46 Hepadnaviruses
Hepatitis B virus infection; hepatitis delta virus infection
Key points
• Hepatitis B virus (HBV) causes acute hepatitis and chronic infection leading to chronic liver disease and hepatocellular carcinoma.
• HBV is a partially double-stranded DNA virus, carrying a reverse transcriptase-like enzyme to replicate viral DNA from an RNA intermediate.
• HBV is transmitted in blood, e.g. through intravenous drug use, by sexual intercourse and from mother to child during childbirth.
• There is considerable geographical variation in exposure; the highest rates are in the Far East, sub-Saharan Africa, Oceania and South America.
• An effective vaccine is available, given in three doses over 6 months. Universal vaccination of young children will reduce the chronic infection rate and long-term sequelae, including hepatocellular carcinoma.
• Treatment of chronic HBV infection includes a course of pegylated interferon or the use of long-term suppressive therapy with nucleos(t)ide analogues.
The hepadnaviridae are a family of hepatotropic DNA viruses with a unique life cycle involving an RNA intermediate and the use of a viral polymerase enzyme with reverse transcriptase activity. There are two recognized genera whose members are species-specific and cause acute and chronic infection of the liver. The genus orthohepadnavirus infects vertebral hosts including humans, great apes, woodchucks and ground squirrels, while the genus avihepadnavirus infects birds such as ducks, herons and storks. Hepatitis B virus (HBV) is the type species of orthohepadnavirus, a major cause of chronic liver disease and hepatocellular carcinoma (HCC) in humans. HBV infection also occurs in the wild in a number of non-human primate species, such as chimpanzees, orangutans and gibbons. Each of these species harbours variants of HBV distinct from those found in man.
Hepatitis B virus
Structure
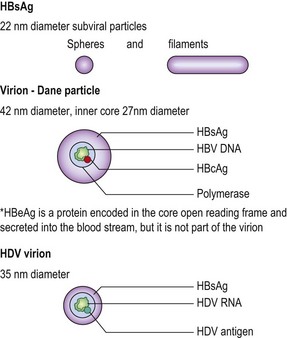
The virion of HBV is a 42 nm double-shelled particle known as the Dane particle. The outer envelope of the virion is formed by hepatitis B surface antigen (HBsAg). The inner core, 27 nm in diameter, consists of hepatitis B core antigen (HBcAg) which encloses the viral genomic DNA and polymerase.
The outer envelope protein, HBsAg, is over-produced by HBV and excess HBsAg is found in abundance in the blood of chronically infected individuals as sub-viral particles. Two different forms of subviral HBsAg can be seen in the blood (Figs 46.1 and 46.2). The predominant form is a small, spherical particle with a diameter of 22 nm. A filamentous form is also present. Both types of particles are composed of lipid, protein and carbohydrate; they are not infectious and consist solely of surplus virion envelope. There may be as many as 1013 of the small particles and filaments per mL. The virions are present in much smaller numbers, usually by a factor of 103 or more.
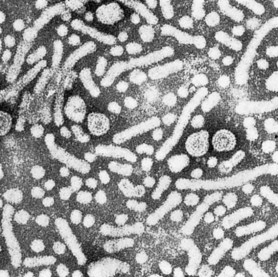
Fig. 46.1 Electron micrograph of the particles in the blood of a patient infected with HBV (original magnification ×130 000).
(Courtesy of Dr A Keen, University of Cape Town.)
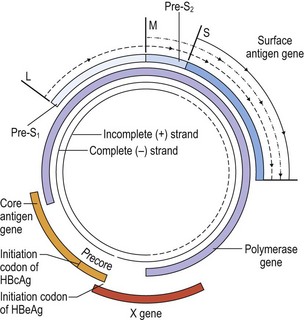
The viral DNA is about 3200 nucleotides long and is circular in configuration (Fig. 46.3). The long negative sense strand is complete, but there is a gap of variable length of about 1000 nucleotides in the complementary positive sense strand. This incomplete strand is closed by the viral polymerase when virus replication starts. There are four overlapping open reading frames on the circular viral DNA coding for the core, surface, polymerase and an X protein which is possibly an activator of transcription. An additional viral protein, the hepatitis B e antigen (HBeAg), is translated from the core open reading frame that encodes the HBcAg protein using an upstream initiating codon. HBeAg is not found in the virion but is secreted from infected cells into the bloodstream, particularly during active viral replication. It is therefore frequently used as a marker indicating significant viral activity. The surface antigen gene is transcribed to produce three messenger RNAs (mRNAs): L, M and S. The S mRNA is the shortest but most abundantly produced. The product of the M mRNA is medium in length and consists of an additional portion of the S reading frame known as pre-S2. The protein from the L mRNA is the longest and comprises an additional pre-S1 segment in addition to pre-S2 and S. The L product is present only in the virions, whereas the M and S proteins are found in the virions as well as the subviral particles.
Genetic variation
HBV can be classified into at least nine genotypes, A–I, based on a sequence divergence of >8% over the entire HBV genome. Some genotypes have a restricted geographical distribution, e.g. genotype E is found predominantly in sub-Saharan Africa, genotypes B and C in the Far East, and genotypes F and H in Central and South America. The HBsAg of all HBV genotypes contains a common ‘a’ determinant which is the main target of the protective antibody response. Immunity induced by infection or immunization with one HBV genotype cross-protects against infection with others.
Stability
There is only limited success culturing HBV using primary hepatocyte cell culture and viral expression is often studied using cell lines with transfected HBV. It is therefore difficult to assess the stability of HBV. Indirect evidence has been obtained from the study of recipients of blood products treated in various ways and from chimpanzee inoculation experiments. Infectivity is lost after autoclaving at 121°C for 20 minutes or dry heat at 160°C for 1 h. HBV remains active after storage at 30–32°C for at least 6 months and when frozen at −15°C for 15 years. HBV in blood can withstand drying for at least 1 week. Effective chemical disinfectants include treatment with hypochlorite (10 000 ppm available chlorine) for 10 min and 2% glutaraldehyde for 5 minutes. In clinical practice, chlorine based disinfectants are the ones most commonly used to disinfect environments contaminated with HBV. Glutaraldehyde was previously used to decontaminate endoscopes but was withdrawn due to toxicity. Effective substitutes include 0.2–0.35% peracetic acid, hypochlorous acid (superoxidised water) and chlorine dioxide.
Replication
The exact cellular receptor and mechanism of entry of HBV into hepatocytes is still unknown. Once in the cytoplasm, the virus uncoats and the nucleocapsid is transported to the nucleus where replication of viral nucleic acid starts. The viral polymerase completes the positive sense strand to form a convalently closed circular (ccc) DNA. This cccDNA forms a mini-chromosome in association with host histone proteins and establishes the basis of the persistent infection. From the cccDNA, mRNAs are transcribed which are then translated to form the various viral proteins including HBsAg and HBcAg. A 3.5 kb mRNA that spans the entire length of the genome is packaged with the viral polymerase and a protein kinase into the core particles. The multi-function viral polymerase then reverse transcribes the packaged RNA into the negative strand genomic DNA. The same viral enzyme then creates the complementary positive strand DNA using the negative strand as template, but this process is left incomplete as the process stops when the nucleocapsid matures. The mature nucleocaspid is then transported to the cytoplasm to be associated with the envelope protein HBsAg in the endoplasmic reticulum to form progeny virions. Some nucleocaspids remain in the nucleus and contribute to the intra-nuclear pool of cccDNA, thus leading to amplification of infection. Integration of the viral DNA genome into a host chromosome can occur during the replication cycle. The position of integration appears to be random and may be important in the mechanism of carcinogenesis. However, unlike other viruses such as HIV, integration is not essential for the replication of HBV.
Acute infection
The incubation period of HBV infection ranges from 6 to 24 weeks, but is often about 2–3 months. Acute infection can be asymptomatic, particularly in children. Symptoms in the prodromal phase include malaise, anorexia, weakness, myalgia, nausea and vomiting. Presence of hepatitis is signalled by the appearance of jaundice and right upper quadrant abdominal pain, accompanied by pale stool and dark coloured urine. Paradoxically, the patient often feels physically better when the jaundice appears. Hepatocellular damage is detectable biochemically with elevated alanine transminase (ALT) levels before the onset of clinical jaundice, and persists after the jaundice has resolved. In some cases, immunological reactions due to circulating immune complexes may manifest as arthralgia, urticarial or maculopapular skin rash, polyarteritis nodosa or glomerulonephritis. Up to 1% of acute hepatitis B infections can become fulminant, resulting in acute liver failure.
Pathology of acute infection
It is not possible to differentiate the various etiological agents of viral hepatitis at the histological level, as the changes are similar. In the acute stage there are signs of inflammation in the portal tracts; the infiltrate is mainly lymphocytic. In the liver parenchyma, infected hepatocytes show ballooning and form acidophilic (Councilman) bodies as they die.
HBV replicates in the hepatocytes, reflected in the detection of viral DNA and HBcAg in the nucleus and HBsAg in the cytoplasm and at the hepatocyte membrane. During the incubation period, high levels of virus are present before the host immune response develops to control the virus. During replication, HBcAg and HBeAg are also present at the cytoplasmic membrane. These antigens induce both B and T cell responses; damage to the hepatocyte can result from antibody-dependent, natural killer (NK) and cytotoxic T cell action. Expression of major histocompatibility complex (MHC) class I antigens is poor in hepatocytes but can be enhanced as interferons are produced in response to the infection. This in turn leads to increased antigen recognition and lysis of the infected hepatocytes. Thus, liver damage in HBV infection is mediated by an immunopathological mechanism.
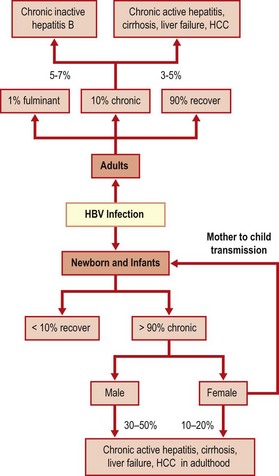
Chronic infection
Most adults with acute HBV infection recover completely. Chronic infection occurs in 5–10% of adults, 20–50 % of children under the age of 6, and in over 90% of newborns who acquire the infection. Four stages of chronic infection are recognized (Fig. 46.4).
3. Inactive chronic infection phase.
4. HBeAg negative chronic hepatitis B – through emergence of hepatitis B variants.
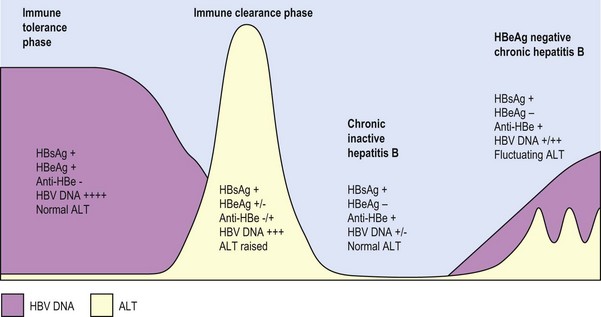
The immune tolerance phase occurs mostly in children who acquired the infection at a young age, but is also seen in immunocompromised individuals. During this phase, the infected individual is immunologically tolerant to the presence of the virus, allowing the virus to replicate to very high levels without showing any symptoms. Serum HBV DNA levels are typically very high (>1 million IU/mL) and HBeAg is present with normal ALT levels. There is little evidence of necroinflammation or fibrosis on liver biopsy. Because of the high level of viraemia, these patients are highly infectious.
The immune clearance phase occurs when immune tolerance is lost. This typically occurs in early adulthood for those who were infected at a young age. This phase is characterized by HBeAg positivity, slightly lower level of HBV DNA (>200 000 IU/mL) compared to the immune tolerance phase, fluctuating levels of ALT and evidence of moderate to severe liver necroinflamamation and rapid progression of fibrosis. This phase may be confused with acute hepatitis due to similarity in symptoms and presence of low levels of IgM anti-HBc. The longer the duration of immune clearance, the more is the resultant liver damage.
For individuals who successfully clear active HBV infection, the phase of inactive chronic infection follows. Seroconversion from HBeAg to anti-HBe occurs and HBV DNA levels become very low (<2000 IU/mL) or undetectable, while ALT levels normalize. These individuals are asymptomatic but most remain HBsAg positive. As a result of successful immunological control of HBV, these patients tend to have a more favourable long-term outcome. Traditionally, these patients have been referred to as ‘healthy hepatitis B carriers’. However, the use of such a term could result in a false sense of security, as between 10–20% may revert to active infection (reactivation) or change to the phase of HBeAg negative chronic hepatitis B through the emergence of viral variants. It is therefore necessary to have life long follow-up of patients with chronic inactive HBV infection.
In some patients, viral activity remains after seroconversion from HBeAg to anti-HBe. This is characterized by periodic fluctuating levels of HBV DNA and ALT. In these patients, a viral variant emerges (see section below) which is HBeAg negative but remains replication competent. The HBV DNA levels of these patients tend to be at a moderate level between 2000–200 000 IU/mL. Due to the fluctuating course, it is sometimes difficult to distinguish between patients in this phase with patients in the inactive phase. Nevertheless, a distinction is important as these patients have active disease and may progress to develop cirrhosis and HCC.
Approximately 0.5% per year of patients with inactive chronic hepatitis B will clear HBsAg. These individuals have resolved HBV infection and the presence of a past HBV infection is indicated by the presence of anti-HBc, with or without anti-HBs. However, in most cases, HBV is not completely cleared and reactivation can occur in the presence of immunosuppression such as after organ transplantation or treatment with immunosuppressive drugs. The risk for development of HCC is significantly reduced in these patients with resolved infection but not completely eliminated, particularly in older patients or those who have already developed cirrhosis. In some individuals, HBV DNA is persistently detectable in peripheral blood in the absence of HBsAg. These patients are said to have ‘occult’ HBV infection.
Pathology and pathogenesis of chronic infection
In chronic hepatitis, damage extends out from the portal tracts, giving a piecemeal necrosis appearance. Some lobular inflammation is also seen. As the disease progresses, fibrosis and, eventually, cirrhosis develops. Chronic liver damage results from continuing, immune-mediated destruction of hepatocytes expressing viral antigens. In addition, autoimmune reactions may contribute to the damage as immune responses are induced to various liver-specific antigens.
Persistence of HBV is indicated by the continued presence of HBsAg and HBV DNA in the blood for more than 6 months. It is not yet clear what determines whether an individual will progress to the chronic state. There may be host genetic factors, but the absence, or relative inefficiency, of the immune response is important, as shown by the increased likelihood of chronic infection in the very young and the immunocompromised. In the neonate, infection occurs in the presence of maternal anti-HBc and HBeAg, which can cross the placenta. It is speculated that this results in tolerance through masking of HBcAg on hepatocyte membranes and thus prevention of its recognition by cytotoxic T cells and other immune mechanisms that could lead to clearance of the virus.
Other factors associated with increased risk of cirrhosis include older age or longer duration of infection, infection with HBV genotype C, high level of HBV DNA, heavy alcohol consumption and concurrent infection with hepatitis C virus (HCV), hepatitis D virus (HDV) or human immunodeficiency virus (HIV).
Hepatocellular carcinoma (HCC)
There is considerable evidence that up to 80% of HCC worldwide is caused by chronic infection with HBV. Thus, the highest rates are found in areas where HBV is endemic and where infection occurs at a very early age. Risk factors for HCC include male gender, a family history of HCC, older age, cirrhosis, infection with HBV genotype C, presence of core promoter variants (see section on variants below) and co-infection with HCV.
While only 5% of patients with cirrhosis develop HCC, between 60–90% of patients with HCC have underlying cirrhosis. Thus, a proportion of HCC occurs in the absence of cirrhosis. The prolonged presence (>40 years) of HBeAg and high levels of HBV DNA are independent risk factors of HCC. Integration of viral DNA is a possible mechanism of carcinogenesis as HBV DNA is often integrated in tumour cells but the site differs in different tumours. HCC can be prevented by vaccination. Experience from Taiwan, which has a high prevalence of both HBV infection and HCC, demonstrated that the introduction of universal childhood vaccination against HBV is effective in reducing the incidence of HCC in young adults.
HBV variants
The frequency with which mutations appear in HBV is dependent on the high rate of replication of the virus and its dependence on replicating DNA via RNA and an RNA-dependent DNA polymerase. In highly viraemic individuals, as many as 1010 mutant genomes may arise each day. Most mutants are defective, but some may explain treatment failure or break-through infection.
HBsAg variants
HBsAg variants arise as an escape mechanism during infection in the presence of anti-HBs. In babies given hyperimmune hepatitis B globulin (HBIG) and active immunization to reduce the risk of infection, but who are exposed to maternal virus during birth, the immune selection pressure from the HBIG and vaccine may result in the selection of HBsAg escape mutants. This is seen particularly in countries where universal childhood HBV vaccination has been practiced for many years, leading to breakthrough infection despite vaccination. Another scenario where immune selection pressure is intense is following liver transplantation for chronic hepatitis B. Immune selection pressure is exerted through the post-transplantation use of HBIG to protect the new liver from being re-infected with HBV.
HBsAg escape mutants mainly affect the ‘a’ determinant of HBsAg, the principal target of anti-HBs. The most common mutation observed is in position 145 of the HBsAg protein with an amino acid change from glycine to arginine (G145R). HBsAg escape mutants are transmissible and the widespread occurrence of HBsAg mutants would create considerable problems for the hepatitis B vaccination programme. In addition, many existing diagnostic assays for HBsAg use specific monoclonal antibodies that detect epitopes associated with the ‘a’ determinant. The presence of such mutations could lead to false negative diagnostic results when the monoclonal antibodies fail to bind to the mutated epitopes. Some cases of so-called ‘occult HBV infection’ are in fact due to false negative HBsAg, with detectable HBV DNA and no detectable antigenaemia.
HBcAg variants
Mutations in the core promoter or pre-core coding regions of the core antigen open reading frame suppress the production of HBeAg, without affecting the synthesis of HBcAg and the assembly of complete virions. In the pre-core mutation variant, a stop codon is introduced between the initiation codons of HBeAg and HBcAg, so that full length HBeAg is no longer produced, but the production of HBcAg is unaffected. Individuals with these mutations are HBeAg negative but positive for HBV DNA. The absence of expression of HBeAg in such mutants is also believed to be an escape phenomenon enabling the virus to survive the cell mediated immune selection pressure from the host during the immune clearance phase. Core promoter mutations have been associated with the development of HCC in some studies.
Polymerase variants
These are detected during therapy with nucleoside analogues and confer drug resistance. Their presence can affect the choice of drugs. The most well recognized mutation affecting the drug lamivudine is a change from methionine to valine or isoleucine at position 204 (M204V/I). This is often accompanied by another change at position 180 (L180M) which helps to maintain viral fitness and continuing replication. In HBV infected patients who receive a liver transplant, anti-viral prophylaxis is often given together with HBIG. Because the polymerase gene and the surface gene in HBV share the same region of the genome, although read in a different reading frame, mutations in the polymerase gene could force a mutation in the surface gene and vice versa. This could lead to complicated selections of mutants affecting both HBsAg and HBV polymerase.
Laboratory diagnosis
The laboratory can test for a wide range of HBV antigens and antibodies, using immnoassays based on enzyme reactivity (EIA) or chemiluminesence (CLIA). HBV DNA can be quantified in serum or plasma using real time polymerase chain reaction (PCR) assays. The standard screening test is for HBsAg, which, if present in the serum, indicates that the patient is currently infected with HBV.
Acute infection
HBV DNA is the first detectable HBV marker in an acute infection followed shortly by HBsAg (Fig. 46.5) and both are present for some weeks before the onset of symptoms. In patients who have rapid viral clearance, antigenaemia is of short duration and may no longer be detectable at the onset of symptoms. If successive serum samples are examined, the development of anti-HBs will confirm recent primary infection, although there is considerable variation in the timing of the appearance of this antibody. HBV DNA may remain positive after disappearance of HBsAg. This creates a so-called ‘window period’ when an individual is HBsAg negative but remains infectious. In such cases, detection of IgM anti-HBc indicates that a primary infection has occurred recently.
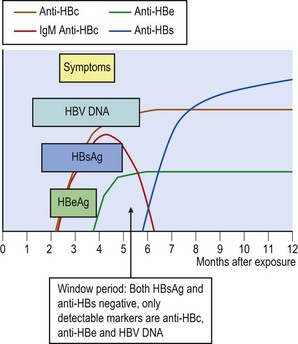
HBeAg is produced when virus is replicating and thus is usually found soon after HBsAg. IgM anti-HBc is a transient response and, if present in high titre, indicates a recent acute infection. Early disappearance of HBeAg and replacement with anti-HBe is a good prognostic sign for recovery. The presence of anti-HBs, occurring in about 90% of individuals after HBsAg clearance, indicates the development of immunity to further infection with HBV.
Chronic infection
Chronic infection with HBV is defined by the presence of HBsAg for more than 6 months. During this time, IgM anti-HBc is replaced with IgG anti-HBc, although in some individuals with chronic active infection, IgM anti-HBc may persist at a low level. HBeAg remains detectable during the immune tolerance and immune clearance phases. It is replaced by anti-HBe either following successful immune clearance into the chronic inactive phase or the development of HBeAg negative mutants during chronic HBeAg negative hepatitis B. These two possible outcomes can be recognized by testing for HBV DNA levels. Chronic inactive HBV infection has a persistently low HBV DNA viral load (Fig. 46.6), whereas chronic HBeAg negative HBV infection often has fluctuating levels of HBV DNA at moderate to high levels.
Treatment
The management of acute hepatitis B is usually supportive. Those with fulminant liver failure following acute infection or end stage liver failure following chronic infection may be candidates for liver transplantation.
The goal of treatment for chronic hepatitis B is to suppress HBV replication, prevent the progression of liver disease and thereby the development of cirrhosis, liver failure and HCC. Patients in the immune tolerance phase with high HBV DNA levels but normal ALT are not candidates for antiviral therapy. Instead, they should be monitored closely at 3–6 monthly intervals. Patients who have chronic inactive infection do not need antiviral treatment. For those who need treatment, the choice is between a course of interferon or long-term suppression with nucleoside or nucleotide analogues.
Interferon alpha (α-IFN) is the first-line treatment option for patients without cirrhosis. Interferon has antiviral, anti-proliferative and immuno-modulatory effects. However, its efficacy is limited and only benefits a small proportion of patients. The current strategy is to use long acting pegylated interferon α for a finite duration of 24–48 weeks. In HBeAg positive individuals, a high pre-treatment ALT level, high histological activity score on liver biopsy, low HBV DNA levels and infection with genotype A or B, rather than C or D, are favourable prognostic indicators of treatment response. Seroconversion from HBeAg to anti-HBe occurs in approximately 30%, but <10% eventually clear HBsAg. Response to HBeAg negative disease is much less predictable and often requires a longer duration of therapy. Side effects are common and include influenza-like symptoms, neutropenia, thrombocytopenia, autoimmune thyroid disorders and psychiatric symptoms such as depression. An exacerbation of hepatitis is common, which is often a favourable indication of a subsequent response. However, in some patients, particularly those with existing cirrhosis, this can lead to hepatic decompensation.
Nucleoside or nucleotide analogues act as a false substrate after phosphorylation in infected hepatocytes and are incorporated into the growing DNA chains resulting in premature chain termination of HBV DNA synthesis. Lamivudine is a first generation nucleoside analogue in widespread use for some years. At a dose of 100 mg daily, lamivudine leads to a marked reduction or elimination of detectable HBV DNA in plasma in about 40% of HBeAg positive and 60–70% of HBeAg negative patients. Between 40–60% of patients have ALT normalization. Although toxicity is low, and in the long term the drug is generally tolerated well, prolonged administration is complicated by the emergence of antiviral resistance, manifested by the reappearance of HBV DNA in plasma and raised ALT levels after initial normalisation. Development of resistance associated mutations in the polymerase enzyme, particularly that of M204V/I, is common and can be detected in 14–32% after 1 year, and increases to 60–70% after 5 years of treatment. Development of resistance is more frequent in those who are immunosuppressed. Due to the rapid development of high-level resistance, lamivudine is now rarely used as a first line therapeutic agent. Instead, it is increasingly being used as a prophylactic agent to prevent HBV reactivation in the immunosuppressed.
Telbuvidine is a L-nucleoside analogue with potent anti-HBV activity. It is similar to lamivudine in mechanism of action and resistance profile but is more potent. However, its use is limited due to a high rate of resistance and cross-resistance with lamivudine. Emtricitabine is another L-nucleoside analogue with similar activity to that of lamivudine.
Adefovir is a nucleotide analogue of deoxyadenosine monophosphate that has demonstrated efficacy in suppressing HBV DNA (20–50%) and normalizing liver function (50–70%). Resistance to adefovir emerges less frequently than with lamivudine. Additionally, adefovir is effective against lamivudine-resistant mutants. Conversely, mutations that arise in response to adefovir therapy are sensitive to lamivudine. These observations led to the use of lamividune–adefovir combination therapy after failing lamivudine monotherapy. However, adefovir is not a very potent agent, and its use is now mostly superseded by newer antiviral agents such as entecavir and tenofovir.
Entecavir is a carbocyclic analogue of 2′ deoxyguanosine. It is a potent suppressor of HBV replication, resulting in loss of serum HBV DNA in 70–90%, and ALT normalization in 70–80% of patients. However, it is partially susceptible to the resistance associated mutations selected by lamivudine. Hence, a higher dose (1 mg instead of 0.5 mg) is required in the presence of lamivudine resistance. Development of full resistance to entecavir requires a two hit mechanism with initial selection of M204V/I followed by several entecavir specific mutations. As a result, treatment failure due to entecavir resistance is rare and is observed only in 3.6% of patients after 96 weeks of treatment. Entecavir resistant HBV is susceptible to adefovir or tenofovir.
Tenofovir is a nucleotide analogue initially approved for the treatment of HIV. It is often used in a co-formulation with emtricitabine. Tenofovir is structurally similar to adefovir, but because tenofovir is less nephrotoxic, a higher dose can be used, thus enhancing the antiviral activity significantly in comparison with adefovir. After 48 weeks of therapy with tenofovir, HBV DNA loss is achieved in 80–90% and ALT normalization in 70–80% of patients. So far, very little resistance against tenofovir has been described. The only limitation of its use is renal toxicity which requires monitoring of renal function and it is contra-indicated in patients with renal insufficiency. The combination of tenofovir with lamivudine or emtricitabine is often used in HIV/HBV co-infection in order to treat both viruses.
Epidemiology
HBV is present in the blood as well as in body fluids such as semen, vaginal secretions and saliva. The presence of HBV in blood underlines the original association of infection with blood transfusion or the use of blood products and infections associated with needle-stick injuries. Sexual transmission is also recognized, as is that which occurs during close contact between family members, siblings, peers and residents in institutions. In these circumstances there will be frequent contact with blood and saliva; the virus gains entry through cuts and abrasions or across mucous membranes. Biting and scratching, sharing of household tools such as toothbrushes and razors could also be important factors. Vertical transmission from mother to child is one of the most important routes. Transmission occurs when maternal blood contaminates the mucous membranes of the baby during birth. Transplacental infection is thought to be quite rare unless maternal viral load is very high.
The World Health Organisation (WHO) has categorised three levels of HBV endemicity – high (≥8%), intermediate (2 to 7%) and low (<2%), based on the local prevalence of HBsAg. High-prevalence regions include sub-Saharan Africa, most of Asia and the Pacific islands. Intermediate-prevalence regions include the Amazon, southern parts of Eastern and Central Europe, the Middle East and the Indian sub-continent. Low-prevalence regions include most of Western Europe and North America. Overall, it is estimated that one-third of the world’s population has been infected with HBV and that 350 million individuals worldwide are chronically infected. Of these, between 15 and 40% will develop serious sequelae in their lifetime. In areas of low endemicity the risk of infection varies widely in different groups according to behaviour. Most cases occur in parenteral drug injectors sharing needles and syringes, and by sexual transmission, both homosexual and heterosexual. Screening of all blood donations using HBsAg and HBV DNA has virtually eliminated transmission by transfusion and blood products.
Healthcare personnel and laboratory workers are at risk of HBV infection through exposure to blood and body fluids of infected patients, although the degree of risk varies with the place and nature of their work, the care with which it is performed and their immune status. High-risk occupations include surgery, dental surgery, and obstetrics and gynaecology, which involve working with sharp instruments, often in restricted spaces. Operators may injure themselves and inoculate patients’ blood. The spilling of a patient’s blood will pose a threat only if there is contamination of unprotected abraded skin (intact skin is resistant to HBV penetration), or mucous membranes.
Patients are also at risk from infected staff, and episodes have been identified in which healthcare workers have transmitted HBV to their patients during invasive procedures, especially in difficult operations where the operator’s hands are hidden and needles and instruments are guided by touch (exposure prone procedures).
Control
Broadly there are two approaches to the prevention of infection with HBV – modification of risk behaviour, and immunisation. Measures for the former include avoiding unprotected sexual contact by the use of condoms, and reducing needle sharing among injecting drug users through needle exchange schemes. Implementation of sensible infection control policies can reduce the risks considerably to healthcare workers and patients. It is essential that blood for transfusion and organ donors for transplantation are screened.
Passive immunization
HBIG is prepared from donors with high titres of anti-HBs. Doses of 200–500 IU in 2–4 mL are given intramuscularly. The use of HBIG is indicated in the following situations:
• after accidental exposure if the victim is not vaccinated, or did not respond to the vaccine
• to babies born to infected mothers (in conjunction with active immunization)
• to prevent infection of a new liver transplanted into a recipient with chronic HBV infection.
HBIG must be given as soon as possible after exposure and preferably within 48 h; a second dose is given 4 weeks later to those who do not respond to current vaccines. Such a regimen does not give absolute protection, but an efficacy of 76% has been reported. In cases of needle-stick injuries, if the victim has not been vaccinated, HBIG should be used and a course of active immunization started, injecting the two materials into different body sites. To prevent mother to child transmission of HBV where the mother is highly infectious, e.g. the mother is HBeAg positive or HBV DNA viral load >1 million IU/mL, a combination of HBIG with active immunization given to the baby within 24 h of birth, is effective in as many as 90% of cases.
Active immunization
Currently available vaccines are produced by cloning the surface antigen gene in yeast cells. The product is particulate and resembles the small particles seen in patients, although it is not glycosylated. The vaccine is administered with alum as adjuvant and injected intramuscularly; care should be taken to avoid injection into fat as this can reduce seroconversion rates. For this reason, injection into the deltoid muscle of the upper arm is recommended. The vaccine is free from major side effects; local swelling and reddening may occur in up to one in five recipients, with a slight fever in only a few cases.
Three doses of vaccine are given at 0, 1 and 6 months. Shorter schedules may be appropriate in some circumstances. The seroconversion rate is influenced by a number of factors, the most important of which are the age and sex of the vaccinee. Rates in excess of 95% are seen in young women, whereas the rate may drop to 80% in older men. Immunosuppressed patients show even lower rates, for example only 50–60% in patients on maintenance dialysis.
The duration of the response to vaccine is variable and dependent on the titre of anti-HBs achieved after completion of the course. A post-vaccination anti-HBs level >10 mIU/mL is considered as protective. However, to ensure a higher level of protection, a booster dose is often recommended if the anti-HBs level taken within 2–3 months after the third dose is <100 mIU/mL. If no response is detected (i.e. <10 mIU/mL), a course of three further doses should be given. A vaccine non-responder is defined as an individual who fails to develop immunity after two vaccine courses. Such individuals are not protected and must seek prophylaxis by passive immunization if they suffer accidental exposure. Those who are known to have responded can be given a booster if they are exposed to the virus. Some individuals show a drop in antibody level with time, but memory B cells will be activated on exposure to the virus and will provide sufficient protection against infection or at least to prevent the development of chronic infection.
Who should be immunized?
Transmission from mother to child is an important route and requires intervention at birth (within 24 h) to protect the child. As most babies infected at birth will become chronically infected, it is essential to target this group. Dependent on the infectiousness of the mother, combined passive and active immunization or active immunization alone can be used. WHO has recommended universal childhood vaccination against HBV. Many countries have now incorporated this into their childhood vaccination programmes. Some countries, such as the UK, have adopted a selective infant vaccination strategy based on the result of antenatal HBV screening.
In the absence of a universal vaccination programme, the following groups should be targeted for HBV vaccination:
• Occupational risk groups – all healthcare workers who may have direct contact with patients’ blood or body fluid, including doctors, nurses, cleaners, laboratory workers and paramedics. Other occupational risk groups such as police, fire and prison service staff, military personnel, morticians and embalmers.
• Babies of chronically infected mothers
• Individuals who change sexual partners frequently
• Close family contacts and sexual partners of a case
• Families adopting children from endemic countries and foster carers
• Individuals with learning difficulties living in long-stay homes
• Patients needing frequent transfusions and/or blood products
• Patients with chronic renal failure
• Patients with chronic liver disease
The delta agent (hepatitis D virus, HDV)
The delta (δ) antigen was first identified in the late 1970s. Initially, it was thought to be an antigen of HBV, but it is part of another virus that cannot replicate without assistance from HBV. The specific helper function provided by HBV is its surface antigen, HBsAg, which also serves as the outer envelope of hepatitis D virus (HDV). HDV is a small (35–37 nm), enveloped particle containing a single, small, circular molecule of RNA of 1.7 kbp with an internal δ antigen enveloped by HBsAg (see Fig. 46.2). The origin of the virus is unknown and it has no homology with HBV. It has some resemblance with the satellite viruses of plants. However, it is also possible that the circular RNA of HDV is derived from an aberrant splicing event in human cells.
Clinical features and pathogenesis
Hepatitis D virus (HDV) can only infect simultaneously with HBV (co-infection) or as a superinfection of an individual chronically infected with HBV. Co-infection has a better prognosis than superinfection, which often results in increase in severity of the chronic hepatitis with increased risk of developing cirrhosis, liver failure and HCC.
Diagnosis
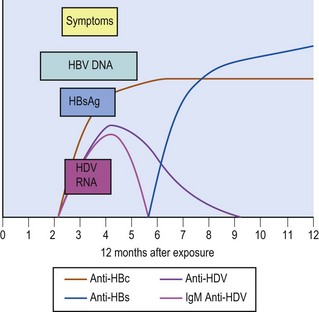
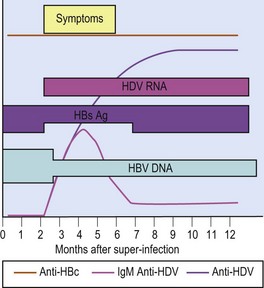
Tests are available for HDV antigen, antibody and RNA. The sequence of appearance of the various markers in a patient co-infected with HBV and HDV is shown in Figure 46.7. The initial antibody response to HDV infection is IgM. In cases of superinfection, the test results are as illustrated in Figure 46.8. During the episode there may be a drop in the HBsAg titre, which, although usually still detectable, may disappear temporarily in a few cases. This can cause some diagnostic confusion. There may also be a reduction in HBV DNA levels which may result in a false assurance that the HBV infection is not active if HDV super-infection is not recognized.
Epidemiology
There are an estimated 25 million HDV infected individuals in the world, but there is considerable geographical variation. Analysis of the RNA from different isolates has shown that there are at least eight different genotypes. Genotype 1 is of worldwide distribution; genotype 3 is found in the Amazon region; genotypes 2 and 4 in the Far East; and genotypes 5–8 in sub-Saharan Africa.
In the 1980s, it was thought that HDV was mainly associated with drug users and that controlling HBV through HBV vaccination or needle exchange schemes would be effective in eliminating HDV infection. There was an initial decrease in the prevalence of HDV in Europe, but it is increasingly recognized that HDV is not disappearing. Intravenous drug use is still an important factor; however it is no longer the sole driver of HDV spread. Immigrants from regions endemic for both HBV and HDV such as Eastern Europe and sub-Saharan Africa now constitute a higher proportion of infected cases in many cities. Despite having the highest prevalence of chronic HBV infection, the prevalence of HDV in South East Asia and China is very low.
Treatment and control
The only proven treatment of HDV infection is a prolonged course of pegylated interferon. However, this is not particularly successful as the sustained response rate after 48 weeks of treatment is less than 30%. There is no benefit in adding ribavirin or nucleos(t)ide analogues against HBV. The same general control measures as for HBV are also relevant for the control of HDV. HBV vaccine will prevent HDV co-infection, but there is no means of protecting against HDV superinfection.
European Association for the Study of the Liver. EASL clinical practice guidelines: management of chronic hepatitis B. Journal of Hepatology. 2009;50:227–242.
Harrison TJ, Dusheiko GM, Zuckerman AJ. Hepatitis viruses. In: Zuckerman AJ, Banatvala JE, Pattison JR, et al. 2009 Principles and Practice of Clinical Virology. ed 6. Chichester: Wiley; 2009:273–320.
Lok ASF, McMahon BJ, Chronic Hepatitis B. AASLD Practice guidelines. Hepatology. 2009;50:1–36.
Seeger C, Zoulim F, Mason WS. Hepadnaviruses. In: Knipe DM, Howley PM. Field’s Virology. ed 5. Philadelphia: Lippincott–Raven; 2007:2977–3030.
Department of Health. Immunisation against infectious disease – ‘The Green Book’. Chapter 18. Hepatitis B. http://www.dh.gov.uk/prod_consum_dh/groups/dh_digitalassets/@dh/@en/documents/digitalasset/dh_108820.pdf.
Health Protection Agency. Hepatitis B. http://www.hpa.org.uk/infections/topics_az/hepatitis_b/menu.htm.