CHAPTER 170 Endocrinology Assessment
CHAPTER 170 Endocrinology Assessment
The endocrine system regulates vital body functions by means of hormonal messengers. Hormones are defined as circulating messengers, with action at a distance from the organ (gland) of origin of the hormone. Hormones can be regulated by nerve cells; endocrine agents can serve as neural messengers. There is also a relationship between the endocrine system and the immune system; autoantibodies may cause an organ to produce an excess or deficiency of a hormone. Manifestations of an endocrine disorder are related to the response of the peripheral tissue to a hormone excess or deficiency.
Hormone action also may be paracrine (acting on adjacent neighboring cells to the cell of origin of the hormone) or autocrine (acting on the cell of origin of the hormone itself); agents acting in these ways are called factors rather than hormones (Fig. 170-1). Hormones generally are regulated in a feedback loop so that the production of a hormone is linked to its effect or its circulating concentration. Endocrine disorders generally manifest from one of four ways:
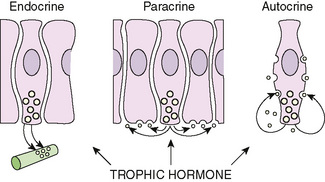
FIGURE 170-1 Schematic representation of mechanisms of action of hormones and growth factors. Although traditional hormones are formed in endocrine glands and transported to distant sites of action through the bloodstream (endocrine mechanism), peptide growth factors may be produced locally by the target cells themselves (autocrine modality of action) or by neighboring cells (paracrine action).
(From Wilson JD, Foster DW [eds]: Williams Textbook of Endocrinology, 8th ed. Philadelphia, WB Saunders, 1992, p 1007.)
Peptide hormones act through specific cell membrane receptors; when the hormone is attached to the receptor, the complex triggers various intracellular second messengers that cause the biologic effects. Peptide hormone receptor number and avidity may be regulated by hormones. Steroid hormones exert their effects by attachment to intranuclear receptors, and the hormone-receptor complex translocates to the nucleus, where it binds with DNA, and leads to further gene activation.
The interpretation of serum hormone levels must be related to their controlling factors. For example, a given value of PTH may be normal in a eucalcemic patient, but inadequate in a hypocalcemic patient with partial hypoparathyroidism or excessive in a hypercalcemic patient with hyperparathyroidism.
HYPOTHALAMIC-PITUITARY AXIS
The hypothalamus controls many endocrine systems either directly or through the pituitary gland. Higher central nervous system (CNS) centers control the hypothalamus. Hypothalamic releasing or inhibiting factors travel through capillaries of the pituitary portal system to control the anterior pituitary gland, regulating the hormones specific for the factor (Fig. 170-2). The pituitary hormones enter the peripheral circulation and exert their effects on target glands, which produce other hormones that feed back to suppress their controlling hypothalamic and pituitary hormones. (Insulin-like growth factor-1 [IGF-1], growth hormone (GH), cortisol, sex steroids, and thyroxine [T4] all feed back on the hypothalamic-pituitary system.) Prolactin is the only pituitary hormone that is suppressed by a hypothalamic factor, prolactin inhibitory factor (dopamine). Hypothalamic deficiency leads to a decrease in most pituitary hormone secretions, but may lead to an increase in prolactin secretion. The hypothalamus is also the location of vasopressin-secreting axons that either terminate in the posterior pituitary gland and exert their effect via vasopressin secretion from this area or terminate in the mediobasal hypothalamus, from which they can exert effects on water balance, even in the absence of the posterior pituitary gland.
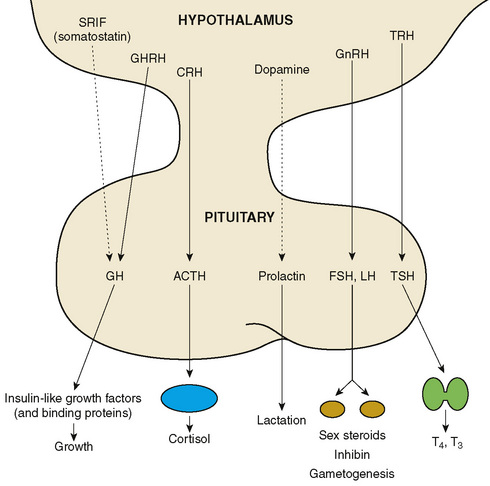
FIGURE 170-2 Hormonal influences of the hypothalamus and pituitary gland. Solid line represents stimulatory influence; dotted line represents inhibitory influence. ACTH, adrenocorticotropic hormone; CRH, corticotropin-releasing hormone or CRF; FSH, follicle-stimulating hormone; GH, growth hormone; GHRH, GH-releasing hormone or GRF; GnRH, gonadotropin-releasing hormone or luteinizing hormone releasing factor, LRF or LHRH; LH, luteinizing hormone; SRIF, somatotropin release–inhibiting factor, somatostatin or SS; TRH, thyrotropin-releasing hormone or TRF; TSH, thyroid-stimulating hormone; T3, triiodothyronine; T4, thyroxine.
In childhood, increased pituitary secretion of various hormones as a result of an adenoma is rare, although cases of pituitary gigantism (growth hormone [GH] excess from a pituitary adenoma) do occur. Destructive lesions of the pituitary gland or hypothalamus are more common in childhood. A craniopharyngioma, a tumor of the Rathke pouch, may descend into the sella turcica, causing erosion of the bone and destruction of pituitary and hypothalamic tissue. Acquired hypopituitarism also may result from pituitary infections; from infiltration, such as with Langerhans cell histiocytosis (histiocytosis X), lymphoma, and sarcoidosis; after radiation therapy or trauma to the CNS; and as a consequence of autoimmunity against the pituitary gland.
Congenital hypopituitarism usually is caused by absence of hypothalamic releasing factors rather than anatomic absence of the pituitary gland itself. Without hypothalamic stimulation, the pituitary gland does not release its hormones but it can be primed for secretion by exogenous hypothalamic releasing factors. However, exogenous administration is of limited clinical value because the end products of the glands themselves can be given to treat hypopituitarism. Congenital defects associated with hypopituitarism range from holoprosencephaly (cyclopia, cebocephaly, orbital hypotelorism), to cleft palate (6% of cases of cleft palate are associated with GH deficiency). Septo-optic dysplasia (optic nerve hypoplasia, absent septum pellucidum, or variations of both, and hypopituitarism) may result in significant visual impairment with pendular nystagmus (to-and-fro nystagmus caused by an inability to focus on a target). The magnetic resonance imaging (MRI) findings of congenital hypopituitarism include an ectopic posterior pituitary gland hot spot and the appearance of a pituitary stalk transection and small pituitary gland.
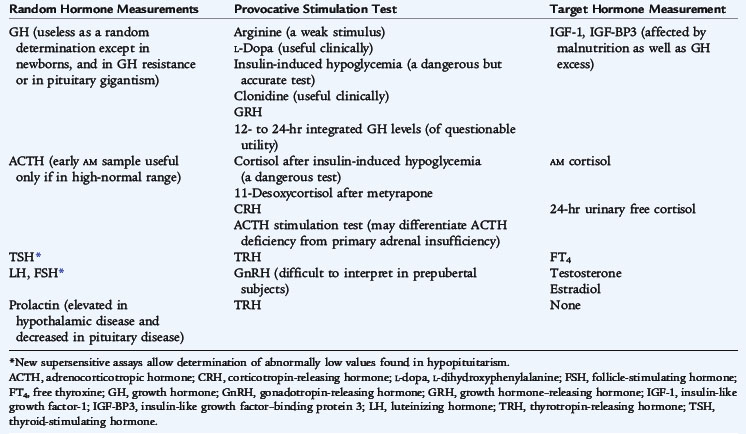
The assessment of pituitary function may be determined by measuring some of the specific pituitary hormone in the basal state; other assessments require the measurement after stimulation. Indirect assessment of pituitary function can be obtained by measuring serum concentrations of the target gland hormones (Table 170-1). Several tests of pituitary function are listed in Table 170-2.
TABLE 170-1 Diagnostic Evaluation of Hypopituitarism
| Manifestation | Cause | Tests* |
|---|---|---|
| Growth failure, hypothyroidism, or both | GH deficiency, TRH/TSH deficiency, or both | Provocative GH tests, free T4, bone age, IGF-1, IGF-BP3 |
| Hypoglycemia | GH deficiency, ACTH insufficiency, or both | Provocative GH tests, test of ACTH secretion, IGF-1, IGF-BP3 |
| Micropenis, pubertal delay or arrest | Hypogonadotropic hypogonadism or GH deficiency | Sex steroids (E2, testosterone), basal LH and FSH (analyzed by ultrasensitive assays) or after GnRH administration, provocative GH tests, IGF-1, IGF-BP3 |
| Polyuria, polydipsia | ADH deficiency | Urine analysis (specific gravity), serum electrolytes, urine and serum osmolality, water deprivation test |
ADH, antidiuretic hormone; CNS, central nervous system; E2, estradiol; FSH, follicle-stimulating hormone; GH, growth hormone; GnRH, gonadotropin-releasing hormone; IGF-1, insulin-like growth factor; IGF-BP3, insulin-like growth factor–binding protein 3; LH, luteinizing hormone; MRI, magnetic resonance imaging; T4, thyroxine; TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormone.
* Each patient with hypopituitarism should have CNS MRI as part of evaluation to determine the etiology of the condition.