 CHAPTER 56 Lysosomal and Peroxisomal Disorders
CHAPTER 56 Lysosomal and Peroxisomal Disorders
PEROXISOMAL DISORDERS
Peroxisomes are subcellular organelles involved in metabolism and biosynthesis of bile acids, membrane phospholipids, and some β-oxidation of long-chain fatty acids. Disorders include conditions caused by abnormal peroxisomal enzyme function and abnormal peroxisomal biogenesis. Clinical symptoms are protean and nearly always include developmental delay and dysmorphic features that can involve the skeleton and the head. Zellweger syndrome, neonatal adrenoleukodystrophy, and infantile Refsum disease are examples of disorders of peroxisome biogenesis. Zellweger syndrome, an autosomal recessive disease (1:100,000 births), is also called cerebrohepatorenal syndrome. Peroxisomes are virtually absent, as are normal peroxisomal functions, which include the oxidation of very long chain fatty acids. Affected infants have high foreheads, flat orbital ridges, widely open fontanelles, hepatomegaly, and hypotonia. Other anomalies are common. Failure to thrive, seizures, and nystagmus develop early, and death occurs within the first year. Refsum disease, neonatal adrenoleukodystrophy, and malonic aciduria are examples of peroxisomal single enzyme disorders. Diagnostic testing includes measurement of very long chain fatty acids in plasma and pipecolic acid in urine. Specific molecular testing, particularly for the disorders involving one in the series of PEX genes, is available for some disorders. Most of these conditions are untreatable; bone marrow transplant can be helpful in X-linked adrenoleukodystrophy.
LYSOSOMAL STORAGE DISORDERS
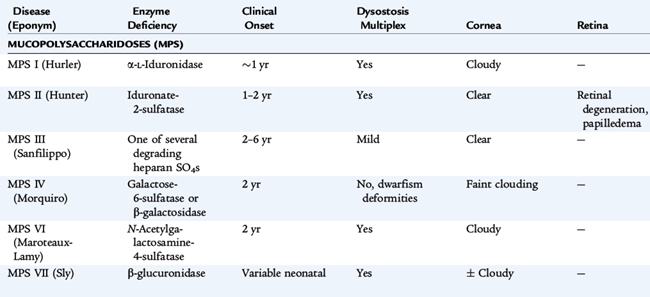
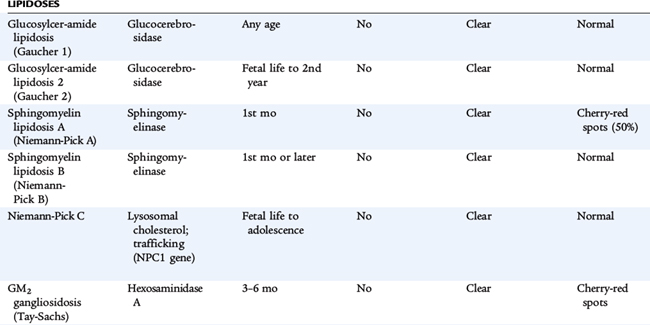
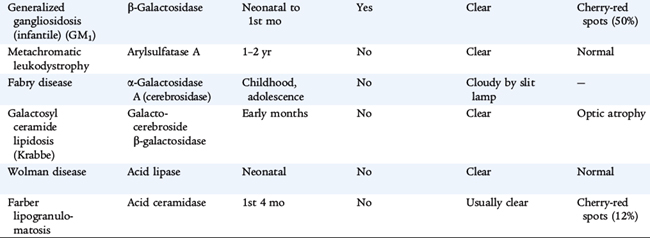
Lysosomes are subcellular organelles that contain degradative enzymes for complex glycosaminoglycans, also called mucopolysaccharides. Glycosaminoglycans are macromolecules that play a number of roles within cells. Genetic disorders result from abnormal formation of the lysosome itself or from deficiency in specific hydrolytic enzymes, in the mechanisms that protect intralysosomal enzymes from hydrolytic destruction, in the transport of materials into the lysosome and of metabolites out of the lysosome. These materials are stored in cells and ultimately result in their destruction, especially in the nervous system. The clinical disorders are diverse, reflecting tissue specificity of lysosomal function and the intrinsic turnover rates of the compounds whose cycling is affected (Table 56-1). Some disorders affect many tissues but spare the brain. Some are apparent only during adult life. Storage in solid organs results in organomegaly. In many of these disorders, developmental delay, corneal clouding, and limitation of joint mobility are common features. Storage in tissues of the upper and lower airways may result in respiratory compromise. Nonimmune hydrops fetalis occurs in several lysosomal disorders.
DIAGNOSTIC TESTING
Diagnostic testing includes measurement of glycosaminoglycans in urine and an enzyme assay for lysosomal enzyme activity in white blood cells. If the urine test is positive, it helps direct specific enzyme measurement. If it is negative, it does not exclude a lysosomal storage disorder, and other testing modalities are needed if clinical signs are convincing. In disorders in which specific mutations are known, molecular testing refines the diagnosis. Specific diagnosis, carrier testing, and prenatal testing require one of these approaches. Enzyme analysis is complex; experienced laboratories are the best source of this testing. If this testing is to be used for prenatal diagnosis, the details about the expression of the enzyme during fetal life must be known. For this reason, molecular testing is more specific for prenatal diagnosis. Making a specific diagnosis is even more important because treatment for some lysosomal disorders is available. Treatment strategies are arduous, so specific diagnosis is crucial.
TREATMENT STRATEGIES
Treatment for many of these conditions is supportive. Careful attention to respiratory status and physical therapy is very important. Specific treatment directed at the metabolic abnormality is available for some lysosomal disorders. For some, bone marrow (stem cell) transplantation can restore lysosomal function. For others, replacement of the missing hydrolytic enzyme by systemic administration of the enzyme allows degradation of stored material. The disorders caused by deficient α-L-iduronidase (Hurler syndrome, Scheie syndrome, and their variants) respond to treatment with intravenous human recombinant α-L-iduronidase (laronidase). Other disorders for which enzyme therapy is available include MPS VI (Maroteaux-Lamy syndrome), Gaucher disease, Fabry disease, and MPS II (Hunter syndrome). Stem cell transplantation has been helpful or is under investigation in the following disorders: MPS type IH (Hurler syndrome), MPS type VI (Marateaux-Lamy syndrome), MPS type VII (Sly syndrome), Krabbe disease, metachromatic leukodystrophy, alpha-fucosidosis, alpha-mannosidosis, Gaucher disease, and Niemann-Pick disease type B.
Treatment needs to begin before clinical signs appear, because central nervous system manifestations are not improved by these approaches.