 CHAPTER 186 Neurocutaneous Disorders
CHAPTER 186 Neurocutaneous Disorders
The skin, teeth, hair, nails, and brain are derived embryologically from ectoderm. Abnormalities of these surface structures may indicate abnormal brain development. The term phakomatosis means mother spots or birthmarks and refers to tuberous sclerosis and neurofibromatosis. Not all of the so-called neurocutaneous disorders have characteristic cutaneous lesions, however, and not all are of ectodermal origin. Neurofibromatosis, tuberous sclerosis, Sturge-Weber disease, von Hippel–Lindau disease, and ataxia-telangiectasia are the most common of the more than 40 neurocutaneous disorders.
NEUROFIBROMATOSIS
Etiology
There are two distinct genetic types of neurofibromatosis. Neurofibromatosis type 1 (NF1), also known as von Recklinghausen disease, is an autosomal-dominant disorder with an incidence of approximately 1 in 3000. It is caused by mutations of the NF1 gene, located at chromosome 17q11.2 and coding for a tumor suppressor gene, neurofibromin. Spontaneous new mutations occur in 30% to 50% of cases. Neurofibromin is a major negative regulator of a key signal transduction pathway in cells, the Ras pathway, which transmits mitogenic signals to the nucleus. Loss of neurofibromin leads to increased levels of activated Ras (bound to guanosine triphosphate [GTP]) and increased downstream mitogenic signaling. Somatic mosaicism, in which an abnormality in one copy of the NF1 gene is present in some cells but not others, indicates a postzygotic mutation, and is called segmental neurofibromatosis. NF2 is an autosomal-dominant disorder with an incidence of 1 in 33,000. Half the cases have no family history. The NF2 gene is a tumor suppressor gene on chromosome 22 that codes for a protein called merlin. Merlin is similar to a family of proteins that serve as links between integral membrane proteins and the cytoskeleton and are involved in Rho-mediated signal transduction.
Clinical Manifestations
The cardinal features of neurofibromatosis are café au lait spots, axillary freckling, cutaneous neurofibromas, and iris hamartomas (Lisch nodules). Café au lait spots are present in more than 90% of patients who have NF1 (Fig. 186-1). They typically appear in the first few years of life and increase in number and size over time. The presence of six or more spots larger than 5 mm suggests the diagnosis. Lisch nodules also increase in frequency with age and are present in more than 90% of adults who have NF1. Approximately 25% of children exhibit these iris nodules.
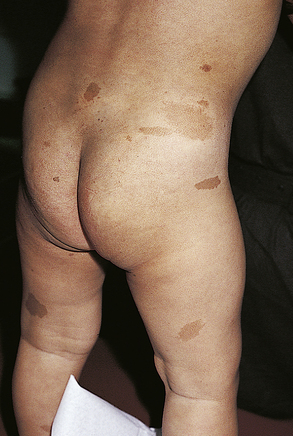
FIGURE 186-1 Café au lait macules.
(From Kliegman RE, Behrman RE, Jenson HB [eds]: Nelson Textbook of Pediatrics, 18th ed. Philadelphia, WB Saunders, 2007 p 2680.)
Neurofibromas are composed of various combinations of Schwann cells, fibroblasts, mast cells, and vascular elements. Dermal neurofibromas are nearly universal and consist of discrete, small, soft lesions that lie within the dermis and epidermis and move passively with the skin. They rarely cause any symptoms. Plexiform neurofibromas are large, occasionally nodular, subcutaneous lesions that lie along the major peripheral nerve trunks. They often cause symptoms including pain, weakness, and invasion of adjacent viscera or spinal cord. Sarcomatous degeneration may occur. Surgical treatment is attempted, but results are often unsatisfactory. Other tumors that occur in NF1 are optic nerve gliomas, astrocytomas of brain and spinal cord, and malignant peripheral nerve tumors.
NF2 predisposes patients to multiple intracranial and spinal tumors, including bilateral acoustic schwannomas, schwannomas of other cranial and spinal nerves, meningiomas, and gliomas. Peripheral nerve tumors, including schwannomas and neurofibromas, are uncommon. The average life span is less than 40 years. Posterior capsular or cortical cataracts are common, but Lisch nodules, café au lait spots, and axillary freckling are not features of the disease.
Common complications are learning disability, scoliosis, macrocephaly, headache, and optic gliomas. Other skeletal complications include sphenoid wing dysplasia and cortical thinning of the long bones with pseudarthrosis. Systemic complications, including peripheral nerve malignancy, pheochromocytoma, renovascular hypertension, and epilepsy, are individually rare. Hyperintense lesions on T2-weighted magnetic resonance images (hamartomas) in the basal ganglia, internal capsule, thalamus, cerebellum, and brainstem are common and distinctive for the disease. They are benign and disappear in adulthood. The average life expectancy of patients with NF1 may be reduced by 10 to 15 years, and malignancy is the most common cause of death. Genetic and psychological counseling are important components of care for this chronic disorder.
TUBEROUS SCLEROSIS
Etiology
Tuberous sclerosis, an autosomal dominant disorder, is characterized by hamartomas in many organs, especially the brain, eyes, skin, kidneys, and heart. The incidence is 1 in 10,000 births. Two thirds of cases are sporadic and thought to represent new mutations. Germline mosaicism is uncommon, but explains how parents who apparently do not have the disease can have multiple children with tuberous sclerosis. Mutations, affecting either of the presumed tumor suppressor genes TSC1 (chromosome 9) or TSC2 (chromosome 16), cause tuberous sclerosis. The TSC1 and TSC2 genes encode distinct proteins, hamartin and tuberin, which are widely expressed in the brain. These proteins may interact as part of a cascade pathway that modulates cellular differentiation, tumor suppression, and intracellular signaling. Tuberin has a GTPase-activating, protein-related domain that may contribute to a role in cell cycle passage and intracellular vesicular trafficking. Somatic loss or intragenic mutation of the corresponding wild-type allele is seen in the associated hamartomas. Among sporadic tuberous sclerosis cases, mutations in TSC2 are more frequent and often accompanied by more severe neurologic deficits.
Clinical Manifestations
The classic clinical features are facial angiofibromas (adenoma sebaceum), mental retardation, and epilepsy. Less than 50% of patients with tuberous sclerosis exhibit all three features. Other major signs are ungual fibromas, retinal hamartomas, hypopigmented macules, shagreen patches, renal angiomyolipoma, cardiac rhabdomyoma, brain tubers, and brain subependymal nodules and astrocytomas. Facial angiofibromas do not develop until 2 to 5 years of age, but hypomelanotic macules, called ash-leaf spots, are present in infancy and best detected with a Wood lamp under ultraviolet light. Shagreen patches are elevated, rough plaques of skin with a predilection for the lumbar and gluteal regions that develop in late childhood or early adolescence. Cardiac rhabdomyomas are largest during prenatal life and infancy and are rarely symptomatic. Occasionally, they may cause arrhythmias or cardiac outflow obstruction. Renal angiomyolipomas may undergo malignant transformation and are the most common cause of death in adults with tuberous sclerosis. Tubers in the cerebral cortex are areas of cerebral dysplasia that, in combination with other microscopic areas of abnormal development, are responsible for the symptoms of mental retardation and epilepsy. Subependymal nodules are hamartomas that may mutate into a growth phase and become subependymal giant cell astrocytomas causing obstruction of cerebrospinal fluid (CSF) outflow and hydrocephalus. These brain lesions can be detected directly by magnetic resonance imaging (MRI). Computed tomography (CT) shows periventricular calcifications within the nodule, especially around the foramen of Monro. Specific DNA probes for TSC1 and TSC2 are available.
Tuberous sclerosis is one of the most common causes of infantile spasms. These children often develop intractable epilepsy, with myoclonic, atonic, partial, and grand mal seizures; mental retardation; autism; and hyperactivity.
STURGE-WEBER SYNDROME
The Sturge-Weber syndrome is characterized by angiomas of the leptomeninges overlying the cerebral cortex in association with an ipsilateral facial port-wine nevus that, at the least, covers part of the forehead and upper eyelid. The nevus may have a much more extensive and even bilateral distribution. This nevus flammeus is an ectasia of superficial venules, not a hemangioma, because it has no endothelial proliferation. Ocular defects of Sturge-Weber syndrome include glaucoma and hemangiomas of the choroid, conjunctiva, and episclera. Glaucoma is present in 30% to 50% of patients and may be progressive. Sturge-Weber syndrome is sporadic and not genetic.
The most common associated neurologic abnormality is seizures. Seizures develop because of ischemic injury to the brain underlying the meningeal angiomas. Angiomas, most commonly located in the posterior parietal, posterior temporal, and anterior occipital lobes, consist of thin-walled veins within the pia mater. They produce venous engorgement and presumably stasis within the involved areas. Positron emission tomography has shown hypoperfusion and hypometabolism in these areas. In some children with Sturge-Weber syndrome, progressive ischemia of the underlying brain develops, resulting in hemiparesis, hemianopia, intractable focal seizures, and dementia. Calcium is detectable in the gyri of the brain underlying the angioma, and, as the intervening sulci are spared, the radiologic picture of tram track or railroad track calcifications is seen in about 60% of cases. Many children with Sturge-Weber syndrome are intellectually normal, and seizures are well controlled with standard anticonvulsants. Hemispherectomy has been proposed for infants whose seizures begin early in life and are difficult to control. Intellectual and motor outcome seems improved, but the surgical risks of the procedure are considerable. Laser surgery is the most promising therapeutic option for cosmetic management of the facial nevus flammeus. Expert ophthalmologic management of glaucoma and choroidal hemangiomas is required.