infect T lymphocytes; indeed, HHV 7 uses the same receptor (CD4) as HIV. The potential significance of these interactions has still to be established.
Laboratory diagnosis
Isolation of HHV 6 or 7 involves co-cultivation of peripheral blood lymphocytes with mitogen-activated cord blood lymphocytes. Very large, refractile, multinucleate cells are produced in culture, and many intact enveloped virions are released into the culture medium. These viruses may be isolated from saliva, particularly HHV 7. Viral DNA detection (by PCR) is now the mainstay of routine diagnosis, with a multiplex assay capable of distinguishing HHV 6A from 6B and 7.
Confirmed laboratory diagnosis is currently available only from specialist laboratories. A particular source of confusion is the ability of HHV 6 to integrate (on occasion) into target cell chromosomes. Antibody tests are becoming more widely used, but results can be confusing. Antibody avidity tests can help to establish whether a primary infection has occurred, as the binding of recently acquired low-avidity IgG is easily disrupted by protein-denaturing agents. Both this method and PCR have proven useful in establishing the correct diagnosis of HHV 6- and 7-associated exanthem subitum in a substantial proportion of suspected cases of infant measles.
Treatment
HHV 6 and 7 are sensitive to ganciclovir, but not to aciclovir. Foscarnet and cidofovir may also be used in serious cases, for example encephalitis in immunocompromised patients. However, treatment is seldom indicated clinically.
Epidemiology
Antibody analysis to date has been based mainly on immunostaining studies with cells infected with either HHV 6 or 7 as antigens. High antibody titres to HHV 6 are found in young children in the first 4 years of life, reflecting recent primary infection in this age group. Primary infection, with viraemia and seroconversion, has also been detected in seronegative transplant recipients of liver or kidney grafts from seropositive donors. Possible transmission by blood transfusion has not been excluded. Infection with HHV 7 occurs, on average, slightly later in childhood. Older persons have lower levels of antibody, but more sensitive EIA tests reveal almost universal HHV 6B and 7 seropositivity. The full spectrum of disease associated with these viruses, and the extent of asymptomatic shedding, remain to be established.
Human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus)
Sequences of DNA representing a completely new human herpesvirus were identified in 1994 in tissues from the epidemic form of Kaposi’s sarcoma in patients with AIDS. The DNA fragments unique to the tumour tissue were found to have some homology with the gammaherpesvirus subfamily, including EBV. This virus, officially named HHV 8, but also referred to as Kaposi’s sarcoma-associated herpesvirus (KSHV), is the latest human tumour virus.
HHV 8 is classified in the rhadinovirus genus of the gammaherpesvirinae along with known viruses of non-human primates. The genome is 165–170 kbp and encodes around 95 proteins, including a group that contains homologues of human proteins involved in cell growth regulation and cytokine production (IL 6; viral IL 6, vIL6). Like other herpesviruses, HHV 8 attaches to cells first via cell surface heparan sulphate and integrins through envelope glycoproteins gpK8.1 and gB. HHV 8 infects dividing B cells. It then proceeds to either a lytic replication cycle, releasing infectious virus, or enters latency, expressing only the latency-associated nuclear antigens (LANAs). LANA 1 maintains the HHV 8 episome whereas LANA 2 suppresses apoptosis via inhibition of p53-mediated transcription. Latency-associated membrane protein (LAMP) has functions reminiscent of EBV’s LMP1 and 2 (see above).
Kaposi’s sarcoma
HHV 8 is strongly associated with all forms of Kaposi’s sarcoma (KS). Classic KS was first described by the Austro-Hungarian dermatologist Moritz Kaposi in 1872 and affects elderly men of Mediterranean, Esatern European or Jewish backgrounds. The endemic form of KS was described in eastern Africa prior to the HIV epidemic, whereas AIDS-associated KS is an aggressive form of the disease that occurs in the context of HIV infection. Lastly, iatrogenic KS develops in immunosuppressed patients (e.g. organ transplant recipients) as a result of medical intervention. HHV 8 is not the sole driver of KS and other factors (e.g. immunosuppression) also play a role; however, KS only develops in individuals infected with HHV 8. The mucocutaneous neoplasm Kaposi described as ‘a multifocal pigmented sarcoma’ was the most common tumour in HIV-infected MSM before the advent of highly active antiretroviral therapy (HAART) and was one of the heralds of the AIDS pandemic in the early 1980s. A transmissible agent was long suspected in KS on epidemiological grounds, transmitted sexually but rarely by blood. Endothelial cells of vascular or lymphatic origin are involved; they have a characteristic spindle shape and are arranged in bundles. Although occurring at multiple sites in skin, lymph glands and the gastrointestinal tract, the tumour itself does not lead to death. Local radiotherapy and systemic chemotherapy have been used in treatment. Lesions on exposed parts of the body are blue-reddish-brown plaques, sometimes raised, and are a cause of considerable concern for the affected individual.
Body cavity-associated B lymphoma/primary effusion lymphoma/multicentric Castleman’s disease
HHV 8 genomes have been found in lymphoma cells from AIDS-related B cell lymphomas termed body cavity-associated B lymphomas (BCBL) or primary effusion lymphomas (PEL). This condition presents as a malignant effusion in the pericardial, pleural or peritoneal spaces. Cell lines established from the tumours can be induced to produce viral proteins and particles, and have been used as a basis for diagnostic tests. A subset (‘plasma cell variant’) of another condition, multicentric Castleman’s disease (MCD), is also strongly associated with HHV 8. MCD is a lymphoproliferative disorder that is often localized in the mediastinum, mesenterium or peripheral lymph nodes.
Laboratory diagnosis
DNA amplification by PCR is used to detect and monitor HHV 8 (viral load in blood). Lytic growth of HHV 8 can be induced in latently infected B cell lymphoma cell lines (many are co-infected with EBV) and, as a result, reagents for antibody testing can be produced – both infected cell lysates for immunoblots, and antigen-containing cells for immunostaining. Recombinant proteins have been synthesized and are useful for EIAs. Now that reagents free of EBV are available, the specific epidemiology of infection with this new herpesvirus is being studied.
Epidemiology
Molecular sequence data have revealed five clades of HHV 8, termed A–E. Clade B predominates in Africa, whereas in Europe and North America clades A and C are more common. Serological surveys have shown that, unlike most of the human herpesviruses, HHV 8 infection is not common, at least in developed countries (fewer than 5% of blood donors in North America and northern Europe have anti-HHV 8 antibodies). Rates of seropositivity are higher in at-risk groups for KS, with up to 30% seropositivity rates in MSM. Confirmation of a high seropositivity rate (up to 60%) in adolescents and adults in most parts of sub-Saharan African countries, and studies on transmission, have shown a link between mothers and their children, and between siblings. Transmission from saliva is considered the most likely route, and the high rates in communities where over-crowding in childhood is seen support this. In Europe, there is increased seropositivity around the Mediterranean, and the presence of HHV 8 DNA in elderly Italians is highly predictive of the development of KS following organ transplantation or HIV infection.
Treatment and control
Ganciclovir has activity against HHV 8, and its use in CMV prophylaxis after organ transplantation may contribute to reducing the risk of reactivation and disease due to HHV 8 in seropositive recipients, or in seronegative recipients who have a seropositive donor. Foscarnet may be an alternative drug. Tumours associated with HHV 8 infection may regress if reduction of immunosuppression is possible (post-transplant) or following improvements in immunity with HAART (in patients with AIDS) – together with local radiotherapy and/or chemotherapy. Rituximab may also have some effect in patients with BCBL/PEL.
Cercopithecine herpesvirus 1 (B virus; herpesvirus simiae)
Description
This virus, antigenically related to HSV, commonly infects Old World (Asiatic) macaque monkeys, causing a mild vesicular eruption on the tongue and buccal mucosa analogous to primary herpetic stomatitis in man. The infection rate in monkeys increases markedly if they are kept in crowded conditions and, although relatively benign in the monkey, this virus is potentially highly pathogenic for man.
Human infection with B virus is rare. It is usually acquired from a monkey bite or from handling infected animals without appropriate personal protective equipment (PPE). In one instance, the wife of an infected monkey handler became infected through contact with her husband’s vesicles, and B virus has also been transmitted in the laboratory from infected monkey cell cultures. Within 5–20 days of exposure, local inflammation may appear at the site of entry, usually on the skin, accompanied by some itching, numbness and vesicular lesions. Ascending myelitis or acute encephalomyelitis may follow, occasionally as long as 5 weeks after exposure, and may not always be recognized. Delay in specific therapy leads to a high mortality rate and serious neurological sequelae in survivors.
Diagnosis and treatment
Diagnosis by isolation of the virus from blood, vesicle fluid, conjunctival swabs or CSF is not always possible and such approaches pose significant problems for the laboratory as a result of safety concerns (ACDP Category 4 organism; see internet reference to ACDP). Herpesvirus particles may be detectable by EM of vesicle fluid. Definitive identification of the virus is available in specialist reference laboratories using immunostaining or DNA analysis by PCR. Demonstration of specific antibodies is complicated by cross-reacting HSV antibody, but new specific antigens based on recombinant glycoproteins offer improved serology. B virus is not as sensitive to aciclovir as HSV, requiring concentrations equivalent to those used for VZV. Treatment needs to be given promptly to be effective, and very high-dose intravenous aciclovir for 2 weeks or longer is recommended. Ganciclovir should be used when there is evidence of CNS involvement. Because of the small number of cases, the best therapeutic regimen is not well established.
Prevention of B virus infection
Guidelines have been issued for the protection of those handling monkeys or monkey tissues. These include:
• screening of macaque colonies when possible
• recommendations for training to prevent exposure
• safe handling and personal protective equipment (PPE) procedures
• immediate risk assessment and care of wounds
• information regarding the risks and nature of the infection.
Prophylaxis in the event of possible exposure involves prompt, rigorous wound washing and cleansing with 10% iodine in alcohol. A course of high-dose oral aciclovir, or valaciclovir, is reserved for prophylaxis of high-risk exposures, namely those on head, neck or torso, deep wounds, and where the source animal has lesions, recent stress or illness, or the material is of CNS or mucosal origin. After prophylaxis, a prolonged observation period is recommended as the onset of infection may be delayed.
Corey L, Wald A. Maternal and neonatal herpes simplex virus infections. The New England Journal of Medicine. 2009;361:1376–1385.
Fishman JA. Infection in solid organ transplant recipients. The New England Journal of Medicine. 2007;357:2601–2614.
Gershon AA, Gershon MD, Breuer J, et al. Advances in the understanding of the pathogenesis and epidemiology of herpes zoster. Journal of Clinical Virology. 2010;48:S2–S7.
Knipe DM, Howley PM. Herpesviridae. In Fields Virology, ed 5, Philadelphia: Lippincott Williams & Wilkins; 2007:2479–2904.
Mahy BWJ, Ter Meulen V. Herpesviridae. In Topley & Wilson’s Microbiology & Microbial Infections: Virology, ed 10, London: Hodder Arnold; 2005:488–577.
Oxman MN. Zoster Vaccine: Current status and future prospects. Clinical Infectious Diseases. 2010;51:197–213.
Parker A, Bowles K, Bradley JA, et al. Management of post-transplant lymphoproliferative disorder in adult solid organ transplant recipients – BCSH and BTS guidelines. British Journal of Haematology. 2010;149:693–705.
Zuckerman AJ, Banatvala JE, Schoub BD, et al. Herpesviridae. In Principles and Practice of Clinical Virology, ed 6, Chichester: Wiley-Blackwell; 2009:95–272.
Advisory Committee on Dangerous Pathogens. Biological agents. The principles, design and operation of Containment Level 4 facilities. http://www.dh.gov.uk/prod_consum_dh/groups/dh_digitalassets/@dh/@en/documents/digitalasset/dh_4135259.pdf.
British Transplantation Society. Active Standards and Guidelines. http://www.bts.org.uk/transplantation/standards-and-guidelines/.
UK Department of Health. Varicella. http://www.dh.gov.uk/prod_consum_dh/groups/dh_digitalassets/@dh/@en/documents/digitalasset/dh_115138.pdf.