Chapter 22 Antimicrobial chemotherapy
Section a – antibiotics
Introduction
Antibiotics are generally products, or modifications thereof, of microbial metabolism that at low concentrations kill or inhibit the growth of other microorganisms. The observation that fungi and yeast could produce substances capable of destroying other bacteria was made by Vuillemin at the end of the nineteenth century and led to the concept of antibiosis (anti – against; bios – life). The microorganisms from which useful antibiotics have been obtained include fungi (Penicillium, Cephalosporium and Micromonospora) and bacteria (Bacillus and Streptomyces). The discovery of penicillin by Alexander Fleming in 1928 marked the beginning of antimicrobial chemotherapy in the modern era. This serendipitous discovery was followed by the introduction of the sulphonamides (1932), cephalosporins (1940s), macrolides (1952), vancomycin (1956), quinolones (1962) fluorinated quinolones (1980s) and quite recently linezolid (2000).
The fundamental basis of antimicrobial chemotherapy is selective toxicity. This is achieved by exploiting structural and biochemical differences between a mammalian host and the causative microorganism. Some of the biochemical and structural differences which have been exploited to produce useful antibiotics include cell metabolism, cell wall synthesis, protein and nucleic acid synthesis. Antibacterial agents may be bacteriostatic or bactericidal and may have either a narrow spectrum (affecting few species or genera) or broad spectrum (affecting both Gram-positive and Gram-negative bacteria) of activity. In this chapter, the antibacterial agents are discussed based on their mechanisms of action, such as the effects on bacterial cell wall synthesis, cell metabolism, protein synthesis and nucleic acid synthesis. Other antibiotics in current use, which exert their action by entirely different mechanisms, are also described.
Inhibition of cell wall synthesis
Unlike eukaryotic cells, prokaryotic cells possess a cell wall which maintains cell integrity and offers protection from the harsh environments in which they exist. The bacterial cell wall conforms to two basic designs which can be distinguished by the Gram stain, i.e. Gram negative and Gram positive. In both designs, the bacterial cell wall is made up of peptidoglycan which consists of the saccharides N-acetylglucosamine (NAG) and N-acetylmuramic (NAM) linked together by peptide bonds to confer mechanical strength. These peptide bonds are formed between the pentaglycine of one saccharide chain with the penultimate D-alanine of another chain, leading to the loss of a terminal alanine. The production of the cell wall in bacteria involves multiple processes such as the partial assembly of cell wall components within the cell, transportation of the partially assembled components across the cell membrane to the cell wall (transglycosylation), assembly into the cell wall and finally cross-linking of the polysaccharide chains. Due the absence of the cell wall in eukaryotes, the interference with the various processes involved in cell wall synthesis serve as useful targets for achieving antibiosis. Commonly used antibiotics such as the β-lactams (penicillins, cephalosporins, carbapenems and monobactams) and vancomycin, which inhibit the synthesis of the cell wall, have a specific effect on one or more of the processes involved in cell wall synthesis.
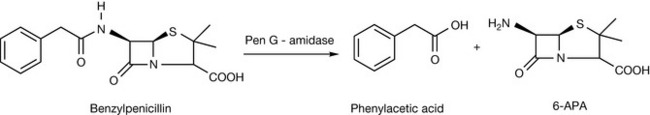
In 1929, Fleming reported the inhibition of the growth of staphylococci by a rare strain of mould, Penicillium notatum. The isolation and identification of the active substance, benzylpenicillin (Penicillin G), was achieved at Oxford University by Florey and Chain towards the end of the Second World War. The penicillin nucleus, with the characteristic β-lactam ring, was isolated in 1959 by Batchelor and co-workers and serves as the precursor for many semi-synthetic penicillin derivatives. The natural penicillins (Penicillin G and phenoxymethylpenicillin (Penicillin V)) are obtained through fermentation processes. The semi-synthetic analogues have mostly been produced using 6-aminopenicillanic acid (6-APA) which is derived from the natural penicillins by selective hydrolysis of the amide side chain while avoiding hydrolysis of the intrinsically more labile β-lactam ring. The selective hydrolysis of the natural penicillins to 6-aminopenicillanic acid has been achieved by the use of penicillin amidases/acylases (Fig. 22.1).
The β-lactam group of antibiotics remains one of the most important and widely used therapeutic agents. The clinically important groups include the penicillins, cephalosporins, carbapenems and monobactams. These compounds derive their classification from the β-lactam moiety which is invariant in their structures and also accounts for their antibacterial activity. The generic structures of the β-lactam antibiotics are shown later on in this chapter.
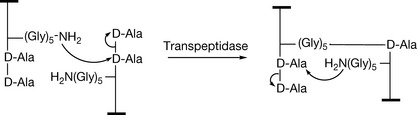
Multiple enzymes are involved in the synthesis of the bacterial cell wall. The β-lactam antibiotics bind to several of these enzymes, which are collectively called penicillin binding proteins (PBP) and inhibit the cross-linking of the peptidoglycan strands. The most important PBP, transpeptidase, is involved in the final cross-linking step in cell wall synthesis. The cross-linking of the bacterial cell wall involves the formation of new peptide bonds between the pentaglycine of one saccharide chain with the penultimate D-alanine of another chain (Fig. 22.2), leading to the loss of the terminal alanine.
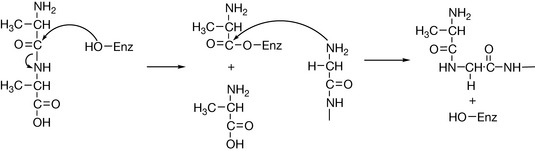
The transpeptidase enzyme participates in the reaction as above and effectively attacks the sensitive amide linkage of the D-alanyl-D-alanine terminated peptides. The transpeptidase enzyme is regenerated after it has facilitated the peptide bond formation between the carbonyl of the penultimate D-alanine group and the N-terminus of glycine (Fig. 22.3).
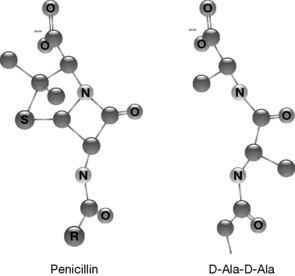
However, the structure of the penicillin (and other β-lactams) resembles the D-alanyl-D-alanine moiety (Fig. 22.4) and can therefore react with the transpeptidase enzyme to form a covalent ester bond.
The acylated enzyme formed by reaction with the β-lactams does not react as readily with nucleophiles as the activated complex formed with D-Ala-D-Ala. Therefore, the transpeptidase enzyme is inactivated, resulting in the inhibition of further cross-linking of the bacterial cell wall. This triggers a sequence of events including the release of bacterial autolysin and eventually results in cell death. The β-lactams are therefore bactericidal.
Structure–activity relationships of the penicillins
It is evident from the spatial orientation of the penicillin molecule relative to the D-alanyl-D-alanine moiety that an intact β-lactam ring is required to maintain structural similarity and therefore competition for the active site of the transpeptidase enzyme. The parent 6-aminopenicillanic acid moiety is invariant in all the penicillins (Fig. 22.5) and allows only two points for chemical modification; the side chain (R) attached to the amino group on C-6 and the carboxyl group on C-3.
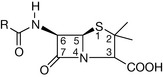
The nature of the side chain (R) determines the hydrophilic/hydrophobic balance of the molecule, the antibacterial spectrum and chemical properties such as instability to acids and β-lactamases – the bacterial defence mechanism. The carboxyl group is a site for salt formation and facilitates the formation of Na, K and Ca salts. The free carboxyl group is required for activity and therefore non-degradable moieties, such as amides and esters, lack antibacterial activity in vitro. Prodrugs, which are rapidly degraded in vivo to release the active penicillin, are formulated to overcome problems with oral bioavailability. Slow-release parenteral dosage forms are achievable by ester formation at the carboxylic acid group or by ion pair formation or by ester formation. Procaine penicillin and benzathine penicillin represent the former type and bacampicillin the latter.
Reactivity of the β-lactam ring
Amide resonance is responsible for the lower susceptibility of carbonyl groups to nucleophilic attack. In a normal amide, the planar arrangement of the O, C and N atoms is generally assumed to be necessary for effective delocalisation of the nitrogen lone pair and therefore exists in the stable canonical forms shown in Figure 22.6.
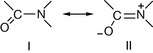
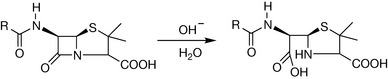
The ring strain caused by the orientation of the β-lactam in the penicillin molecule affects this resonance stabilisation and results in a greater probability of canonical form I. The corollary is that the electron-deficient carbon is more susceptible to nucleophilic attack. Such attack leads to disruption of the C–N bond, dissimilarity of the resulting molecule to D-alanyl D-alanine, and subsequently loss of antibacterial action. For example, in dilute alkali solutions, the nucleophilic attack on the electro-deficient C-7 carbon in the penicillin molecule leads to rapid hydrolysis to penicilloic acids (Fig. 22.7).
Other important reactions of the β-lactam antibiotics, such as attack by bacterial lactamases and reaction with penicillin-binding proteins (e.g. transpeptidase enzyme), proceed via an identical mechanism of nucleophilic attack on the C-7 carbon.
Penicillins are also unstable in acidic solutions although the degradation proceeds by a different mechanism from that of alkaline hydrolysis. In aqueous acid, the oxygen of the C-7 carbonyl becomes protonated (Fig. 22.8). The neighbouring carbon (C-7) is rendered more electron deficient and therefore more susceptible to nucleophilic attack. This leads to the formation of penicilloic acids. The acylation of the 6-amino group of the penicillins is an essential feature for antibacterial activity. However, the oxygen in the amide side chain can interact with the electro-deficient carbon of the β-lactam ring in what is referred to as neighbouring group participation. This yields an intermediate product which leads to the formation of penillic and penicillenic acids (Fig. 22.8).
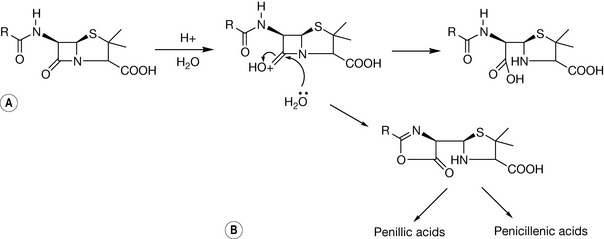
This mechanism explains the instability of penicillins such as Penicillin G in the acidic environment of the stomach. Substituents at the C-6 position in penicillins affect both the carbonyl carbon and the nitrogen of the β-lactam ring inductively. The attachment of an electron withdrawing group such as an amino group results in the reduction of the net negative charge on the oxygen in the β-lactam ring. This subsequently slows down the conversion from state A to B. In addition, the use of an electron withdrawing group at the R position reduces the effect of neighbouring group participation since the net negative charge of the oxygen is reduced, rendering its interaction with the C-7 carbonyl improbable.
Classification of the penicillins
The penicillins can be classified on the basis of their antibacterial activity (broad spectrum, narrow spectrum), method of production (natural or semi-synthetic) and based on their stability/instability to bacterial β-lactamases.
Naturally occurring penicillins
Benzylpenicillin ((6R)-6-(2-phenylacetamido) penicillanic acid, Penicillin G (Fig. 22.9)) and phenoxymethyl penicillin ((6R)-6-(2-phenoxyacetamido) penicillanic acid, Penicillin V) are naturally occurring penicillins produced by strains of Penicillium notatum. The β-lactam ring in Penicillin G is susceptible to hydrolysis by bacterial β-lactamases and is also unstable in acidic or alkaline environments, as explained before. The instability in an acidic environment makes Penicillin G unsuitable for oral administration and it is therefore formulated as the freely soluble sodium and potassium salts for parenteral administration.
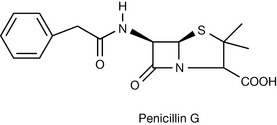
Penicillin G is administered either as an intramuscular injection or as a slow intravenous infusion. Due to the high water solubility of the potassium and sodium salts of Penicillin G, it is rapidly absorbed from intramuscular sites and frequent dosing is required to maintain therapeutic concentrations. The adsorption of benzylpenicillin can be delayed by producing derivatives of reduced solubility. Procaine penicillin, an equimolar salt of procaine and penicillin, occurs as white crystals or microcrystalline powder with reduced water solubility. It is therefore suitable for use in intramuscular depot preparations, providing therapeutic concentrations of Penicillin G to inhibit sensitive organisms for up to 24 h. Benzathine penicillin, which has poorer aqueous solubility, is also used in intramuscular depot preparations. It is prepared by the reaction of dibenzylethylene diamine with penicillin G in a 1:2 ratio and provides therapeutic concentrations for up to 120 h when administered by deep intramuscular injection.
In Penicillin V (Fig. 22.10), the additional electro-withdrawing effect of the phenoxy substituent, as opposed to the phenyl in Penicillin G, reduces the propensity of the β-lactam ring to undergo acid-catalysed hydrolysis. This makes it possible to administer Penicillin V orally.
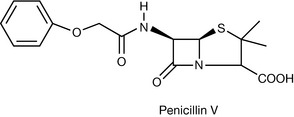
Penicillin G and Penicillin V are both too lipophilic to penetrate the Gram-negative bacterial cell wall, labile to β-lactamase inactivation and are therefore classified as narrow-spectrum antibiotics susceptible to β-lactamase inactivation. The natural penicillins are generally indicated for infections caused by Gram-positive organisms including most anaerobes. They are ineffective against Pseudomonas aeruginosa.
β-Lactamase stable penicillins
Bacteria produce hydrolytic enzymes called β-lactamases which are serine hydrolyase enzymes (penicillinases and cephalosporinases) which hydrolyse the β-lactam ring. The subsequent loss of resemblance to the D-alanyl-D-alanine results in the loss of antibacterial activity. The naturally occurring penicillins are hydrolysed by Gram-positive β-lactamases and this led to the design of compounds resistant to β-lactamases. The addition of a bulky carbocyclic or heterocyclic aromatic ring directly to the carbonyl moiety, substituting the amino group on 6-aminopenicillanic acid, causes steric hindrance around the sensitive carbonyl of the β-lactam ring. This effect either slows down the rate of hydrolysis or results in the loss of affinity for the active site of the β-lactamase enzyme. The design of β-lactamase stable penicillins led to methicillin (Fig. 22.11).
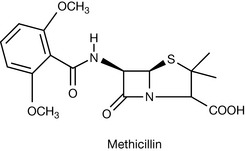
Methicillin has lost its clinical significance due to the high incidence of resistant Staphylococcus aureus infections. The use of heterocyclic substituents, based on isoxazolyl, led to the development of the penicillins cloxacillin and flucloxacillin (Fig. 22.12).
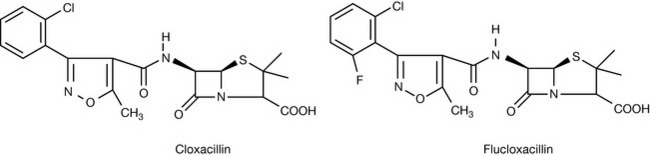
The isoxazolyl penicillins are highly lipophilic and therefore do not penetrate the Gram-negative bacterial cell wall. They are classified as narrow-spectrum penicillins stable to Gram-positive β-lactamases. These penicillins can be administered orally because of their stability in acid. Members of this class of antibiotics are preferred for treatment of staphylococcal infections of the skin and soft tissue. The isoxazolyl penicillins are prepared as sodium salts and available as capsules, intramuscular and slow intravenous infusions, elixirs and syrups.
Broad-spectrum penicillins
The antibacterial spectrum of the penicillins depends on their ability to cross the bacterial cell wall. However, structural differences exist in the composition of the cell wall in Gram-negative and Gram-positive bacteria and result in marked differences in permeability. The naturally occurring penicillins are too lipophilic to penetrate the outer lipoprotein/liposaccharide layers of the cell walls of Gram-negative bacteria and are therefore classified as narrow-spectrum (active against mostly Gram-positive bacteria) antibiotics. The Gram-negative bacterial cell wall contains hydrophilic protein channels called porin trimers that allow the passage of hydrophilic substances of molecular weight not exceeding 600 Daltons. This observation led to the synthesis of the aminopenicillins, such as ampicillin and amoxicillin (Fig. 22.13), which are active against both Gram-negative and Gram-positive bacteria.
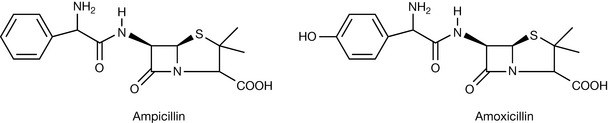
The general design of the aminopenicillins is based on the substitution of an amino group on the α-carbon of Penicillin G. This lowers the lipophilicity of the naturally occurring penicillins and facilitates penetration of the Gram-negative cell wall via the porin trimer. The electron withdrawing property of the amino group confers acid stability on the aminopenicillins and hence the successful design of oral dosage forms. Amoxicillin is prepared as the sodium salt for parenteral use (i.m./i.v.) and as the trihydrate for capsules and paediatric suspensions. Ampicillin is used as the sodium salt in parenteral preparations, trihydrate for capsules, but also as the unmodified compound for capsules and oral suspensions.
The aminopenicillins are, however, not stable to bacterial β-lactamases. Several steps have been taken to extend their activity to β-lactamase-producing organisms and these include co-formulation with β-lactamase stable penicillins and the use of β-lactamase inhibitors.
Combination treatments often involve the use of broad-spectrum penicillin and β-lactamase-stable penicillin. Clinically useful combinations include ampiclox® (ampicillin and cloxacillin) and magnapen® (ampicillin and flucloxacillin). These combinations are usually indicated for infections caused by susceptible organisms where a mixed infection is present and includes penicillin-resistant staphylococci.
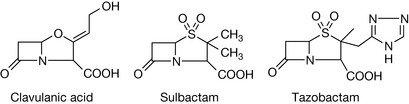
The alternative to combination treatments is the use of β-lactamase enzyme inhibitors such as clavulanic acid, sulbactam and tazobactam (Fig. 22.14), which bear structural resemblance to the β-lactam antibiotics and subsequently compete for the active site of the β-lactamase enzyme.
The initial interaction between the hydrolytic enzyme and the inhibitor involves the attack on the electro-deficient β-lactam carbon. With the β-lactam antibiotics, the attack on the carbon leads to the disruption of the C–N bond and subsequent loss of antibacterial activity. The β-lactamase enzyme is regenerated after disruption of the β-lactam ring. With the β-lactamase inhibitors, a subsequent attack by the nucleophiles at the active site of the β-lactamase enzyme results in the formation of a second bond between the enzyme and inhibitor. This increased interaction prevents the regeneration of the β-lactamase enzyme, leading to its inactivation (Fig. 22.15).
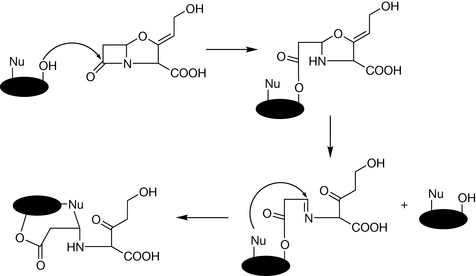
Figure 22.15 Irreversible inactivation of β-lactamases by formation of two covalent bonds between the enzyme and inhibitor.
This concept has been applied successfully to the development of broad-spectrum antibiotics suitable for use in infections involving β-lactamase-producing organisms. A clinically useful example is co-amoxiclav which is a mixture of amoxicillin (as the trihydrate or sodium salt) and clavulanic acid (as potassium clavulanate) and is available as oral (tablet, oral suspension) and parenteral (intravenous infusion and injection) preparations.
The oral bioavailability of ampicillin (approx. 30%) is less than that of amoxicillin (approx. 70–90%). The absorption of ampicillin following oral administration has been enhanced by the formation of suitable prodrugs. At intestinal pH, ampicillin exists as a zwitterion. The conversion of ampicillin into an ester (R = degradable ester group, Fig. 22.16) suppresses zwitterion formation and presents ampicillin as a simple base, which would be 10–50% un-ionised at intestinal pH and subsequently undergo passive absorption.
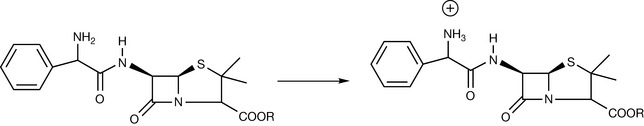
However, the penicillins require a free carboxylic acid moiety at C-3 and therefore ester groups used in suppressing ionisation must be readily degradable to facilitate activity. Suitable prodrugs have been developed using acyloxymethyl esters (bacampicillin and pivampicillin) and phthalidyl esters (talampicillin). Prodrugs of ampicillin based on acyloxymethyl ester formation undergo rapid hydrolysis in vivo after or during passage through the intestinal mucosa, leading to the liberation of free active ampicillin (Fig. 22.17).
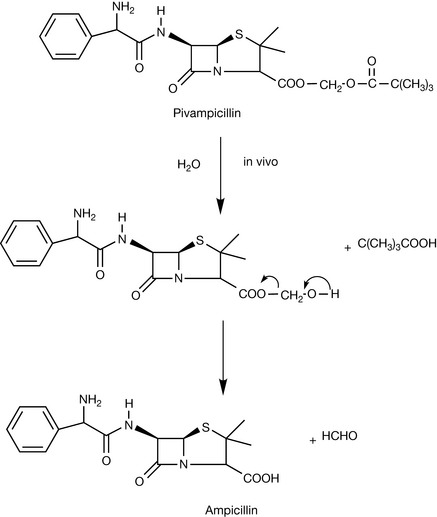
Talampicillin, which is based on a phthalidyl ester, liberates the active compound (ampicillin) with the formation of phthaldehydic acid (Fig. 22.18).
Antipseudomonal penicillins
The Gram-negative bacteria Pseudomonas aeruginosa stands out as a difficult organism due to its resistance to the activity of antibiotics. Modification of the structure of the 6-APA to achieve improved permeability of the Gram-negative cell wall and specificity for pseudomonal penicillin binding proteins (PBP) has been achieved with the synthesis of the carboxypenicillins and the acylureido penicillins.
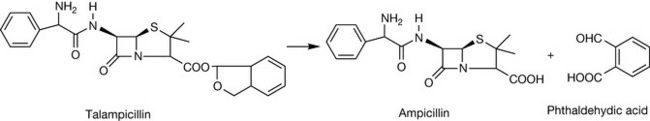
The addition of carboxylic, sulfanic or sulphonic acid groups to the α-carbon of the amide side chain improves the antipseudomonas activity in vitro. The first clinically useful antipseudomonal carboxypenicillin was carbenicillin (Fig. 22.19).
Carbenicillin is used by itself or in combination with gentamicin for the treatment of infections by sensitive strains of Pseudomonas aeruginosa. The replacement of the phenyl moiety with 3-thienyl groups to produce ticarcillin leads to an increase in antipseudomonal activity. However, the carboxy penicillins show diminished activity against streptococci and enterococci due to reduced binding to the PBPs.
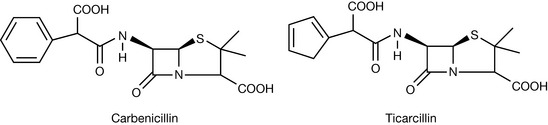
The acylureido penicillins are derivatives of ampicillin designed to improve the penetration of the outer membrane of the Pseudomonas cell. The clinically useful ones are azlocillin and piperacillin (Fig. 22.20).
Although the carboxy and acylureido penicillins are active against Pseudomonas, they are still susceptible to inhibition by bacterial β-lactamases. Most of the antipseudomonal penicillins are now used in combination with β-lactamase inhibitors to improve their activity against susceptible β-lactamase-producing organisms. Some clinical examples are Tazocin® (Tazobactam (sodium salt) + Piperacillin (sodium salt)), Timetin® (Clavulanic acid (potassium salt) + Ticarcillin (sodium salt)). The antipseudomonal penicillins are presented as parenteral preparations because of their instability in acidic conditions.
Cephalosporins
The introduction of the cephalosporins was a result of investigations in the mid-1940s by Giuseppe Brotzu who discovered that Cephalosporium acremonium, isolated for a sewage outfall, inhibited the growth of several bacterial species including Salmonella typhus. The prototype of the cephalosporins, cephalosporin C, was subsequently isolated by Abraham and Newton. The enzymatic removal of the side chain in cephalosporin C, analogous to the removal of the benzyl group in Penicillin G, produces 7-aminocephalosporanic acid (7-APA; Fig. 22.21). 7-APA is the key intermediate in the synthesis of most of the present-day cephalosporins and can also be produced through chemical synthesis.
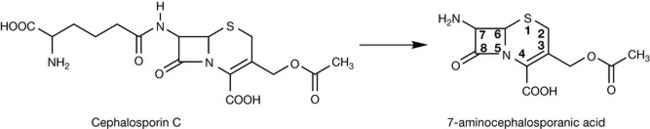
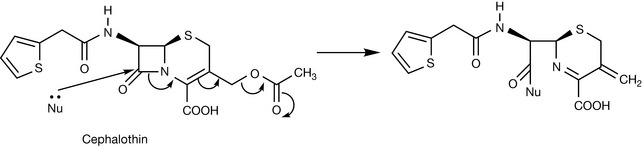
Although the cephalosporins have a similar chemical mechanism of action as the penicillins, the presence of a six-membered ring rather than a five-membered ring in the penicillins relieves ring strain and renders the cephalosporins less chemically reactive. However, upon attack of the C-8 carbonyl by nucleophiles, suitable substitutions at C-3 can act as an electron sink (Fig. 22.22) and increase the reactivity of the β-lactam ring. The acetoxy substituent extending from the dihydrothiazine ring in cephalosporin C and cephalothin is a classical example.
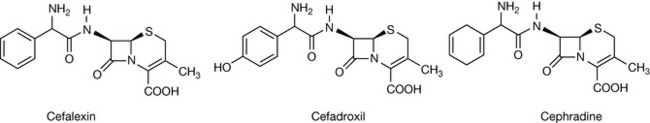
The cephalosporins are classified as first-, second- or third-generation agents. They differ in terms of antibacterial spectrum, stability to bacterial β-lactamases and pharmacokinetics. As a therapeutic group, the cephalosporins are not absorbed from the gut unless they possess a phenylglycine-like moiety on the α-carbon of the side chain. Examples of cephalosporins with this feature are cefalexin, cefadroxil and cefaclor. Cephradine is an exception and features a cyclohexadienyl substituent.
First-generation cephalosporins
First-generation agents possess potent activity against Gram-positive bacteria but only moderate potency against Gram-negative bacteria. They have no activity against Pseudomonas aeruginosa. The first-generation agents suitable for oral administration include cefalexin, cefadroxil and cephradine (Fig. 22.23).
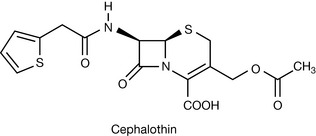
Cephalothin (Fig. 22.24) represents a first-generation agent which is suitable for parenteral administration. These parenteral forms are characterised by the presence of an acetoxymethyl group extending from the dihydrothiazine ring which, as discussed above, increases the reactivity of the lactam ring.
Second-generation cephalosporins
The second generation of cephalosporins possess an extended spectrum of antibacterial activity compared to the first-generation agents. They are active against Gram-negative pathogens such as Escherichia coli and some species of Klebsiella, Proteus and Neisseria gonorrhoea. They are, however, ineffective against Pseudomonas. Cefaclor represents an example of an orally active second-generation agent. Most of the second-generation cephalosporins are parenteral agents with a nitrogen-rich heterocyclic moiety extending from the dihydrothiazine ring. A clinically useful example is cefamandole (Fig. 22.25).
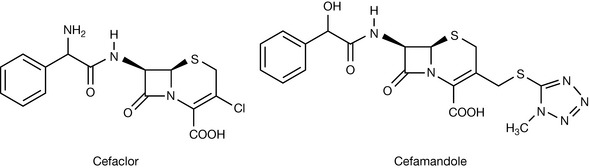
The cephamycins, a class of cephalosporins obtained from Streptomyces sp., display broad-spectrum antibacterial activity and stability to β-lactamases. The cephamycins contain a methoxyl substituent on the β-lactam ring and the shielding effect makes the ring less susceptible to lactamase attack. A clinically useful semi-synthetic analogue of cephamycin is Cefoxitin (Fig. 22.26).
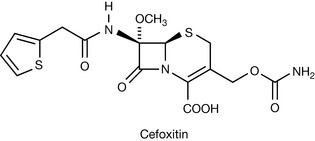
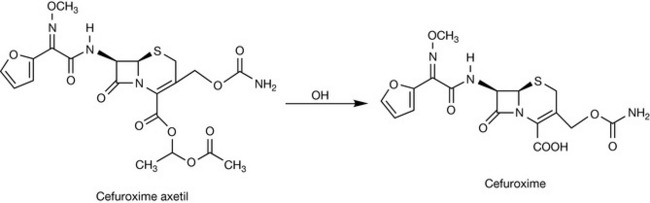
Perhaps one of the most successful second-generation cephalosporins is cefuroxime (Zinacef®) (Fig. 22.27). Cefuroxime is stable to bacterial β-lactamases and can be administered either parenterally or orally as its acetoxyethoxy ester cefuroxime axetil (Zinnat®).
Third-generation cephalosporins
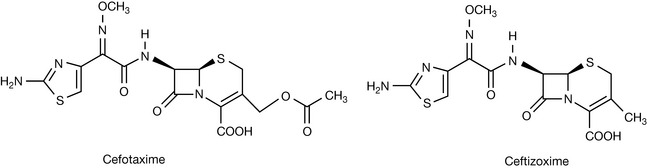
The third-generation agents display a broader spectrum of antibacterial activity compared to the first- and second-generation cephalosporins. Although these agents display enhanced activity against Gram-negative bacteria they are generally not clinically effective against Pseudomonas. The third-generation agents have high β-lactamase resistance and this is attributed to the acylamino side chain. The useful clinical examples are cefotaxime and ceftizoxime (Fig. 22.28).
Ceftriaxone has a long elimination half-life and therefore allows once-a-day dosing. Some third-generation agents are active against Pseudomonas aeruginosa and include the clinically useful agent ceftazidime (Fig. 22.29).
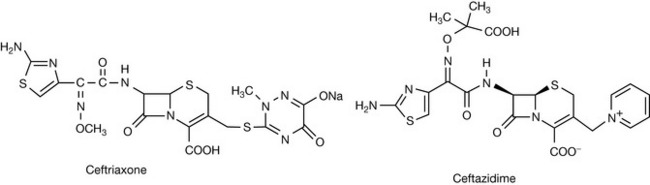
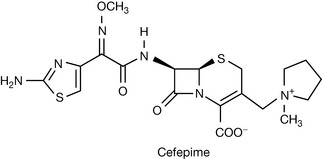
Newer agents such as cefepime referred to as fourth-generation cephalosporins (Fig. 22.30), possess a quaternary nitrogen and have improved activity against Pseudomonas.
The complete structure activity evaluation of all the existing cephalosporins is beyond the scope of this chapter. However, there are some generalisations:
Carbapenems
The prototype of the carbapenems, thienamycin, was isolated in 1976 by Kahan and co-workers from Streptomyces cattlea. Unlike the other β-lactam antibiotics, the carbapenems possess an exocyclic sulphur atom. The series allows chemical modification at the position marked R in Figure 22.31.
Thienamycin (Fig. 22.32) is a potent broad-spectrum antibiotic with β-lactamase inhibiting properties. This compound could not be marketed because of chemical and biological instability. The molecule was prone to inactivation in concentrated solutions due to the nucleophilic attack of the terminal primary amine group on the β-lactam ring.
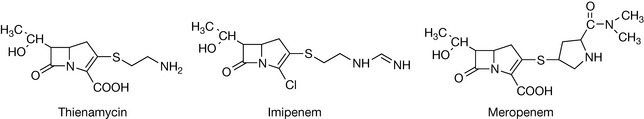
In 1985, the self-inactivating property of thienamycin was overcome by the use of a terminal imino functionality which is less nucleophilic. Imipenem (Fig. 22.32) has a broad spectrum of activity and is administered by deep intramuscular injection or as an intravenous infusion. Renal dehydropeptidase 1, an enzyme present in the kidney, attacks and inactivates imipenem. This problem has been overcome by the co-administration of imipenem with cilastatin, a renal dehydropeptidase 1 inhibitor. Meropenem (Fig. 22.32), an analogue introduced in 1996, is stable to the action of renal dehydropeptidase 1 and is also administered by deep intramuscular injection or as an intravenous infusion. Since the discovery of thienamycin in 1976, only parenteral analogues have been successfully used clinically because the carbapenem structure is unstable in both the stomach and intestine.
Monocyclic β-lactam (monobactams)
The monobactams are β-lactam antibiotics which contain only one cyclic ring (Fig. 22.33). Unlike the other β-lactams, the monobactams are active almost entirely against Gram-negative bacteria. The general monobactam structure allows for structural alteration at a single locus (R). The only clinically useful example is aztreonam (Fig. 22.34).
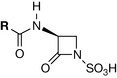
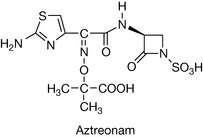
Although the monobactams bear a structural similarity to the penicillins, clinical hypersensitivity reactions in penicillin-sensitive patients is rare. It is indicated for Gram-negative infections including Pseudomonas aeruginosa, Haemophilus influenzae and Neisseria meningitides and administered by deep intramuscular injection or as an intravenous infusion.
Other inhibitors of cell wall synthesis
Vancomycin, a mucopeptide produced by Streptomyces orientalis, was first isolated from soil samples in 1956. It is bactericidal and acts by inhibiting cell wall replication. Vancomycin interacts with D-alanine and prevents the reaction of transpeptidase with the latter in the final stages of cross-linking. Due to its large size, vancomycin cannot enter the porin channels in Gram-negative bacteria and is therefore only effective in Gram-positive infections. It remains one of the few agents active against MRSA.
Inhibition of bacterial cell metabolism
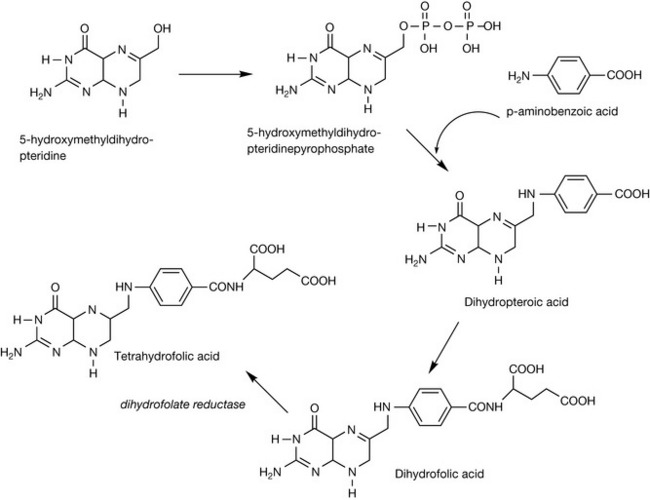
The exploitation of subtle differences between the cellular metabolism in bacteria and mammalian host presents a niche for achieving selective toxicity. One such difference which has been exploited successfully to produce a useful class of antibiotics is the difference in the synthesis of tetrahydrofolate. Both bacterial and mammalian cells require folate in the form of tetrahydrofolate as a co-factor for thymidylate synthesis. Mammalian cells are incapable of synthesising folate and have specialised mechanisms for transporting folate from the diet into cells. On the contrary, most bacterial cells do not possess a similar transport mechanism and are incapable of utilising preformed folate. Bacteria therefore have to synthesise their own dihydrofolate and require endogenous p-aminobenzoic acid as a precursor (Fig. 22.35).
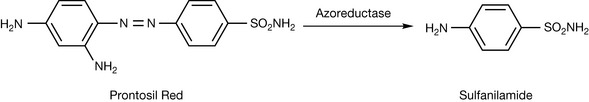
In 1931, Gerhard Domagk and co-workers found that the azo dye sulfamidochrysoidine (Prontosil Red®) was effective against virulent strains of Streptococci pyogenes in infected mice. The antimicrobial activity was later found to be due to its reduced metabolite, sulfanilamide (Prontosil white; Fig 22.36). Sulfanilamide forms the prototype of this class of synthetic antibacterial agents known as the sulphonamides.
Sulphonamides
Following the introduction of the sulphonamides in clinical use, the antibacterial activity was found to be inhibited by the presence of pus. This inhibitory effect of pus was found to be due to the presence of the structurally similar p-aminobenzoic acid (PABA): a compound which is also present in folic acid. This led to the observation that the sulphonamides competed with PABA, leading to the disruption of folate synthesis and cessation of bacterial growth. The importance of the sulphonamides in present-day therapeutics has decreased due to the increase in bacterial resistance. Also, the typical side effects of sulphonamide therapy such as Stevens-Johnson syndrome, renal failure and blood dyscrasias are reasons why less toxic and more active alternatives such as the penicillins are preferred in practice.
The evaluation of the antibacterial activity of the sulphonamides stems from a general structure (the nitrogen of the sulphonamido group is designated N1 and the nitrogen of the p-amino substituent is designated N4) with certain provisions (Fig. 22.37):
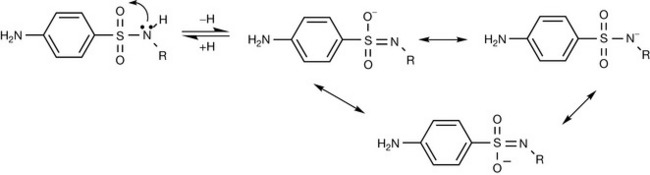
The first generation of sulphonamides was characterised by the tendency to crystallise in the kidney due to their insolubility. This is the result of strong intermolecular hydrogen bonding. This problem was aggravated by the metabolism of the N4-amino moiety to the less soluble acetyl derivative. The precipitation of the low solubility N4-acetyl metabolites results in crystalluria. Fortunately, the R group in the general structure of the sulphonamides can be modified to overcome the problem of insolubility. The sulphonamides are acidic (proton donor) compounds and this is due to the electron withdrawing sulphonyl moiety that stabilises the anion formed after the loss of hydrogen (Fig. 22.38).
The prototype sulphonamide, sulfanilamide has a pKa of 10.4. Substitution of an electron withdrawing group at the R position increases the acidity (decreased pKa) of the sulphonamido N–H. Subsequently, compounds with a pKa of 6–7 will be ionised at physiological pH (7.4) and can be useful in the treatment of systemic infection with decreased risk of crystalluria. This principle has been applied in the design of sulfadimidine (pKa 7.4) and sulfadiazine (pKa 6.5) (Fig. 22.39) by the addition of the electron withdrawing 4,6,-dimethylpyrimidine and pyrimidine, respectively.
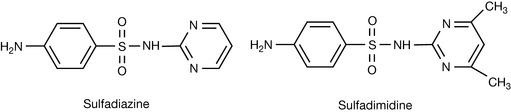
Sulfadimidine is indicated for the treatment of upper and lower respiratory infections, urinary tract and genital tract infections. It is formulated as the sodium salt for parenteral administration. Sulfadiazine is indicated for the prevention of rheumatic fever recurrence and is formulated as the sodium salt for injection or as sulfadiazine tablets.
Although the addition of an electron withdrawing group results in increased ionisation at physiological pH, the downside is that complete ionisation of a sulphonamide will result in rapid excretion, requiring frequent administration, and therefore rendering it unsuitable for systemic administration.
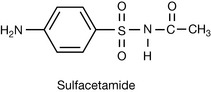
A typical example is sulfacetamide (Fig. 22.40) which possesses a carbonyl substituent directly attached to the sulphonamido nitrogen. The reduced pKa (5.4) means it is 99.9% ionised at physiological pH. The potentially rapid excretion of sulfacetamide has limited its use to the treatment of ocular infections, such as conjunctivitis, caused by susceptible organisms. It is formulated as the sodium salt and used as a 10–30% w/v ophthalmic solution.
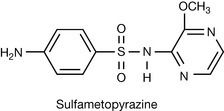
Even at optimal pKa, regular dosing is required to maintain therapeutic concentrations because of the rapid excretion of sulphonamides. The renal excretion of the sulphonamides can be reduced by increasing the degree of protein binding. The substituent at R can be selected to increase the degree of protein binding and therefore slow down the rate of excretion since only unbound drug is filtered through the glomerulus. Substitution of a methoxyl group on the heterocyclic ring results in an increase in protein binding as in sulfametopyrazine (Fig. 22.41) and yields longer-acting sulphonamides with the advantage of less frequent administration. Sulfametopyrazine remains useful in the treatment of urinary tract infections and chronic bronchitis and may be administered at a dose of 2 g once weekly. However, toxic effects due to accumulation are more likely to occur with these longer-acting sulphonamides.
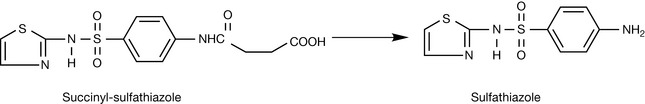
The sulphonamides have been used in the treatment of intestinal infections. Substitution of the aniline N4 with a succinyl group produces an acidic compound which is fully ionised at intestinal pH and is therefore not appreciably absorbed into the bloodstream. Slow enzymatic hydrolysis of the product in the intestine releases the free primary amine group which is essential for activity. This concept of prodrug design has been exploited in the synthesis of succinyl-sulfathiazole (pKa 4.5) from sulfathiazole (pKa 7.1) (Fig. 22.42).
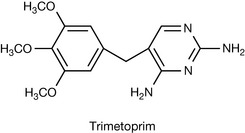
The importance of the sulphonamides has decreased as a result of increased bacterial resistance and they have been replaced by compounds with higher therapeutic indices and activity. The biosynthetic pathway for tetrahydrofolate provides an additional point for selective attack on bacteria. The conversion of dihydrofolic acid to tetrahydrofolate (coenzyme F) requires dihydrofolate reductase. Trimetoprim (Fig. 22.43), a sulfone, is structurally similar to dihydrofolate (see Fig. 22.35) and therefore competitively inhibits the formation of tetrahydrofolate, ultimately affecting the biosynthesis of proteins and nucleic acids.
Due to a mutation in the human genome, trimetoprim inhibits bacterial dihydrofolate reductase but not mammalian dihydrofolate reductase. Trimetoprim has therefore been used in combination with the sulphonamides to provide synergism in activity while reducing the toxicity potential of the individual compounds. A useful clinical example is co-trimoxazole, which is a compound formulation of trimetoprim and sulfamethoxazole.
Bacterial protein synthesis inhibitors
The mechanism(s) through which both eukaryotes and prokaryotes synthesise protein for various cell activities are generally universal. However, differences exist in the ribosomes, the multi-macromolecular complexes in which decoding of the genetic information for the synthesis of proteins occurs. Bacterial ribosomes have a sedimentation coefficient of 70S and dissociate reversibly into 50S and 30S subunits. Mammalian ribosomes exhibit a sedimentation coefficient of 80S and are composed of 60S and 40S subunits. Most antibiotics which affect bacterial protein synthesis have an affinity or specificity for the 70S and or its subunits. The major class of antibiotics which selectively interfere with protein synthesis on 70S ribosomes include the aminoglycosides, macrolides, tetracycline, licosamides, chloramphenicol and the aminocyclitols.
Aminoglycosides
The aminoglycosides are products of actinomycetes or a semi-synthetic derivative. The first aminoglycoside, streptomycin, was isolated by Selman Walksman in 1944 from Streptomyces griseus. This group of broad-spectrum antibiotics includes kanamycin, gentamicin, amikacin, netilmicin, tobramycin and neomycin and are used primarily in the treatment of Gram-negative infections. The aminoglycosides have specificity for the 30S subunit of the bacterial 70S ribosome. Binding to the 30S subunit results in interference with the initiation of protein synthesis and misreading of mRNA.
The work on the mode of action of these drugs is largely concentrated on streptomycin, which has been shown to bind to a specific protein in the 30S subunit of bacterial ribosomes. This event inhibits protein synthesis and/or causes the production of defective proteins which results in cell death.
Macrolides
These antimicrobial agents are derived from the actinomycetes. The prototype macrolide, erythromycin, was isolated in 1952 by McGuire and co-workers from Streptomyces erythreus. Other clinically useful examples which are semi-synthetic analogues of erythromycin are clarithromycin and azithromycin. The compounds are so named due to the presence of a 14-member macrocyclic lactone ring. The large lactone rings are linked through glycoside bonds with amino-sugars. The macrolides bind to the 23S mRNA molecule in the 50S subunit and inhibit the translocation step in bacterial protein synthesis. The modification of erythromycin to clarithromycin and azithromycin improves acid stability and therefore facilitates improved absorption in the stomach.
Tetracyclines
The tetracyclines are derived from Streptomyces and have a broad spectrum of activity which includes rickettsial and chlamydial infections. The first tetracycline, chlortetracycline (Aureomycin) was discovered by Benjamin Duggar in 1948. They are characterised by 4-fused rings with a vast array of substitutions (Fig. 22.44). The commonly used tetracyclines which bear the general structure below include chlortetracycline (7-chlorotetracycline, R1=Cl, R2=OH, R3=H, R4=H), tetracycline (R1=H, R2=OH, R3=H, R4=CH3), and doxycyline (R1=H, R2=H, R3=OH, R =CH3).
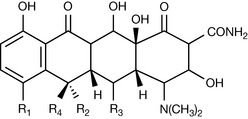
These differ little in antibacterial activity and are distinguished by their pharmacokinetic behaviour. Minocycline (R1=N(CH3)2, R2=H, R3=OH, R4=H) shows a broader spectrum of antibacterial activity. The tetracyclines act by blocking the binding of aminoacyl tRNA to the A site on the ribosome. The tetracyclines subsequently inhibit protein synthesis at the small ribosomal subunits of both the 70S (prokaryote) and 80S (eukaryote) ribosome. However, an active transport system for the tetracyclines in bacteria means effective concentrations are achieved in the bacterial cells but not in mammalian cells. The tetracyclines are presented as capsules and tablets for oral use.
Oxazolidiones
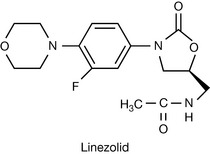
The oxazolidiones, of which linezolid is the prototype, are the synthetic antibiotics indicated for the treatment of methicillin resistant Staphylococcus aureus (MRSA) infections.
Linezolid (Fig. 22.45) exerts its antibacterial action by binding to the 23S ribosomal RNA of the 50S subunit. The oxazolidione represent a fairly recent addition to the arsenal of antibacterial agents, and research to produce useful analogues is in progress.
Bacterial nucleic acid synthesis inhibitors
Chemotherapeutic agents which selectively affect the synthesis of DNA and/or RNA in bacterial cells can block the growth of these organisms, leading to rapid death. The quinolones and the rifamycin derivatives are nucleic acid synthesis inhibitors which are selectively active against prokaryotes.
Quinolones
The quinolones are synthetic antibiotics which trace their origin to a concerted effort by scientists to synthesise novel antimalarial agents. In 1962, nalidixic acid (Fig. 22.46) was isolated from the by-products of chloroquine synthesis.
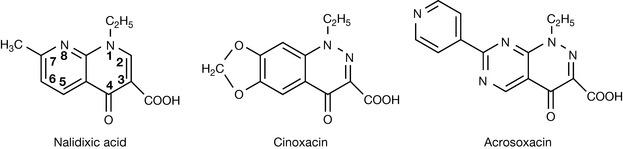
Nalidixic acid is indicated for the treatment of urinary tract infections. The success of nalidixic acid in the 1960s led to the synthesis of other analogues of comparable activity such as cinoxacin and acrosoxacin. Information gleaned from the activity of several synthesised quinolones indicates that the following structural requirements are essential for activity:
Several important observations have been made regarding the substitution pattern(s) in the A-ring (pharmacophore) and in the B-ring. In general, the A-ring facilitates entry into the bacteria, fluorine substitution at C-6 increases antibacterial activity while a piperazine at C-7 confers antipseudomonal properties. Substitutions on the B-ring have little effect on antibacterial activity. Based on the increased activity due to fluorine substitution at C-6, Bayer produced the first marketed fluoroquinolone, ciprofloxacin. This was followed by other clinically useful analogues such as ofloxacin, norfloxacin, perfloxacin (Fig. 22.47), levofloxacin and grepafloxacin.
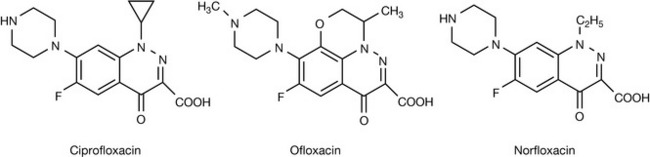
The fluorinated quinolones are generally more potent and less toxic than the first-generation agents. Agents containing a piperazine group at the C-7 position, such as ciprofloxacin and norfloxacin, are useful in the treatment of pseudomonal infections. The fluoroquinolones inhibit bacterial topoisomerase II (predominantly) and topoisomerase IV.
Rifamycins
The rifamycins belong to a class of naturally occurring highly substituted derivatives of naphthalene called ansamycins. These compounds are the products of the Streptomyces. The prototype rifamycin B was isolated from Streptomyces mediterranei and a series of chemical modifications led to rifampicin, which is active against Gram-positive bacteria (including Mycobacterium tuberculosis, see Section B) and some Gram-negative bacteria. Rifampicin acts specifically on the bacterial RNA polymerase and is inactive against mammalian RNA polymerase and DNA polymerase. This class of compounds inhibits bacterial mRNA synthesis by binding to the beta subunit of the polymerase and subsequently blocking the entry of the nucleotide required to activate the polymerase.
Chloramphenicol
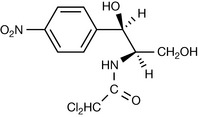
Chloramphenicol was originally obtained from Streptomyces venezuela but is now produced by chemical synthesis. Although it has a broad spectrum of activity, it is rarely used systemically due to a high incidence of aplastic anaemia.
The compound has two asymmetric centres (Fig. 22.48) but only diastereoisomer the R,R-isomer demonstrates significant antibacterial activity. Chloramphenicol binds to the 50S subunit and inhibits the bacterial enzyme peptidyl transferase. This prevents the growth of the polypeptide chain during protein synthesis.
Clindamycin and lincomycin
These antibiotics have limited use because of their serious side effects. They bind exclusively to the 50S subunit of 70S ribosomes and the binding site is related in some way to those for erythromycin and chloramphenicol since erythromycin can displace bound lincomycin, and lincomycin inhibits chloramphenicol binding.
A major adverse effect associated with the penicillins is the development of IgE type 1 or accelerated immunological reactions. These allergic reactions develop rapidly after administration and the manifestations may range from harmless exanthemas to anaphylactic reactions. Anaphylactic reactions occur in about 0.015–0.045% of patients and can be fatal in 10% of these cases. The β-lactams such as penicillins and cephalosporins may form drug–protein conjugates directly due to their inherent reactivity (Fig. 22.49). The β-lactams can react with lysine groups on proteins to form the penicilloyl/cephalosporyl group, which is the major antigenic determinant.
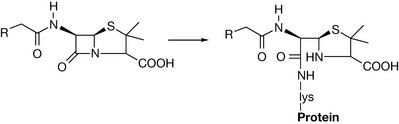
Once they are covalently bound to a macromolecular carrier, they exist in the form of a hapten, and the bound penicillins subsequently participate in immunological reactions. Penicillenic acid, which is one of the degradation products of the penicillins, is also highly reactive and is much more immunogenic than penicillin itself. The hypersensitivity reactions occur when patients with preformed specific IgE antibodies to the penicilloyl/cephalosporyl group, which are bound to mast cells and circulating basophils, are exposed to the haptenic structures. This leads to the release of inflammatory mediators. It is estimated that 8% of patients develop allergic reactions to penicillin. On the contrary, 5% develop reactions to the cephalosporins and this has been attributed to the lower chemical reactivity of the parent cephalosporanic acid molecule. The penicilloyl/cephalosporyl moiety may be introduced into humans as a result of previous therapy, contamination, exposure to residues of penicillins/cephalosporins in vaccines and meat from drug-treated livestock. Mouldy food can serve as a good source of traces of penicillins. In addition, immunologically competent macromolecular impurities in penicillin preparations formed by polymerisation react with proteinaceous material in the fermentation medium or amidases from the cleavage of the side chain.
Section b – antituberculosis drugs
Tuberculosis (TB) is a disease that has plagued mankind for centuries, dating as far back as ancient Egyptian times, and is caused by a slow-growing type of bacterium called Mycobacterium tuberculosis. The disease is spread by coughing, talking, spitting and sneezing, which spread the mycobacteria through the air in tiny aerosolised droplets of water which are inhaled into the lungs. The bacteria are then engulfed by macrophages in the air sacks of the lungs, called the alveoli, where they replicate and can spread throughout the body. Although 95% of the bacteria are killed in the lung, the remaining bacteria can continue to grow if untreated, or remain dormant for years before being reactivated. The primary location of the disease called pulmonary TB is in the lungs, where bacterial growth destroys tissue, making it very hard for the patient to breathe, resulting in death. It is in the lungs where the disease is most contagious but symptoms can also include meningitis, legions on the skin and degradation of the heart, bones and intestines, which makes treating the disease and choosing the correct drug regimens very difficult.
TB was much more prevalent in the past than it is today and has caused the deaths of around one billion people over the last two hundred years. Improvement of sanitation and living conditions have been significant factors in reducing cases of TB, but the introduction of chemotherapy in the 1950s has been instrumental in fighting this age-old adversary. In recent years, however, the disease has made a strong resurgence and now infects approximately one-third of the world’s population and causes 8 million new cases of TB each year, resulting in around 2 million deaths worldwide. The resurgence has been caused due to three main reasons. First, the chemotherapy used to attack the complex and robust bacteria is not very efficacious by modern drug standards and has to be given as a precise combination over a period of months in order for it to be effective. Second, poor compliance leads to drug-resistant strains of the mycobacterium which are not susceptible to the current cocktail of drugs available. This leaves the pharmacist and doctor faced with the option of administering other non-tuberculosis-specific and potentially toxic antibiotics to treat the disease as a last resort, the failure of which will almost certainly result in the death of the patient. Last, there is also a strong epidemiological coexistence between TB and HIV, as patients who are immunocompromised readily contract the disease which has led to approximately one-third of all AIDS-related deaths resulting from TB infections.
The disease, and the bacterium which is the cause, are vastly complex and symptoms can vary enormously, which makes its treatment difficult. For example, a patient may have open cavities or lesions that are teaming with the bacteria which the drugs can not easily eradicate. There are other problems associated with killing the bacterium using the available drugs, which mostly kill the bacteria only when they are growing. In this case, the drugs have to penetrate a very complicated mycobacterial cell wall which is sometimes described as ‘waxy’ because of its complex fatty acid barrier which makes getting a drug molecule into the cytoplasm extremely difficult. Additionally, after treatment, the bacteria that are not killed outright can lie in a dormant state, waiting to be reactivated. These bacteria are called ‘persistors’ and the reason for reactivation is not understood, which makes destroying them very difficult. It is therefore vitally important that all of the growing bacteria are killed in the first instance to destroy as many bacteria as possible, which will also help prevent drug resistance from occurring, which is proving to be a major problem in developing countries.
TB is treated in two phases. There is an initial phase and a continuation phase and depending upon the patient’s ability to comply with the drug regimen, these phases may be carried out either unsupervised or supervised. The latter is known as DOT (directly observed treatment), where the drugs are given to the patient three times per week in order to maintain the strict regimen that must be observed.
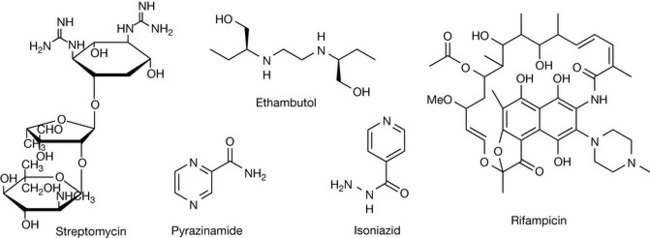
During the initial phase there is the concurrent use of at least three drugs for 2 months, which is designed to reduce the bacterial population as rapidly as possible in order to prevent resistance. These drugs are generally given as a combination preparation, or ‘triple therapy’, unless one of the component drugs cannot be given because of intolerance or potential drug resistance. The three drugs normally used in combination are isoniazid (INH), rifampicin (RIF) and pyrazinamide (PZA) which in combined tablet form are known as Rifater®. Ethambutol (EMB) is now also used during the initial phase. Streptomycin (SM) may be used in cases where resistance to INH has been established before treatment begins but is rarely used in the UK where, due to its potentially toxic nature, it is only available for named patients. Treatment may be extended but only in cases of meningitis or for resistant strains, which may also require modification of the regimen.
Traditionally, TB drugs are generally considered to be either first-line or second-line. The first-line drugs exhibit superior efficacy with acceptable toxicity, whereas the second-line drugs have either less efficacy, increased side effects or both. Discussed below are the main drugs used to treat TB, although others such as kanamycin, amikacin and thioacetazone may also be used. It is important to note that no new drug has been specifically designed and introduced to treat TB since rifampicin in 1962, which indicates the difficulty in discovering new agents and highlights how, by modern standards, these drugs are relatively poor.
First-line drugs
As discussed earlier, one of the main reasons it is hard for drugs to destroy M. tuberculosis is because it has a complex and almost impermeable cell wall. In recent years, the sequencing of the genome of M. tuberculosis has furnished medicinal chemists with a plethora of new, specific and validated targets which may be exploited to design more drugs to fight growing and drug-resistant bacteria. Moreover, the mechanism of action of many of the existing drugs designed in the 1940s and 1950s has been established, giving us more information on how best to use the current medicines.
Isoniazid (INH) (isonicotinic acid hydrazide, Fig. 22.50) was discovered via a purely synthetic strategy in the quest to find a new agent to combat the disease. It is bacteriostatic at concentrations of 0.025–0.05 μg/ml in vitro but at higher concentrations is bactericidal. This is a prodrug and requires activation before destroying the bacteria, which it does by inhibiting a ketoenoylreductase enzyme known as InhA. This enzyme catalyses the last of four steps involved in the 2-carbon elongation cycle of fatty acid biosynthesis. INH is first activated by the catalase-peroxidase enzyme katG which oxidises the isonicotinic acyl group of the molecule to either an isonicotinic acyl anion or radical. These forms then react with either an NADH radical or anion whilst in the active site of InhA to form an isonicotinic acyl-NADH complex. This new molecule is then tightly bound to the active site of the enzyme, thus preventing access of the natural enoyl-AcpM substrate which stops the production of mycolic acids and hence the biosynthesis of the cell wall of the bacteria.
Resistance to INH generally occurs when the drug is administered alone for 3 months and is caused mainly by the absence of the gene encoding the catalase-peroxidase katG, which prevents activation of the drug. Mutations in InhA have also been identified and postulated as a reason for resistance to the drug.
INH is well absorbed orally or intramuscularly and distributes well throughout the body, and its metabolism occurs initially by liver N-acetyltransferase. This means that patients who are poor acetylators can experience toxicity problems, as acetylation of INH is first required before the hydrolysis can occur and the drug cleared by the kidneys.
Ethambutol (EMB, Fig. 22.50), chemical name (S,S′)-(+)-2,2′-(ethylenediimino)di-1-butanol, exhibits bacteriostatic activity against M. tuberculosis in vitro or in macrophages at 1 μg/mL. Stepwise resistance of the drug occurs when the drug is administered alone but cross-resistance with other antimicrobials is rare, so its principle role is as a companion drug to prevent resistance.
EMB inhibits the polymerisation of the cell wall arabinan component of the mycolylarabinogalactan and lipoarabinomannan, which are integral structural polysaccharides. It is believed that it may specifically target the arabinosyl transferase enzyme embB with only the (S,S′)-diasteromer binding to the active site, which is supported by the fact that the (R,R′)-diastereomer is inactive.
Approximately 80% of EMB is absorbed orally and is well distributed throughout the body, including the central nervous system, before being metabolised to inactive metabolites which are readily excreted in urine. The major toxic effect of the drug is neuropathy resulting in visual problems when a patient has insufficient renal activity and in some cases infrequent hypersensitivity reactions occur, which include dermatitis, arthralgias and fever.
Pyrzinamide (PZA, Fig. 22.50) is a synthetic analogue of nicotinamide, is bactericidal against growing bacteria, and is unconventional in the fact that it has high sterilising activity in vivo and is responsible for shortening the therapy to 6 months. It is most active at acidic pH in vitro, which is similar to the environment found intracellularly in phagolysosomes, which may account for its high sterilising activity, but it shows no activity at physiological pH. The exact mechanism of action of this is unclear. However, it is known that PZA is a prodrug which requires activation to pyrazinoic acid (POA) by the PZase/nicotinamidase enzyme. In this form, and at low pH, the protonated POA can bring protons into the cell, which causes cytoplasmic acidification resulting in the de-energising of the cell because of a collapse in proton motive force which effects membrane transport. It is also known that resistance to PZA rapidly evolves if the drug is used alone and is associated with mutation in the pncA gene which encodes the PZase/nicotinamidase enzyme.
PZA is well absorbed orally and distributes well throughout the body, reaching concentration levels above that needed to kill the tubercle bacilli. The peak plasma concentrations are around 50 μg/mL, resulting in a half-life of the drug of 12–24 hours, which makes once-daily dosing practical. It is metabolised by the liver with metabolites, mainly POA, being excreted by the kidneys. The most common side effects with PZA are nausea and vomiting, and early trials with the drug resulted in the dose of the drug being lowered from 40–50 mg/kg/day to 20–35 mg/kg/day, indicating that dosages of the drug may need changing if liver and renal function are compromised.
Rifampicin (RIF, Fig. 22.50) is a broad-spectrum semi-synthetic derivative of the complex macrocyclic antibiotic rifamycin B, produced by Streptomyces mediterranei, and is known as rifampin outside the UK. An important feature about RIF is that it is active against both actively growing and slowly metabolising non-growing bacilli. The latter feature is thought to be important in reducing the treatment from 12–18 months to 9 months.
RIF is bactericidal against M. tuberculosis at 0.005–0.2 μg/mL and works by inhibiting RNA synthesis by binding to the bacterial DNA-dependent RNA polymerase β-subunit encoded by the rpoB gene. Resistance occurs rapidly when used alone and arises because of a mutation in a defined 81-bp region of the rpoB gene and can cause cross-resistance to other rifamycins such as rifabutin and rifapentine, which have very similar structures. RIF is well absorbed orally, giving peak plasma concentrations of 7–8 μg/mL after a dose of 600 mg and distributes well throughout the body. Its lipophilic nature allows it to distribute throughout the central nervous system and penetrate phageosomes, and this makes it a good candidate to treat the meningitis form of the disease. It is deacylated to an active form which undergoes biliary excretion and enterohepatic recirculation and, due to autoinduction of RIF metabolism, biliary excretion increases with continued therapy. This means that plasma concentrations become maximal after six doses whether administered daily or twice weekly, whereby subsequent excretion of the drug takes place primarily to the gastrointestinal tract, with lesser amounts in urine.
Streptomycin (SM) is an aminoglycoside antibiotic isolated from the bacteria Streptomyces griseus and was introduced in the 1940s as the first drug to reduce TB mortality. Importantly, SM is traditionally known as a first-line drug. However, because it is available only as a named drug, it may more accurately be classed a second-line drug. Due to its hydrophilic nature owing to the presence of several hydroxyl groups and two guanidine groups, this drug is poorly bioavailable orally and has to be administered by an intramuscular injection and is virtually excluded from the central nervous system. A dose of 1 g yields peak plasma concentrations of 25–45 μg/mL, which is more than enough for the drug to be inhibitory, which can be achieved at levels of 0.4–10 μg/mL. It is bactericidal against M. tuberculosis in vitro but is inactive against intracellular tubercle bacilli, and resistance develops rapidly when it is given alone.
The drug works by interfering with protein synthesis, as it inhibits the initiation of mRNA translation which leads to misreading of the genetic code, ultimately damaging the cell membrane. The main site of action is in the small 30S subunit of the ribosome, specifically at ribosomal protein S12 (rpsL) and 16S rRNA (rrs) in the protein synthesis. The mutations in rpsL and rrs give rise to SM resistance and, as other aminoglycoside antibiotics such as kanamycin, amikacin, viomycin and capreomycin are inhibitors of protein synthesis, cross-resistance can be a problem. The toxicity of SM is like that of the other aminoglycosides but with less renal and acoustic toxicity (affects the hearing).
Second-line drugs
Second-line anti-TB drugs are generally used when the first-line drugs are ineffective due to resistance or patient intolerance. They are all less efficacious than the first-line drugs and generally exhibit more toxic effects to the patient.
Ethionamide and protionamide (Fig. 22.51) are very similar in structure and action to INH and they are also prodrugs that undergo catalase-oxygenase activation to inhibit the enzyme inhA, thus preventing the synthesis of mycolic acids and consequently inhibiting cell wall biosynthesis. Resistance also occurs by mutation of the oxygenase enzymes and inhA and it is tuberculostatic at 0.6–2.5 μg/mL, making it less potent than INH. It is well absorbed orally and well distributed throughout the body, and is metabolised by the liver with metabolites excreted via the kidney. However, ETH and PTH also interfere with INH acetylation, which can cause gastrointestinal side effects leading to poor compliance.
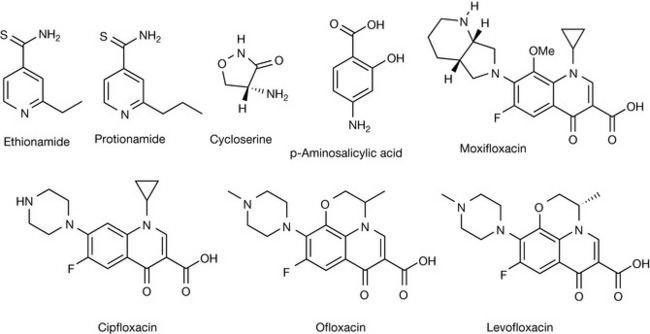
Cycloserine (Fig. 22.51) possesses a broad range of antimycobacterial activity and inhibits M. tuberculosis at 5–20 μg/mL, again by preventing cell wall biosynthesis. It works by mimicking D-alanine which is the natural substrate for the enzyme D-alanine racemase (Alr) and D-alanine: D-alanine ligase (Ddl), thus preventing the synthesis of the mycolyl peptidoglycan. Alr serves to convert L-aniline to D-alanine and mutations in the active site of this enzyme have been proposed to be responsible for the resistance of CS. CS is readily absorbed orally and widely distributed amongst tissues before being excreted by the kidneys, with little of the drug being metabolised. It is one of several alternatives for re-treatment regimens or for treatment of primary drug-resistant TB, although it has poor activity against MDR-TB strains.
Para-aminosalicylic acid (Fig. 22.51) is bacteriostatic against M. tuberculosis and is not completely absorbed orally due to its hydrophilic nature, as dictated by the functional groups attached to the benzene ring. A 4-g dose will yield plasma concentrations of 70–80 μg/mL, from which about 85% of the absorbed drug will be excreted in the urine as various degradation products. It is thought that PAS works by inhibiting a dihydrofolate reductase (DHFR) or the salicylate kinase enzyme Dhbe, which is responsible for synthesising iron-regulating molecules called siderophores which are necessary for the bacteria to survive, although it has been suggested that they also interfere with folic acid metabolism.
The first of the ‘quinolone’ drugs, nalidixic acid (Fig. 22.51), was obtained in the early 1960s during the manufacture of chloroquine and since then many fluorinated derivatives have been synthesised and evaluated as anti-TB drugs showing bactericidal activity. Ciprofloxacin, ofloxacin and levofloxacin (the S(−) form of ofloxacin) are the best studied of the quinolones and work by inhibiting DNA synthesis targeting the DNA gyrase A and B subunits at concentrations well within the achievable plasma levels. It can be seen that the structures of the quinolones are very similar, with only small differences based around a central quinolone pharmacophore which have been designed to increase binding to the gyrase enzyme and improve the druglike properties of the molecule or reduce toxic effects. Resistance to the FQs has been found to occur when they are administered on their own. However, ofloxacin, in combination with other second-line drugs, may be used as a treatment for resistant TB. The use of these drugs is still being investigated and, indeed, new drug molecules are being evaluated and tested to treat the disease in the future, with the best example being that of the diarylquinolone R207910.