 CHAPTER 62 Anemia and Hyperbilirubinemia
CHAPTER 62 Anemia and Hyperbilirubinemia
ANEMIA
Embryonic hematopoiesis begins by the 20th day of gestation and is evidenced as blood islands in the yolk sac. In midgestation, erythropoiesis occurs in the liver and spleen; the bone marrow becomes the predominant site in the last trimester. Hemoglobin concentration increases from 8 to 10 g/dL at 12 weeks to 16.5 to 18 g/dL at 40 weeks. Fetal red blood cell (RBC) production is responsive to erythropoietin, and the concentration of this hormone increases with fetal hypoxia and anemia.
After birth, hemoglobin levels increase transiently at 6 to 12 hours, then decline to 11 to 12 g/dL at 3 to 6 months. A premature infant (<32 weeks’ gestational age) has a lower hemoglobin concentration and a more rapid postnatal decline of hemoglobin level, which achieves a nadir 1 to 2 months after birth. Fetal and neonatal RBCs have a shorter life span (70 to 90 days) and a higher mean corpuscular volume (110 to 120 fL) than adult cells. In the fetus, hemoglobin synthesis in the last two trimesters of pregnancy produces fetal hemoglobin (hemoglobin F), composed of two alpha chains and two gamma chains. Immediately before term, the infant begins to synthesize beta-hemoglobin chains; the term infant should have some adult hemoglobin (two alpha chains and two beta chains). Fetal hemoglobin represents 60% to 90% of hemoglobin at term birth, and the levels decline to adult levels of less than 5% by 4 months of age.
For a term infant, blood volume is 72 to 93 mL/kg, and for a preterm infant, blood volume is 90 to 100 mL/kg. The placenta and umbilical vessels contain approximately 20 to 30 mL/kg of additional blood that can increase neonatal blood volume and hemoglobin levels transiently for the first 3 days of life if clamping or milking (stripping) of the umbilical cord is delayed at birth. Delayed clamping increases the risk of polycythemia, increased pulmonary vascular resistance, hypoxia, and jaundice, but it improves glomerular filtration. Early clamping may lead to anemia, a cardiac murmur, poor peripheral perfusion but lower pulmonary vascular pressures, and less tachypnea. Hydrostatic pressure affects blood transfer between the placenta and the infant at birth, and an undesired fetal-to-placental transfusion occurs if the infant is situated above the level of the placenta.
The physiologic anemia noted at 2 to 3 months of age in term infants and at 1 to 2 months of age in preterm infants is a normal process that does not result in signs of illness and does not require any treatment. It is a physiologic condition believed to be related to several factors, including increased tissue oxygenation experienced at birth, shortened RBC life span, and low erythropoietin levels.
Etiology
Symptomatic anemia in the newborn period (Fig. 62-1) may be caused by decreased RBC production, increased RBC destruction, or blood loss.
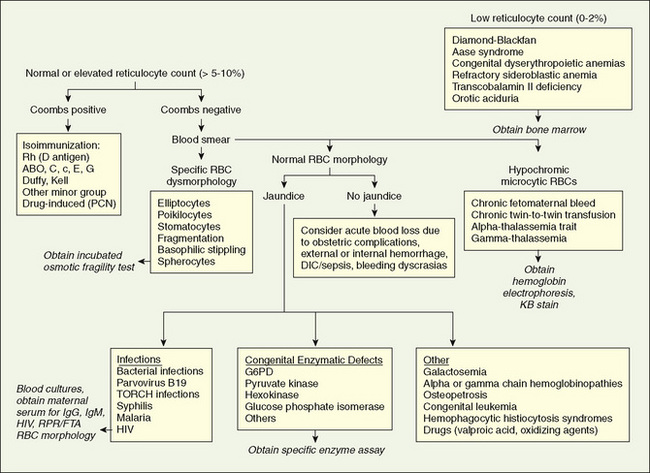
FIGURE 62-1 Differential diagnosis of neonatal anemia. The physician obtains information from the family, maternal and labor and delivery histories, and laboratory tests, including hemoglobin, reticulocyte count, blood type, direct Coombs test, peripheral smear, red blood cell (RBC) indices, and bilirubin concentration. DIC, disseminated intravascular coagulation; FTA, fluorescent treponemal antibody test; G6PD, glucose-6-phosphate dehydrogenase; KB, Kleihauer-Betke; PCN, penicillin; RPR, rapid plasma reagin tests.
(From Ohls RK: Anemia in the neonate. In Christensen RD, editor: Hematologic Problems of the Neonate, Philadelphia, 2000, Saunders, p 162.)
Decreased Red Blood Cell Production
Anemia caused by decreased production of RBCs appears at birth with pallor, a low reticulocyte count, and absence of erythroid precursors in the bone marrow. Potential causes of neonatal decreased RBC production include bone marrow failure syndromes (congenital RBC aplasia [Diamond-Blackfan anemia]), infection (congenital viral infections [parvovirus, rubella], acquired bacterial or viral sepsis), nutritional deficiencies (protein, iron, folate, vitamin B12), and congenital leukemia.
Increased Red Blood Cell Destruction
Immunologically mediated hemolysis in utero may lead to erythroblastosis fetalis, or the fetus may be spared, and hemolytic disease may appear in the newborn. Hemolysis of fetal erythrocytes is a result of blood group differences between the sensitized mother and fetus, which causes production of maternal IgG antibodies directed against an antigen on fetal cells.
ABO blood group incompatibility with neonatal hemolysis develops only if the mother has IgG antibodies from a previous exposure to A or B antigens. These IgG antibodies cross the placenta by active transport and affect the fetus or newborn. Sensitization of the mother to fetal antigens may have occurred by previous transfusions or by conditions of pregnancy that result in transfer of fetal erythrocytes into the maternal circulation, such as first trimester abortion, ectopic pregnancy, amniocentesis, manual extraction of the placenta, version (external or internal) procedures, or normal pregnancy.
ABO incompatibility with sensitization usually does not cause fetal disease other than extremely mild anemia. It may produce hemolytic disease of the newborn, however, which is manifested as significant anemia and hyperbilirubinemia. Because many mothers who have blood group O have IgG antibodies to A and B before pregnancy, the firstborn infant of A or B blood type may be affected. In contrast to Rh disease, ABO hemolytic disease does not become more severe with subsequent pregnancies. Hemolysis with ABO incompatibility is less severe than hemolysis in Rh-sensitized pregnancy, either because the anti-A or anti-B antibody may bind to nonerythrocytic cells that contain A or B antigen or because fetal erythrocytes have fewer A or B antigenic determinants than they have Rh sites. With the declining incidence of Rh hemolytic disease, ABO incompatibility has become the most common cause of neonatal hyperbilirubinemia requiring therapy—currently accounting for approximately 20% of clinically significant jaundice in the newborn.
Erythroblastosis fetalis classically is caused by Rh blood group incompatibility. Most Rh-negative women have no anti-Rh antibodies at the time of their first pregnancy. The Rh antigen system consists of five antigens: C, D, E, c, and e; the d type is not antigenic. In most Rh-sensitized cases, the D antigen of the fetus sensitizes the Rh-negative (d) mother, resulting in IgG antibody production during the first pregnancy. Because most mothers are not sensitized to Rh antigens at the start of pregnancy, Rh erythroblastosis fetalis is usually a disease of the second and subsequent pregnancies. The first affected pregnancy results in an antibody response in the mother, which may be detected during antenatal screening with the Coombs test and determined to be anti-D antibody. The first affected newborn may show no serious fetal disease and may manifest hemolytic disease of the newborn only by the development of anemia and hyperbilirubinemia. Subsequent pregnancies result in an increasing severity of response because of an earlier onset of hemolysis in utero. Fetal anemia, heart failure, elevated venous pressure, portal vein obstruction, and hypoalbuminemia result in fetal hydrops, which is characterized by ascites, pleural and pericardial effusions, and anasarca (see Chapter 60). The risk of fetal death is high.
The management of a pregnancy complicated by Rh sensitization depends on the severity of hemolysis, its effects on the fetus, and the maturity of the fetus at the time it becomes affected. The severity of the hemolysis can be assessed by the quantity of bilirubin transferred from the fetus to the amniotic fluid, quantified by spectrophotometric analysis of the optical density (at 450 nm) of amniotic fluid.
Three zones of optical densities with decreasing slopes toward term gestation have been developed to predict the severity of the illness. The high optical density zone 3 is associated with severe hemolysis. Fetuses in the lower zones probably are not affected. If a fetus’ optical density measurement for bilirubin falls into zone 3, and the fetus has pulmonary maturity as determined by the lecithin-to-sphingomyelin ratio, the infant should be delivered and treated in the neonatal intensive care unit. If the lungs are immature and the fetus is between 22 and 33 weeks of gestational age, an ultrasound-guided intrauterine transfusion with O-negative blood into the umbilical vein is indicated and may have to be repeated until pulmonary maturity is reached or fetal distress is detected. Indications for fetal intravascular transfusion in sensitized fetuses between 22 and 32 weeks of gestational age include a fetal hematocrit of less than 25% to 30%, fetal hydrops, and fetal distress too early in gestation for delivery. Intravascular intrauterine transfusion corrects fetal anemia, improves the outcome of severe hydrops, and reduces the need for postnatal exchange transfusion, but is associated with neonatal anemia as a result of continued hemolysis plus suppressed erythropoiesis.
Prevention of sensitization of the mother carrying an Rh-positive fetus is possible by treating the mother during gestation (>28 weeks’ gestational age) and within 72 hours after birth with anti-Rh-positive immune globulin (RhoGAM). The dose of RhoGAM (300 μg) is based on the ability of this amount of anti-Rh-positive antibody to bind all the possible fetal Rh-positive erythrocytes entering the maternal circulation during the fetal-to-maternal transfusion at birth (approximately 30 mL). RhoGAM may bind Rh-positive fetal erythrocytes or interfere with maternal anti-Rh-positive antibody production by another, unknown mechanism. RhoGAM is effective only in preventing sensitization to the D antigen. Other blood group antigens that can cause immune hydrops and erythroblastosis include Rh C, E, Kell, and Duffy. Anti-Kell alloimmunity produces lower amniotic bilirubin levels and a lower reticulocyte count because in addition to hemolysis it inhibits erythropoiesis.
Nonimmune causes of hemolysis in the newborn include RBC enzyme deficiencies of the Embden-Meyerhof pathway, such as pyruvate kinase or glucose-6-phosphate dehydrogenase deficiency. RBC membrane disorders are another cause of nonimmune hemolysis. Hereditary spherocytosis is inherited as a severe autosomal recessive form or less severe autosomal dominant form and is the result of a deficiency of spectrin, a protein of the RBC membrane. Hemoglobinopathies, such as thalassemia, are another cause of nonimmunologically mediated hemolysis.
Blood Loss
Anemia from blood loss at birth is manifested by two patterns of presentation, depending on the rapidity of blood loss. Acute blood loss after fetal-maternal hemorrhage, rupture of the umbilical cord, placenta previa, or internal hemorrhage (hepatic or splenic hematoma; retroperitoneal) is characterized by pallor, diminished peripheral pulses, and shock. There are no signs of extramedullary hematopoiesis and no hepatosplenomegaly. The hemoglobin content and serum iron levels initially are normal, but the hemoglobin levels decline during the subsequent 24 hours. Newborns with chronic blood loss caused by chronic fetal-maternal hemorrhage or a twin-to-twin transfusion present with marked pallor, heart failure, hepatosplenomegaly with or without hydrops, a low hemoglobin level at birth, a hypochromic microcytic blood smear, and decreased serum iron stores. Fetal-maternal bleeding occurs in 50% to 75% of all pregnancies, with fetal blood losses ranging from 1 to 50 mL; most blood losses are 1 mL or less, 1 in 400 are approximately 30 mL, and 1 in 2000 are approximately 100 mL.
The diagnosis of fetal-maternal hemorrhage is confirmed by the Kleihauer-Betke acid elution test. Pink fetal RBCs are observed and counted in the mother’s peripheral blood smear because fetal hemoglobin is resistant to acid elution; adult hemoglobin is eluted, leaving discolored maternal cells (patients with sickle cell anemia or hereditary persistence of fetal hemoglobin may have a false-positive result, and ABO incompatibility may produce a false-negative result).
Diagnosis and Management
Hemolysis in utero resulting from any cause may produce a spectrum of clinical manifestations at birth. Severe hydrops with anasarca, heart failure, and pulmonary edema may prevent adequate ventilation at birth, resulting in asphyxia. Infants affected with hemolysis in utero have hepatosplenomegaly and pallor and become jaundiced within the first 24 hours after birth. Less severely affected infants manifest pallor and hepatosplenomegaly at birth and become jaundiced subsequently. Patients with ABO incompatibility often are asymptomatic and show no physical signs at birth; mild anemia with jaundice develops during the first 24 to 72 hours of life.
Because hydrops, anemia, or jaundice are secondary to many diverse causes of hemolysis, a laboratory evaluation is needed in all patients with suspected hemolysis. A complete blood count, blood smear, reticulocyte count, blood type, and direct Coombs test (to determine the presence of antibody-coated RBCs) should be performed in the initial evaluation of all infants with hemolysis. Reduced hemoglobin levels, reticulocytosis, and a blood smear characterized by polychromasia and anisocytosis are expected with isoimmune hemolysis. Spherocytes commonly are observed in ABO incompatibility. The determination of the blood type and the Coombs test identify the responsible antigen and antibody in immunologically mediated hemolysis.
In the absence of a positive Coombs test and blood group differences between the mother and fetus, other causes of nonimmune hemolysis must be considered. RBC enzyme assays, hemoglobin electrophoresis, or RBC membrane tests (osmotic fragility, spectrin assay) should be performed. Internal hemorrhage also may be associated with anemia, reticulocytosis, and jaundice when the hemorrhage reabsorbs; ultrasound evaluation of the brain, liver, spleen, or adrenal gland may be indicated when nonimmune hemolysis is suspected. Shock is more typical in patients with internal hemorrhage, whereas in hemolytic diseases heart failure may be seen with severe anemia. Evaluation of a possible fetal-maternal hemorrhage should include the Kleihauer-Betke test.
The treatment of symptomatic neonatal anemia is transfusion of crossmatched packed RBCs. If immune hemolysis is present, the cells to be transfused must be crossmatched against maternal and neonatal plasma. Acute volume loss may necessitate resuscitation with nonblood products, such as saline if blood is not available; packed RBCs can be given subsequently. To correct anemia and any remaining blood volume deficit, 10 to 15 mL/kg of packed RBCs should be sufficient. Cytomegalovirus-seronegative blood should be given to cytomegalovirus-seronegative infants, and all blood products should be irradiated to reduce the risk of graft-versus-host disease; blood should be screened for HIV, hepatitis B and C, and syphilis. Recombinant erythropoietin may improve the hematocrit in infants with a hyporegenerative anemia after in utero transfusion.
HYPERBILIRUBINEMIA
Hemolytic disease of the newborn is a common cause of neonatal jaundice. Nonetheless, because of the immaturity of the pathways of bilirubin metabolism, many newborn infants without evidence of hemolysis become jaundiced.
Bilirubin is produced by the catabolism of hemoglobin in the reticuloendothelial system. The tetrapyrrole ring of heme is cleaved by heme oxygenase to form equivalent quantities of biliverdin and carbon monoxide. Because no other biologic source of carbon monoxide exists, the excretion of this gas is stoichiometrically identical to the production of bilirubin. Biliverdin is converted to bilirubin by biliverdin reductase. One gram of hemoglobin produces 35 mg of bilirubin. Sources of bilirubin other than circulating hemoglobin represent 20% of bilirubin production; these sources include inefficient (shunt) hemoglobin production and lysis of precursor cells in bone marrow. Compared with adults, newborns have a twofold to threefold greater rate of bilirubin production (6 to 10 mg/kg/24 hr vs. 3 mg/kg/24 hr). This increased production is caused in part by an increased RBC mass (higher hematocrit) and a shortened erythrocyte life span of 70 to 90 days compared with the 120-day erythrocyte life span in adults.
Bilirubin produced after hemoglobin catabolism is lipid soluble and unconjugated and reacts as an indirect reagent in the van den Bergh test. Indirect-reacting, unconjugated bilirubin is toxic to the central nervous system and is insoluble in water, limiting its excretion. Unconjugated bilirubin binds to albumin on specific bilirubin binding sites; 1 g of albumin binds 8.5 mg of bilirubin in a newborn. If the binding sites become saturated or if a competitive compound binds at the site, displacing bound bilirubin, free bilirubin becomes available to enter the central nervous system. Organic acids such as free fatty acids and drugs such as sulfisoxazole can displace bilirubin from its binding site on albumin.
Bilirubin dissociates from albumin at the hepatocyte and becomes bound to a cytoplasmic liver protein Y (ligandin). Hepatic conjugation results in the production of bilirubin diglucuronide, which is water soluble and capable of biliary and renal excretion. The enzyme glucuronosyltransferase represents the rate-limiting step of bilirubin conjugation. The concentrations of ligandin and glucuronosyltransferase are lower in newborns, particularly in premature infants, than in older children.
Conjugated bilirubin gives a direct reaction in the van den Bergh test. Most conjugated bilirubin is excreted through the bile into the small intestine and eliminated in the stool. Some bilirubin may undergo hydrolysis back to the unconjugated fraction by intestinal glucuronidase, however, and may be reabsorbed (enterohepatic recirculation). In addition, bacteria in the neonatal intestine convert bilirubin to urobilinogen and stercobilinogen, which are excreted in urine and stool and usually limit bilirubin reabsorption. Delayed passage of meconium, which contains bilirubin, also may contribute to the enterohepatic recirculation of bilirubin.
Bilirubin is produced in utero by the normal fetus and by the fetus affected by erythroblastosis fetalis. Indirect, unconjugated, lipid-soluble fetal bilirubin is transferred across the placenta and becomes conjugated by maternal hepatic enzymes. The placenta is impermeable to conjugated water-soluble bilirubin. Fetal bilirubin levels become only mildly elevated in the presence of severe hemolysis, but may increase when hemolysis produces fetal hepatic inspissated bile stasis and conjugated hyperbilirubinemia. Maternal indirect (but not direct) hyperbilirubinemia also may increase fetal bilirubin levels.
Etiology of Indirect Unconjugated Hyperbilirubinemia
Physiologic jaundice is a common cause of hyperbilirubinemia among newborns. It is a diagnosis of exclusion, made after careful evaluation has ruled out more serious causes of jaundice, such as hemolysis, infection, and metabolic diseases. Physiologic jaundice is the result of many factors that are normal physiologic characteristics of newborns: increased bilirubin production resulting from an increased RBC mass, shortened RBC life span, and hepatic immaturity of ligandin and glucuronosyltransferase. Physiologic jaundice may be exaggerated among infants of Greek and Asian ancestry.
The clinical pattern of physiologic jaundice in term infants includes a peak indirect-reacting bilirubin level of no more than 12 mg/dL on day 3 of life. In premature infants, the peak is higher (15 mg/dL) and occurs later (fifth day). The peak level of indirect bilirubin during physiologic jaundice may be higher in breast milk–fed infants than in formula-fed infants (15 to 17 mg/dL vs. 12 mg/dL). This higher level may be partly a result of the decreased fluid intake of infants fed breast milk. Jaundice is unphysiologic or pathologic if it is clinically evident on the first day of life, if the bilirubin level increases more than 0.5 mg/dL/hr, if the peak bilirubin is greater than 13 mg/dL in term infants, if the direct bilirubin fraction is greater than 1.5 mg/dL, or if hepatosplenomegaly and anemia are present.
Crigler-Najjar syndrome is a serious, rare, autosomal recessive, permanent deficiency of glucuronosyltransferase that results in severe indirect hyperbilirubinemia. Type II responds to enzyme induction by phenobarbital, producing an increase in enzyme activity and a reduction of bilirubin levels. Type I does not respond to phenobarbital and manifests as persistent indirect hyperbilirubinemia, often leading to kernicterus. Gilbert disease is caused by a mutation of the promoter region of glucuronosyltransferase and results in a mild indirect hyperbilirubinemia. In the presence of another icterogenic factor (hemolysis), more severe jaundice may develop.
Breast milk jaundice may be associated with unconjugated hyperbilirubinemia without evidence of hemolysis during the first to second week of life. Bilirubin levels rarely increase to more than 20 mg/dL. Interruption of breastfeeding for 1 to 2 days results in a rapid decline of bilirubin levels, which do not increase significantly after breastfeeding resumes. Breast milk may contain an inhibitor of bilirubin conjugation or may increase enterohepatic recirculation of bilirubin because of breast milk glucuronidase.
Jaundice on the first day of life is always pathologic, and immediate attention is needed to establish the cause. Early onset often is a result of hemolysis, internal hemorrhage (cephalhematoma, hepatic or splenic hematoma), or infection (Table 62-1). Infection also is often associated with direct-reacting bilirubin resulting from perinatal congenital infections or from bacterial sepsis.
TABLE 62-1 Etiology of Unconjugated Hyperbilirubinemia
| Hemolysis Present | Hemolysis Absent | |
|---|---|---|
| Common | Physiologic jaundice, breast milk jaundice, internal hemorrhage, polycythemia, infant of diabetic mother | |
| Rare | Mutations of glucuronyl transferase enzyme (Crigler-Najjar syndrome, Gilbert disease), pyloric stenosis, hypothyroidism, immune thrombocytopenia |
Physical evidence of jaundice is observed in infants when bilirubin levels reach 5 to 10 mg/dL (vs. 2 mg/dL in adults). When jaundice is observed, the laboratory evaluation for hyperbilirubinemia should include a total bilirubin measurement to determine the magnitude of hyperbilirubinemia. Bilirubin levels greater than 5 mg/dL on the first day of life or greater than 13 mg/dL thereafter in term infants should be evaluated further with measurement of indirect and direct bilirubin levels, blood typing, Coombs test, complete blood count, blood smear, and reticulocyte count. These tests must be performed before treatment of hyperbilirubinemia with phototherapy or exchange transfusion. In the absence of hemolysis or evidence for either the common or the rare causes of nonhemolytic indirect hyperbilirubinemia, the diagnosis is either physiologic or breast milk jaundice. Jaundice present after 2 weeks of age is pathologic and suggests a direct-reacting hyperbilirubinemia.
Etiology of Direct Conjugated Hyperbilirubinemia
Direct-reacting hyperbilirubinemia (defined as a direct bilirubin level >2 mg/dL or >20% of the total bilirubin) is never physiologic and should always be evaluated thoroughly according to the diagnostic categories (Table 62-2). Direct-reacting bilirubin (composed mostly of conjugated bilirubin) is not neurotoxic to the infant but signifies a serious underlying disorder involving cholestasis or hepatocellular injury. The diagnostic evaluation of patients with direct-reacting hyperbilirubinemia involves the determination of the levels of liver enzymes (aspartate aminotransferase, alkaline phosphatase, alanine aminotransferase, and γ-glutamyl transpeptidase), bacterial and viral cultures, metabolic screening tests, hepatic ultrasound, sweat chloride test, and occasionally liver biopsy. Additionally, the presence of dark urine and gray-white (acholic) stools with jaundice after the second week of life strongly suggests biliary atresia. The treatment of disorders manifested by direct bilirubinemia is specific for the diseases that are listed in Table 62-2. These diseases do not respond to phototherapy or exchange transfusion.
TABLE 62-2 Etiology of Conjugated Hyperbilirubinemia
COMMON
Hyperalimentation cholestasis
CMV infection
Other perinatal congenital infections (TORCH)
Inspissated bile from prolonged hemolysis
Neonatal hepatitis
Sepsis
UNCOMMON
Hepatic infarction
Inborn errors of metabolism (galactosemia, tyrosinemia)
Cystic fibrosis
Biliary atresia
Choledochal cyst
α1-Antitrypsin deficiency
Neonatal iron storage disease
Alagille syndrome (arteriohepatic dysplasia)
Byler disease
CMV, cytomegalovirus; TORCH, toxoplasmosis, other, rubella, cytomegalovirus, herpes simplex.
Kernicterus (Bilirubin Encephalopathy)
Lipid-soluble, unconjugated, indirect bilirubin fraction is toxic to the developing central nervous system, especially when indirect bilirubin concentrations are high and exceed the binding capacity of albumin. Kernicterus results when indirect bilirubin is deposited in brain cells and disrupts neuronal metabolism and function, especially in the basal ganglia. Indirect bilirubin may cross the blood-brain barrier because of its lipid solubility. Other theories propose that a disruption of the blood-brain barrier permits entry of a bilirubin-albumin or free bilirubin–fatty acid complex.
Kernicterus usually is noted when the bilirubin level is excessively high for gestational age. It usually does not develop in term infants when bilirubin levels are less than 20 to 25 mg/dL, but the incidence increases as serum bilirubin levels exceed 25 mg/dL. Kernicterus may be noted at bilirubin levels less than 20 mg/dL in the presence of sepsis, meningitis, hemolysis, asphyxia, hypoxia, hypothermia, hypoglycemia, bilirubin-displacing drugs (sulfa drugs), and prematurity. Other risks for kernicterus in term infants are hemolysis, jaundice noted within 24 hours of birth, and delayed diagnosis of hyperbilirubinemia. Kernicterus has developed in extremely immature infants weighing less than 1000 g when bilirubin levels are less than 10 mg/dL because of a more permeable blood-brain barrier associated with prematurity.
The earliest clinical manifestations of kernicterus are lethargy, hypotonia, irritability, poor Moro response, and poor feeding. A high-pitched cry and emesis also may be present. Early signs are noted after day 4 of life. Later signs include bulging fontanelle, opisthotonic posturing, pulmonary hemorrhage, fever, hypertonicity, paralysis of upward gaze, and seizures. Infants with severe cases of kernicterus die in the neonatal period. Spasticity resolves in surviving infants, who may manifest later nerve deafness, choreoathetoid cerebral palsy, mental retardation, enamel dysplasia, and discoloration of teeth as permanent sequelae. Kernicterus may be prevented by avoiding excessively high indirect bilirubin levels and by avoiding conditions or drugs that may displace bilirubin from albumin. Early signs of kernicterus occasionally may be reversed by immediately instituting an exchange transfusion (see later).
Therapy of Indirect Hyperbilirubinemia
Phototherapy is an effective and safe method for reducing indirect bilirubin levels, particularly when initiated before serum bilirubin increases to levels associated with kernicterus. In term infants, phototherapy is begun when indirect bilirubin levels are between 16 and 18 mg/dL. Phototherapy is initiated in premature infants when bilirubin is at lower levels, to prevent bilirubin from reaching the high concentrations necessitating exchange transfusion. Blue lights and white lights are effective in reducing bilirubin levels.
Under the effects of phototherapy light with maximal irradiance in the 425- to 475-nm wavelength band, bilirubin is transformed into isomers that are water soluble and easily excreted. Unconjugated bilirubin (IX) is in the 4Z, 15Z configuration. Phototherapy causes a photochemical reaction producing the reversible, more water-soluble isomer 4Z, 15E bilirubin IX. This isomer can be excreted easily, bypassing the liver’s conjugation system. Another photochemical reaction results in the rapid production of lumirubin, a more water-soluble isomer than the aforementioned isomer, which does not spontaneously revert to unconjugated native bilirubin and can be excreted in urine.
Complications of phototherapy include an increased insensible water loss, diarrhea, and dehydration. Additional problems are macular-papular red skin rash, lethargy, masking of cyanosis, nasal obstruction by eye pads, and potential for retinal damage. Skin bronzing may be noted in infants with direct-reacting hyperbilirubinemia. Infants with mild hemolytic disease of the newborn occasionally may be managed successfully with phototherapy for hyperbilirubinemia, but care must be taken to follow these infants for the late occurrence of anemia from continued hemolysis.
Exchange transfusion usually is reserved for infants with dangerously high indirect bilirubin levels who are at risk for kernicterus. As a rule of thumb, a level of 20 mg/dL for indirect-reacting bilirubin is the exchange number for infants with hemolysis who weigh more than 2000 g. Asymptomatic infants with physiologic or breast milk jaundice may not require exchange transfusion, unless the indirect bilirubin level exceeds 25 mg/dL. The exchangeable level of indirect bilirubin for other infants may be estimated by calculating 10% of the birth weight in grams: the level in an infant weighing 1500 g would be 15 mg/dL. Infants weighing less than 1000 g usually do not require an exchange transfusion until the bilirubin level exceeds 10 mg/dL.
The exchange transfusion usually is performed through an umbilical venous catheter placed in the inferior vena cava or, if free flow is obtained, at the confluence of the umbilical vein and the portal system. The level of serum bilirubin immediately after the exchange transfusion declines to levels that are about half of those before the exchange; levels rebound 6 to 8 hours later as a result of continued hemolysis and redistribution of bilirubin from tissue stores.
Complications of exchange transfusion include problems related to the blood (transfusion reaction, metabolic instability, or infection), the catheter (vessel perforation or hemorrhage), or the procedure (hypotension or necrotizing enterocolitis [NEC]). Unusual complications include thrombocytopenia and graft-versus-host disease. Continuation of phototherapy may reduce the necessity for subsequent exchange transfusions.
Polycythemia (Hyperviscosity Syndrome)
Polycythemia is an excessively high hematocrit (≥65%) and leads to hyperviscosity that produces symptoms related to vascular stasis, hypoperfusion, and ischemia. As the hematocrit increases from 40% to 60%, there is a small increase in blood viscosity. When the central hematocrit increases to greater than 65%, the blood viscosity begins to increase markedly, and symptoms may appear. Neonatal erythrocytes are less filterable or deformable than adult erythrocytes, which further contributes to hyperviscosity. A central venous hematocrit of 65% or greater is noted in 3% to 5% of infants. Infants at special risk for polycythemia are term and post-term small for gestational age infants, infants of diabetic mothers, infants with delayed cord clamping, and infants with neonatal hyperthyroidism, adrenogenital syndrome, trisomy 13, trisomy 18, trisomy 21, twin-to-twin transfusion syndrome (recipient), and Beckwith-Wiedemann syndrome. In some infants, polycythemia may reflect a compensation for prolonged periods of fetal hypoxia caused by placental insufficiency; these infants have increased erythropoietin levels at birth.
Polycythemic patients appear plethoric or ruddy and may develop acrocyanosis. Symptoms are a result of the increased RBC mass and of vascular compromise. Seizures, lethargy, and irritability reflect abnormalities of microcirculation of the brain, whereas hyperbilirubinemia may reflect the poor hepatic circulation or the increased amount of hemoglobin that is being broken down into bilirubin. Additional problems include respiratory distress and primary pulmonary hypertension of the newborn (PPHN) that result in part from elevated pulmonary vascular resistance. The chest radiograph often reveals cardiomegaly, increased vascular markings, pleural effusions, and interstitial edema. Other problems are NEC, hypoglycemia, thrombocytopenia, priapism, testicular infarction, hemiplegic stroke, and feeding intolerance. Many of these complications also are related to the primary condition associated with polycythemia (small for gestational age infants are at risk for hypoglycemia and PPHN after periods of hypoxia in utero).
Long-term sequelae of neonatal polycythemia relate to neurodevelopmental abnormalities that may be prevented by treatment of symptomatic infants with partial exchange transfusion after birth. A partial exchange transfusion removes whole blood and replaces it with normal saline. The equation used to calculate the volume exchanged is based on the central venous hematocrit because peripheral hematocrits may be falsely elevated:
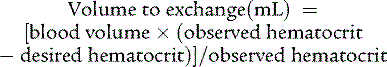
The desired hematocrit is 50%, and the blood volume 85 mL/kg.
COAGULATION DISORDERS
Disorders of coagulation are common in the neonatal period. Hemorrhage during this time may be a result of trauma, inherited permanent deficiency of coagulation factors, transient deficiencies of vitamin K–dependent factors, disorders of platelets, and disseminated intravascular coagulation (DIC) seen in sick newborns with shock or hypoxia. Thrombosis also is a potential problem in the newborn because of developmentally lower circulating levels of antithrombin III, protein C (a vitamin K–dependent protein that inhibits factors VIII and V), and the fibrinolytic system.
Coagulation factors do not pass through the placenta to the fetus, and newborn infants have relatively low levels of the vitamin K–dependent factors II, VII, IX, and X. Contact factors XI and XII, prekallikrein, and kininogen also are lower in newborns than in adults. Fibrinogen (factor I); plasma levels of factors V, VIII, and XIII; and platelet counts are within the adult normal range.
Because of the transient, relative deficiencies of the contact and vitamin K–dependent factors, the partial thromboplastin time (PTT), which is dependent on factors XII, IX, VIII, X, V, II, and I, is prolonged in the newborn period. Preterm infants have the most marked prolongation of the PTT (50 to 80 seconds) compared with term infants (35 to 50 seconds) and older, more mature infants (25 to 35 seconds). The administration of heparin and the presence of DIC, hemophilia, and severe vitamin K deficiency prolong the PTT.
The prothrombin time (PT), which is dependent on factors VII, X, V, II, and I, is a more sensitive test for vitamin K deficiency. The PT is only slightly prolonged in term infants (13 to 20 seconds) compared with preterm infants (13 to 21 seconds) and more mature patients (12 to 14 seconds). Abnormal prolongations of the PT occur with vitamin K deficiency, hepatic injury, and DIC. Levels of fibrinogen and fibrin degradation products are similar in infants and adults. The bleeding time, which reflects platelet function and number, is normal during the newborn period in the absence of maternal salicylate therapy.
Vitamin K is a necessary cofactor for the carboxylation of glutamate on precursor proteins, converting them into the more active coagulation factors II, VII, IX, and X; γ-carboxyglutamic acid binds calcium, which is required for the immediate activation of factors during hemorrhage. There is no congenital deficiency of hepatic synthesis of these precursor proteins, but in the absence of vitamin K their conversion to the active factor is not possible. Levels of protein induced by vitamin K absence increase in vitamin K deficiency and are helpful diagnostic markers; vitamin K administration rapidly corrects the coagulation defects, reducing protein induced by vitamin K absence to undetectable levels.
Although most newborns are born with reduced levels of vitamin K–dependent factors, hemorrhagic complications develop only rarely. Infants at risk for hemorrhagic disease of the newborn have the most profound deficiency of vitamin K–dependent factors, and these factors decline further after birth. Because breast milk is a poor source of vitamin K, breastfed infants are at increased risk for hemorrhage that usually occurs between days 3 and 7 of life. Bleeding usually ensues from the umbilical cord, circumcision site, intestines, scalp, mucosa, and skin, but internal hemorrhage places the infant at risk for fatal complications, such as intracranial bleeding.
Hemorrhage on the first day of life resulting from a deficiency of the vitamin K–dependent factors often is associated with administration to the mother of drugs that affect vitamin K metabolism in the infant. This early pattern of hemorrhage has been seen with maternal warfarin or antibiotic (e.g., isoniazid or rifampin) therapy and in infants of mothers receiving phenobarbital and phenytoin. Bleeding also may occur 1 to 3 months after birth, particularly among breastfed infants. Vitamin K deficiency in breastfed infants also should raise suspicion about the possibility of vitamin K malabsorption resulting from cystic fibrosis, biliary atresia, hepatitis, or antibiotic suppression of the colonic bacteria that produce vitamin K.
Bleeding associated with vitamin K deficiency may be prevented by administration of vitamin K to all infants at birth. Before routine administration of vitamin K, 1% to 2% of all newborns have hemorrhagic disease of the newborn. One intramuscular dose (1 mg) of vitamin K prevents vitamin K–deficiency bleeding. Treatment of bleeding resulting from vitamin K deficiency involves intravenous administration of 1 mg of vitamin K. If severe, life-threatening hemorrhage is present, fresh frozen plasma also should be given. Unusually high doses of vitamin K may be needed for hepatic disease and for maternal warfarin or anticonvulsant therapy.
Clinical Manifestations and Differential Diagnoses of Bleeding Disorders
Bleeding disorders in a newborn may be associated with cutaneous bleeding, such as cephalhematoma, subgaleal hemorrhage, ecchymosis, and petechiae. Facial petechiae are common in infants born by vertex presentation, with or without a nuchal cord, and usually are insignificant. Mucosal bleeding may appear as hematemesis, melena, or epistaxis. Internal hemorrhage results in organ-specific dysfunction, such as seizures, accompanied by intracranial hemorrhage. Bleeding from venipuncture or heel-stick sites, circumcision sites, or the umbilical cord also is common.
The differential diagnosis depends partly on the clinical circumstances associated with the hemorrhage. In a sick newborn, the differential diagnosis should include DIC, hepatic failure, and thrombocytopenia. Thrombocytopenia in an ill neonate may be secondary to consumption by trapping of platelets in a hemangioma (Kasabach-Merritt syndrome) or may be associated with perinatal, congenital, or bacterial infections; NEC; thrombotic endocarditis; PPHN; organic acidemia; maternal preeclampsia; or asphyxia. Thrombocytopenia also may be due to peripheral washout of platelets after an exchange transfusion. Treatment of a sick infant with thrombocytopenia should be directed at the underlying disorder, supplemented by infusions of platelets, blood, or both.
The etiology of DIC in a newborn includes hypoxia, hypotension, asphyxia, bacterial or viral sepsis, NEC, death of a twin while in utero, cavernous hemangioma, nonimmune hydrops, neonatal cold injury, neonatal neoplasm, and hepatic disease. The treatment of DIC should be focused primarily on therapy for the initiating or underlying disorder. Supportive management of consumptive coagulopathy involves platelet transfusions and factor replacement with fresh frozen plasma. Heparin and factor C concentrate should be reserved for infants with DIC who also have thrombosis.
Disorders of hemostasis in a well child are not associated with systemic disease in a newborn but reflect coagulation factor or platelet deficiency. Hemophilia initially is associated with cutaneous or mucosal bleeding and no systemic illness. If bleeding continues, hypovolemic shock may develop. Bleeding into the brain, liver, or spleen may result in organ-specific signs and shock.
In a well child, thrombocytopenia may be part of a syndrome such as Fanconi anemia syndrome (involving hypoplasia and aplasia of the thumb), radial aplasia-thrombocytopenia syndrome (thumbs present), or Wiskott-Aldrich syndrome. Various maternal drugs also may reduce the neonatal platelet count without producing other adverse effects. These drugs include sulfonamides, quinidine, quinine, and thiazide diuretics.
The most common causes of thrombocytopenia in well newborns are transient isoimmune thrombocytopenia and transient neonatal thrombocytopenia in well infants born to mothers with idiopathic thrombocytopenic purpura (ITP). Isoimmune thrombocytopenia is caused by antiplatelet antibodies produced by the HPLA1-negative mother after her sensitization to specific paternal platelet antigen (HPA-1a and HPA-5b represent 85% and 10% of cases) expressed on the fetal platelet. The incidence is 1:1000 to 1:2000 births. This response to maternal-sensitized antibodies that produce isoimmune thrombocytopenia is analogous to the response that produces erythroblastosis fetalis. The maternal antiplatelet antibody does not produce maternal thrombocytopenia, but after crossing the placenta this IgG antibody binds to fetal platelets that are trapped by the reticuloendothelial tissue, resulting in thrombocytopenia. Infants with thrombocytopenia produced in this manner are at risk for development of petechiae, purpura, and intracranial hemorrhage (an incidence of 10% to 15%) before or after birth. Vaginal delivery may increase the risk of neonatal bleeding; cesarean section may be indicated.
Specific treatment for severe thrombocytopenia (<20,000 platelets/mm3) or significant bleeding is transfusion of ABO-compatible and RhD-compatible, HPA-1a-negative and HPA-5b-negative maternal platelets. Because the antibody in isoimmune thrombocytopenia is directed against the fetal rather than the maternal platelet, plateletpheresis of the mother yields sufficient platelets for performing a platelet transfusion to treat the affected infant. After one platelet transfusion, the infant’s platelet count dramatically increases and usually remains in a safe range. Without treatment, thrombocytopenia resolves during the first month of life as the maternal antibody level declines. Treatment of the mother with intravenous immunoglobulin or the thrombocytopenic fetus with intravascular platelet transfusion (cordocentesis) is also effective. Cesarean section reduces the risk of intracranial hemorrhage.
Neonatal thrombocytopenia in infants born to women with ITP also is a result of placental transfer of maternal IgG antibodies. In ITP, these autoantibodies are directed against all platelet antigens, and mother and newborn may have low platelet counts. The risks of hemorrhage in an infant born to a mother with ITP may be lessened by cesarean section and by treatment of the mother with corticosteroids.
Treatment of an affected infant born to a mother with ITP may involve prednisone and intravenous immunoglobulin. In an emergency, random donor platelets may be used and may produce a transient increase in the infant’s platelet count. Thrombocytopenia resolves spontaneously during the first month of life as maternal-derived antibody levels decline. Elevated levels of platelet-associated antibodies also have been noted in thrombocytopenic infants with sepsis and thrombocytopenia of unknown cause who were born to mothers without demonstrable platelet antibodies.
The laboratory evaluation of an infant (well or sick) with bleeding must include a platelet count, blood smear, and evaluation of PTT and PT. Isolated thrombocytopenia in a well infant suggests immune thrombocytopenia. Laboratory evidence of DIC includes a markedly prolonged PTT and PT (minutes rather than seconds), thrombocytopenia, and a blood smear suggesting a microangiopathic hemolytic anemia (burr or fragmented blood cells). Further evaluation reveals low levels of fibrinogen (<100 mg/dL) and elevated levels of fibrin degradation products. Vitamin K deficiency prolongs the PT more than the PTT, whereas hemophilia resulting from factors VIII and IX deficiency prolongs only the PTT. Specific factor levels confirm the diagnosis of hemophilia.