 CHAPTER 57 Mitochondrial Disorders
CHAPTER 57 Mitochondrial Disorders
MITOCHONDRIAL FUNCTION
Mitochondria generate energy from oxidative phosphorylation to produce ATP by transferring electrons formed by glycolysis and the Krebs cycle to a cascade that generates NADH and FADH2 (Fig. 57-1). Mitochondria reside in most organs, so signs and symptoms of mitochondrial dysfunction can affect multiple organ systems. The more dependent on energy production an organ is, the more profound the symptoms of deficiency of mitochondrial function in that organ will be. Taken together, mitochondrial disorders may affect as many as 1 in 5000 people.
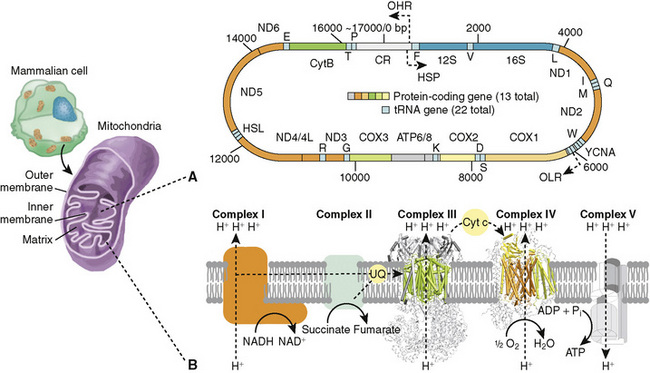
FIGURE 57-1 The mammalian mitochondrial genome and its protein-coding gene repertoire involved in the oxidative phosphorylation pathway. (A) Schematic representation of genes within mammalian mitochondrial genome (~7000 bp). Genes on the outer circle are transcribed from the light-strand. Location of the tRNAs (red boxes) conform to the canonical placental mammalian arrangement. (B) Simplified view of the mitochondrial oxidative phosphorylation machinery. Complexes I (NADH dehydrogenase) and II (succinate dehydrogenase) receive electrons from either NADH or FADH2. Electrons are then carried between complexes by the carrier molecules coenzyme Q/ubiquinone (UQ) and cytochrome c (CYC). The potential energy of these electron transfer events is used to pump protons against the gradient, from the mitochondrial matrix into the intermembrane space [complexes I and III (cytochrome bc1) and IV (cytochrome c oxidase)]. ATP synthesis by complex V (ATP synthase) is driven by the proton gradient and occurs in the mitochondrial matrix. HSP, putative heavy-strand promoter; IM, intermembrane space; MM, mitochondria matrix; OHR, origin of heavy-strand replication; OLR, origin of light-strand replication.
(From da Fonseca RR, Johnson WE, O’Brien SJ, et al: The adaptive evolution of the mammalian mitochondrial genome. BMC Genomics 9:119, 2008.)
SIGNS AND SYMPTOMS OF GENETIC DISORDERS OF MITOCHONDRIAL FUNCTION
The signs and symptoms of mitochondrial disorders are protean. Symptoms depend on how an organ is affected by energy deficiency. Muscle function that is compromised will result in muscle pain, fatigue, and weakness. Myopathy is common and may show ragged red fibers on a muscle biopsy (Table 57-1). Rhabdomyolysis can occur. Brain dysfunction may be expressed as seizures, loss of intellectual function, headache, or signs consistent with stroke. Spastic paraplegia may occur. Ataxia and basal ganglia symptoms are features of some disorders. Vision and eye muscle movement may be compromised. Cardiomyopathy is frequent, and cardiac rhythm disturbances occur. Liver dysfunction may be expressed as both synthetic deficiencies and liver failure. Diabetes may signal pancreatic involvement. Renal tubular abnormalities and renal failure both occur. Gastrointestinal symptoms include both diarrhea and constipation that are difficult to treat. Alper disease (cerebral degeneration and liver disease) and Leigh disease (subacute necrotizing encephalomyelopathy) show similar brain lesions but in distinctly different areas of the brain. Because the signs and symptoms involve multiple organs and may seem nonspecific, physicians may not suspect a mitochondrial disorder until significant progression has occurred.
TABLE 57-1 Signs and Symptoms of Mitochondrial Disorders
| Mitochondrial Complex | Clinical Presentation |
|---|---|
| Complex I | Leigh, Alpers, leukodystrophy, cardiomyopathy, lactic acidosis |
| Complex II | Neurodegeneration, Leigh syndrome, paraganglioma |
| Complex III | Tubulopathy and encephalopathy, GRACILE syndrome |
| Complex IV (COX) | Leigh syndrome, infantile cardiomyopathy, neonatal liver failure |
| Complex V | Malformation syndrome, neonatal lactic acidosis |
| Mitochondrial translation | Macrocystic leukodystrophy, lactic acidosis, myopathy |
| mtDNA maintenance | mtDNA depletion syndromes: Alpers, Leigh, hepatopathy, ataxia |
GRACILE syndrome, growth retardation, aminoaciduria, cholestasis, iron overload, lactic acidosis, early death; mtDNA, mitochondrial DNA.
BIOCHEMICAL ABNORMALITIES IN MITOCHONDRIAL FUNCTION
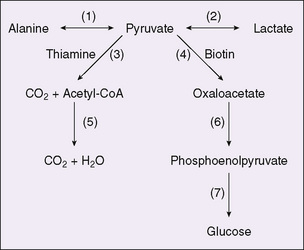
Defects in the mitochondrial respiratory chain produce lactic acidosis. Given the complexity of the respiratory chain, it is not surprising that the described defects are varied as to cause, intensity, and tissues affected. However, not all patients with mitochondrial disorders exhibit lactic acidosis. The metabolism of glucose to carbon dioxide and water, with pyruvate as an intermediate (Fig. 57-2), occurs as part of the energy cycle in many tissues. Interference with mitochondrial oxidative metabolism results in the accumulation of pyruvate. Because lactate dehydrogenase is ubiquitous, and because the equilibrium catalyzed by this enzyme greatly favors lactate over pyruvate, the accumulation of pyruvate results in lactic acidosis. The most common cause of such lactic acidosis is oxygen deficiency caused by anoxia or poor perfusion. Lactic acidosis also occurs when specific reactions of pyruvate are impaired. In the liver, pyruvate undergoes carboxylation to form oxaloacetate using the enzyme pyruvate carboxylase; deficiency in this enzyme causes severe lactic acidosis. In many tissues, lactate is catabolized to form acetyl coenzyme A (CoA) by the pyruvate dehydrogenase complex; deficiency in pyruvate dehydrogenase also can cause lactic acidosis. Because these reactions also play a role in gluconeogenesis, hypoglycemia can be a feature of these disorders. These disorders comprise forms of primary lactic acidosis. They frequently present as intractable, lethal acidosis in the first days or weeks of life and are difficult to treat. Some of the enzymes in this pathway can be measured and specific diagnosis can be made. This may require white blood cells or tissue biopsy.
GENETICS OF MITOCHONDRIAL DISORDERS
Mitochondrial function is carried out by proteins that are coded for by both nuclear and mitochondrial genes. These enzymes are extremely complicated, and several are quite large. The mitochondrial genome encodes 13 subunits of the enzymes involved in mitochondrial oxidative phosphorylation. More than 85 autosomal genes code for the rest of the subunits of these enzymes. In children, only about 20% of cases of mitochondrial disease are caused by mutations in mitochondrial DNA (mtDNA); the rest are due to mutations in nuclear genes. Depletion of mtDNA itself often results from mutations in nuclear genes that maintain mtDNA (e.g., polymerase [DNA directed], gamma [POLG]. Most disorders show autosomal recessive inheritance. A few are passed on by maternal mtDNA mutation (Table 57-2).
TABLE 57-2 Inheritance in Mitochondrial Disorders
| Mitochondrial Complex | Genes | Inheritance |
|---|---|---|
| Complex I | 43 subunits, 7 mtDNA mtDNA maintenance genes | Autosomal recessive (>90%) |
| Complex II | SDH | Autosomal recessive and dominant (rare) |
| Complex III | Assembly factor for complex III | Autosomal recessive |
| Complex IV (COX) | 23 genes; autosomal and mitochondrial and assembly factors | Autosomal recessive |
| Complex V | ATP synthase | Maternally inherited |
| Mitochondrial translation | Myopathy and anemia | Autosomal recessive |
| mtDNA maintenance | mtDNA depletion, Alpers, Leigh, failure to thrive, and myopathy | Autosomal recessive |
mtDNA, mitochondrial DNA; SDH, succinate dehydrogenase.
TREATMENT OF MITOCHONDRIAL DISORDERS
Repairing the basic energy deficit and getting the appropriate drugs and cofactors to the appropriate location within the mitochondrion are difficult. Nevertheless, a number of strategies are used, including judicious physical therapy and exercise with adequate rest, adequate nutrition, and cofactors for the deficient pathway. Treatment is limited for most mitochondrial defects. Vitamin cofactors for the respiratory chain, such as riboflavin and vitamin E, and pharmaceutical forms of coenzyme Q are often used. When a single organ bears most of the damage, organ transplant may be effective. Identification of family members at risk may allow earlier diagnosis and treatment.
de Baulny H.O., Benoist J.F., Rigal O., et al. Methylmalonic and propionic acidaemias: management and outcome. J Inherit Metab Dis. 2005;28(3):415-423.
Debray F.G., Lambert M., Mitchell G. Disorders of mitochondrial function. Curr Opin Pediatr. 2008;20(4):471-482.
Heese B.A. Current strategies in the management of lysosomal storage diseases. Semin Pediatr Neurol. 2008;15(3):119-126.
Kayser M.A. Inherited metabolic diseases in neurodevelopmental and neurobehavioral disorders. Semin Pediatr Neurol. 2008;15(3):127-131.
Kliegman R.M., Behrman R.E., Jenson H.B., et al. Nelson Textbook of Pediatrics, 18th ed. Philadelphia: Saunders, 2007.
Koeberl D.D., Kishnani P.S., Chen Y.T. Glycogen storage disease types I and II: treatment updates. J Inherit Metab Dis. 2007;30(2):159-164.
Kompare M., Rizzo W.B. Mitochondrial fatty-acid oxidation disorders. Semin Pediatr Neurol. 2008;15(3):140-149.
Longo N. Inborn errors of metabolism: new challenges with expanded newborn screening programs. Am J Med Genet C Semin Med Genet. 2006;142C(2):61-63.
Maillot F., Lilburn M., Baudin J., et al. Factors influencing outcomes in the offspring of mothers with phenylketonuria during pregnancy: the importance of variation in maternal blood phenylalanine. Am J Clin Nutr. 2008;88(3):700-705.
Stea T.H., Mansoor M.A., Wandel M., et al. Changes in predictors and status of homocysteine in young male adults after a dietary intervention with vegetables, fruits and bread. Eur J Nutr. 2008;47(4):201-209.
Tuchman M., Lee B., Lichter-Konecki U., et al. Cross-sectional multicenter study of patients with urea cycle disorders in the United States. Mol Genet Metab. 2008;94(4):397-402.
Vockley J., Ensenauer R. Isovaleric acidemia: new aspects of genetic and phenotypic heterogeneity. Am J Med Genet C Semin Med Genet. 2006;142C(2):95-103.