Chapter 12 Drugs interacting with mammalian enzymes
Introduction
There are a number of systems within the body that are controlled by enzymatic systems and which function in the control of disease states. In certain cases these systems are involved in a relatively narrow area of activity, e.g. HMG-CoA reductase, whilst in other situations the enzyme systems are involved in a variety of functions, e.g. phosphodiesterases. The following sections detail aspects of lipid-lowering drugs including the action of statins with HMG-CoA reductase, the sulphonamide diuretics and carbonic anhydrase activity and their involvement with the Na/Cl/K symporter, ACE inhibitors, a number of different phosphodiesterase inhibitors involved in erectile dysfunction and chronic obstructive pulmonary disease and the proton pump inhibitors used in the treatment of gastric ulcers.
Lipid-lowering drugs
Cholesterol is an important steroid in various aspects of the function of human cells. It is a precursor of a number of important steroids, e.g. corticosteroids and the sex hormones. It is also important for the maintenance of cell wall integrity. Unfortunately, cholesterol can also cause problems when present in high levels in the body due to its involvement in atherosclerosis. Atherosclerosis results from a build-up of cholesterol in various forms in the arteries, thereby reducing the flow of blood. Blood clots can be produced and these can result in either heart attacks or strokes. Cholesterol is taken into the body either via the diet or is synthesised in the liver. Cholesterol is absorbed into the bloodstream; however, it is insoluble in its free form in the blood and is transported by means of lipoproteins. Lipoproteins are particles that have a hydrophilic outer shell comprising phospholipids, free cholesterol and apolipoproteins and an inner hydrophobic core containing the lipids such as cholesteryl esters and triglycerides. Although there are a number of lipoproteins associated with transport of cholesterol, two in particular are often discussed in relation to heart disease: low-density lipoproteins (LDLs) and high-density lipoproteins (HDLs). LDL cholesterol is often described as bad cholesterol whilst HDL cholesterol is good cholesterol. LDL cholesterol is bad cholesterol since it is associated with plaque formation, which is a fundamental cause of atherosclerosis. HDL, or good cholesterol, is associated with the removal of excess cholesterol in the body since it transports cholesterol to the liver for disposal. Since high levels of cholesterol in the blood (hypercholesterolaemia) is considered a major risk factor for atherosclerosis, its reduction is obviously of great importance given the level of this disease state in the population. High levels of triglycerides are also linked to heart disease, although the precise relationship is less clear than with cholesterol.
There are presently a number of drugs that may be applied to the lowering of lipids in the body and these include:
These different classes of drugs work on a variety of different pathways in the body.
Anion exchange resins
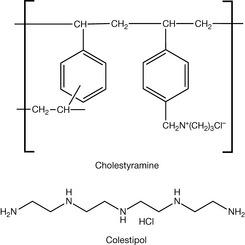
Anion exchange resins such as Colestyramine and Colestipol (Fig. 12.1) have been used for some time as lipid-lowering agents. These two compounds act as bile acid sequestrating agents, binding to bile acids in the intestine via an electrostatic mechanism where the negatively charged acids bind to the positively charged resin. Through this process insoluble complexes are formed which are then excreted. As a result of this process plasma bile acid levels are reduced and subsequently cholesterol, which is the biosynthetic precursor of bile acids, is converted into bile acids and thus the levels of cholesterol in the body are reduced.
Fibrates
Fibrates are a class of lipid-lowering drugs that are all related to fibric acid (Fig. 12.2). It is reported that their discovery was serendipitous. In the 1950s, available evidence suggested that androsterone was capable of reducing high levels of lipids in the blood. Formulation scientists trying to overcome the administration difficulties of androsterone used clofibrate as a solvent for the steroid. This product, subsequently known as Atromid, was shown to be effective as a lipid-lowering drug. Further studies eventually demonstrated that the lipid-lowering effect of this formulated product was due not to the androsterone but to the solvating agent clofibrate. Since the original discovery, a number of ‘fibrates’ have been introduced and several are still available clinically, however, the use of these drugs is less common nowadays due to the introduction of the newer lipid-lowering agents. Fibrates are, however, indicated as first-line therapy for patients with serum triglyceride levels greater than 10 mm/L. The fibrates that are clinically used at present include ciprofibrate, bezafibrate, fenofibrate and gemfibrozil (Fig.12.2).
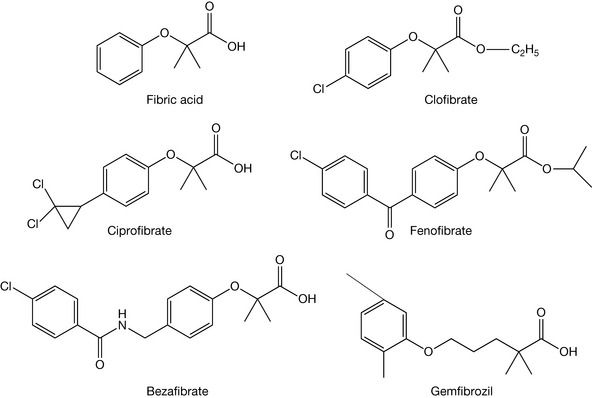
Fibrates act as peroxisome proliferator activated receptors (PPAR) agonists and in particular PPARα agonists. PPARs, amongst other things, play an important role in the regulation of metabolism, including lipid metabolism. The action of the fibrates results in lowering of elevated cholesterol levels via increasing HDL levels and decreasing LDL (and very low-density lipoproteins VLDLs) levels as well as reducing triglyceride levels. The reduction in triglyceride levels is through reduced production in the liver and an increase in the rate at which they are removed from the blood stream.
Ezetimibe
Ezetimibe (Fig. 12.3) is a drug that works by reduction of blood cholesterol by inhibiting the absorption of cholesterol by the small intestine. As indicated above, cholesterol in the body is derived from dietary sources or is synthesised in the body. Acyl Coenzyme A: Cholesterol A Transferase (ACAT) is a membrane protein that catalyses the synthesis of cholesteryl esters from cholesterol. Inhibition of ACAT has therefore been investigated as a route to reducing blood cholesterol levels and thus a means of preventing atherosclerosis. Whilst investigating potential ACAT inhibitors, investigators at Schering-Plough discovered a group of azetidinon-2-ones (Fig. 12.4) that were inhibitors of cholesterol absorption.
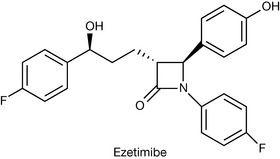
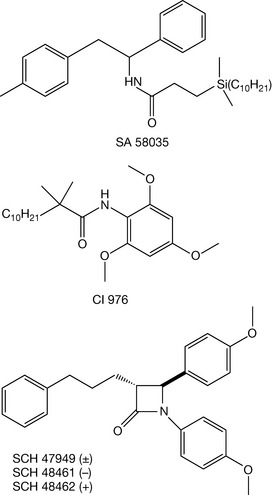
This work involved a study of conformationally restricted forms of SA 58035 and CI 976, which are examples of two classes of compounds that act as ACAT inhibitors. During this work it was demonstrated that monocyclic β-lactams such as those shown in Figure 12.4 were inhibitors of cholesterol absorption. However, it was also discovered that they did not act as ACAT inhibitors, but via a different mechanism. From this work the now clinically used drug ezetimibe has been developed and it is now known that this drug acts by inhibiting cholesterol absorption in the intestines. It is believed that ezetimibe undergoes phase II metabolism to form the glucuronide of the phenolic hydroxy group. This metabolite appears to become localised in the brush border of the small intestine, thereby preventing cholesterol uptake. The precise mode of action of ezetimibe is still unclear. It has recently been reported that NPC1L1 (Niemann-Pick C1-Like 1) is the major target for ezetimibe; however, this has been contradicted.
Statins
The statins are a group of compounds that were discovered about forty years ago. During the 1960s, researchers had shown that cholesterol could be obtained from the diet or it could be synthesised in the body. It was observed that if humans consumed a diet rich in cholesterol then the body stopped synthesising it. This suggested that there was some form of feedback mechanism in the body that resulted in the inhibition of cholesterol synthesis. It was proposed that the enzyme system HMG CoA reductase was the enzyme system that was inhibited in this process.
The biosynthesis of cholesterol occurs in three main phases: stage one is the formation of mevalonic acid, stage two is conversion of mevalonate in farnesylpyrophosphate, and the final stage involves the head-to-tail condensation of two farnesylpyrophosphate units to yield squalene which subsequently yields cholesterol via lanosterol. The mevalonic acid (mevalonate) sequence is shown in Figure 12.5. In this sequence two molecules of acetyl coenzyme A condense to generate acetoacetyl CoA, which reacts with a further molecule of acetyl CoA to generate 3-hydroxy-3-methylglutaryl CoA (HMG CoA). HMG CoA is reduced by HMG CoA reductase to yield mevalonic acid. Note this last step is most important since it is inhibition of this enzyme system by statins which is pivotal in their lipid-lowering activity.
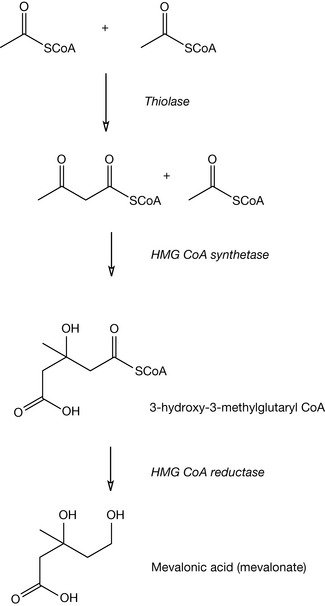
In the early 70s, in Japan, Drs Endo and Kuroda began studies to find chemical entities that would inhibit the activity of HMG CoA reductase and thus interfere with the synthesis of cholesterol in the human body. They investigated many microorganisms to find such inhibitors since they proposed that such organisms might produce inhibitors in order to combat attack on the mevalonic acid biosynthesis, which is prevalent in many organisms.
The action of the statins is related to their structural similarities to HMG CoA. In Figure 12.5, we can see the conversion of HMG CoA into mevalonic acid via the enzyme HMG CoA reductase. This process occurs via the intermediate mevalonyl CoA. The structure of both and the statin pharmacophore are shown in Figure 12.6. It is the dihydroxyheptanoic acid portion of the statins (circled in the diagram of the pharmacophore) that closely resembles the endogenous substrate for the HMG CoA reductase that allows the statins to act as inhibitors of the reductase. In some cases, such as with simvastatin (Fig. 12.7), the dihydroxyheptanoic acid portion exists as the lactone form and, as such, the statin is in fact a prodrug form of the parent molecule. Under physiological conditions the lactone form of the drug is in equilibrium with open chain form.
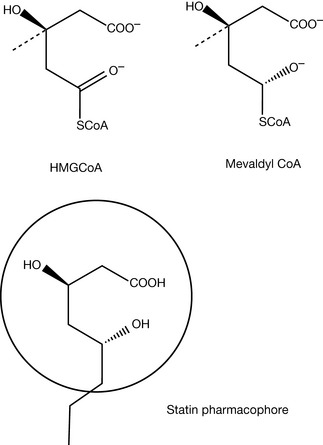
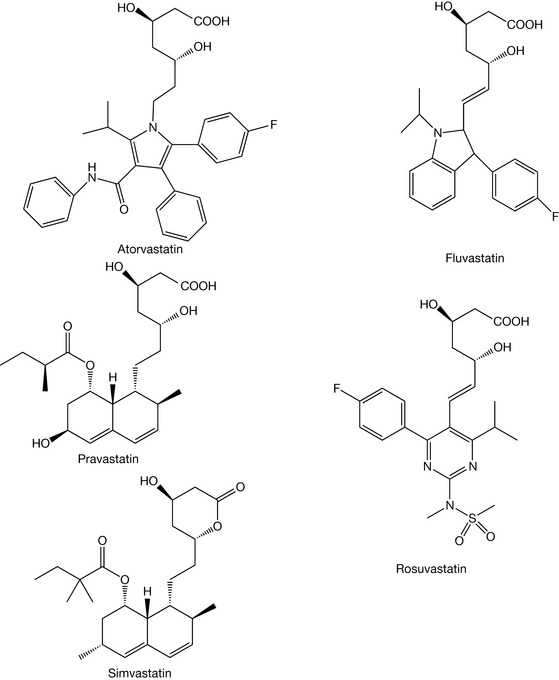
Statins act to reduce the production of cholesterol by interfering in the biosynthesis of cholesterol. The therapeutically used statins are shown in Figure 12.7.
Compounds of the nicotinic acid group
Nicotinic acid and various analogues have been reported to produce lipid-modifying effects. At present, nicotinic acid (including modified release forms) and acipimox are available for clinical use (Fig. 12.8).
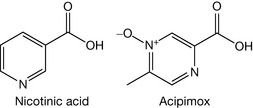
These two compounds have been reported to act by lowering levels of LDLs and VLDLs whilst raising levels of HDLs. It is believed that this action arises from the blocking of the breakdown of adipose tissue fats, and that the recently reported nicotinic acid receptors may be involved.
Angiotensin-converting enzyme inhibitors
Angiotensin-converting enzyme (ACE) inhibitors are indicated for the treatment of high blood pressure, heart failure and for the prevention of strokes. ACE inhibitors act on the renin-angiotensin-aldosterone system (Fig. 12.9). This system is involved in the regulation of various biological systems relating to hypotension, reduced blood volume, decreased sodium and sympathetic stimulation. In response to these factors renin, which is a proteolytic enzyme, is released into the circulation by the kidneys. Renin acts on circulating angiotensinogen and converts it to angiotensin I which is subsequently converted to angiotensin II by ACE.
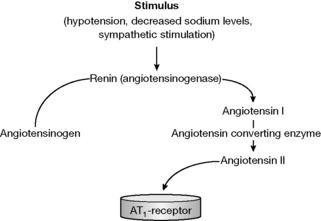
The renin-angiotensin system is used clinically to treat high blood pressure by the use of ACE inhibitors. Thus we can see that in the system above angiotensin I is converted into angiotensin II via the action of ACE, which increases blood pressure. Therefore, if we can inhibit the action of this enzyme system there will be the opportunity to reduce high blood pressure in hypertensive patients.
The discovery and development of ACE inhibitors
Studies undertaken in the 1960s were based on the assumption that ACE was involved in hypertension. During this work, an extract from the venom of the Brazilian pit viper Bothrops jararcara was investigated and shown to be a potent inhibitor of ACE. From the one extract that demonstrated high activity, a number of peptides were isolated and the amino acid sequence was elucidated. One of these, the nonapeptide teprotide (Glu-Trp-Pro-Arg-Pro-Gln-Ile-Pro-Pro) was shown to be highly active but unfortunately it did not possess oral activity. The search then began for compounds with this ACE inhibitory activity that were also orally active. Following many investigations, captopril (Fig. 12.10) was introduced as the first potent orally active ACE inhibitor that was clinically effective.
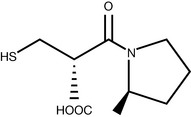
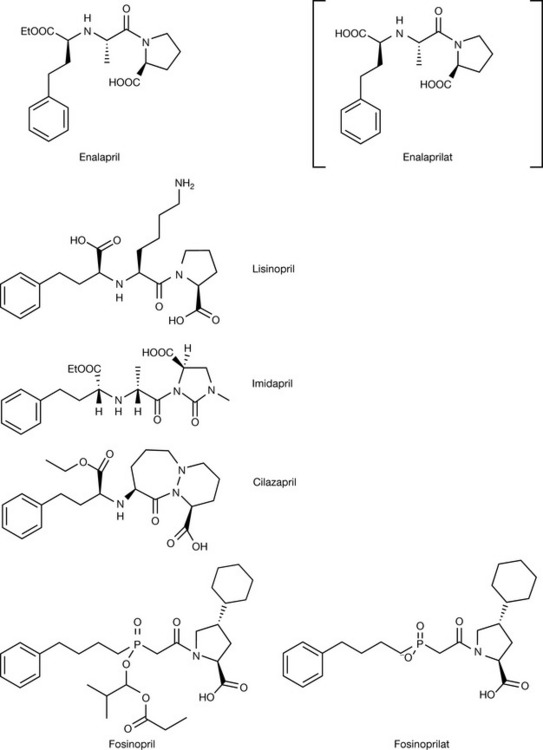
When the drug was introduced into the clinic it was most efficacious, although a number of adverse side effects were observed including skin rash and loss of taste, and thus the search for better ACE inhibitors began. This has resulted in a large number of compounds presently available as ACE inhibitors. Some of these are: captopril, cilazapril, enalapril (maleate), fosinopril, imidapril, lisinapril, moexipril (hydrochloride), perindopril (erbumine, [t-butyl amine]), quinapril, ramipril and trandolapril (Figs 12.11, 12.12).
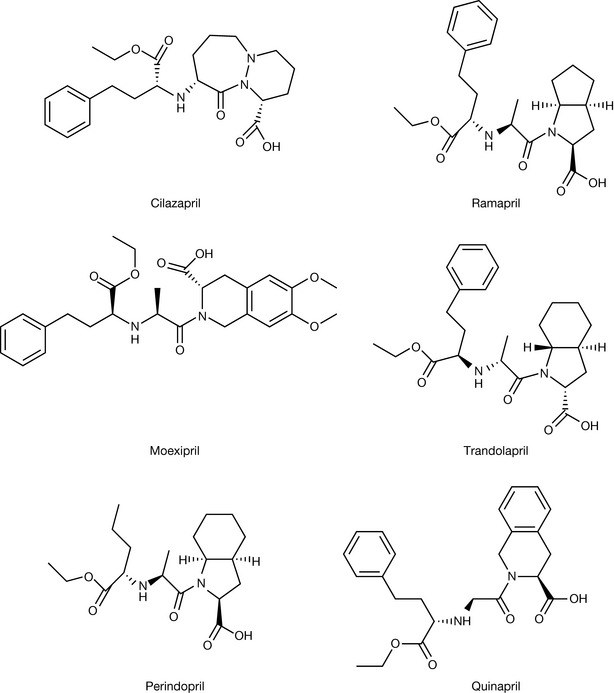
The side effects associated with captopril were thought to be associated with the sulphydryl moiety since similar observations had been made in the case of penicillamine. The search for improved ACE inhibitors led to the development of a number of compounds from an understanding of the nature of ACE and its substrates. Throughout the initial work on the development of ACE inhibitors it was known that:
Enalaprilat was the first member of the dicarboxylate class of ACE inhibitors introduced. In these compounds the sulphydryl group is replaced by a carboxylic acid. When this compound was developed it was found that, because of the structural modifications introduced to deliver activity similar to captopril, the compound became orally inactive. In order to overcome these problems the ethyl ester prodrug enalapril was developed. This is rapidly metabolised in the body to release the active form. The quantitative structure–activity relationship (QSAR) studies that were undertaken around this time also indicated that there was a hydrophobic pocket present in ACE close to where the proline groups would sit and that by introducing a variety of modified ring systems, including bicyclic and spiro systems in place of the proline system, compounds with higher potency could be made (Figs 12.11, 12.12).
Another aspect that arose from these studies was the possibility that phosphinic acid groups could be incorporated into the inhibitors. Fosinopril (Fig. 12.11) is an example of such a product. However, once again note that this compound is a prodrug that is hydrolysed in vivo to the active form fosfinoprilat.
Phosphodiesterase inhibitors
Cyclic nucleotides such as cyclic adenosine 3’,5’-monophosphate (cAMP) and cyclic guanosine 3’,5’-monophosphate (cGMP) play an important role in cellular signalling as second messengers in response to, for instance, neurotransmitters or hormones. The initial action involves the (hormone) interacting with a G protein-coupled receptor that activates the enzyme systems adenyl cyclase or guanyl cyclase, which are able to convert adenosine triphosphate (ATP) or guanosine triphosphate (GTP) into their cyclic forms (Figs 12.13, 12.14). cAMP and cGMP, as second messengers, are then involved in binding to, and activation of, protein kinases that are themselves involved in a variety of actions via phosphorylation. These protein kinases are involved in wide ranging regulatory processes such as control of sugar, glycogen and lipid metabolism as well as the flow of Ca2+ through ion channels. Phosphodiesterases act by conversion of cGMP or cAMP into 5’-AMP or 5’-GMP which are inactive as shown in Figures 12.13 and 12.14. As such, these phosphodiesterases (PDEs) may be considered as regulators of the activity of these second messengers. Phosphodiesterase inhibitors are drugs that interfere with the action of the phosphodiesterases thereby modifying the inactivation of cGMP or cAMP.
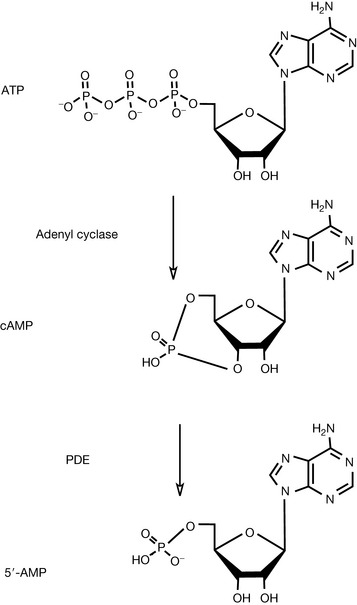
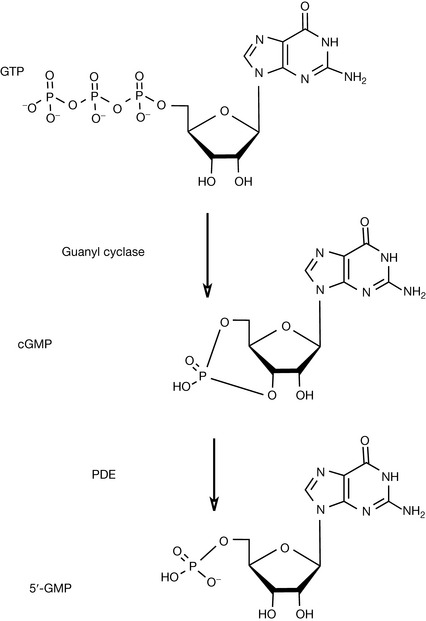
There are at present 11 families of mammalian PDEs reported and within each family there are subgroups that have been classified. It is recognised that some PDEs are non-specific in that they can inactivate both cAMP and cGMP whilst others are selective for the inactivation of one or other of these cyclic nucleotides. In terms of PDE inhibitors, it has been discovered that some compounds may act as non-selective phosphodiesterase inhibitors whilst other compounds may act in a very specific fashion with useful clinical activity.
Non-selective phosphodiesterase inhibitors
A series of xanthine derivatives including theophylline, caffeine, theobromine and pentoxifylline (Fig. 12.15) that have bronchodilator properties have been shown to be non-selective phosphodiesterase inhibitors.
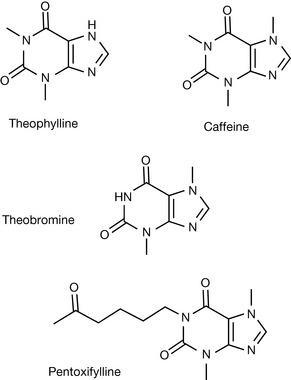
It has been proposed that these bronchodilator effects of xanthines result from a relaxant effect on bronchial smooth muscle and that the xanthines regulate the cAMP and cGMP in the smooth muscle via PDE inhibition.
Selective phosphodiesterase inhibitors
Within the last 10 years there have been a number of important developments in terms of selective PDE inhibitors with the licensing of phosphodiesterase 5 (PDE5) inhibitors in the late 1990s paving the way for a range of new clinical applications.
Phosphodiesterase 5 selective inhibitors
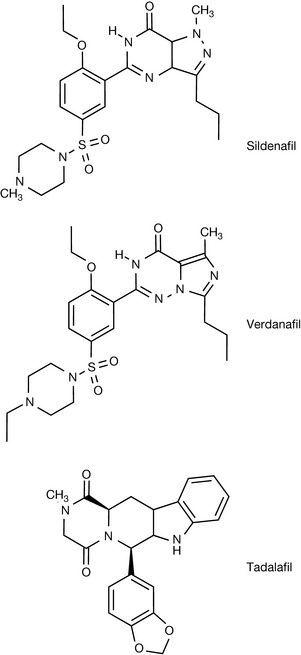
Researchers at Pfizer who were investigating compounds that possessed antianginal and antihypertensive properties discovered this group of compounds. The researchers were interested in atrial natriuretic factor (ANF), because of its vasodilator and natriuretic properties, in cGMP and PDEs. They were working on the hypothesis that because ANF produced its effects via the stimulation of guanyl cyclase, which resulted in an increase in cGMP, and that this was degraded by PDEs. They proposed that compounds that inhibited the action of the PDEs would reduce the degradation of cGMP and hence potentiate the vasodilator and natriuretic properties of ANF. This group began by studying zaprinast (Fig. 12.16). This drug had been shown to have antiallergy drug properties but also had vasodilator properties resulting in smooth muscle relaxation allowing increased flow of blood. Zaprinast was found to be a poorly selective PDE inhibitor but from this basic work information was obtained that led to investigations concerned with improving the selectivity towards PDE5 and also the potency. Investigations of dipole moments of zaprinast and cGMP showed similarities. Computational studies were able to show that cGMP could adopt a syn conformation and that modification of the skeleton of zaprinast would enable the inclusion of isosteric moieties for the cyclic phosphate portion of the cGMP. X-ray studies indicated co-planarity via intramolecular hydrogen bonding within the molecule; therefore, this portion of the molecule was left intact. Following several investigations the pyrazolopyrimidinone lead compound shown in Figure 12.16 was synthesised and shown to have significant activity and selectivity towards PDE5. Further studies led to the discovery of sildenafil, which had a 100-fold increase in PDE5 inhibitory activity compared to zaprinast, and exceptional selectivity for PDE5 versus other PDEs. When the drug was taken forward into clinical trials in a study of patients with coronary heart disease, the results were somewhat less promising than had been hoped for. A number of side effects were noted during the trial, one of which was an increase in erectile function, and thus the research then focused on the possibility of using the drug for patients suffering from erectile dysfunction.
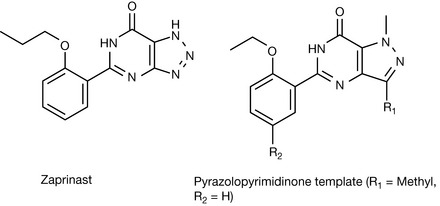
The mechanism by which sildenafil produced this change in erectile function was investigated. It was found that through sexual stimulation nitric oxide (NO) is released in the penis, which causes guanyl cyclase to increase the levels of cGMP in the corpus cavernosum. The cGMP causes relaxation of the smooth muscle, increased blood flow and this results in an erection; then, through the action of PDE5, the cGMP is converted to GMP and the erection stops. Sildenafil was shown to inhibit the breakdown of cGMP by interaction with the active site of the PDE5 system thus reducing the hydrolysis and thereby improving erectile function. Since the introduction of sildenafil, other agents that are also capable of treating erectile dysfunction have been reported, e.g. vardenafil and tadalafil (Fig. 12.17).
Other selective phosphodiesterase inhibitors
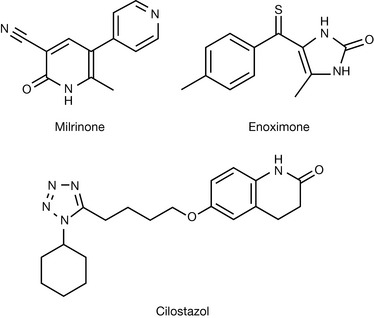
Following on from the introduction of sildenafil, a number of other selective PDE inhibitors have been introduced, including PDE3 and PDE4 inhibitors. The PDE3 selective inhibitors milrinone and enoximone (Fig. 12.18) are indicated for congestive heart failure, whilst cilostazol, also a selective PDE3 inhibitor, is used in the treatment of intermittent claudication.
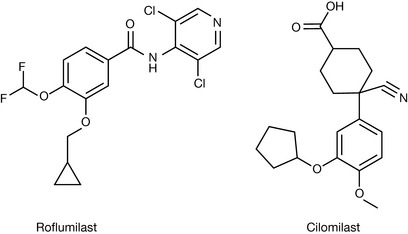
The PDE4 selective inhibitors being studied at present are roflumilast and cilomilast (Fig. 12.19); these compounds are being investigated for use in chronic obstructive pulmonary disease because of their possible effects on inflammatory mediators.
Sulphonamide diuretics
Diuretics are drugs that promote the formation/secretion of urine in the kidney. There are a number of different classes of drugs that are used to achieve this promotion of urine formation, and a variety of mechanisms by which this may occur including mercurial diuretics, which are no longer used, osmotic diuretics, carbonic anhydrase inhibitors, thiazide and thiazide-like diuretics, loop diuretics and potassium-sparing diuretics. Diuretic drugs are used to aid medical situations where there is a build-up of fluid in the body that occurs, for instance in high blood pressure, congestive heart failure, liver and kidney disease. Additionally some diuretics are also used in the treatment of glaucoma. In this section we will concentrate on those compounds that contain a sulphonamide functional group, i.e. thiazide/thiazide-like and loop diuretics, and we shall consider the mechanisms of action of these compounds, including consideration of their carbonic anhydrase inhibitory activity and their involvement with the Na/Cl/K symporter.
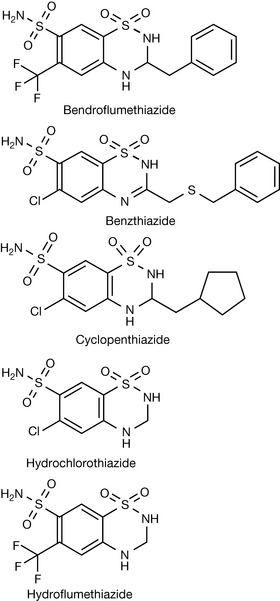
There are a relatively large number of compounds that might be grouped together under the heading of sulphonamide diuretics. Within the group of compounds containing this functionality, we have a variety of different classes of diuretics based on their mechanism of action. These compounds are considered active as carbonic anhydrase inhibitors (acetazolamide), inhibitors of the reabsorption of Na+ Cl− (thiazide and thiazide-like diuretics [benzthiazide, hydrochlorothiazide]) and inhibitors of the Na K Cl symporter (furosemide).
The development of sulphonamide diuretics arose from early observations concerning the use of sulfanilamide as an antibacterial agent. Sulfanilamide was shown to be the active metabolite of prontosil (Fig. 12.20) when used in vivo.
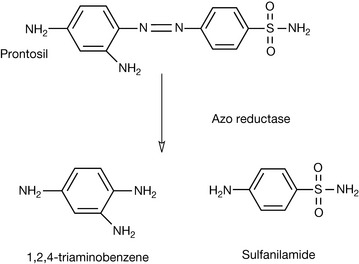
It was observed that this compound produced a diuretic effect although the diuresis was somewhat limited. Attempts were made to synthesise a range of compounds related to sulfanilamide to improve the diuretic effect. The basic premise in this work appears to have been based on synthesising compounds, which contained the -SO2NH2 moiety, attached to an aromatic or a heterocyclic ring system. From this early work based on the investigation of heterocyclic ring usage, acetazolamide (Fig. 12.21) was developed.
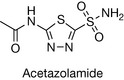
Sulfanilamide and acetazolamide were shown to be carbonic anhydrase inhibitors.
In addition to the development of acetazolamide, work was also undertaken on the substitution of the benzene ring of sulfanilamide. Many compounds were prepared including a number with sulphonamide groups present.
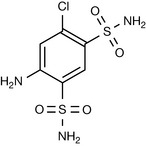
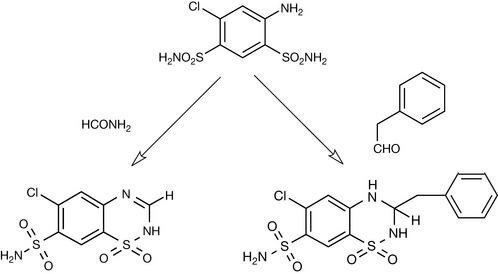
One of the compounds that resulted from these studies was 4-amino-6-chloro-3-benzenedisulfonamide (Fig. 12.22). This compound displayed some diuretic properties; however, it was never used as a routine clinical diuretic. Further modifications of this compound were attempted, involving the derivatisation of the 4-amino functionality. Treatment of this compound with, for instance, formamide, resulted in cyclisation with the formation of thiazides, whilst reaction with aldehydes and ketones resulted in the preparation of hydrothiazides (Fig. 12.23).
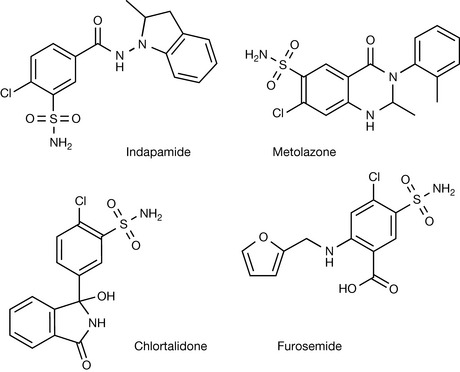
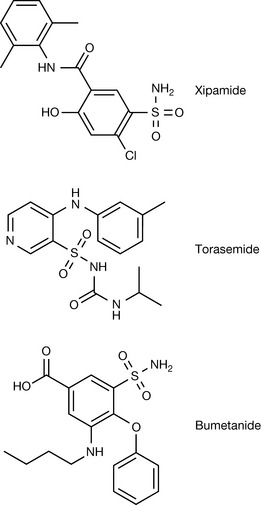
Further investigations in this area led to the preparation of a range of compounds classified as thiazide-like diuretics, e.g. indapamide, metolazone and chlortalidone, and other compounds such as furosemide (Fig. 12.24).
The BNF lists a number of other compounds that can be classified as sulphonamide diuretics and these compounds are detailed in Figures 12.25 and 12.26.
Mechanism of action
The functioning unit of the kidney is the nephron, and blood is brought into the nephron via the afferent arteriole where it enters the renal corpuscle, which consists of the glomerulus and Bowman’s capsule. About one-fifth of the blood (plasma) passing through the nephron in any given time is filtered by this system. The filtrate that passes into Bowman’s capsule contains water, nutrients, minerals (salts) and waste material, but red and white blood cells, platelets and large molecules such as proteins will not pass into Bowman’s capsule. On leaving the renal corpuscle, the blood enters a set of capillaries known as the peritubular capillaries, whilst the filtrate enters the proximal convoluted tubule and then to the proximal straight tubule, the loop of Henle, the distal convoluted tubule and thence to collecting tubule. During this process there is the possibility for a series of exchanges of both liquid and solutes between capillaries and the tubules. Throughout the passage through the nephron there are changes in the osmolarity of the plasma via exchanges that can occur through concentration gradients within the various sections; these are controlled by different mechanisms. Through these processes liquid, electrolytes and nutrients (salts, amino acids, glucose) are reabsorbed whilst the waste material (e.g. urea) is taken on to the collecting duct and thence to the bladder. In those disease states where diuretics are employed, the diuretics act by reducing the reabsorption of water and salts, thereby increasing urine production. As explained in the earlier section, the majority of the sulphonamide diuretics were developed from the initial idea that sulfanilamide and acetazolamide were carbonic anhydrase inhibitors. It is now well known that the thiazide and thiazide-like diuretics act by different mechanisms. Thiazides act in the distal tubule by interfering with the reabsorption of sodium and chloride ions; this action results from binding with the Na/Cl/K symporter.
The loop diuretics such as furosemide, torasemide and bumetanide act in the thick ascending loop of Henle, inhibiting the reabsorption of sodium, potassium and chloride ions.
Section B – proton pump inhibitors
Observations on the effect of the potential antviral drug pyridylthioacetamide (Fig. 12.27) in reducing gastric acid secretion (GAS) led to the lead compound H124/26 which was highly effective in reducing GAS. It was found that this compound was metabolically converted to timoprazole (Fig. 12.27) which was the active form of the drug. The sulphoxide group was thus found to be an important element in the drug action and picoprazole was developed and found to be free from the antithyroid effects exhibited by timoprazole.
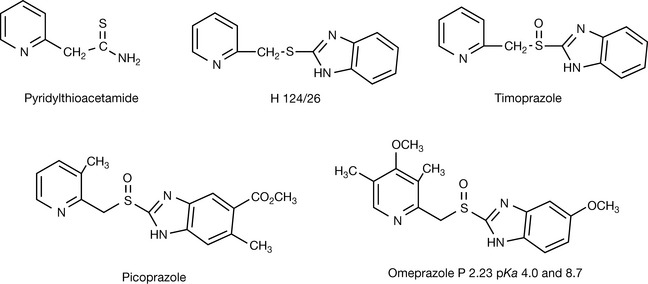
More potent analogues were achieved by increasing the basicity of the pyridine, eventually resulting in omeprazole (Fig. 12.27). The addition of a 4-methoxy group to a pyridine ring raises its pKa from 5.2 to 7.1 due to the electron-rich substituent increasing the electron density in the ring;1 the methyl groups also increase the electron density in the ring. Thus it would seem that the measured pKa value of 8.7 for omeprazole2 refers to the nitrogen in the pyridine ring which is thus a strong nucleophile. Radiolabelled 3H omeprazole was given i.v. and found to accumulate in parietal cells in the gastric mucosa. Parietal cells contain the membrane channel protein H+K+ATPase which is involved in exporting H+ (and Cl−) out of the cell in exchange for K+ (and 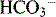 ) into the cell. Figure 12.28 shows a proposed mechanism for the export of H+ out of the parietal cells via phosphorylation of the membrane channel protein.
) into the cell. Figure 12.28 shows a proposed mechanism for the export of H+ out of the parietal cells via phosphorylation of the membrane channel protein.
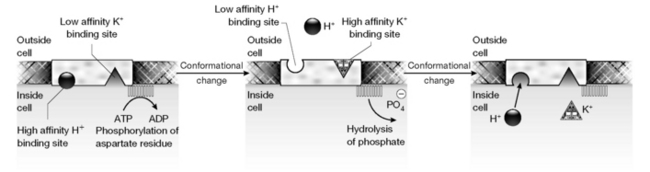
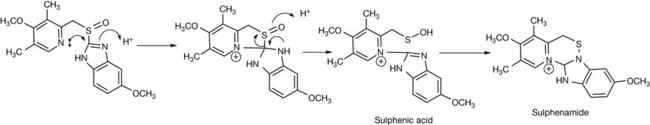
Omeprazole becomes completely ionised and thus trapped in secretory channels of the parietal cells due to an acid-catalysed rearrangement which converts it into a quaternary amine. The mechanism of the structural rearrangement is not known exactly. It seems likely that it is the sulphenic acid (Fig. 12.29) intermediate that reacts with the H+K+ATPase and rather than the sulphenamide (Fig. 12.29), which is a more stable end product of the rearrangement.2
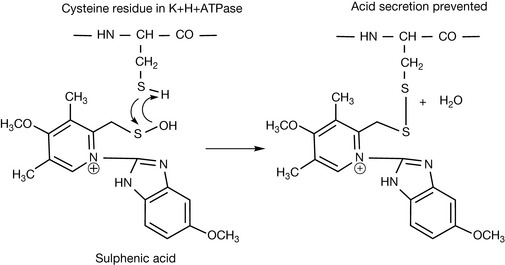
The sulphenic acid intermediate reacts with a cysteine residue in the H+K+ATPase (Fig. 12.30) preventing it from functioning properly and thus preventing acid secretion by the parietal cell. Regeneration of the enzyme takes 3–5 days. There is a cysteine residue one amino acid removed from the phosphorylation site of the protein which may be the target of the drug. Since the drug undergoes rearrangement when exposed to acidic conditions it has to be administered in the form of capsules or enterically coated tablets which are resistant to the stomach acid and pass through the stomach, releasing the drug in the small intestine. The drug can also be given intravenously.
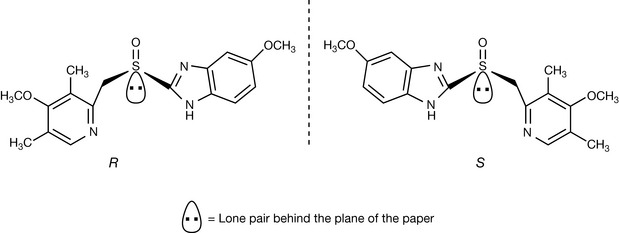
Sulphoxides have a tertrahedral geometry and, like carbon atoms, can exhibit chirality, with the lone pair on the sulphur atom taking lowest priority in the same manner as a proton attached to a carbon atom has lowest priority. Omeprazole can exist as R and S enantiomers (Fig. 12.31). The S isomer contibutes 90% inhibition of gastric acid production compared with 20% for R-isomer, and greater efficacy has been claimed for it. The S enantiomer is marketed as esomeprazole.
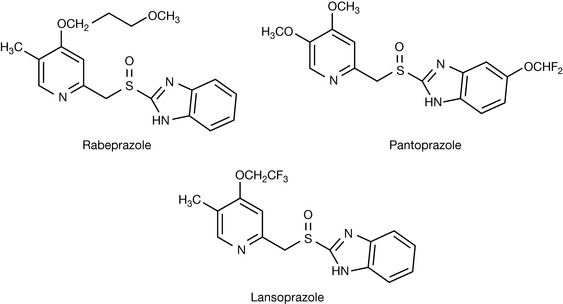
Since omeprazole reached the market, a number of compounds with similar actions have been marketed (Fig. 12.32). They all act via the same mechanism, as can be seen from the essential pharmacophore which is present in all the structures, and there is no particularly strong argument for using one over another.
Formulations of proton pump inhibitors in the BNF
1 Grandberg I., Faizova G.K., Kost A.N. Comparative basicities of substituted pyridines and electronegativity series for substituents in the pyridine series. Khimiya Geterotsiklicheskikh Soedinenii. 1966;2:561-566.
2 Qaisi A.M., Tutunji M.F., Tutunji L.F. Acid Decomposition of omeprazole in the absence of thiol: A differential pulse polarographic study at the static mercury drop electrode (SMDE). J Pharm Sci. 2006;95:385-391.