 CHAPTER 73 Lymphocyte Disorders
CHAPTER 73 Lymphocyte Disorders
Disorders that affect lymphocyte development or function result in significant immunodeficiency because lymphocytes provide antigen specificity and memory responses. Lymphocytes develop from hematopoietic stem cells through a series of stages that culminate in a common lymphoid progenitor, which gives rise to T lymphocytes, B lymphocytes, and natural killer (NK) lymphocytes. B cells complete their development in the bone marrow, and T cells develop in the thymus from bone marrow–derived precursors (Fig. 73-1). Isolated B-cell disorders result in antibody deficiency diseases, whereas T-cell disorders usually cause combined immunodeficiency because they are necessary for cell-mediated immunity, immunity to intracellular pathogens, and antibody synthesis by B cells. NK cells are an important component of the innate immune response and can kill virus-infected cells and tumor cells. Antibodies can enhance NK cell function by antibody-mediated cellular cytotoxicity.
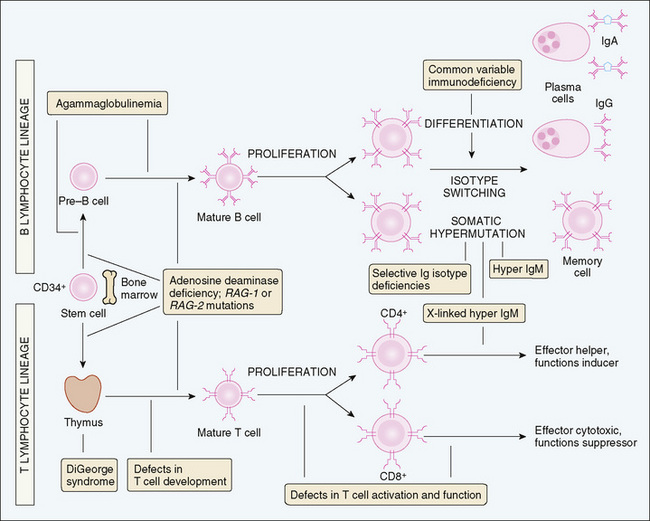
FIGURE 73-1 Sites of cellular abnormalities in congenital immunodeficiencies. In primary immunodeficiency diseases, the maturation or activation of B or T lymphocytes may be blocked at different stages. B, B lymphocyte; T, T lymphocyte.
(Adapted from Abbas AK, Lichtman AH, Pober JS: Cellular and molecular immunology, 3rd ed, Philadelphia, Saunders, 1997.)
ETIOLOGY AND CLINICAL MANIFESTATIONS
Antibody Deficiency Diseases
B cells synthesize antibodies that can kill pathogens in conjunction with complement proteins, facilitate uptake of pathogens by phagocytic cells, and neutralize toxins secreted by pathogens. Disorders of B cells result in an increased susceptibility to infections by encapsulated bacteria because these pathogens resist uptake by phagocytic cells and require antibodies for their clearance. A variety of defects can affect the development or function of B cells.
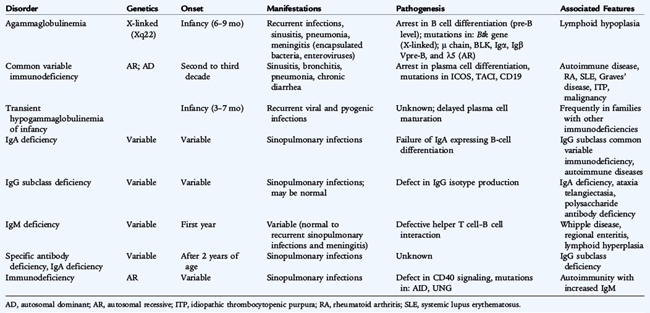
Agammaglobulinemia results from the absence of B cells with subsequent absence or severe decrease in immunoglobulin levels and a total absence of specific antibody. X-linked agammaglobulinemia affects males and is characterized by a profound deficiency of B cells, severe hypogammaglobulinemia, and absence of lymphoid tissue (Table 73-1; see Fig. 73-1). The defect is caused by mutations in a gene encoding for the tyrosine kinase Btk on chromosome Xq22 that is involved in signaling via the pre–B-cell receptor and the B-cell antigen receptor. The B-cell antigen receptor complex consists of an immunoglobulin molecule in association with several transmembrane and cytoplasmic molecules that function in signal transduction. Defects in any of the genes that encode for structural or signaling molecules associated with the pre–B-cell and B-cell receptors can lead to agammaglobulinemia. These gene defects are inherited in an autosomal recessive manner and include mutations in the μ heavy chain gene on chromosomes 14 and λ5, and Vpre-B surrogate light chains, Igα, Igβ, and BLNK. X-linked agammaglobulinemia is more common than the autosomal recessive forms because, in affected males, only one copy of the gene needs to be defective to express the disease. Other unknown gene defects can also lead to agammaglobulinemia.
Patients with agammaglobulinemia usually present during the first 6 to 12 months of life, as maternally derived antibodies wane, although some patients present with symptoms at 3 to 5 years of age. These patients develop infections with Streptococcus pneumoniae, Haemophilus influenzae type b, Staphylococcus aureus, and Pseudomonas, organisms for which antibody is an important opsonin. They also have increased susceptibility to giardiasis and enteroviral infections, leading to chronic enteroviral meningoencephalitis and vaccine–associated poliomyelitis (if immunized with oral live, attenuated poliovirus vaccine).
Common variable immunodeficiency (CVID) is a heterogeneous disorder characterized by hypogammaglobulinemia developing after an initial period of normal immune function, most commonly in the second and third decades of life (see Table 73-1). Serum IgG levels are less than 500 mg/dL (usually <300 mg/dL) with IgA levels less than 10 mg/dL and low IgM levels. Antibody titers to protein antigens, such as tetanus and diphtheria, and to polysaccharide antigens, such as pneumococcus, are absent. T-cell function is variable; patients may have decreased numbers of CD4 T cells, decreased lymphocyte proliferation to mitogens and antigens, or normal T-cell numbers and function. B cells may be present at low or normal numbers. Patients exhibit normal-sized or enlarged tonsils and lymph nodes and may have splenomegaly. They are susceptible to frequent respiratory tract infections; bronchiectasis; autoimmune diseases such as hemolytic anemia, thrombocytopenia, and neutropenia; gastrointestinal disease with malabsorption, chronic diarrhea, liver dysfunction, and Helicobacter pylori infection; granulomatous disease; and cancer, especially lymphoma. CVID is observed frequently in families with IgA deficiency. The gene defects leading to the majority of cases of CVID are unknown. Some patients have a defect in the gene encoding for ICOS, the “inducible costimulator” on activated T cells, TACI (transmembrane activator and calcium-modulating cyclophilin ligand interactor), or CD19. These patients have normal T-cell numbers and function but have a decrease in naive and memory B cells. Some patients who were thought to have CVID were found by genetic studies to have X-linked agammaglobulinemia, X-linked lymphoproliferative disease, or hyper-IgM syndrome. It is important to exclude these disorders and other causes of hypogammaglobulinemia, such as hypogammaglobulinemia associated with thymoma or secondary to immunoglobulin loss (intestinal loss), before making the diagnosis of CVID.
Selective IgA deficiency is defined as serum IgA levels less than 10 mg/dL with normal or increased levels of other immunoglobulins. The diagnosis cannot be confirmed until the patient is at least 4 years of age when IgA levels should reach adult levels. Selective IgA deficiency occurs in approximately 1 in 500 individuals. Many patients with selective IgA deficiency are asymptomatic. In others, it is associated with recurrent sinopulmonary infections, IgG2 subclass deficiency, specific antibody deficiency, food allergy, autoimmune disease, or celiac disease. IgA deficiency occurs in families, suggesting autosomal inheritance. It also is seen in families with CVID.
IgG subclass deficiency occurs when the level of antibodies in one or more of the four IgG subclasses is selectively decreased while total IgG levels are normal or only slightly decreased. The IgG2 subclass is the most prevalent of the IgG subclasses. Deficiency in IgG1 usually is reflected as hypogammaglobulinemia. IgG2 subclass deficiency is the most common of the IgG subclass deficiencies and often is associated with IgA deficiency, ataxia-telangiectasia, and reduced capacity to produce antibody against polysaccharide antigens. IgG3 subclass deficiency also has been associated with recurrent infections. Absence of IgG4 is not considered to be clinically significant. An inability to synthesize specific antibody titers to protein or polysaccharide antigens is the best marker of IgG subclass deficiency associated with recurrent infections and requiring therapy.
Transient hypogammaglobulinemia of infancy is a temporary condition characterized by delayed immunoglobulin production. The pathogenesis of this disorder is unknown but is thought to result from a prolongation of the physiologic hypogammaglobulinemia of infancy. The immunoglobulin nadir at 6 months of age is accentuated, with immunoglobulin levels less than 200 mg/dL. Immunoglobulin levels remain diminished throughout the first year of life and usually increase to normal, age-appropriate levels, generally by 2 to 4 years of age. Occasionally, low levels persist longer. The incidence of sinopulmonary infection is increased in some patients. The diagnosis is supported by normal levels of both B and T cells, and normal antibody responses to protein antigens such as diphtheria and tetanus toxoids. The transient nature of this disorder cannot be confirmed, however, until immunoglobulin levels return to normal ranges.
Antibody deficiency syndrome is characterized by recurrent infections with normal immunoglobulin levels and normal lymphocyte numbers and subsets, but an inability to synthesize specific antibody to polysaccharide antigens, such as to the 23-valent pneumococcal vaccine. The pathogenesis of this disorder is unknown. Lack of specific antibody titers explains the recurrent infections and justifies therapy.
Combined Immunodeficiency Diseases
Disorders that affect T-cell development or function usually result in combined immunodeficiency because T cells provide necessary signals for B-cell differentiation. Hyper-IgM syndrome is usually classified under antibody deficiency diseases or B-cell disorders. However, the most common form of hyper-IgM, X-linked hyper-IgM, is a combined immunodeficiency disease with deficient T cell function. Hyper-IgM syndrome is characterized by a failure of immunoglobulin isotype switching from IgM and IgD to IgG, IgA, or IgE, and a lack of memory responses. Affected patients have normal or elevated serum levels of IgM with low or absent levels of IgG, IgA, and IgE. Immunoglobulin isotype switching allows a B cell to maintain antigen specificity while altering immunoglobulin function and is directed by cytokines, requiring direct interaction between CD40 ligand on a CD4 T cell and CD40 on a B cell (Fig. 73-2). Subsequent signal transduction via CD40 activates several signaling molecules and transcription factors, including nuclear factor κB (NF-κB) and two enzymes, activation-induced cytidine deaminase (AID), and uracil DNA-glycosylase (UNG). In addition, signaling via CD40 on B cells and other antigen-presenting cells leads to upregulation of costimulatory molecules important for promoting T cell differentiation.
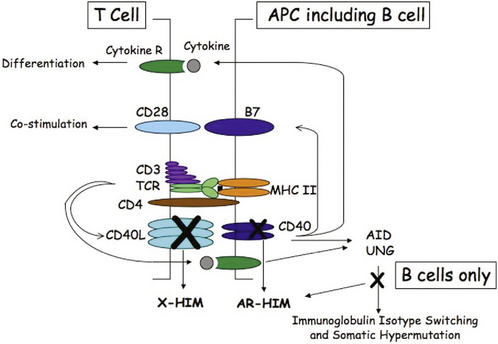
FIGURE 73-2 Schematic representation of the interaction between a CD4 T cell and a B cell. T cell activation follows T cell receptor (TCR) recognition of peptide antigen presented via MHC class II molecules resulting in CD40 ligand (CD40L) expression and cytokine synthesis. CD40L stimulates the B cell via CD40, resulting in expression of costimulatory molecules (B7) that are important in T cell priming and cytokine synthesis that drives T cell differentiation. CD40 signaling and signaling by cytokines in B cells activate activation-induced cytidine deaminase (AID) and uracil DNA-glycosylase (UNG) to promote immunoglobulin isotype switching and somatic hypermutation. Defects in CD40L cause X-linked hyper-IgM (X-HIM), and defects in CD40, AID, or UNG cause autosomal recessive hyper-IgM (AR-HIM). Defects in either CD40L or CD40 affect T cell costimulation and priming, leading to T cell defects, whereas defects in AID and UNG maintain normal T cell costimulation and function.
X-linked hyper-IgM results from defects in the CD40 ligand gene, inhibiting the capacity of the ligand to bind to CD40. As a result, no signal reaches the B cell for immunoglobulin isotype switching or antigen-presenting cells for upregulation of costimulatory molecules necessary for T-cell priming. Defects in CD40, inherited in an autosomal recessive manner, lead to a similar immunodeficiency due to a lack of signaling via CD40 (Fig. 73-2 and Table 73-2). Other forms of autosomal recessive hyper-IgM result from defects in B-cell AID or UNG and present with a failure of immunoglobulin isotype switching without any abnormality in T-cell function. These forms of hyper-IgM are an antibody deficiency disease and not a combined immunodeficiency disease (see Table 73-1). Patients with defects in CD40 ligand or CD40 have small lymph nodes with no germinal centers, whereas patients with defects in AID or UNG have large germinal centers and lymphadenopathy.
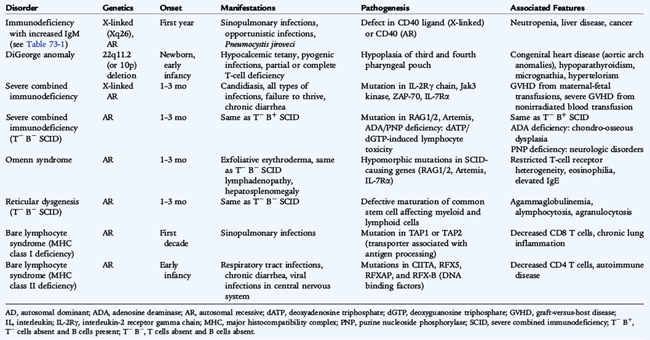
All of these patients have increased susceptibility to sinopulmonary infections; however, patients with defects in CD40 ligand or CD40 also are susceptible to opportunistic infections, such as P. jiroveci (carinii) and Cryptosporidium parvum. Autosomal recessive forms of hyper-IgM appear to increase susceptibility to autoimmune disease. The hyper-IgM phenotype is also found in an X-linked disorder associated with ectodermal dysplasia, resulting from the defects in the gene encoding the NF-κB essential modulator (NEMO) and results in low IgG, IgA, and IgE levels with normal or elevated IgM levels. Patients with defects in NEMO are susceptible to a wider spectrum of infectious organisms, especially meningitis and infection with atypical mycobacteria, because NF-κB signaling is important for several functions of both innate and adaptive immune systems.
Severe combined immunodeficiency (SCID) is characterized by a profound lack of T-cell numbers or function and B-cell dysfunction resulting from absence of B cells from the gene defect itself or secondary to lack of T-cell function (see Fig. 73-1). T cells develop from bone marrow–derived precursors in the thymus (see Fig. 73-2), where they undergo several stages of development characterized by recombination of the variable region of T-cell antigen receptor chains α and β (or γ and δ). In addition, thymocytes differentiate into CD4 or CD8 T cells if they interact with major histocompatibility complex (MHC) class II or class I molecules, respectively. The T-cell antigen receptor recognizes peptide fragments only from antigens that are presented via MHC molecules: CD4 T cells by MHC class II molecules and CD8 T cells by MHC class I molecules. Because the variable region of the T-cell antigen receptor rearranges randomly to provide as much variability as possible, not all T-cell antigen receptors can interact with the MHC molecules present in the individual. A process of positive selection occurs in the thymus to select thymocytes with antigen receptors that can interact with the expressed MHC molecules (see Fig. 73-2), while all other thymocytes in the thymus die. Some of the positively selected thymocytes have antigen receptors, however, that recognize self-antigens presented by MHC molecules in the thymus. These cells are deleted in the thymus by a process of negative selection. Positive selection and negative selection in the thymus ensure that the mature T cells that leave the thymus for the peripheral lymphoid tissues can function with the individual’s MHC molecules and do not mount autoimmune responses. SCID can result from any specific genetic mutation that interferes with T cell development in the thymus or T cell function in the periphery.
X-linked SCID, the most common form, is caused by mutations in the gene on chromosome Xq13.1 coding for the common gamma chain of the interleukin -2 (IL-2), IL-4, IL-7, IL-9, IL-15, and IL-21 receptors. Affected patients have no T cells or NK cells in the peripheral blood but have normal numbers of B cells. Immunoglobulin levels are low or undetectable. Lymph nodes and tonsils are absent. The defect in T cell and NK cell development results from a failure of signaling via the IL-7 and IL-15 receptors, respectively. Defects in the IL-7 receptor alpha chain also result in absent T cells but normal numbers of B cells and NK cells, whereas IL-15 is necessary for NK cell development.
Autosomal recessive SCID results from defects in signaling molecules, such as Janus tyrosine kinase 3 (Jak3), which signals downstream of the common gamma chain, or ZAP-70 kinase, necessary for CD4 T-cell signaling. In Jak3 deficiency, T cells and NK cells are absent, whereas nonfunctional B cells are present in normal numbers. In ZAP-70 deficiency, there is a marked decrease in CD8 T cells with normal numbers of nonfunctional CD4 T cells.
Recombination of the variable regions of the T-cell receptor and immunoglobulin molecules requires two genes called recombinase activating gene (RAG) 1 and 2 as well as other genes important in DNA excision and repair such as Artemis. Defects in any of the genes that abolish its function result in autosomal recessive SCID with no T cells or B cells present. Mutations in RAG1, RAG2, or Artemis that preserve limited function result in Omenn syndrome, a variant form of SCID that is characterized by exfoliative erythroderma, lymphadenopathy, hepatosplenomegaly, marked eosinophilia, elevated serum IgE, and impaired T-cell function. Patients with Omenn syndrome have T cells in the periphery, but these T cells have a limited repertoire.
Bare lymphocyte syndrome results from defects in transcription factors that regulate expression of class II molecules or genes that affect transport of antigen peptides, which leads to absence of either MHC class I or MHC class II molecules. Lymphoid tissue and B cells may be present in normal amounts, but CD4 T cells are decreased or absent in class II deficiency, whereas CD8 cells are decreased or absent in class I deficiency. Some patients may have normal numbers of CD4 or CD8 T cells, but none of the T cells are functional because peptide antigens cannot be presented to T cells.
Deficiencies in adenosine deaminase (ADA) and purine nucleoside phosphorylase, two enzymes involved in the purine salvage pathway, also result in SCID. These deficiencies are caused by mutations in the ADA gene on chromosome 20q12-q13.1 or the purine nucleoside phosphorylase gene on chromosome 14q13. Accumulation of nucleoside substrates or their metabolic products in the plasma and urine is toxic to lymphocytes. T cells are particularly sensitive to the accumulation of toxic purine metabolites, especially deoxyadenosine triphosphate and deoxyadenosine, which results in absent or markedly reduced T-cell function, often with absent B cells or B-cell function. Most patients exhibit severe infection early in life, although the diagnosis in patients with partial immune function is not established until after 5 years of age or, occasionally, in adulthood. Patients with late-onset diagnosis are generally lymphopenic; they may have B cells and normal total immunoglobulin levels but little functional antibody (Nezelof syndrome). All patients with ADA or purine nucleoside phosphorylase deficiency SCID have lymphopenia and loss of immune function over time.
Clinical manifestations of SCID include failure to thrive, severe bacterial infection in the first month of life, chronic candidiasis, infection with P. jiroveci (carinii) and other opportunistic organisms, and intractable diarrhea. Patients often have skin disease similar to eczema, possibly related to graft-versus-host disease (GVHD) from engraftment of maternal lymphocytes, which is not usually fatal. Patients with SCID are extremely susceptible to fatal GVHD, however, from lymphocytes in blood transfusions. Patients with T-cell disorders always should receive irradiated blood products.
DiGeorge syndrome is the classic example of T-cell deficiency and is the result of dysmorphogenesis of the third and fourth pharyngeal pouches, resulting in hypoplasia of the thymus required for T-cell maturation. Most, but not all, patients with DiGeorge syndrome have a field defect on chromosome 22q11.2, including the velocardiofacial syndrome and CATCH 22 syndrome (cardiac anomalies, abnormal facies, thymic hypoplasia, cleft palate, and hypocalcemia). DiGeorge syndrome is classically characterized by hypocalcemic tetany, conotruncal and aortic arch anomalies, and increased infections. The diagnosis of CATCH 22 is established by fluorescent in situ hybridization with a DNA probe to detect deletions in chromosome 22q11.2. Most patients have partial immune defects with low T-cell numbers and function that improve with age. Severe T-cell deficiency is rare, but it results in SCID.
Wiskott-Aldrich syndrome is an X-linked disorder characterized by thrombocytopenia, eczema, defects in cell-mediated and humoral immunity, and a predisposition to lymphoproliferative disease (Table 73-3). It is caused by mutations of the gene on chromosome Xp11.22 coding for the 53-kD Wiskott-Aldrich syndrome protein, expressed in lymphocytes, platelets, and monocytes. Deficiency of this protein results in elevated levels of IgE and IgA, decreased IgM, poor responses to polysaccharide antigens, waning T-cell function, and profound thrombocytopenia. Opportunistic infections and autoimmune cytopenias become problematic in older children. Isolated X-linked thrombocytopenia also results from mutations of the identical gene. One third of patients with Wiskott-Aldrich syndrome die as a result of hemorrhage, and two thirds die as a result of recurrent infection caused by bacteria, cytomegalovirus, P. jiroveci (carinii), or herpes simplex virus. Stem cell transplantation has corrected the immunologic and hematologic problems in some patients.
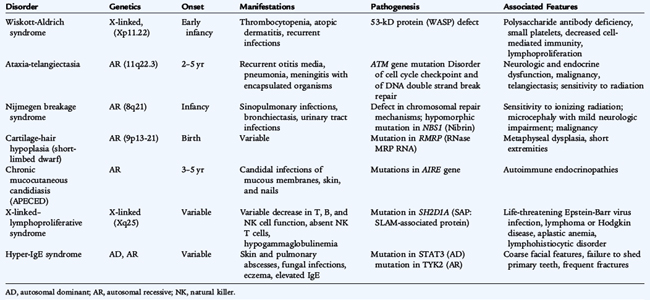
Ataxia-telangiectasia is a syndrome caused by the ATM (ataxia-telangiectasia, mutated) gene on chromosome 11q22.3, which codes for a phosphatidylinositol-3 kinase (see Table 73-3). Patients have cutaneous and conjunctival telangiectasias and progressive cerebellar ataxia with degeneration of Purkinje cells. IgA deficiency, IgG2 subclass deficiency of variable severity, low IgE levels, and variably depressed T-cell function may be seen. The normal function of the ATM gene is not clear but it seems to be involved in detecting DNA damage, blocking cell growth division until the damage is repaired, or both. Ataxia-telangiectasia cells are exquisitely sensitive to irradiation. Leukemias, lymphomas, and diabetes also may be present. Sexual maturation is delayed. There is no uniformly effective therapy, but antimicrobial therapy and intravenous immunoglobulin (IVIG) replacement therapy may be helpful.
Chronic mucocutaneous candidiasis (autoimmune-polyendocrinopathy-candidiasis-ectodermal dystrophy [APECED]) is characterized by chronic or recurrent candidal infections of the mucous membranes, skin, and nails (see Table 73-3). There is normal antibody production but significantly decreased or absent lymphocyte proliferation and delayed skin reactivity to Candida. Patients usually do not respond to topical antifungal therapy and must be treated with oral antifungal agents. In most patients, an autoimmune endocrine disorder, such as hypoparathyroidism and Addison disease, develops by early adulthood. Other autoimmune disorders have been reported. The insidious onset requires the need for frequent evaluation for autoimmune endocrine disorders. This disease results from a defect in the gene for the transcription factor autoimmune regulator (AIRE), which is necessary for expression of peripheral tissue antigens in the thymus, resulting in tolerance to these tissues in normal individuals.
Patients with X-linked lymphoproliferative disease (see Table 73-3) have a defect in immune responsiveness to Epstein-Barr virus (EBV). Boys with this disease are normal until infected with EBV, which is acutely fatal in 80% of patients. The disease is caused by a mutation in the gene called SH2D1A at chromosome Xq25, which codes for an adapter protein that normally inhibits signal transduction in proliferating T cells. With EBV infection, the mutation results in extensive expansion of CD8 T cells, hepatic necrosis, and death. Boys who survive initial EBV infection have significant hypogammaglobulinemia and are at high risk for developing aplastic anemia and lymphoma. Treatment of the acute EBV infection with prednisone and acyclovir may be helpful; VP-16 and anti-CD20 monoclonal antibody also may be of use. IVIG for hypogammaglobulinemia is indicated. Stem cell transplantation can prevent disease progression and provide a cure.
Hyper-IgE syndrome is characterized by markedly elevated serum IgE levels, a rash that resembles atopic dermatitis, eosinophilia, and staphylococcal abscesses of the skin, lungs, joints, and viscera (see Table 73-3). Infections with H. influenzae type b, Candida, and Aspergillus also may occur. These patients have coarse facial features, develop osteopenia, and may have giant pneumatoceles in the lungs after staphylococcal pneumonias. Although serum IgG, IgA, and IgM levels are normal, humoral immune responses to specific antigens are reduced, as is cell-mediated immunity. Long-term treatment with antistaphylococcal medications is indicated, and immunoglobulin replacement therapy may be helpful. Most patients have an autosomal dominant form of inheritance, whereas some patients appear to have an autosomal recessive inheritance. Defects in the TYK2 gene have recently been found in a patient with an autosomal recessive hyper-IgE syndrome, and defects in the STAT3 (signal transducer and activator of transcription 3) gene were identified in one with autosomal dominant hyper-IgE syndrome. Both of these genes are involved in IL-6 signaling. Patients with STAT3 defects have a deficiency of Th17 cells, proinflammatory cells that require IL-6 for their differentiation, secrete IL-17, and appear to be involved in immunity to bacterial and fungal pathogens as well as in the pathogenesis of autoimmune disease.
TREATMENT
The approach to therapy of lymphocyte disorders depends on the diagnosis, clinical findings, and laboratory findings (Table 73-4). When immunodeficiency is suspected, and while the evaluation is in process, all blood products need to be irradiated and negative for cytomegalovirus. Lymphocytes present in blood products can cause fatal GVHD in patients with SCID. Cytomegalovirus infection can be fatal in an immunodeficiency patient undergoing stem cell transplantation. Live virus vaccines should be withheld from patients and household members until a diagnosis is established.
TABLE 73-4 General Management of Patients with Immunodeficiency
Infections should be treated with appropriate antibiotics. Prevention of recurrent infections provides a better quality of life and decreases possible consequences. Patients with milder forms of antibody deficiency diseases may benefit from vaccination with protein-conjugate vaccines to H. influenzae type b and S. pneumoniae, assaying postvaccination titers at least 1 month later. These vaccines are administered routinely to young children; however, older children and adults with antibody deficiency syndrome also should receive them to generate protective antibody levels. Other microorganisms, including nonvaccine S. pneumoniae serotypes, can cause infections in susceptible patients. In addition to immunization, antibiotic prophylaxis or IVIG may prevent recurrent infections.
Antibiotic prophylaxis can be provided with once-daily administration of trimethoprim-sulfamethoxazole or amoxicillin at one half of the total daily therapeutic dose, especially if the patient does not have a severe antibody deficiency and has not had severe or complicated infections. Therapy with antibiotics may be complicated by the development of immediate, autoimmune, immune complex, and delayed-type hypersensitivity drug reactions. Alternating prophylactic antibiotics on a monthly basis may help reduce the incidence of drug-resistant microorganisms.
Immunoglobulin replacement therapy, intravenously or subcutaneously, is a lifesaving therapy for patients with severe antibody deficiency diseases. It provides passive immunity against common microorganisms and reduces the frequency and severity of infection in most patients with antibody deficiency diseases. Immunoglobulin replacement therapy is indicated for patients with agammaglobulinemia, hyper-IgM syndrome, other antibody deficiency diseases and combined immunodeficiency diseases; for patients with infections requiring hospitalization, especially in intensive care units, or with infections that affect growth as well as hearing or speech development; and for patients who have failed antibiotic prophylaxis. Immunoglobulin replacement therapy is usually administered at a total monthly dose of 400 to 600 mg/kg of body weight administered every 3 to 4 weeks intravenously, or every 1 to 2 weeks subcutaneously by infusion pump. IVIG therapy should be monitored by regularly measuring trough immunoglobulin levels, antibody titers to H. influenzae type b, and, most importantly, the patient’s clinical course. Patients who continue to have recurrent infections, especially in the last week before the administration of IVIG, may require higher doses or more frequent administration. A combination of prophylactic antibiotics and immunoglobulin replacement therapy may be indicated in patients who continue to have recurrent infections. Other causes of recurrent infections should be reconsidered, however. Complications of IVIG therapy include transfusion reactions with chills, fever, and myalgias. They usually can be prevented in subsequent infusions by pretreatment with an antihistamine and an antipyretic and by a slower rate of infusion. Headache from aseptic meningitis can develop after IVIG therapy, usually in the first 24 hours. It most often responds to treatment with ibuprofen. Thrombosis and acute renal failure have also been reported, usually in high-risk patients. Allergic reactions to IVIG can occur in patients with absent IgA. Allergic reactions are rare and should not occur in patients who have detectable serum IgA levels or patients who cannot synthesize any antibodies. Subcutaneous administration of immunoglobulin has fewer adverse effects but may be complicated by local reactions at the site(s) of infusion. The risk of transmission of infectious agents, despite preparation from large numbers of selected donors, is extremely low.
Therapy for severe T-cell disorders is stem cell transplantation, preferably from an human leukocyte antigen–matched sibling (see Chapter 76). Immunoglobulin replacement provides passive antibody-mediated immunity. Some patients continue to have poor B-cell function after stem cell transplantation and require immunoglobulin replacement therapy. GVHD, in which the transplanted cells initiate an immune response against the host tissues, is the main complication of stem cell transplantation. Patients with the ADA deficiency form of SCID who lack histocompatible sibling donors can receive repeated intramuscular replacement doses of ADA, stabilized by coupling to polyethylene glycol. Gene therapy has been performed in several patients with the common gamma chain deficiency form and ADA deficiency form of SCID by transfer of a normal gene into bone marrow stem cells, which are infused into the patient. Gene therapy for common gamma chain deficiency was successful in most of the treated patients; however, it was complicated by the development of leukemia in some patients.
PREVENTION AND NEWBORN SCREENING
Prenatal diagnosis is possible for all immunodeficiency diseases with an identified gene defect. Testing can be performed by gene sequencing, especially when the mutation in the index case has been identified. Polymerase chain reaction amplification of T-cell receptor excision circles that are present in recent thymic emigrants is currently being tested as a newborn screening method to identify patients with SCID, which would allow the initiation of therapy before any serious infections or complications develop.