 CHAPTER 175 Thyroid Disease
CHAPTER 175 Thyroid Disease
THYROID PHYSIOLOGY AND DEVELOPMENT
Thyrotropin-releasing hormone (TRH), a tripeptide synthesized in the hypothalamus, stimulates the release of pituitary thyroid-stimulating hormone (TSH). Pituitary TSH is a glycoprotein that stimulates the synthesis and release of thyroid hormones by the thyroid gland. Thyroid function develops in three stages:
Thyroxine (T4), triiodothyronine (T3), and TSH do not cross the placenta in significant amounts. Concentrations in fetal serum reflect primarily fetal secretion and metabolism. Maternal thyroid antibodies, iodides (including radioactive iodides), and medications given to mothers to treat hyperthyroidism (e.g., propylthiouracil, methimazole) cross the placenta and affect fetal thyroid function. An infant born prematurely or with intrauterine growth restriction may have an interruption of the normal maturational process and appear to have hypothyroidism by standard tests.
The thyroid gland (1) concentrates iodine and (2) binds it (organifies it) to tyrosine molecules to produce either monoiodotyrosine or diiodotyrosine, with subsequent (3) coupling of two tyrosines, T4 or T3. The major fraction of circulating T3 (approximately two thirds) is derived from peripheral deiodination of T4 to T3, but some is produced by the thyroid gland itself. In Graves disease, a larger fraction originates in the thyroid gland. The conversion of T4 to T3 requires the removal of one iodine from the outer ring of tyrosine; removing an iodine from the inner ring results in reverse T3, which has little biologic effect. Preferential conversion of T4 to reverse T3 rather than T3 occurs in utero and in all forms of severe illness, including respiratory distress syndrome, fevers, anorexia, cachexia, and starvation. Conversion from T4 to T3 increases immediately after birth and throughout life. T4 and T3 are noncovalently bound to a specific serum carrier protein, T4-binding globulin, and, to a lesser extent, albumin. Only small (<0.02%) fractions of T4 and T3 are not bound; free T4 (as it is converted to free T3) and free T3 are biologically active. Free T3 exerts metabolic effects and negative feedback on TSH release (Fig. 175-1).
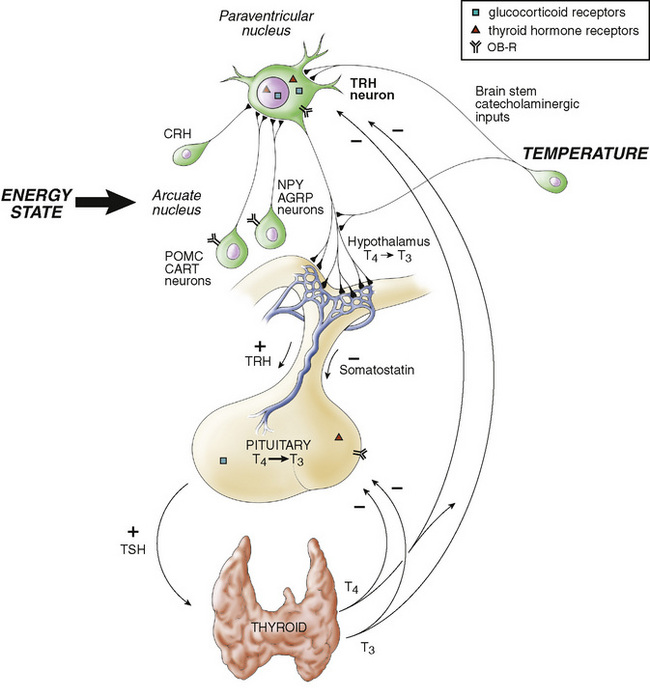
FIGURE 175-1 Interrelationships of the hypothalamic-pituitary-thyroid axis. Thyroid-stimulating hormone (TSH) from the pituitary gland stimulates the secretion of thyroxine (T4) and triiodothyronine (T3) from the thyroid gland. These act at the pituitary gland level to control secretion of TSH by a negative feedback mechanism. In addition, T4 is metabolized to the potent T3 within the pituitary gland by a monoiodinase. Secretion of TSH is stimulated by thyrotropin-releasing hormone (TRH) from the hypothalamus and inhibited by somatostatin. Thyroid hormone acts at the hypothalamus to stimulate secretion of somatostatin (somatostatin acts as a negative signal to the pituitary secretion of TSH). CRH, corticotropin-releasing hormone; OB-R, leptin receptor.
(From Melmed S, Polonsky K, Kronenberg H, Larsen R [eds]: Williams Textbook of Endocrinology, 10th ed. Philadelphia, Elsevier, 2003, p 101.)
Serum TSH increases just after birth, but soon decreases to lower values considered normal for later life. T4 secretion increases after birth, partially as a result of the peak in TSH and partially because of maturation of thyroid metabolism. Serum thyroid hormone concentrations decrease, but only slowly reach values routinely found in adults. It is important to refer to age-adjusted normative data to interpret thyroid function tests properly, whether relative to making diagnoses of hyperthyroidism or hypothyroidism or when adjusting therapy. Free T4 is the test of choice because it eliminates the effects of variation in protein binding, which can be substantial.
Table 175-1 summarizes laboratory test results in various types of thyroid abnormalities. In usual circumstances, plasma concentrations of TSH above the normal range indicate primary hypothyroidism, and concentrations below the normal range most often indicate the presence of hyperthyroidism. Although a thyroid scan rarely is indicated in the evaluation of pediatric thyroid disease, thyroid agenesis, ectopic thyroid tissue and the diagnoses of hyperfunctioning “hot” nodules or of nonfunctioning “cold” nodules may be detected by this test. A thyroid scan performed with the short-lived isotope radioactive iodine (123I) indicates the size, shape, and location of the thyroid gland and iodine concentrating ability. A solitary nodule is a source of concern for the possibility of cancer, especially if it is solid and nonfunctional. Ultrasound may determine whether it is cystic or solid. If the nodule is solid, a 123I scan indicates its functional status. Excisional biopsies usually are performed on solitary nodules. Scans are rarely indicated in the diagnosis of Hashimoto thyroiditis or thyrotoxicosis.

THYROID DISORDERS
Hypothyroidism
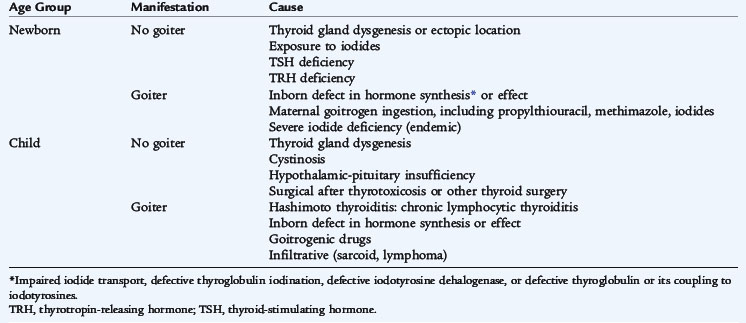
Hypothyroidism is diagnosed by a decreased serum free T4 and may be the result of diseases of the thyroid gland (primary hypothyroidism), abnormalities of the pituitary gland (secondary) or abnormalities of the hypothalamus (tertiary). Hypothyroidism is congenital or acquired and may be associated with a goiter (Table 175-2).
Congenital Hypothyroidism
Congenital hypothyroidism occurs in approximately 1 in 4000 live births and is caused by dysgenesis, disorders of embryogenesis (agenesis, aplasia, ectopia), far more often than by dyshormonogenesis disorders (e.g., enzyme defects). Thyroid tissue usually is not palpable in these sporadic nongoitrous conditions. Dyshormonogenesis, disorders of intrathyroid metabolism or goitrous congenital hypothyroidism, occurs in about 1 in 30,000 live births. The goiter reflects an inborn error of metabolism in the pathway of iodide incorporation or thyroid hormone biosynthesis or reflects the transplacental passage of antithyroid drugs given to the mother. The free T4 concentration is low, and the TSH level is elevated. Routine neonatal screening programs to measure cord blood or heel-stick TSH values occur in every state in the United States. An immediate confirmatory serum sample should be obtained from any infant having a positive result on a screening test. A low T4 and high TSH confirm the finding.
Isolated secondary or tertiary hypothyroidism is rare, occurring in 1 in 100,000 live births; the free T4 is normal to low. When tertiary or secondary hypothyroidism is detected, assessment of other pituitary hormones and investigation of pituitary-hypothalamic anatomy via magnetic resonance imaging (MRI) are indicated. Although not a hypothyroid condition, congenital T4-binding globulin deficiency occurs in about 1 in 10,000 live births and is associated with a low serum total T4 concentration, a normal TSH and serum free T4, and a euthyroid status. This entity does not require treatment with thyroid hormone because it is merely a binding protein abnormality. It is commonly X-linked dominant.
Clinical manifestations of congenital hypothyroidism in the immediate newborn period usually are subtle but become more evident weeks or months after birth. By then, it is too late to ensure that there is not a detriment to the infant’s cognitive development. Newborn screening is crucial to make an early diagnosis and initiate thyroid replacement therapy by younger than 1 month of age. Findings at various stages after birth include gestation greater than 42 weeks, birth weight greater than 4 kg, hypothermia, acrocyanosis, respiratory distress, large posterior fontanelle, abdominal distention, lethargy and poor feeding, jaundice more than 3 days after birth, edema, umbilical hernia, mottled skin, constipation, large tongue, dry skin, and hoarse cry. Thyroid hormones are crucial for maturation and differentiation of tissues such as bone (the bone age is often delayed at birth because of intrauterine hypothyroidism) and brain (most thyroid-dependent brain maturation occurs 2 to 3 years after birth) (Table 175-3).
TABLE 175-3 Symptoms and Signs of Hypothyroidism
ECTODERMAL
CIRCULATORY
NEUROMUSCULAR
SKELETAL
METABOLIC
CK, creatine kinase; ECG, electrocardiographic.
When treatment is initiated within 1 month or less after birth, the prognosis for normal intellectual development is excellent; screening programs usually offer therapy within 1 to 2 weeks of birth. If therapy is instituted after 6 months, when the signs of severe hypothyroidism are present, the likelihood of normal intellectual function is markedly decreased. Growth improves after thyroid replacement, even in late diagnosed cases. The dose of T4 changes with age; 10 to 15 μg/kg of T4 is used for a newborn, but about 3 μg/kg is used later in childhood. In neonatal hypothyroidism, the goal is to bring the serum free T4 rapidly into the upper half of the range of normal. Suppression of TSH is not seen in all cases and is not necessary in all cases because such suppression may lead to excessive doses of T4.
Acquired Hypothyroidism
The etiology of acquired hypothyroidism is presented in Table 175-2. The clinical manifestations may be subtle. Hypothyroidism should be suspected in any child who has a decline in growth velocity, especially if not associated with weight loss (see Table 175-3). The most common cause of acquired hypothyroidism in older children in the United States is lymphocytic autoimmune thyroiditis (Hashimoto thyroiditis). In many areas of the world, iodine deficiency is the etiology of endemic goiter (endemic cretinism). The failure of the thyroid gland may be heralded by an increase of TSH before T4 levels decrease. In contrast to untreated congenital hypothyroidism, acquired hypothyroidism is not a cause of permanent developmental delay.
Hashimoto Thyroiditis
Also known as autoimmune or lymphocytic thyroiditis, Hashimoto thyroiditis is a common cause of goiter and acquired thyroid disease in older children and adolescents. A family history of thyroid disease is present in 25% to 35% of patients. The etiology is an autoimmune process targeted against the thyroid gland with lymphocytic infiltration and lymphoid follicle and germinal center formation preceding fibrosis and atrophy.
Clinical manifestations include a firm, nontender euthyroid, hypothyroid, or, rarely, hyperthyroid (hashitoxicosis) diffuse goiter with a pebble-like feeling; an insidious onset after 6 years of age (the incidence peaks in adolescence, with a female predominance); and sometimes a pea-sized delphian lymph node above the thyroid isthmus. Associated autoimmune diseases include diabetes mellitus type 1 (DM1), adrenal insufficiency (Schmidt syndrome), and hypoparathyroidism. Autoimmune polyglandular syndrome type I consists of hypoparathyroidism, Addison disease, mucocutaneous candidiasis, and often hypothyroidism. Autoimmune polyglandular syndrome type II consists of Addison disease, DM1, and frequently autoimmune hypothyroidism. Trisomy 21 and Turner syndrome predispose to the development of autoimmune thyroiditis.
The diagnosis may be confirmed by serum antithyroid peroxidase (previously antimicrosomal) and antithyroglobulin antibodies. Neither biopsy nor thyroid scan is indicated in Hashimoto thyroiditis, although the thyroid scan with reduced uptake may differentiate hashitoxicosis from Graves disease.
Treatment with thyroid hormone sufficient to normalize TSH and free T4 is indicated for hypothyroidism in Hashimoto thyroiditis. Patients without manifestation of hypothyroidism require periodic thyroid function testing (serum TSH and free T4) every 6 to 12 months to detect the later development of hypothyroidism. Goiter with a normal TSH usually is not an indication for treatment.
Hyperthyroidism
Graves Disease
Most children with hyperthyroidism have Graves disease, the autonomous functioning of the thyroid caused by autoantibodies stimulating the thyroid stimulating immunoglobulins (TSIs). The resulting excessive synthesis, release, and peripheral metabolism of thyroid hormones produce the clinical features. Hashimoto thyroiditis and thyrotoxicosis are on a continuum of autoimmune diseases; there is overlap in their immunologic findings. Antimicrosomal and antithyroglobulin antibodies may be present in thyrotoxicosis, although the values are usually lower than in Hashimoto thyroiditis. Exceptionally high titers of antibodies may indicate the thyrotoxic phase of Hashimoto thyroiditis with the subsequent evolution toward permanent hypothyroidism. In Graves disease, serum T4 or T3 or both levels are elevated, whereas TSH is suppressed. Rare causes of hyperthyroidism include McCune-Albright syndrome, thyroid neoplasm, TSH hypersecretion, subacute thyroiditis, and iodine or thyroid hormone ingestion.
Clinical Manifestations
Graves disease presents as hyperthyroidism (Table 175-4) and is about five times more common in girls than in boys, with a peak incidence in adolescence. Personality changes, mood instability, and poor school performance are common initial problems. Tremor, anxiety, inability to concentrate, and weight loss may be insidious and confused with a psychological disorder until thyroid function tests reveal the elevated serum free T4 level. Serum T4 may be near-normal, whereas serum T3 is selectively elevated (T3 toxicosis, a rare entity). A firm, homogeneous goiter is usually present. Many patients complain of neck fullness and, in older subjects, a change in the size of their shirt collars. Thyroid gland enlargement is best visualized with the neck only slightly extended and with the examiner lateral to the patient; palpation of the thyroid gland is best performed with the examiner’s hands around the neck from the back. The patient swallows so that the examiner can feel and examine the size, consistency, nodularity, and motion of the gland. The examiner should watch the patient swallow to note any discernible enlargement or asymmetry of the thyroid lobes. The thickness is estimated, and the dimensions of each lobe are measured vertically and laterally. Auscultation may reveal a bruit over the gland, which needs to be differentiated from a carotid bruit.
TABLE 175-4 Clinical Manifestations of Hyperthyroidism
| Associated Change | Sign/Symptom |
|---|---|
| Increased catecholamine effects | |
| Hypermetabolism | |
| Myopathy | |
| Miscellaneous |
* Unusual except in subacute thyroiditis with hyperthyroid phase.
Treatment
Three treatment choices are available: pharmacologic, surgical, and radioactive iodine.
Drugs
Medical therapy to block thyroid hormone synthesis consists of propylthiouracil (5–7 mg/kg/24 hours orally in divided doses every 8 hours) or methimazole (0.5–0.7 mg/kg/24 hours orally in divided doses every 8–12 hours). Both medications are equally effective; methimazole may be easier to manage and titrate. Propranolol is started if symptoms are severe (2–3 mg/kg/24 hours orally) to control cardiac manifestations and is tapered as the propylthiouracil or methimazole takes effect. Propylthiouracil usually is continued for 1 to 2 years because the remission rate is approximately 25% per year. In patients complying with the treatment regimen, the 2-year course of treatment can be repeated. Propylthiouracil should suppress thyroid function to normal, without the need to add thyroid hormone replacement to normalize serum free T4. Complications of propylthiouracil are lupus-like syndrome, rash, granulocytopenia, and jaundice. The granulocytopenia is an idiosyncratic complication of rapid onset, which is observed only in the early months after institution of antithyroid medication and which must be attended to promptly by monitoring the complete blood count (CBC). If a suppressed white blood cell count is observed, antithyroid therapy must be discontinued; this is potentially lethal (more rarely, thrombocytopenia or aplastic anemia) and is a rare complication that affects 3 in 10,000 users a year. After resolution, the other of the two antithyroid medications can be started because there is usually less than a 50% chance of a similar reaction from the other medication. These side effects are sometimes severe and usually reversible after discontinuation of antithyroid therapy; failure to monitor and then discontinue propylthiouracil when complications arise may be fatal. Iodine administration, which may suppress thyroid function but becomes ineffective in a few weeks, is sometimes used as a preparation for surgery but never for long-term therapy.
Surgery
Surgical treatment consists of partial or complete thyroidectomy. Risks associated with thyroidectomy include the use of anesthesia and the possibility that the thyroid removal will be excessive, causing hypothyroidism, or that it will be inadequate, resulting in persistent hyperthyroidism. In addition, keloid formation, recurrent laryngeal nerve palsy, and hypoparathyroidism (transient postoperative or permanent) may occur. Thyroid storm caused by the release of large amounts of preformed hormone is a serious but rare complication. Even with optimal immediate postoperative results, patients may become hypothyroid within 10 years.
Radioiodine
Radioiodine (131I) is slower in exerting therapeutic effects, may require repeated dosing, and is likely to cause permanent hypothyroidism. Ultimately, hypothyroidism is the desired outcome because it is easier and safer to treat than continued hyperthyroidism. Although studies reveal no long-term consequences, concern remains about possible sequelae in children. This method of treatment is entering the mainstream for adolescents and adults. Radioiodine given to a pregnant teenager renders the fetus hypothyroid and is contraindicated.
Thyroid Storm
Thyroid storm (see Table 175-4) is a rare medical emergency consisting of tachycardia and hyperthermia. Treatment includes reducing the hyperthermia with a cooling blanket and administering propranolol to control the tachycardia, hypertension, and autonomic hyperfunction symptoms. Iodine may be given to block thyroid hormone release. Hydrocortisone may be indicated for relative adrenal insufficiency, and therapy for heart failure includes diuretics and digoxin.
Congenital Hyperthyroidism
This disorder results from transplacental passage of maternal TSIs and may be masked for several days until the short-lived effects of transplacental maternal antithyroid medication wear off (assuming the mother was receiving such medication), at which time the effects of TSIs are observed. Irritability, tachycardia (often with signs of cardiac failure simulating cardiomyopathy), polycythemia, craniosynostosis, bone age advancement, poor feeding and, later, failure to thrive are the clinical hallmarks. This condition may be anticipated if the mother is known to be thyrotoxic in pregnancy. Cure of hyperthyroidism before pregnancy (surgery or radioiodine treatment) limits or curtails T4 production but not the underlying immune disturbance producing TSIs; thus, the infant still may be affected, at least transiently.
Treatment for a severely affected neonate includes oral propranolol, 2 to 3 mg/kg/24 hours in divided doses, and propylthiouracil, approximately 5 mg/kg/24 hours orally in three divided doses. Because the half-life of the immunoglobulin is several weeks, spontaneous resolution of neonatal thyrotoxicosis resulting from transplacental passage of TSIs usually occurs by 2 to 3 months of age. Observation without treatment is indicated in patients who are minimally affected.
TUMORS OF THE THYROID
Carcinoma of the thyroid is rare in children, but papillary and follicular carcinomas represent 90% of children’s thyroid cancers. A history of therapeutic head or neck irradiation or radiation exposure from nuclear accidents predisposes a child to thyroid cancer. Carcinoma usually presents as a firm to hard, painless, nonfunctional solitary nodule and may spread to adjacent lymph nodes. Rapid growth, hoarseness (recurrent laryngeal nerve involvement), and lung metastasis may be present. If the nodule is solid on ultrasound, is cold on radioiodine scanning, and feels hard, the likelihood of a carcinoma is high. Excisional biopsy usually is performed, but fine needle aspiration biopsy also may be diagnostic.
Treatment includes total thyroidectomy, selective regional node dissection, and radioablation with 131I for residual or recurrent disease. The prognosis is usually good if the disease is diagnosed early.
Medullary carcinoma of the thyroid may be asymptomatic except for a mass. Diagnosis is based on the presence of elevated calcitonin levels, either in the basal state or after pentagastrin stimulation, and histologic examination. This tumor most often occurs with multiple endocrine neoplasia 2a or 2b, pheochromocytoma, or alone, possibly in a familial pattern. The presence of mutations of the RET proto-oncogene is predictive of the development of medullary carcinoma of the thyroid in some families. Screening the other members of the family is indicated after a proband is recognized. Prophylactic thyroidectomy is indicated for the family members with the same allele.