 CHAPTER 147 Cardiomyopathies
CHAPTER 147 Cardiomyopathies
ETIOLOGY
A cardiomyopathy is an intrinsic disease of the heart muscle and is not associated with other forms of heart disease (Table 147-1). There are three types of cardiomyopathy based on anatomic and functional features:
TABLE 147-1 Etiology of Myocardial Disease
FAMILIAL/HEREDITARY
INFECTIOUS (MYOCARDITIS)
METABOLIC/NUTRITIONAL/ENDOCRINE
CONNECTIVE TISSUE/GRANULOMATOUS DISEASE
DRUGS/TOXINS
CORONARY ARTERIES
OTHER DISORDERS/CONDITIONS
SLE, systemic lupus erythematosus.
Dilated cardiomyopathies are the most common. They are often idiopathic, but may be due to infection (echovirus or Coxsackie B virus) or be postinfectious, familial, or secondary to systemic disease or to cardiotoxic drugs. Hypertrophic cardiomyopathies are usually familial with autosomal dominant inheritance, but may occur sporadically. Restrictive cardiomyopathies are rare; they may be idiopathic or associated with systemic disease (Table 147-2).
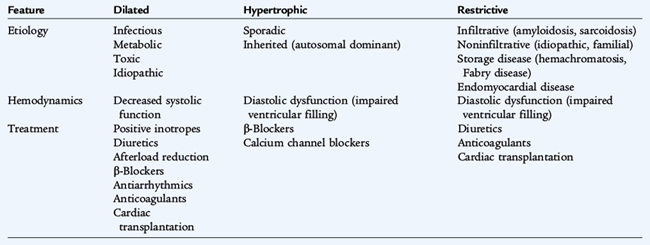
CLINICAL MANIFESTATIONS
Dilated cardiomyopathies result in enlargement of the left ventricle only or both ventricles. Myocardial contractility is variably decreased. Clinically, children with dilated cardiomyopathy present with signs and symptoms of inadequate cardiac output and heart failure. Tachypnea and tachycardia are present on examination. Peripheral pulses are often weak because of a narrow pulse pressure. Rales may be audible on auscultation. The heart sounds may be muffled and an S3 is often present. Concurrent infectious illness may result in circulatory collapse and shock in children with dilated cardiomyopathies.
Hypertrophic cardiomyopathy is initially difficult to diagnose. Infants, but not older children, frequently present with signs of heart failure. Sudden death may be the initial presentation in older children. Dyspnea, fatigue, chest pain, syncope or near-syncope, and palpitations may be present. A murmur is heard in more than 50% of children referred after identification of an affected family member. Restrictive cardiomyopathies are relatively rare in pediatrics. Presenting symptoms usually include dyspnea exacerbated by a respiratory illness, syncope, hepatomegaly, and an S4 heart sound on examination.
IMAGING STUDIES
Cardiomegaly usually is seen on chest radiographs for all three types of cardiomyopathies. The electrocardiogram (ECG) in dilated cardiomyopathy may have nonspecific ST-T wave changes and left ventricular hypertrophy. ECG evidence of right ventricular hypertrophy is present in 25% of children with cardiomyopathy. The ECG with hypertrophic cardiomyopathy is universally abnormal, but changes are nonspecific. Primary hypertrophic cardiomyopathy is associated with a prolonged QT interval. Children with restrictive cardiomyopathies may show atrial enlargement on the ECG. Echocardiography features vary by type of cardiomyopathy. Dilated cardiomyopathies result in left atrial and ventricular dilation, a decreased shortening fraction, and globally depressed contractility. Asymmetrical septal hypertrophy and left ventricular outflow tract obstruction are seen in hypertrophic cardiomyopathies. Massive atrial dilation is seen in restrictive cardiomyopathies. Endomyocardial biopsy specimens, obtained while the patient is hemodynamically stable, identify histologic type and allow tests for mitochondrial or infiltrative diseases.
TREATMENT
Supportive therapy, including diuretics, inotropic medications, and afterload reduction, is provided for all three types of cardiomyopathy and depends on the degree of symptoms and severity of cardiovascular compromise. If a specific etiology can be identified, treatment is directed at the etiology. Symptomatic therapy with close monitoring and follow-up is crucial. Because of the high mortality rate associated with all forms of cardiomyopathy, cardiac transplantation must be considered.