 CHAPTER 75 Complement System
CHAPTER 75 Complement System
The complement system consists of several plasma and membrane proteins that function in the innate immune response as well as in adaptive immunity by facilitating antibody-mediated immunity. Complement proteins can kill pathogens with or without antibody, opsonize pathogens to facilitate their uptake by phagocytes, or mediate inflammation. The complement system can be activated through three pathways—the classic, alternative, or lectin pathways—that involve a cascade-like, sequential activation of complement factors resulting in an amplified response (Fig. 75-1). Disorders of the complement system predispose to recurrent infection, autoimmunity, and angioedema (Table 75-1).
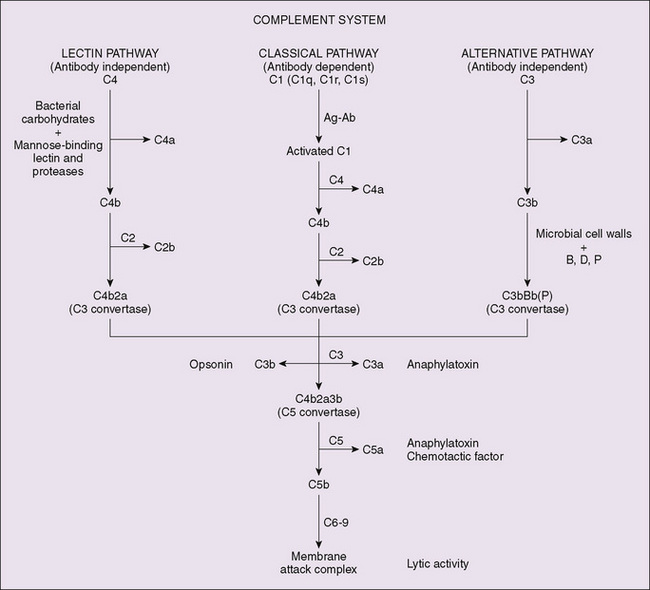
Figure 75-1 Complement component cascade involving the classic, alternate, and lectin pathways. The initiating events for the pathways differ, but result in production of C3 cleaving enzyme activity, which is the pivotal step as the three pathways converge to the terminal activation sequences. Ag-Ab, antigen-antibody complex; B, factor B; D, factor D (factor B clearing enzyme); P, properdin.
TABLE 75-1 Deficiency of Complement and Associated Disease
| Deficient Protein | Associated Disease |
|---|---|
| C1q, C1r | SLE, glomerulonephritis; occasional pneumococcal infection |
| C2 | SLE, arthritis, JRA, recurrent infections in some patients, rare glomerulonephritis |
| C3 | Recurrent infections, rare glomerulonephritis, or SLE |
| C4 | SLE-like disease, pyogenic infection |
| C5 | Recurrent meningococcal infections, rare glomerulonephritis, or SLE |
| C6 | Recurrent meningococcal, rare glomerulonephritis, or SLE |
| C7 | Recurrent meningococcal, Raynaud phenomenon |
| C8 | Recurrent infections |
| C9 | Occasional meningococcal infection, autoimmune disease in some patients |
| Properdin | Recurrent infections, meningococcal infection (often fatal) |
| Factor H | Glomerulonephritis, meningococcal infection |
| Factor I | Recurrent infections |
| C4-binding protein | Collagen vascular disease |
| C5a inhibitor | Familial Mediterranean fever |
| C3b receptor | SLE |
| C1 inhibitor | Hereditary angioedema |
JRA, juvenile rheumatoid arthritis; SLE, systemic lupus erythematosus.
ETIOLOGY
The three pathways for complement activation are initiated by different mechanisms. The classic pathway is activated by antigen-antibody complexes or by C-reactive protein and trypsin-like enzymes. The alternative pathway may be activated by C3b generated through classic complement activation or by endotoxin or fungal antigens (zymogen). The lectin pathway is initiated by the interaction of mannose-binding lectin with microbial carbohydrate. The three pathways lead to the generation of a C3 convertase, which activates the complement protein C3 and initiates a cascade common to the three pathways, culminating in the formation of a membrane attack complex (MAC), which can lyse pathogens and other target cells (see Fig. 75-1). The MAC is a complex of C5b, C6, C7, C8, and several C9 molecules. The MAC generates pores in the cell membrane, leading to lysis of the cells. The MAC without C9 can form small pores; however, the addition of C9 leads to a large transmembrane pore that enhances lysis of the target cell. C3a and C5a can release histamine from mast cells and basophils, leading to increased vascular permeability and smooth muscle contraction. In addition, C5a has chemotactic activity, attracting phagocytes to the site of complement activation, and it can cause degranulation of phagocytic cells. C3b acts as an opsonin when attached to the surface of a pathogen by binding to phagocytes via complement receptor 1 (CR1). Its degradation product, iC3b, can bind to CR3 on the surface of phagocytes. Activation of the phagocyte leads to ingestion of the C3b-coated or iC3b-coated pathogen.
Activation of the classic pathway by an antigen-antibody complex is initiated by the binding of C1q to the Fc portion of an antibody molecule in the immune complex. C1r autoactivates and cleaves C1s, which cleaves C4 and then C2, forming the C3 convertase, C4b2a. C4b2a is activated by the lectin pathway when lectins, such as mannose-binding protein, bind to sugar residues on the surface of pathogens, and mannose-binding protein–associated proteases cleave C4 and C2. The alternative pathway is always active at a low level and is amplified when active C3 binds to a surface that lacks regulatory proteins. C3b generated from C3 binds to factor B, which is cleaved by factor D to form the alternative pathway C3 convertase, C3bBb. Properdin binds to and stabilizes the C3 convertase (see Fig. 75-1).
The complement system is under tight regulation because it has potent inflammatory activity and the potential to cause significant damage to host cells. The complement cascade is inherently regulated by a short half-life of the sites on C4b and C3b that allow them to bind to cell surfaces and by instability of the C3 convertases, C4b2a and C3bBb. In addition, C1-inhibitor regulates the cascade at an early stage by blocking active sites on C1r, C1s, and the proteases associated with the lectin pathway. Factor I destabilizes C3 convertase complexes and degrades the active fragments. Other inhibitors include membrane proteins, such as decay accelerating factor, CR1, membrane cofactor protein, and plasma proteins such as C4b-binding protein and factor H. Formation of the MAC can be blocked by cell surface CD59, protein S, and other plasma proteins. Deficiency of any of these regulatory proteins can result in an inflammatory response, tissue damage, or excessive complement consumption.
Disorders of complement proteins can result from inherited deficiency or can be secondary to increased consumption. The consequences of decreased complement depend on the affected factor (see Table 75-1). Deficiencies of early components of the classic pathway (C1, C2, or C4) are not usually associated with severe infections, although patients with C2 deficiency may present with milder recurrent infections. Patients with C1, C2, or C4 deficiency are susceptible to autoimmune diseases, especially systemic lupus erythematosus. The exact mechanism of this susceptibility is not known but is thought to arise from the role of these early components in clearing immune complexes.
Deficiency of properdin, C3, or the terminal components predisposes patients to severe recurrent infections. Deficiency of C3, the major opsonin, due to a genetic defect or secondary to excessive consumption from a deficiency in factor H, factor I, or the presence of C3 nephritic factor predisposes patients to infections, especially with encapsulated organisms. Deficiency of one of the terminal components that compose the MAC predisposes patients to infection with Neisseria meningitidis. Complement deficiency may be found in 40% of patients presenting with recurrent neisserial infections. Deficiency of mannose-binding lectin also is associated with an increased frequency of bacterial infections, including sepsis.
Congenital deficiency of C1-inhibitor results in hereditary angioedema, characterized by recurrent episodes of nonpruritic angioedema lasting 48 to 72 hours, which occur spontaneously or after minor trauma, stress, or anxiety. Angioedema can occur in any tissue. Abdominal edema can cause acute abdominal pain; edema of the upper airway can be life-threatening and may necessitate emergency tracheostomy. The disorder is inherited as an autosomal dominant disease and results from a heterozygous deficiency of C1-inhibitor leading to serum levels less than 30% of normal values. Some mutations (type II hereditary angioedema) result in normal or elevated antigenic levels of C1-inhibitor with defective function. An acquired form of angioedema results from autoantibodies to C1-inhibitor in lymphoid malignancies or autoimmune disorders but is uncommon in childhood. C1-inhibitor is a regulator of Hageman factor (clotting factor XIIa), clotting factor XIa, plasma kallikrein, and plasmin in addition to C1r and C1s. Lack of inhibition of the contact system, Hageman factor and plasma kallikrein, is responsible for the development of angioedema.
LABORATORY STUDIES
The CH50 test is a widely available test of classic complement pathway function based on an antibody-dependent hemolytic assay, which measures the serum dilution that results in lysis of 50% of sheep red blood cells. The CH50 test depends on the function of all nine complement proteins, C1 through C9, and is the best screen for complement function. The AP50 test, which measures complement activation using red blood cells from different species (e.g., rabbit) that can activate the alternative pathway, is less widely available than the CH50 test. Abnormal results of both tests indicate a deficiency in a terminal component common to both pathways, whereas an abnormal result of one or the other test indicates a deficiency of an early component of the respective pathway. If the CH50 or AP50 levels are abnormal, individual components can be analyzed in specialized laboratories.
Determination of C1-inhibitor levels and function is needed to diagnose hereditary angioedema. Some functional tests miss rare mutations that allow C1-inhibitor to bind C1s, but not one or more of the other enzymes with which it interacts. Low C1-inhibitor levels or function results in chronically decreased C4 levels and decreased C2 levels during acute attacks. Low C1q levels are found in acquired C1-inhibitor deficiency, which distinguishes it from hereditary angioedema. Tests for autoantibodies to C1-inhibitor and C1q can be performed by enzyme-linked immunosorbent assay.
TREATMENT
Specific treatment of complement deficiencies with component replacement is not available. Long, frequent courses of antibiotics constitute the primary therapy. Immunization of patients and close contacts with pneumococcal and meningococcal vaccines may be useful, but infections may still occur in immunized complement-deficient patients. Replacement of complement proteins by plasma transfusion has been used in some patients with C2 deficiency or factor H deficiency.
Patients with C1-inhibitor deficiency and frequent episodes of angioedema respond to prophylactic use of an oral attenuated androgen (stanozolol or danazol), which increases serum concentrations of C1-inhibitor. Adverse effects, including masculinization in females, growth arrest, and hepatitis, limit their use. Care must be taken in using plasma transfusions for angioedema. Prophylactic administration of fresh frozen plasma before surgery can prevent angioedema, but administration during an acute episode may exacerbate the episode. Angioedema of the airway can present as an acute emergency, necessitating tracheostomy because administration of epinephrine, antihistamines, or corticosteroids is ineffective in reversing this type of angioedema. Purified C1-inhibitor is now available and can be used prophylactically (before surgery) and during acute episodes of angioedema. Angiotensin-converting enzyme inhibitors, such as captopril, should be avoided in patients with C1-inhibitor deficiency because they can precipitate episodes of angioedema by inhibiting degradation of kinins that mediate edema formation. Novel therapeutic agents, including a kallikrein inhibitor and a bradykinin receptor 2 antagonist, are being investigated as potential therapy for hereditary angioedema.