 CHAPTER 52 Carbohydrate Disorders
CHAPTER 52 Carbohydrate Disorders
GLYCOGEN STORAGE DISEASES
Many glycogen storage diseases are characterized by hypoglycemia and hepatomegaly (Table 52-1). Glycogen, the storage form of glucose, is found most abundantly in the liver, where it modulates blood glucose levels, and in muscles, where it facilitates anaerobic work. Glycogen is synthesized from uridine diphosphoglucose through the concerted action of glycogen synthetase and brancher enzyme (Fig. 52-1). The accumulation of glycogen is stimulated by insulin. Glycogenolysis occurs through a cascade initiated by epinephrine or glucagon. It results in rapid phosphorolysis of glycogen to yield glucose-1-phosphate, accompanied by a lesser degree of hydrolysis of glucose residues from the branch points in glycogen molecules. In the liver and kidneys, glucose-1-phosphate is converted to glucose-6-phosphate through the actions of phosphoglucomutase; glucose-6-phosphatase hydrolyses glucose-6-phosphate to produce glucose. The latter enzyme is not present in muscles. Glycogen storage diseases fall into the following four categories:
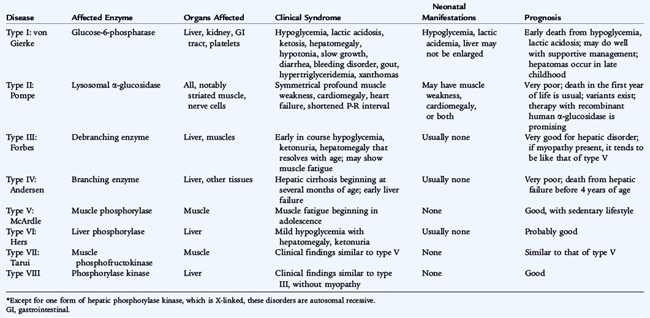
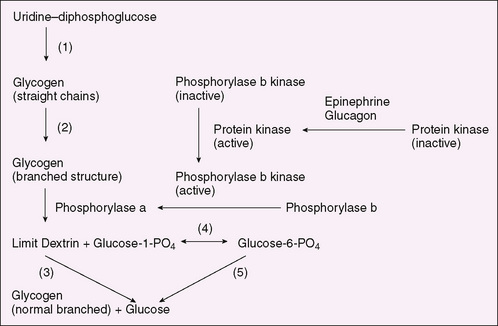
FIGURE 52-1 Glycogen synthesis and degradation. (1) Glycogen synthetase, (2) brancher enzyme, (3) debrancher enzyme, (4) phosphoglucomutase, (5) glucose-6-phosphatase.
The diagnosis of glycogen storage disease can often be confirmed by DNA mutation testing in blood cells. When this is feasible, invasive procedures, such as muscle and liver biopsy, can be avoided. When mutation testing is not available, enzyme measurements in the tissue suspected to be affected confirm the diagnosis. If the diagnosis cannot be established, metabolic challenge and exercise testing may be needed. Treatment of hepatic glycogen storage disease is aimed at maintaining satisfactory blood glucose levels or supplying alternative energy sources to muscle. In glucose-6-phosphatase deficiency (type I), the treatment usually requires nocturnal intragastric feedings of glucose during the first 1 or 2 years of life. Thereafter, snacks or nocturnal intragastric feedings of uncooked cornstarch may be satisfactory; hepatic tumors (sometimes malignant) are a threat in adolescence and adult life. No specific treatment exists for the diseases of muscle that impair skeletal muscle ischemic exercise. Enzyme replacement early in life is effective in Pompe disease (type II), which involves cardiac and skeletal muscle.
GALACTOSEMIA
Galactosemia is an autosomal recessive disease caused by deficiency of galactose-1-phosphate uridyltransferase (Fig. 52-2). Clinical manifestations are most striking in a neonate who, when fed milk, generally exhibits evidence of liver failure (hyperbilirubinemia, disorders of coagulation, and hypoglycemia), disordered renal tubular function (acidosis, glycosuria, and aminoaciduria), and cataracts. The neonatal screening test must have a rapid turnaround time because affected infants may die in the first week of life. Affected infants are at increased risk for severe neonatal Escherichia coli sepsis. Major effects on liver and kidney function and the development of cataracts are limited to the first few years of life; older children have learning disorders. Girls usually develop premature ovarian failure despite treatment.
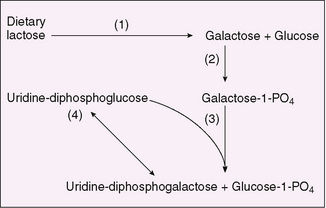
FIGURE 52-2 Pathway of galactose metabolism. (1) Lactase (intestinal), (2) galactokinase, (3) galactose-1-phosphate uridyltransferase, (4) uridine diphosphoglucose 4-epimerase.
Laboratory manifestations of galactosemia depend on dietary galactose intake. When galactose is ingested (as lactose), levels of plasma galactose and erythrocyte galactose-1-phosphate are elevated. Hypoglycemia is frequent, and albuminuria is present. Galactose frequently is present in the urine and can be detected by a positive reaction for reducing substances (Clinitest tablets) without a reaction with glucose oxidase on urine strip tests. The absence of urinary reducing substances cannot be relied on to exclude the diagnosis. The diagnosis is made by showing extreme reduction in erythrocyte galactose-1-phosphate uridyltransferase. DNA testing for the mutations in galactose-1-phosphate uridyltransferase confirms the diagnosis. Renal tubular dysfunction may be evidenced by a normal anion gap hyperchloremic metabolic acidosis. Treatment by the elimination of dietary galactose results in rapid correction of abnormalities, but infants who are extremely ill before treatment may die before therapy is effective. The concentration of galactose-1-phosphate rarely returns to normal even after treatment is begun.
Galactokinase deficiency, an autosomal recessive disorder, also leads to the accumulation of galactose in body fluids (see Fig. 52-2), which results in the formation of galactitol (dulcitol) through the action of aldose reductase. Galactitol, acting as an osmotic agent, can be responsible for cataract formation and, rarely, for increased intracranial pressure. These are the only clinical manifestations. Individuals homozygous for galactokinase deficiency usually develop cataracts after the neonatal period, whereas heterozygous individuals may be at risk for cataracts as adults. Treatment consists of lifelong elimination of galactose from the diet.
Hereditary fructose intolerance, in many ways, is analogous to galactosemia. When fructose is ingested, deficiency of fructose-1-phosphate aldolase leads to the intracellular accumulation of fructose-1-phosphate with resultant emesis, hypoglycemia, and severe liver and kidney disease. Elimination of fructose and sucrose from the diet cures the clinical disease. Affected patients sometimes spontaneously avoid fructose-containing foods and thus have no dental caries. Fructosuria is analogous to galactokinase deficiency in that it is caused by fructokinase deficiency, but the defect in fructosuria is harmless.