 CHAPTER 137 Cystic Fibrosis
CHAPTER 137 Cystic Fibrosis
ETIOLOGY AND EPIDEMIOLOGY
Cystic fibrosis (CF) is an autosomal recessive disorder that is the most common life-limiting genetic disease in whites. In the United States, the incidence of CF is approximately 1 in 3200 for whites, 1 in 15,000 African Americans, and 1 in 31,000 persons of Asian heritage. The gene for CF, which is located on the long arm of chromosome 7, encodes for a polypeptide, the cystic fibrosis transmembrane regulator (CFTR). CFTR is a chloride channel located on the apical surface of epithelial cells. CFTR is important for the proper movement of salt and water across cell membranes and maintaining the appropriate composition of various secretions, especially in the airway, liver, and pancreas. The most common mutation is a deletion of three base pairs resulting in the absence of phenylalanine at the 508 position (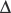 F508). More than 1000 mutations of the CFTR gene have been identified.
F508). More than 1000 mutations of the CFTR gene have been identified.
The secretory and absorptive characteristics of epithelial cells are affected by abnormal CFTR, resulting in the clinical manifestations of CF. The lack of normal CFTR function alters chloride ion conductance in the sweat gland, resulting in excessively high sweat sodium and chloride levels. This is the basis of the sweat chloride test, which is still the standard diagnostic test for this disorder. It is positive (elevated sweat chloride > 60 mEq/L) in 99% of patients with CF. It is not completely understood how the abnormal chloride conductance accounts for all of the clinical manifestations of CF, but it is known that abnormal airway secretions make the airway more prone to colonization with bacteria. Defects in CFTR may also reduce the function of airway defenses and promote bacterial adhesion to the airway epithelium. This all leads to chronic airway infections and eventually to bronchial damage (bronchiectasis).
CLINICAL MANIFESTATIONS
CF is a chronic progressive disease that can present with protein and fat malabsorption (failure to thrive, hypoalbuminemia, steatorrhea), liver disease (cholestatic jaundice), or chronic respiratory infection (Table 137-1). Infants often present with failure to thrive, while pulmonary manifestations predominate in older children. The respiratory epithelium of patients with CF exhibits marked impermeability to chloride and an excessive reabsorption of sodium. These alterations in the bioelectrical properties of the epithelium lead to a relative dehydration of airway secretions, which results in airway obstruction and impaired mucociliary transport. This, in turn, leads to endobronchial colonization with bacteria, especially Staphylococcus aureus and Pseudomonas aeruginosa. Pseudomonas aeruginosa predominates in older patients with advanced disease. Chronic bronchial infection results in persistent or recurrent cough that is often productive of sputum, especially in older children. Chronic airway infection leads to airway obstruction and bronchiectasis and eventually to pulmonary insufficiency and premature death. The median age of survival (years) is currently in the mid-30s. Digital clubbing is common in patients with CF, even in those without significant lung disease. Chronic sinusitis and nasal polyposis are common. Any child with nasal polyps, especially if younger than 12 years of age, should be evaluated for CF.
TABLE 137-1 Complications of Cystic Fibrosis
RESPIRATORY COMPLICATIONS
GASTROINTESTINAL COMPLICATIONS
OTHER COMPLICATIONS
Pulmonary infections with virulent strains of Burkholderia cepacia are difficult to treat and may be associated with accelerated clinical deterioration. Allergic bronchopulmonary aspergillosis (ABPA) is a hypersensitivity reaction to Aspergillus in the CF airways. It causes airway inflammation/obstruction and aggravates CF lung disease. The treatment for ABPA is parenteral corticosteroids (prednisone) and antifungal agents (itraconazole). Minor hemoptysis is usually due to airway infection, but major hemoptysis is caused by bleeding from bronchial artery collateral vessels in damaged/chronically infected portions of the lung. Pneumothoraces can occur in patients with advanced lung disease.
Ninety percent of patients with CF are born with exocrine pancreatic insufficiency. The inspissation of mucus and subsequent destruction of the pancreatic ducts result in the inability to excrete pancreatic enzymes into the intestine. This leads to malabsorption of proteins, sugars (to a lesser extent), and especially fat. Fat malabsorption manifests clinically as steatorrhea (large foul-smelling stools), deficiencies of fat-soluble vitamins (A, D, E, and K), and failure to thrive. Protein malabsorption can present early in infancy as hypoproteinemia and peripheral edema. Approximately 10% of patients with CF are born with intestinal obstruction caused by inspissated meconium (meconium ileus). In older patients, intestinal obstruction may result from thick inspissated mucus in the intestinal lumen (distal intestinal obstruction syndrome). In adolescent or adult patients, progressive pancreatic damage can lead to enough islet cell destruction to cause insulin deficiency. This initially presents as glucose intolerance, but true diabetes that requires insulin therapy (CF-related diabetes) may develop. The failure of the sweat ducts to conserve sodium and chloride may lead to hyponatremia and hypochloremic metabolic alkalosis, especially in infants. Inspissation of mucus in the reproductive tract leads to reproductive dysfunction in both males and females. In males, congenital absence of the vas deferens and azoospermia are nearly universal. In females, secondary amenorrhea is often present as a result of chronic illness and reduced body weight. Fertility is also diminished by abnormal secretions in the fallopian tubes and cervix, but women with CF can conceive.
The diagnosis of CF should be seriously considered in any infant presenting with failure to thrive, cholestatic jaundice, chronic respiratory symptoms, or electrolyte abnormalities (hyponatremia, hypochloremia, metabolic alkalosis). CF should be in the differential diagnosis of children with chronic respiratory or gastrointestinal symptoms, especially if there is digital clubbing. Any child with nasal polyps should be evaluated for CF.
DIAGNOSTIC STUDIES
There are readily available commercial DNA tests that detect up to 100 CF mutations, but because there are more than 1000 identified mutations and some that have yet to be identified, DNA analysis will not detect all cases of CF. Many states now have newborn screening for CF, which is based either on elevated immunoreactive trypsinogen (IRT) levels or DNA tests. Newborn screening identifies the majority of infants with CF, but there are both false positive and false negative results. Therefore, the diagnostic test of choice is still the sweat test. Indications for performing a sweat test are listed in Table 137-2. The following criteria should be met to establish the diagnosis of CF:
TABLE 137-2 Indications for Sweat Testing
RESPIRATORY
GASTROINTESTINAL
MISCELLANEOUS
Although the sweat test is both specific and sensitive for CF, it is subject to technical problems, and there are false positive and false negative results (Table 137-3). Other supportive tests include the measurement of bioelectrical potential differences across nasal epithelium (not widely available) and measurement of fecal elastase levels. Low fecal elastase levels indicate exocrine pancreatic insufficiency. Detection of a known CF mutation by DNA analysis is useful but not foolproof. CF genotyping done by commercial laboratories identifies approximately 95% of patients with CF, but there are mutations that are not identified by standard testing.
TABLE 137-3 Causes of False Positive and False Negative Results on Sweat Testing
FALSE POSITIVE
FALSE NEGATIVE
Identification of carriers (heterozygotes) and prenatal diagnosis of children with the F508 and other common mutations is currently offered at most medical centers. Present testing techniques can identify more than 90% of carriers. Prenatal detection of a known CF genotype may be accomplished by amniotic fluid or chorionic villus sampling.
TREATMENT
The treatment of CF is multifactorial, but it is primarily directed toward the gastrointestinal and pulmonary complications. Presently there is no effective treatment for the underlying defect (abnormal salt and water transit across epithelial cells due to the abnormal CFTR chloride channel). However, medications that improve the function of defective CFTR and gene replacement therapy are both being actively investigated. The current management of pulmonary complications is directed toward facilitating clearance of secretions from the airways and minimizing the effects of chronic bronchial infection. Airway secretion clearance techniques (chest physiotherapy) help remove mucus from the airways, and aerosolized DNAse and 7% hypertonic saline, both delivered by nebulizer, decrease the viscosity of mucus. Antibiotic therapy is important in controlling chronic infection. Monitoring pulmonary bacterial flora via airway cultures and providing aggressive therapy with appropriate antibiotics (oral, aerosolized, and parenteral) help to slow the progression of lung disease. Patients often require 2- to 3-week courses of high-dose intravenous antibiotics and aggressive chest physiotherapy to treat pulmonary exacerbations. Antibiotics are selected based on organisms identified by sputum culture. If patients are unable to provide sputum, then a throat culture for CF pathogens can be used to direct therapy. Common infecting organisms are Pseudomonas aeruginosa and Staphylococcus aureus.
Exocrine pancreatic insufficiency is treated with enteric-coated pancreatic enzyme capsules, which contain lipase and proteases. Patients with CF are encouraged to follow high-calorie diets, often with the addition of nutritional supplements. Even with optimal pancreatic enzyme replacement, stool losses of fat and protein may be high. Fat should not be withheld from the diet, even when significant steatorrhea exists. Rather, pancreatic enzyme doses should be titrated to optimize fat absorption, although there is a limit to the doses that should be used. Lipase dosages exceeding 2500 U/kg/meal are contraindicated because they have been associated with fibrosing colonopathy. Fat-soluble vitamins (A, E, D, and K) are recommended, preferably in a water-miscible form.
Newborns with meconium ileus may require surgical intervention, but some can be managed with contrast (Gastrografin) enemas. Intestinal obstruction in CF patients beyond the neonatal period is often due to distal intestinal obstruction syndrome (DIOS), which may need to be treated with courses of oral laxatives (polyethelyne glycol) or, in more refractory cases, with balanced intestinal lavage solutions (GoLYTELY). Pancreatic enzyme dosage adjustment, adequate hydration, and dietary fiber may help prevent recurrent episodes. Patients with CF-related diabetes are treated with insulin, primarily to improve nutrition and prevent dehydration, as ketoacidosis is rare. Although transaminase elevation is common in patients with CF, only 1 to 3% of patients have progressive cirrhosis resulting in portal hypertension. Cholestasis is treated with the bile salt, ursodeoxycholic acid. Portal hypertension and esophageal varices due to cirrhosis of the liver are managed, when necessary, with portal vein shunting procedures or liver transplantation. Patients with symptomatic sinus disease and nasal polyps may require sinus surgical procedures.
The management of CF is complex and is best coordinated by personnel at accredited CF centers. As with other severe chronic diseases, management of patients with CF requires a multidisciplinary team. Physicians and other care providers, including nurses, respiratory therapists, nutritionists, and social workers must work with patients and their families to maintain an optimistic, comprehensive, and aggressive approach to treatment.