 CHAPTER 54 Organic Acid Disorders
CHAPTER 54 Organic Acid Disorders
DISORDERS OF ORGANIC ACID METABOLISM
Organic acid disorders result from a block in the pathways of amino acid catabolism. Occurring after the amino moiety has been removed, they result in the accumulation of specific organic acids in the blood and urine. Treatment is directed at the specific abnormality, with restriction in precursor substrates and administration of enzyme cofactors, when available. Outcome is influenced by frequency and severity of ketoacidotic crises and is optimal when diagnosis is made before the onset of the first episode. Liver transplantation has been used in some patients, but long-term outcome after liver transplantation has not been well studied. Confirmatory testing begins with a urine organic acid profile and plasma amino acid profile. When abnormal results confirm the specific disorder, DNA testing may identify the mutations involved. More specific testing if a mutation is not found requires enzyme measurements in appropriate tissues.
PROPIONIC ACIDEMIA AND METHYLMALONIC ACIDEMIA
Propionic acidemia and methylmalonic acidemia result from defects in a series of reactions called the propionate pathway (Fig. 54-1). Defects in these steps produce ketosis and hyperglycinemia. Propionic acidemia and methylmalonic acidemia are identified by neonatal screening with tandem mass spectometry methods. The clinical manifestations of both of these disorders in the neonatal period consist of tachypnea, vomiting, lethargy, coma, intermittent ketoacidosis, hyperglycinemia, neutropenia, thrombocytopenia, hyperammonemia, and hypoglycemia. If these disorders are not identified by neonatal screening, intermittent episodes of metabolic acidosis occur. Crises occur during periods of catabolic stress, such as fever, vomiting, and diarrhea; they also may occur without an apparent precipitating event. During periods of neutropenia, the risk of serious bacterial infection is increased. Failure to thrive and impaired development are common.
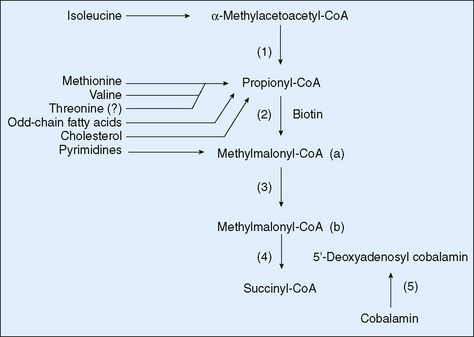
FIGURE 54-1 The propionate pathway. (1) β-Ketothiolase, (2) propionyl-CoA carboxylase, (3) methylmalonyl-CoA isomerase, (4) methylmalonyl-CoA mutase, (5) cobalamin metabolic pathway. CoA, coenzyme A.
Propionic acidemia results from deficiency in propionyl CoA-carboxylase, an enzyme that has two pairs of identical subunits. All forms of propionic acidemia are inherited in an autosomal recessive manner and are due to mutations in one of the subunits. Methylmalonic acidemia results from deficiency in methylmalonyl mutase; this may be caused by mutations in the gene for the mutase protein itself or in one of the steps of the synthesis of the cobalamin cofactors for the enzyme. A complex set of defects in cobalamin metabolism results in other forms of methylmalonic acidemia, some of which are associated with hyperhomocystinemia. Treatment with massive doses of hydroxycobalamin (the active form of vitamin B12) is helpful in some cases of methylmalonic acidemia.
For propionic acidemia and the vitamin B12–unresponsive forms of methylmalonic acidemia, management includes the restriction of dietary protein and addition of a medical food deficient in the specific amino acid precursors of propionyl-CoA (isoleucine, valine, methionine, and threonine). Carnitine supplementation is often needed because it is lost in the urine as acylcarnitines. Intestinal bacteria produce a significant quantity of propionate; thus, antibacterial treatment to reduce the population of bacteria in the gut has some beneficial effect in propionic acidemia and vitamin B12–unresponsive methylmalonic acidemia.
ISOVALERIC ACIDEMIA
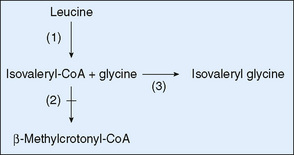
Isovaleric acidemia results from a block in the catabolism of leucine. Its clinical manifestations are similar to those of defects in the propionate pathway. The strong odor of isovaleric acid results in sweaty feet odor in untreated infants. Restricting the intake of leucine, glycine therapy enhances the formation of isovalerylglycine, a relatively harmless conjugate of isovaleric acid (Fig. 54-2), which is excreted in the urine. Patients identified by newborn screening may have a milder phenotype; genotype-phenotype correlation studies are in progress to determine whether some forms do not need aggressive treatment.
GLUTARIC ACIDEMIA I
Glutaric acidemia I results from a deficiency at the end of the lysine catabolic pathway. It is an autosomal recessive disease produced by deficiency of glutaryl-CoA dehydrogenase activity (Fig. 54-3). Clinical manifestations include macrocephaly, which may be present at birth, and dystonia, which characteristically develops after the first 18 months of life, after an episode of intercurrent illness associated with fever and metabolic distress. Clinical features include metabolic strokelike episodes associated with infarction of the basal ganglia. Treatment includes a protein-restricted diet accompanied by a medical food deficient in lysine. Despite this treatment, as many as one third of children still develop brain damage. Improved outcome may occur when episodes of metabolic distress are managed aggressively.
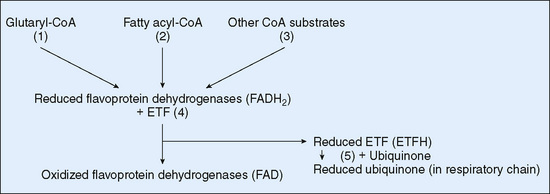
FIGURE 54-3 Scheme of flavoprotein metabolism with reference to glutaric aciduria types I and II. (1) Glutaryl-CoA dehydrogenase (deficient in glutaric aciduria type1), (2) fatty acyl-CoA dehydrogenases, (3) other flavoprotein dehydrogenases, (4) ETF (deficiency results in glutaric aciduria type II), (5) ETF-ubiquinone oxidoreductase (deficiency results in glutaric aciduria type II). CoA, coenzyme A; ETF, electron transfer flavoprotein.
BIOTINIDASE DEFICIENCY AND HOLOCARBOXYLASE DEFICIENCY
Biotin is a ubiquitous vitamin that is covalently linked to many carboxylases and cannot be recycled from its attachment to the carboxylases. Thus, inherited biotinidase deficiency greatly increases the dietary requirement for biotin. Affected individuals become biotin deficient while consuming normal diets. Clinical disease can appear in the neonatal period or be delayed until later infancy, depending on the degree of deficiency.
Clinical manifestations of biotin deficiency vary greatly (seizures, hypotonia, sensory neural deafness, alopecia, skin rash, metabolic acidosis, and immune deficits) and, undoubtedly, depend on which enzymes in which tissues have the most biotin depletion. Carboxylation is a crucial reaction in the metabolism of organic acids; most patients with biotinidase deficiency excrete abnormal amounts of several organic acids, among which β-methylcrotonylglycine is prominent. In addition to biotinidase deficiency, an inherited deficiency of holocarboxylase synthetase gives rise to severe disease and to similar patterns of organic aciduria. Both conditions respond well to treatment with large doses of biotin (10 to 40 mg/day). Confirmatory testing is accomplished with quantitative measurement of biotinidase activity.