 CHAPTER 53 Amino Acid Disorders
CHAPTER 53 Amino Acid Disorders
DISORDERS OF AMINO ACID METABOLISM
Disorders of amino acid metabolism are the result of the inability to catabolize specific amino acids derived from protein. Usually a single amino acid pathway is involved. This amino acid accumulates in excess and is toxic to various organs, such as the brain, eyes, skin, or liver. Treatment is directed at the specific pathway, and usually involves dietary restriction of the offending amino acid and supplementation with special medical foods (formulas) that provide the other amino acids and other nutrients. Confirmatory testing includes quantitative specific plasma amino acid profiles along with specific mutation testing and sometimes enzymology.
PHENYLKETONURIA
Phenylketonuria (PKU), an autosomal recessive disease, primarily affects the brain and occurs in 1:10,000 persons. Classic PKU is the result from a defect in the hydroxylation of phenylalanine to form tyrosine (Fig. 53-1); the activity of phenylalanine hydroxylase in the liver is absent or greatly reduced. Affected infants are normal at birth, but severe mental retardation (IQ 30) develops in the first year of life. The clinical syndrome in the untreated child classically includes blond hair, blue eyes, eczema, and mousy odor of the urine but is rarely seen because of universal neonatal screening for PKU. A positive newborn screening test must be followed up by performing quantitative plasma amino acid analysis, measuring phenylalanine and tyrosine. A plasma phenylalanine value of greater than 360 μM (6 mg/dL) is consistent with the diagnosis of one of the hyperphenylalaninemias and demands prompt evaluation and treatment. Untreated, classic PKU is characterized by blood phenylalanine concentrations higher than 600 μM. Milder forms of hyperphenylalaninemia are indicated by values of plasma phenylalanine lower than this but higher than 360 μM. A significant percentage of premature infants and a few full-term infants have transient elevations in phenylalanine. Short-term follow-up usually identifies these infants promptly. Mutation testing of the PAH gene reveals more than 400 mutations. Some mutations are associated with mild hyperphenylalaninemia.
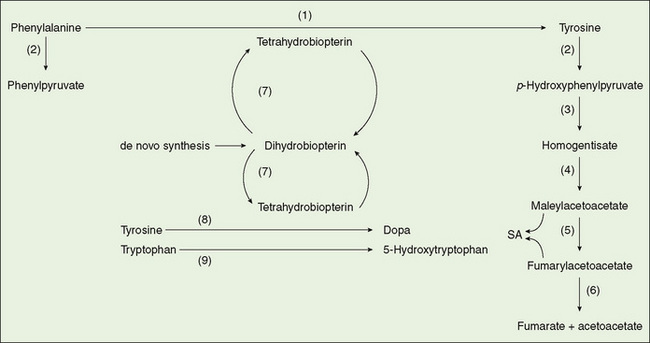
FIGURE 53-1 Metabolism of aromatic amino acids. (1) Phenylalanine hydroxylase, (2) transaminase, (3) p-hydroxyphenylpyruvate oxidase, (4) homogentisate oxidase, (5) maleylacetoacetate isomerase, (6) fumarylacetoacetate hydrolase, (7) dihydrobiopterin reductase, (8) tyrosine hydroxylase, (9) tryptophan hydroxylase. SA, succinylacetone.
Treatment is designed to maintain plasma phenylalanine values in the therapeutic range of 120 to 360 μM, at least for the first 10 years of life. However, many persons have difficulty achieving this level of control consistently, particularly in adolescence and adulthood.
A small percentage of infants diagnosed with PKU (≤2% in the United States) have a defect in the synthesis or metabolism of tetrahydrobiopterin, the cofactor for phenylalanine hydroxylase and for other enzymes involved in the intermediary metabolism of aromatic amino acids. In these infants, a progressive, lethal central nervous system disease develops, reflecting abnormalities in other neurotransmitter metabolism for which tetrahydrobiopterin is necessary. Disorders in biopterin metabolism are diagnosed by measuring dihydrobiopterin reductase in erythrocytes and by analyzing biopterin metabolites in urine. This testing is carried out in all hyperphenylalaninemic infants.
Outcome of treatment in classic PKU is excellent. Most infants with classic PKU who are treated using a diet specifically restricted in phenylalanine and begun within the first 10 days of life achieve normal intelligence. Learning problems and problems with executive function are reported; early and consistent dietary control provides the best chance for optimal outcome. In the disorders of biopterin biosynthesis, restriction of dietary phenylalanine reduces plasma phenylalanine, but does not ameliorate the clinical symptoms. This condition must be treated by replacement of the cofactor or by neuropharmacologic agents. Outcome is less predictable than in classic PKU.
Sustained control is difficult to achieve after the first 10 years of life. The safe concentration of phenylalanine in older children and adults with PKU has not been clearly established. Reversible cognitive dysfunction is associated with acute elevations of plasma phenylalanine in adults and children with PKU. If the elevated level has been sustained, the dysfunction may not be reversible. Treatment with modified preparation of tetrahydrobiopterin has shown good responses in some individuals with PKU. Maternal hyperphenylalaninemia requires rigorous management before conception and throughout pregnancy to prevent fetal brain damage, congenital heart disease, and microcephaly.
TYROSINEMIAS
Tyrosinemia is identified in neonatal screening programs using tandem mass spectrometry methods. Elevated tyrosine levels also occur as a nonspecific consequence of severe liver disease or transient tyrosinemia of the newborn, which responds to ascorbic acid treatment. The inherited disorders of tyrosine metabolism are the target of neonatal screening. Tyrosinemia I, which is due to fumarylacetoacetate hydrolase deficiency (see Fig. 53-1), is a rare disease in which accumulated metabolites produce severe liver disease associated with bleeding disorder, hypoglycemia, hypoalbuminemia, elevated transaminases, and defects in renal tubular function. Hepatocellular carcinoma may occur eventually. Quantitative measurement of plasma tyrosine and blood or urine succinylacetone is performed after a positive neonatal screening test for hypertyrosinemia. The diagnosis of tyrosinemia I is confirmed by an increased concentration of succinylacetone; DNA testing is available for some mutations. Treatment with nitisinone (NTBC) (an inhibitor of the oxidation of parahydroxyphenylpyruvic acid) effectively eliminates the production of the toxic succinylacetone. A low-phenylalanine, low-tyrosine diet may also play a role. These treatments have supplanted liver transplantation in many children identified by neonatal screening. Whether they completely eliminate the occurrence of hepatocellular carcinoma is unknown.
Tyrosinemias II and III are more benign forms of hereditary tyrosinemia. Blocked metabolism of tyrosine at earlier steps in the pathway is responsible, and succinylacetone is not produced. The clinical features include hyperkeratosis of palms and soles and keratitis, which can cause severe visual disturbance. Treatment with a phenylalanine-restricted and tyrosine-restricted diet is effective.
HOMOCYSTINURIA
Homocystinuria, an autosomal recessive disease (1:200,000 live births) involving connective tissue, the brain, and the vascular system, is caused by a deficiency of cystathionine β-synthase. In the normal metabolism of the sulfur amino acids, methionine gives rise to cystine; homocysteine is a pivotal intermediate (Fig. 53-2). When cystathionine β-synthase is deficient, homocysteine accumulates in the blood and appears in the urine. Another result is enhanced reconversion of homocysteine to methionine, resulting in an increase in the concentration of methionine in the blood. The neonatal screening test most commonly used measures methionine. An excess of homocysteine produces a slowly evolving clinical syndrome that includes dislocated ocular lenses; long, slender extremities; malar flushing; and livedo reticularis. Arachnodactyly, scoliosis, pectus excavatum or carinatum, and genu valgum are skeletal features. Mental retardation, psychiatric illness, or both may be present. Major arterial or venous thromboses are a constant threat.
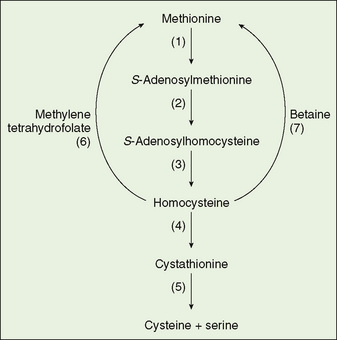
FIGURE 53-2 Metabolism of methionine and homocysteine. (1) Methionine adenosyltransferase, (2) S-methyltransferase, (3) S-adenosylhomocysteine hydrolase, (4) cystathionine β-synthase, (5) cystathionase, (6) homocysteine methyltransferase, (7) betaine-homocysteine methyltransferase.
Homocystinuria has no neonatal manifestations. Confirmation of the diagnosis requires demonstration of elevated total homocysteine in the blood. A plasma amino acid profile reveals hypermethioninemia. Measurement of cystathionine β-synthase is not clinically available, but numerous mutations in the gene are known and can be tested.
There are two clinical forms of homocystinuria. In one form, activity of the deficient enzyme can be enhanced by the administration of large doses of pyridoxine (100 to 1000 mg/day). Folate supplementation is added to overcome folate deficiency if folate is trapped in the process of remethylation of homocysteine to methionine. This pyridoxine-responsive form comprises about 50% of cases and is the more likely form to be missed by neonatal screening because the methionine concentrations are not always above the screening cutoff. The second form is not responsive to pyridoxine therapy. The accumulation of homocysteine is controlled with a methionine-restricted diet and cystine and folate supplementation. The use of supplemental betaine (trimethylglycine), a donor of methyl groups for remethylation of homocysteine to methionine, also has a role in the management of pyridoxine-unresponsive patients. Sometimes diet and betaine are required to control plasma homocysteine, even in pyridoxine-responsive patients. The prognosis is good for infants whose plasma homocysteine concentration is controlled.
MAPLE SYRUP URINE DISEASE
Maple syrup urine disease (MSUD) is an autosomal recessive disease, more properly named branched chain ketoaciduria. A deficiency of the decarboxylase initiates the degradation of the ketoacid analogs of the three branched chain amino acids—leucine, isoleucine, and valine (Fig. 53-3). MSUD is rare (1:250,000) in the general population but much more common in some population isolates (Pennsylvania Mennonites 1:150). Neonatal screening programs commonly include MSUD.
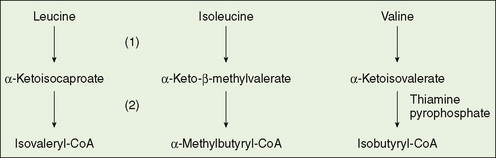
FIGURE 53-3 Metabolism of the branched chain amino acids. (1) Aminotransferases, (2) α-ketoacid dehydrogenase complex.
Although MSUD does have intermittent-onset and late-onset forms, clinical manifestations of the classic form typically begin within 1 to 4 weeks of birth. Poor feeding, vomiting, and tachypnea commonly are noted, but the hallmark of the disease is profound depression of the central nervous system, associated with alternating hypotonia and hypertonia (extensor spasms), opisthotonos, and seizures. The urine has the odor of maple syrup.
Laboratory manifestations of MSUD include hypoglycemia and a variable presence of metabolic acidosis, with elevation of the undetermined anions; the acidosis is caused, in part, by plasma branched chain organic acids and, in part, by the usual ketone bodies, β-hydroxybutyrate and acetoacetate. Branched chain ketoacids (but not β-hydroxybutyrate or acetoacetate) react immediately with 2,4-dinitrophenylhydrazine to form a copious, white precipitate.
The definitive diagnosis of MSUD generally is made by showing large increases in plasma leucine, isoleucine, and valine concentrations and identification of alloisoleucine in the plasma in excess. The urinary organic acid profile is usually abnormal and shows the ketoacid derivatives of the branched chain amino acids.
Provision of adequate calories and protein, excluding branched chain amino acids, is also crucial. The intake of branched chain amino acids (all three are essential amino acids) is restricted to the amounts required for growth in severely affected infants; restriction must be continued for life. Hemodialysis, hemofiltration, or peritoneal dialysis can be lifesaving during acidotic crises. Ordinary catabolic stresses, such as moderate infections or labor and delivery in a pregnant mother with MSUD, can precipitate clinical crises. Liver transplantation effectively treats MSUD.
DISORDERS OF AMMONIA DISPOSAL
Inherited enzymatic deficiencies have been described for each of the steps of urea synthesis (Fig. 53-4). Neonatal screening does not currently detect all of the disorders in the urea cycle.
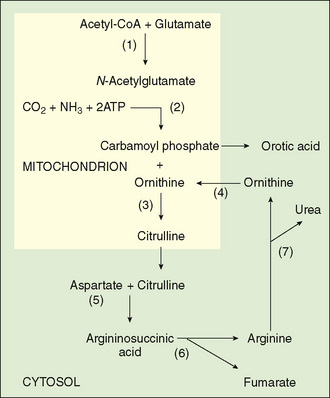
FIGURE 53-4 The urea cycle. (1) N-acetylglutamate synthase, (2) carbamoylphosphate synthetase, (3) ornithine carbamoylphosphate synthetase, (3) ornithine carbamoyltransferase, (4) ornithine translocator, (5) argininosuccinic acid synthetase, (6) argininosuccinic acid lyase, (7) arginase.
Ornithine Carbamoyltransferase Deficiency: Ornithine carbamoyltransferase (OTC) deficiency is unique among the defects in the urea cycle in that it is X-linked. The number of known gene mutations is quite large and includes both deletions and point mutations. If the enzyme is nonfunctional, there is no OTC activity in affected males, who are likely to die in the neonatal period. Affected females are heterozygous and, because of lyonization, may have a significant degree of enzyme deficiency and may be clinically affected. Clinical manifestations range through lethal disease in the male (coma, encephalopathy) to clinical normalcy in a high percentage of females. Late-onset forms in males also occur. Manifestations in clinically affected females include recurrent emesis, lethargy, seizures, developmental delay, and mental retardation. Affected females may spontaneously limit their protein intake.
Confirmatory testing for OTC includes a plasma amino acid profile, which may show deficient citrulline and arginine concentrations. A urine organic acid profile shows increased excretion of orotic acid. Mutation testing and sequencing of the entire coding region of the related genes are available as clinical tests.
Treatment of Hyperammonemia
During episodes of symptomatic hyperammonemia, protein intake is eliminated, and intravenous glucose is given in sufficient quantity to suppress catabolism of endogenous protein. Ammonia can be eliminated by use of the ammonia scavenger agents, sodium benzoate and sodium phenylacetate, which are excreted in the urine as conjugates of glycine and glutamine. Arginine, which is usually deficient, is supplied. Sterilization of the intestine and/or treatment with lactulose can provide brief benefit during acute episodes of hyperammonemia. When hyperammonemia is extreme, direct removal of ammonia, usually using hemodialysis or hemofiltration, is more effective than exchange transfusion or peritoneal dialysis. Despite successful management of hyperammonemic crises, the long-term outcome for males with severe neonatal hyperammonemia is guarded. Early liver transplantation has increased survival, especially in males with severe OTC deficiency.
A reduction of dietary protein intake is the mainstay of ongoing treatment for hyperammonemia. Crystalline essential amino acids can be supplied in amounts just sufficient to support new protein synthesis. Arginine is an essential amino acid when arginine synthesis via the urea cycle is grossly impaired, thus arginine must be supplied. Citrulline needs to be supplied for some urea cycle disorders. For OTC deficiency and carbamoyl phosphate synthase deficiency, treatment with phenylbutyrate (which is metabolized to phenylacetate) prevents accumulation of ammonia.
DISORDERS OF AMINO ACID TRANSPORT THAT AFFECT SPECIFIC TRANSPORT MECHANISMS IN THE KIDNEY AND INTESTINE
Cystinuria is a disorder of renal tubular transport of cystine, lysine, arginine, and ornithine. Although intestinal transport is affected in some genetic forms, the symptoms are largely due to the renal transport abnormality. The concentration of cystine exceeds its solubility product and results in significant renal stones. Evaluation and diagnosis is based on the pattern of amino acid excretion in the urine. Mutation testing can be done. Treatment is based on increasing the solubility of cystine by complexing it with compounds such as penicillamine.
Intestinal transport of tryptophan is impaired in Hartnup syndrome; pellagra-like symptoms result from this deficiency. Diagnosis is based on the amino acid pattern in urine, and treatment with tryptophan is successful.