60 Prion diseases (transmissible spongiform encephalopathies)
Creutzfeldt–Jakob disease; Gerstmann–Sträussler–Scheinker syndrome; fatal familial insomnia; iatrogenic Creutzfeldt–Jakob disease; kuru; variant Creutzfeldt–Jakob disease; bovine spongiform encephalopathy; scrapie
Key points
• Prion diseases are fatal neurodegenerative disorders with very lengthy incubation periods caused by unconventional agents. They are transmitted by inoculation or ingestion; the intracerebral route of transmission results in the shortest incubation period.
• Prion diseases occur in mammals, including scrapie in sheep, bovine spongiform encephalopathy (BSE) in cattle and Creutzfeldt–Jakob disease (CJD) in man.
• The infectious agents are known as prions, which are thought to lack nucleic acid and appear to consist entirely of a modified host-encoded protein, the prion protein.
• Prion diseases do not elicit conventionally detectable immune responses. Asymptomatic or subclinical infections are very difficult to detect.
• Human prion diseases occur in sporadic, familial and acquired forms, all of which are rare diseases. In 1996, a new form of human prion disease known as variant CJD was identified, which results from infection with the BSE agent, probably via the oral route.
Prion diseases, or transmissible spongiform encephalopathies (TSEs), are a unique group of fatal neuro-degenerative disorders occurring in human beings and animals that possess two major characteristics:
1. All are transmissible to a variety of mammals, either experimentally or by natural exposure. The precise nature of the transmissible agents involved is unknown (see below), but they possess physical and chemical properties that are quite distinct from those of conventional viruses and bacteria. There is increasing evidence to support the prion hypothesis, which states that the infectious agent is composed entirely of protein, without any nucleic acid, for which the term prion (proteinaceous infectious particle) is used. No evidence of a conventional host immune reaction has been found in prion diseases.
2. The diseases caused by these agents are characterized in all species by neurodegeneration in the central nervous system (CNS), usually with spongiform change. This consists of numerous small vacuoles (10–200 µm) that are formed within neuronal cell bodies and their processes (Fig. 60.1A), probably through dilatation of neuronal lysosomal, Golgi and endoplasmic reticulum structures. Spongiform change may be reversible in its early stages. Ultimately, neuronal death occurs accompanied by reactive proliferation of astrocytes and microglia. The normal cellular form of the prion protein (PrPC) is converted by misfolding into an abnormal disease-associated form (PrPSc) and accumulates within the CNS, usually as diffuse deposits, but occasionally in the form of amyloid plaques.
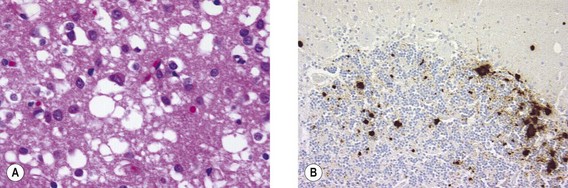
Fig. 60.1 (A) Spongiform change in the cerebral cortex in a case of sporadic CJD consists of numerous small cyst-like spaces that tend to coalesce in the neuropil and around neurones (centre). Haematoxylin and eosin stain, original magnification ×400. (B) In this case of sporadic CJD, prion protein (PrP) accumulation in the cerebellum has occurred in the form of numerous amyloid plaques that stain intensely on immunohistochemistry for PrP. Original magnification ×200.
The transmissible agent: a prion?
Although the precise nature of prions is uncertain, they are subviral in size and notoriously resistant to inactivation by many physical and chemical agents, including:
• exposure to ionizing or ultraviolet radiation
Prions have been studied by experimental transmission to mice and hamsters; this has identified around 20 strains of scrapie, a naturally occurring TSE in sheep. These strains are defined by their biological properties, particularly the disease incubation period and the nature and distribution of the pathology in the CNS. Incubation periods range from 60 days to more than 2 years, which is close to the natural lifespan of mice and hamsters, and cases of asymptomatic infection have been recorded in inoculated animals dying from unrelated causes after a prolonged period. The incubation period is influenced by:
• the route of inoculation – intracerebral inoculation is the most efficient mode of infection; oral or parenteral routes are often less efficient
• the dose of the injected inoculum and its infective titre
• host genetic factors, particularly polymorphisms in the prion protein gene.
PrPC is expressed in a variety of cells, including neurones in the CNS, where it may act as a copper-binding protein, and is thought to play a role in synaptic function. This protein has a molecular weight of 33–35 kDa and is highly conserved through a wide range of species, but its amino acid sequence varies from one species to another. This variation is thought to influence the species barrier, a factor that regulates the success of disease transmission from one species to another under experimental and natural conditions.
During prion infection, PrPC appears to undergo a conformational change to convert to PrPSc. This abnormal isoform has a relatively high beta-pleated sheet conformation, which renders it partially resistant to digestion with proteinase K and allows it to aggregate as amyloid fibrils in the brain. In the prion hypothesis, conversion of the PrPC to PrPSc occurs by a direct interaction, which sparks off a catalytic conversion. This has been replicated experimentally using recombinant prion protein, which has been misfolded in a cell-free system to produce infectious PrPSc. These findings support the prion hypothesis; however, it is not known whether this biochemical change alone accounts for all the biological properties of prions. For example, the mechanisms by which the various strains of scrapie agent have different biological properties are unclear, and some research groups believe that a second (as yet unidentified) molecule determines the strain of the infectious agent.
Pathogenesis
Transmission of prions to experimental hamsters and mice can be achieved by inoculation of infected brain tissue into a number of sites. After oral inoculation, spread of infection may occur along either the vagus nerve to the brainstem, or along sympathetic nerve fibres that connect via the splanchnic nerve complex to the spinal cord. Infection then spreads to the brain at a rate of around 1 mm/day, probably within neurones. Peripheral inoculation of the agent may be followed by a phase of accumulation and replication in the lymphoid tissues, mainly in follicular dendritic cells. In the spleen, there is a postulated ‘neuro-immune’ connection that allows access to the splanchnic plexus and then to the CNS.
Human prion diseases
The most common form of human prion disease was first described in the 1920s and is known as Creutzfeldt–Jakob disease (CJD) after the authors of the early reports. Since then, an ever-widening spectrum of human prion diseases has been identified (Table 60.1), with three main subgroups, comprising:
• idiopathic disorders, where the cause is unknown
• familial disorders, occurring as autosomal dominant disorders
• acquired disorders, following accidental infection by inoculation or ingestion.
Table 60.1 Human spongiform encephalopathies classified by aetiology
| Aetiology | Encephalopathy |
|---|---|
| Idiopathic disorders | Sporadic CJD (around 90% of all cases) |
| Familial disorders | Familial CJD (around 10% of all cases) Gerstmann–Sträussler–Scheinker syndrome Fatal familial insomnia |
| Transmitted from person-to-person | Kuru Iatrogenic CJD (less than 1% of all cases) |
| Transmitted from bovines to man | Variant CJD |
CJD, Creutzfeldt–Jakob disease.
The identification of the prion protein and sequencing of the human prion protein gene on chromosome 20 have greatly increased our understanding of human transmissible spongiform encephalopathies.
Diagnosis
The diagnosis of human prion diseases requires a range of clinical, biochemical, genetic and pathological studies. Careful assessment of the clinical features (see below) by an experienced neurologist allows a presumptive diagnosis of CJD to be made with a high level of accuracy (at least 70%). Analysis of the prion protein gene is essential to identify cases of familial CJD associated with a pathogenic mutation. Genetic studies have also demonstrated a naturally occurring polymorphism at codon 129 in the prion protein gene, which is important in determining disease susceptibility (see below). At present, there is no form of screening test for human prion diseases and no specific treatment is available. A definitive diagnosis depends on examination of the brain at autopsy. Brain biopsy is not a routine investigation, as the procedure can compromise an already ill patient and the neurosurgical instruments used have to be destroyed if the diagnosis is confirmed, to prevent accidental iatrogenic infection via subsequent neurosurgical procedures.
Neuropathology
It is recommended that all suspected CJD cases should be investigated by autopsy, with appropriate permission for retention and examination of the brain to allow confirmation of the clinical diagnosis and accurate subclassification of the case. The principal neuropathological features of human prion diseases are:
• spongiform change (Fig. 60.1A)
• amyloid plaque formation (Fig. 60.1B).
A range of techniques is now available to detect PrPSc accumulation within the CNS. PrPSc can be detected by immunocytochemistry in the CNS in a variety of patterns, including amyloid plaques (see Fig. 60.1B), perivacuolar and perineuronal and axonal deposition. PrPSc can also be detected by western blot techniques, which allow further study of the PrPSc isotype in terms of its glycosylation and molecular weight following digestion with proteinase K (Fig. 60.2).
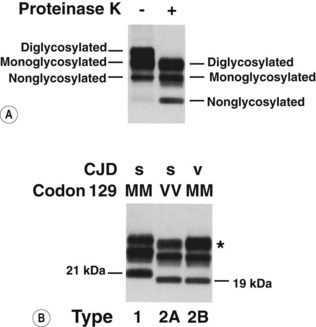
Fig. 60.2 Western blot analysis of prion protein (PrP) in post-mortem CJD brain. PrP from CJD brain occurs in three glycoforms resulting in diglycosylated, monoglycosylated or non-glycosylated PrP, which can be separated by western blotting into distinct bands. Proteinase K treatment (+) destroys the normal form of the prion protein, but in CJD it only partly denatures the disease-associated protein, which has an increased mobility (shown in A). Two main subtypes of the abnormal protein can be identified: (B) types 1 and 2 are found in sporadic CJD (s) irrespective of the genotype at codon 129. Examples of two types (MM1 and VV2) are shown here. All cases of variant CJD (v) thus far tested are methionine homozygotes (MM) and have had a type 2 PrP characterized by the predominance of the diglycosylated glycoform (*), termed type 2B. The type 2 cases found in sporadic CJD, in which the monoglycosylated glycoform predominates, are termed type 2A.
Sporadic CJD
CJD occurs most commonly as a sporadic disorder, affecting around one person per million population per year worldwide. Sporadic CJD usually presents as a rapidly progressive dementia of less than 1 year’s duration, often accompanied by other neurological abnormalities, including cerebellar dysfunction, pyramidal and extrapyramidal signs, cortical blindness and akinetic mutism. Many of these features evolve as the disease progresses, and electro-encephalography often shows characteristic diffuse, periodic, synchronous discharges. The peak incidence is in the seventh decade of life, but the disease has been described in teenagers and even in the ninth decade. The disease is untreatable and invariably fatal; most patients survive for only 4 months after the onset of major symptoms.
The naturally occurring methionine/valine polymorphism at codon 129 in the prion protein gene is of major influence in determining susceptibility to sporadic CJD (Table 60.2). The mechanism for this influence is unclear. The precipitating event leading to development of sporadic CJD remains unknown; it might possibly reflect selective exposure to an unidentified ubiquitous agent, perhaps causing disease only in genetically susceptible individuals, or a somatic mutation involving the prion protein in the CNS (Fig. 60.3). No occupational or dietary risk factors exist for sporadic CJD. The global distribution of sporadic CJD is markedly different from that of scrapie, particularly in Australia, where scrapie has been eradicated for many decades, but sporadic CJD occurs at a similar frequency to that in the UK, where scrapie is endemic.
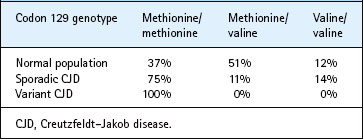
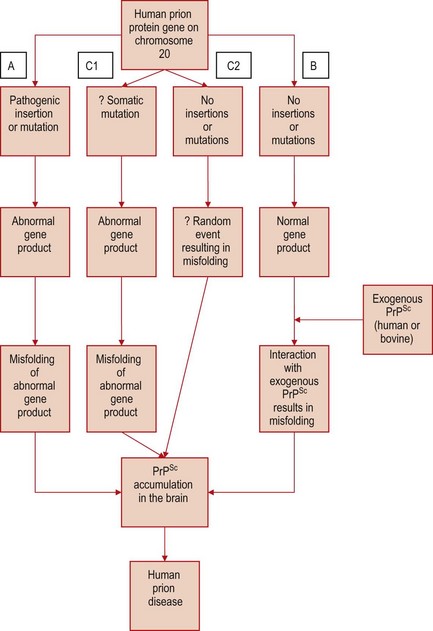
Fig. 60.3 Postulated mechanisms of PrPSc accumulation in CJD. (A) An inherited PrP gene mutation results in an unstable form of prion protein that misfolds to cause familial CJD. (B) Exogenous PrPSc interacts with the normal host prion protein, resulting in PrPSc accumulation. This mechanism is thought to operate in iatrogenic CJD (exposure to human PrPSc) and variant CJD (exposure to bovine PrPSc). The cause of sporadic CJD is unknown, but proponents of the prion hypothesis have suggested that a somatic mutation in a neurone (C1) might initiate the process of PrPSc accumulation in the brain, or alternatively a rare random event results in spontaneous misfolding of the normal prion protein (C2). These ‘rare events’ might also explain the low incidence of sporadic CJD.
Familial prion diseases
Around 10% of cases of CJD occur as autosomal dominant inherited disorders; these are associated with mutations or insertions in the open reading frame of the human prion protein gene on chromosome 20 (see Fig. 60.3). Gerstmann–Sträussler–Scheinker syndrome (GSS) is the best known example of an inherited human prion disease. In this very rare disorder, which affects middle-aged adults, cerebellar ataxia, nystagmus and gait abnormalities are prominent clinical features, with dementia occurring only towards the end of the illness. Numerous multicentric PrP amyloid plaques are present throughout the CNS. GSS was the first human disease linked to a mutation in the PrP gene; a codon 102 proline-to-leucine mutation was identified in 1989 and has subsequently been found in several GSS families, including the original kindred described in 1936. Fatal familial insomnia is an extremely rare inherited disorder, characterized clinically by disturbances of sleep and autonomic function with relative intellectual preservation in middle age. This disorder is associated with a unique PrP genotype (codon 178 asparagine, 129 methionine/methionine), and neuropathological changes mainly in the thalamus. The relationship between the genotype, neuropathology and clinical features of this enigmatic disorder await further study.
Acquired prion diseases
Iatrogenic CJD
CJD can occur by accidental transmission from one person to another (see Fig. 60.3). Iatrogenic CJD was first described in 1974 in a corneal graft recipient. Other examples of iatrogenic CJD have involved accidental inoculation of contaminated CNS tissue from a CJD patient to another patient, for example following the implantation of inadequately decontaminated intracerebral electrodes, or via human dura mater grafts. In the UK, the most common form of iatrogenic CJD occurs in recipients of human growth hormone derived from cadaveric pituitary glands, at an approximate incidence of 1 in 10 000 at-risk individuals, with an average age at death of 25 years and an incubation period of up to 20 years.
Kuru
Kuru was a disease occurring in the Fore tribe of Papua New Guinea, apparently in association with practices related to ritualistic cannibalism. The brain was handled and apparently eaten, particularly by females and children, who went on to develop this disorder. The name kuru means shivering or trembling in the local dialect, and reflects the predominant clinical manifestations of a progressive cerebellar syndrome with dementia. The incubation period was variable, ranging from around 5 to over 40 years. There was no evidence of mother-to-child transmission. Its increasing prevalence made it the primary cause of death in most individuals within the tribes in the 1960s. The disease is now extinct since cannibalistic practices were abandoned.
Variant CJD
The emergence of bovine spongiform encephalopathy (BSE) as a new epidemic in UK cattle raised the possibility that this disorder might be transmitted to man through the food chain. A National CJD Surveillance Unit was established for the UK in May 1990, based in Edinburgh. In 1996, the Unit identified a new form of human prion disease, now known as variant CJD, which by 2010 has affected more than 170 people in the UK, and 47 in other countries including France, Ireland, the Netherlands, Spain, Portugal, USA, Canada, Japan and Saudi Arabia.
• The age of onset is unusually young (mean 28 years), with a range from 12–74 years.
• The clinical illness is prolonged, with an average duration of 14 months (range 6–39 months).
• The clinical features are also unusual, with psychiatric and sensory symptoms at onset, followed by ataxia and myoclonus, with dementia only in the final stages of the illness.
• So far, all patients with definite variant CJD are methionine homozygotes at codon 129 in the prion protein gene (see Table 60.2).
The neuropathological features are relatively uniform and consist of massive accumulations of PrPSc throughout the brain, with the formation of multiple amyloid plaques surrounded by spongiform change, known as florid plaques (Fig. 60.4). Variant CJD differs from other forms of human prion disease in that PrPSc can be detected in lymphoid tissues (in follicular dendritic cells) both during the clinical illness (Fig. 60.5) and in the late preclinical illness. This is consistent with variant CJD being acquired by the oral route (see below) and has implications for the possible transmission of the variant CJD agent by surgical instruments used on lymphoid tissues (e.g. in tonsillectomies).
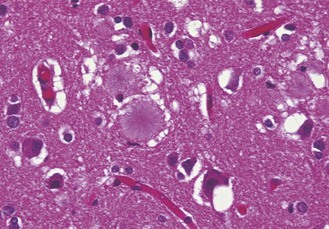
Fig. 60.4 The cerebral cortex in variant CJD contains numerous aggregates of PrPSc in the form of fibrillary amyloid plaques (centre), surrounded by spongiform change. Haematoxylin and eosin stain, original magnification ×200.
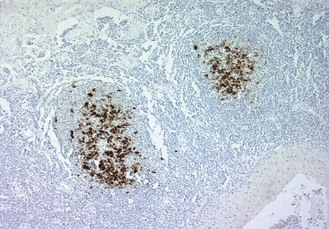
Fig. 60.5 Section of a tonsil from a patient with variant CJD stained to show the prion protein by immunohistochemistry. The dark stain demonstrates accumulation of the prion protein in the follicular dendritic cells within germinal centres of the tonsil. Accumulation of prion protein in lymphoid tissues does not occur in sporadic CJD. Original magnification ×100.
The variant CJD prion is also present in the blood of individuals in the preclinical incubation phase of the illness. Four cases of variant CJD transmission by blood transfusion have been reported in the UK. Many steps have been taken in the UK to prevent further transmission by this route, including filtration of whole blood to remove white cells (which may carry the highest levels of infectivity), sourcing of plasma products from overseas, and banning recipients of blood transfusions from themselves becoming blood donors.
Experimental strain typing studies in mice have shown that the transmissible agent in variant CJD is identical to the BSE agent. Furthermore, biochemical analysis of PrPSc from cases of variant CJD shows a similar banding pattern in western blot preparations to PrPSc from BSE in cattle, which is distinct from sporadic CJD (see Fig. 60.2). These findings provide strong support for the hypothesis that variant CJD is causally linked to BSE, and there is epidemiological evidence that dietary exposure through beef products is the most likely route of infection.
Recently, a decrease in the incidence and death rate for variant CJD has been reported in the UK. However, continuing surveillance is required, since the incubation period for variant CJD is unknown and future cases may emerge in other genetic subgroups (relating to the codon 129 polymorphism in the PRNP gene) in the UK population.
Bovine spongiform encephalopathy and other animal prion diseases
Prion diseases have been recorded in an increasing number of captive or domesticated animals (Table 60.3), the most significant of which is BSE. This was identified in 1985 in the UK and spread rapidly throughout the cattle population to affect around 1% of all adult cattle by 1992. It appears that this epidemic resulted from:
• oral ingestion of a prion (possibly a novel strain of scrapie) via contaminated meat and bone meal in cattle feed
• changes in the rendering processes by which this feed was produced in the 1980s, allowing the BSE agent to persist as a contaminant
• recycling of contaminated cattle carcasses in the meat and bone meal fed to cattle appears to have fuelled the epidemic.
Table 60.3 Animal diseases caused by scrapie and related agents
| Disease | Occurrence | Hosts |
|---|---|---|
| Scrapie | Common in many countries worldwide | Sheep and goats |
| Transmissible mink encephalopathy | Very rare, mostly in farms in the USA | Mink |
| Chronic wasting disease | Spreading from Colorado and Wyoming | Mule deer and elk |
| Bovine spongiform encephalopathy | Widespread epidemic in dairy cattle in the UK; smaller outbreaks in other countries | Domestic cattle |
| Unnamed spongiform encephalopathy | Small number of exotic ungulates in zoos | Nyala, gemsbok, greater kudu, Arabian oryx, eland |
| Feline spongiform encephalopathy | Small numbers of domestic cats in the UK and a few large cats in zoos | Domestic cats, cheetah, lion, puma, ocelot, tiger |
The incidence of BSE is now declining markedly as a result of a ban on the use of bovine organs as foodstuffs for other animals. Experimental studies on BSE indicate that it represents a single prion strain. This strain was also transmitted by foodstuff to domestic cats, large cats and exotic ungulates in zoos (see Table 60.3). There is good evidence that the BSE agent has also spread to man, giving rise to variant CJD (see above).
Scrapie
The first animal spongiform encephalopathy to be described was scrapie, an endemic disorder of sheep and goats that has been present in Europe for at least two centuries. Affected animals become ataxic and wasted, and often rub or scrape the fleece off the sides of their bodies, hence the name. The first experimental transmission of scrapie from affected to healthy sheep by intra-ocular injection of spinal cord homogenate was reported in 1936. Natural transmission is thought to occur at parturition, or by scarification or via the oral (alimentary) route. The placenta from an infected ewe is one known source of infection that can contaminate the farm environment. There is no epidemiological evidence that scrapie is pathogenic to man.
Bruce ME, Will RG, Ironside JW, et al. Transmissions to mice indicate the ‘new variant’ CJD is caused by the BSE agent. Nature. 1997;389:498–501.
Collinge J, Clarke A. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936.
Ironside JW, Ghetti B, Head MW, et al. Prion Diseases, ed 8. Love S, Louis DN, Ellison DW, eds. Greenfield’s Neuropathology, vol 2. London: Hodder Arnold. 2008:1197–1273.
Ironside JW, Head MW, Bell JE, et al. Laboratory diagnosis of variant Creutzfeldt–Jakob disease. Histopathology. 2000;37:1–9.
Peden AH, Head MW, Ritchie DL, et al. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozgous patient. Lancet. 2004;364:527–529.
Prusiner SB. Prions. Proceedings of the National Academy of Sciences of the USA. 1998;95:13363–13383.
Wadsworth JD, Collinge J. Update on human prion disease. Biochimica et Biophysica Acta. 2007;1772:598–609.
Ward HJT, Everington D, Cousens SN, et al. Risk factors for variant Creutzfeldt–Jakob disease: A case-control study. Annals of Neurology. 2006;59:111–120.
Will RG, Ironside JW, Zeidler M, et al. A new variant of Creutzfeldt–Jakob disease in the UK. Lancet. 1996;347:921–925.
Health and Safety Executive. BSE – Occupational guidance. http://www.hse.gov.uk/pubns/web22.pdf.
Health Protection Agency. CJD Incidents Panel. http://www.hpa.org.uk/web/HPAweb&Page&HPAwebAutoListName/Page/1204031511121.
Department of Health. Guidance from the ACDP TSE Risk Management Subgroup. http://www.dh.gov.uk/ab/ACDP/TSEguidance/index.htm?ssSourceSiteId=en.
Medical Research Council Prion Unit. http://www.prion.ucl.ac.uk/.
UK Creutzfeldt–Jakob Disease Surveillance Unit. http://www.cjd.ed.ac.uk/.