104 Hematologic Disorders
Many hematologic disorders, both hereditary and acquired, manifest during the first week of life. Early recognition of disease processes, an understanding of their pathogenesis, and prompt institution of necessary (and often lifesaving) therapies are vital.
RBC Disorders
Hemolytic Anemias
Etiology and Pathogenesis
ABO incompatibility primarily occurs in blood group O mothers with fetuses who have blood group A or B. All group O individuals have anti-A and anti-B antibodies that are produced as a result of immune stimulation by the A or B antigens contained in food and bacteria. Interactions between these maternal isoantibodies and fetal red blood cells (RBCs) result in hemolysis. Fifteen percent of pregnancies are ABO incompatible, yet evidence of ABO incompatibility disease is found in only 3% of pregnancies and necessitates exchange transfusion (ET) in fewer than 1% of pregnancies. This is because ABO hemolytic disease tends to occur in newborns whose mothers have high levels of immunoglobulin G (IgG) antibody. Although anti-A and anti-B antibodies are found in the plasma as IgA, IgM, and IgG, only the latter can cross the placenta and interact with fetal RBCs.
Rh incompatibility affects one of every 15 pregnancies and causes a wide variety of symptoms in the fetus, ranging from mild to severe hemolytic anemia and hydrops fetalis. Sensitization to the Rh (D) antigen is the result of exposure of an Rh-negative mother to Rh-positive blood. Possible exposures include prior pregnancy with an Rh-positive fetus, fetomaternal hemorrhage, and obstetric procedures (e.g., amniocentesis, chorionic villus sampling, abortion). Unlike A or B antigens, which are expressed on a number of different tissues, Rh antigens are expressed only on RBCs. Thus, maternal anti-Rh (anti-D) IgG antibodies (Rh-negative mother) cross the placenta and interact with a greater number of fetal RBCs (Rh-positive infant), resulting in significant fetal hemolysis.
Clinical Presentation and Differential Diagnosis
Both ABO incompatibility and Rh disease are associated with jaundice within the first 24 hours of life. In cases of severe hemolytic disease (i.e., erythroblastosis fetalis), infants also present with signs of hydrops fetalis (ascites, pleural or pericardial effusions, edema), pallor (secondary to anemia), petechiae or purpura (caused by thrombocytopenia), and hepatosplenomegaly (result of extramedullary hematopoiesis and splenic sequestration) (Figure 104-1). Each manifestation has a long list of possible causes, but the combination of jaundice and anemia with any of the above findings should focus clinical attention on diseases associated with hemolysis.
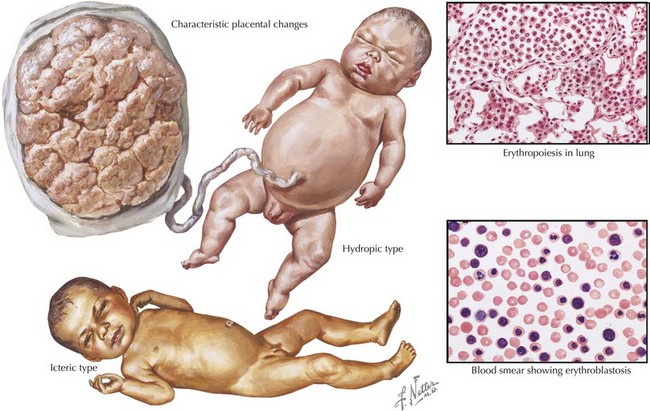
The differential diagnosis of neonatal anemia includes chronic or acute blood loss, congenital disorders of erythrocyte production (e.g., Fanconi’s anemia, Diamond-Blackfan), erythrocyte membrane defects (e.g., hereditary spherocytosis, hereditary elliptocytosis), congenital enzyme deficiencies (e.g., G6PD [glucose-6-phosphate dehydrogenase], pyruvate kinase), infection, and hemoglobin disorders (Figure 104-2). Of the hemoglobinopathies, α-thalassemias are the most common and severe. The switch from fetal (α2γ2) to adult (α2β2) hemoglobin occurs during the first year of life. As a result, defects in α-globin synthesis manifest in utero, whereas defects in β-globin synthesis become apparent in late infancy. Deletion of three (hemoglobin H disease) or four (hemoglobin Barts) α-globin genes can cause significant hemolytic anemia and present as hydrops fetalis. Newborn screening enables early detection and treatment of infants with major hemoglobinopathies and therefore reduces the mortality and morbidity associated with these conditions.
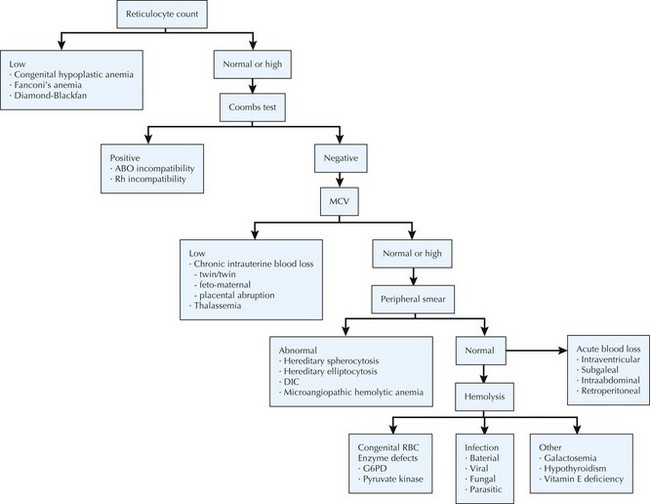
Diagnostic Approach
Hemolytic disease of newborns is associated with rapidly progressive or prolonged indirect hyperbilirubinemia, signs of hemolysis on peripheral blood smear (e.g., schistocytes and spherocytes), an elevated reticulocyte count, and anemia. Blood group testing reveals evidence of ABO or Rh incompatibility between the mother and newborn. Direct and indirect Coombs’ testing is positive in Rh disease but only weakly positive in ABO incompatibility. Infants with Rh disease should be monitored for thrombocytopenia (caused by liver dysfunction or disseminated intravascular coagulation [DIC]), hypoglycemia (secondary to islet cell hyperplasia of the pancreas), and direct hyperbilirubinemia (may be the result of hepatocellular damage).
Management and Therapy
Phototherapy and ET are the primary modes of treatment for infants with hemolytic disease. In Rh incompatibility, intensive phototherapy should be started immediately after birth. Prompt initiation of phototherapy might prevent the need for ET.
Treatment of neonatal hyperbilirubinemia with ET was introduced in the early 1950s. Although ET is proven to reduce mortality and the risk of kernicterus, it is associated with serious complications, including hemodynamic instability, apnea, coagulopathies, electrolyte imbalance, vascular thromboses, sepsis, arrhythmias, and necrotizing enterocolitis (NEC) (see Chapter 100). Efforts to reduce perinatal mortality and the need for ET have led to the development of several prenatal care strategies, including RhoGAM, use of Doppler ultrasound to detect fetal anemia, and intrauterine blood transfusions.
The use of RhoGAM (anti-D prophylaxis) in Rh-negative women has led to a marked decline in Rh sensitization and hemolytic disease of the newborn. Studies have demonstrated that of all Rh-sensitized pregnancies with Rh-positive fetuses, 51% require no treatment, 31% require treatment after full-term delivery, 10% are delivered early and need ET, and 9% require intrauterine fetal transfusion. In developed countries, a high proportion of clinically significant hemolytic disease is now caused by antibodies to antigens other than D (i.e., anti-C, anti-E, or anti-Kell) and therefore is not preventable with RhoGAM.
Intravenous immunoglobulin (IVIG) is a supplemental therapy that may be effective in reducing the need for ET in infants with immune-mediated hemolytic disease. In isoimmune hemolysis, RBCs are destroyed by an antibody-dependent cytotoxic process directed by Fc receptor–bearing cells of the reticuloendothelial system. IVIG’s mechanism of action is postulated to be attributable to nonspecific blockade of Fc receptors. Potential benefits of IVIG over ET include relative ease of administration, reduced invasiveness, and improved safety profile. Preliminary studies have demonstrated lower maximum bilirubin levels and shorter durations of hospitalization among patients receiving IVIG treatment.
Polycythemia
Etiology and Pathogenesis
Polycythemia can be subdivided into three categories based on the underlying etiology: (1) increased RBC mass and plasma volume secondary to maternal diabetes or “blood transfusion” (e.g., delayed cord clamping, twin–twin, or maternal–fetal transfusion); (2) increased RBC mass and normal plasma volume related to a congenital syndrome (trisomies 13, 18, and 21); and (3) increased RBC mass and normal or decreased plasma volume caused by intrauterine growth retardation, placental insufficiency, maternal hypertension, or smoking. The incidence of polycythemia is 1% to 5% in all neonates versus 10% to 15% in neonates who are small for gestational age.
Clinical Presentation and Differential Diagnosis
Polycythemic infants appear ruddy and plethoric with sluggish capillary refill and poor peripheral perfusion. Hyperviscosityof blood results in increased resistance to blood flow and decreased oxygen delivery. Although most neonates with polycythemia are asymptomatic, it can cause abnormalities in central nervous system function (lethargy, apnea, tremors or jitteriness, poor feeding, and hypotonia), decreased renal function (oliguria, proteinuria, and hematuria), cardiorespiratory distress (tachypnea, cyanosis, and cardiomegaly), and coagulation disorders. Rare complications include strokes, seizures, congestive heart failure, renal vein thrombosis, DIC, and NEC. Studies have also noted associations between hyperviscosity and long-term motor and cognitive neurodevelopmental disorders.
Diagnostic Approach
Polycythemia is defined as a venous hematocrit above 65%. In neonates, hematocrit (Hct) levels peak at 2 hours of life and then progressively decrease, stabilizing by 6 to 24 hours of life. Capillary Hct values are significantly higher than venous values, and arterial and umbilical vessel values are generally lower. Infants should also be monitored for hypoglycemia, hypocalcemia, hyperbilirubinemia, and thrombocytopenia.
Management and Therapy
Partial ET (PET) is used as a method to lower Hct and treat hyperviscosity. Studies have shown that PET reduces pulmonary vascular resistance and increases cerebral blood flow velocity. PET should be performed in symptomatic infants with Hct greater than 65% or asymptomatic infants with Hct greater than 70%. Either crystalloid (normal saline) or colloid (albumin) solution may be used. The volume to be exchanged is based on the observed and desired Hct (generally 50%–55%). Exchange volume (mL) = Blood volume × (Observed Hct – Desired Hct)/Observed Hct. Blood volume is estimated to be 80 to 90 mL/kg. During this procedure, blood is removed using a central venous or arterial line and replaced with fluid infused via a peripheral intravenous line. Complications of PET are thought to be similar to a single- or double-volume ET (Figure 104-3).

Platelet Disorders and Coagulopathies
Thrombocytopenia
Etiology and Pathogenesis
Thrombocytopenia is diagnosed in 1% to 5% of newborns and 22% to 35% of infants admitted to neonatal intensive care units. The most frequent cause of early-onset thrombocytopenia (<72 hours of life) is reduced megakaryopoiesis secondary to chronic fetal hypoxia from maternal diabetes, pregnancy-induced hypertension (PIH), or intrauterine growth restriction. Late-onset thrombocytopenia (>72 hours of life) is caused by sepsis (e.g., bacterial infection with group B β-hemolytic streptococci, Escherichia coli, Enterococcus spp.) or NEC in more than 80% of cases. Less common disorders that present with thrombocytopenia at birth include congenital viral infections (e.g., cytomegalovirus [CMV]), perinatal asphyxia, aneuploidy (e.g., trisomies 13, 18, and 21), neonatal alloimmune thrombocytopenia (NAIT), and neonatal autoimmune thrombocytopenia. NAIT results from the transplacental passage of maternal IgG antibodies directed against fetal platelet antigens inherited from the father and absent on maternal platelets. Neonatal autoimmune thrombocytopenia affects one or two per 1000 pregnancies and is associated with the transplacental passage of maternal platelet autoantibodies in mothers with idiopathic thrombocytopenic purpura (ITP) or systemic lupus erythematosus.
Clinical Presentation and Differential Diagnosis
Affected neonates may be asymptomatic, present with petechiae or purpura, or have symptoms of bleeding. Early-onset thrombocytopenia is generally mild to moderate and self-limiting (usually resolves within 10 days). Late-onset thrombocytopenia develops rapidly over 1 to 2 days, can be severe (platelets <30 × 109/L) depending on the cause, and may take several weeks to recover. NAIT often occurs in the first pregnancy (almost 50% of cases), is extremely severe (platelets <20 × 109/L), and may result in major bleeding (intracranial, pulmonary, renal). Intracranial hemorrhage (ICH) is seen in 10% to 20% of pregnancies with untreated NAIT versus fewer than 1% of mothers with ITP. Bleeding risk is highest in patients with NAIT followed by sepsis or NEC and chronic fetal hypoxia.
Diagnostic Approach
Thrombocytopenia is defined as a platelet count below 150 × 109/L. Neonates with thrombocytopenia secondary to chronic fetal hypoxia have additional hematologic abnormalities including transient neutropenia, increased circulating nucleated RBCs with or without polycythemia, elevated erythropoietin levels, and evidence of hyposplenism (spherocytes, target cells, Howell-Jolly bodies). The diagnosis of NAIT is made by demonstrating platelet antigen incompatibility between the mother and baby serologically or by polymerase chain reaction.
Management and Therapy
Platelet transfusion is the only specific therapy for neonatal thrombocytopenia. Clear indications for transfusion include active bleeding in association with thrombocytopenia, neonates in the first week of life with platelets below 50 × 109/L, and severe thrombocytopenia (platelets <30 × 109/L). Infants of mothers with autoimmune disease should have their platelet counts determined at birth. In those with thrombocytopenia, a platelet count should be repeated after 2 to 3 days (time of platelet nadir). If severe thrombocytopenia develops, treatment with IVIG (1 g/kg) for 2 days is usually effective.
Management of pregnancies with NAIT remains controversial, although most centers now rely on noninvasive strategies. A recent study of women with known human platelet antigen incompatibility treated with IVIG alone at a dose of 1 g/kg weekly (beginning at 16 weeks of gestation if the previous sibling had an ICH and 32 weeks if not) resulted in live births, no ICH, and no neonatal deaths. Because of the high risk of ICH in neonates with NAIT, platelet counts should be maintained above 50 × 109/L for the first 2 weeks of life, and all infants with severe thrombocytopenia should undergo a head ultrasound to look for evidence of ICH. Transfusion of PLA1-negative platelets is usually required to achieve an increase in platelet count.
Hemorrhagic Disease of the Newborn
Etiology and Pathogenesis
Hemorrhagic disease of the newborn (HDN) is caused by low plasma levels of vitamin K–dependent clotting factors (II, VII, IX, X), which are synthesized in the liver. Concentrations of these factors in neonates are 30% to 60% of those in adults. In the absence of prophylactic vitamin K, HDN occurs in 1 in 200 to 400 infants. Although placental transfer of vitamin K does occur, it is not always adequate. As a result, infants with insufficient enteral intake of vitamin K can quickly become deficient. Breast milk contains lower amounts of vitamin K than formula, thus increasing the risk of vitamin K deficiency in breastfed infants.
Clinical Presentation and Differential Diagnosis
Classic HDN is observed on days 1 to 7 of life and associated with bleeding from the gastrointestinal tract, cutaneous sites (e.g., circumcision), and nasal passages. Late HDN occurs during weeks 2 to 12. Common sites of bleeding include intracranial, cutaneous, and gastrointestinal.
Diagnostic Approach
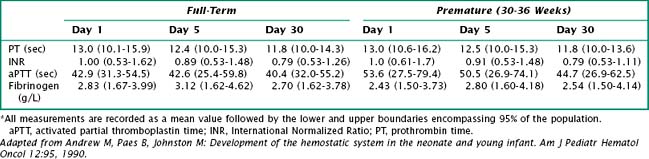
Vitamin K deficiency results in a prolonged prothrombin time (PT) and an International Normalized Ratio (INR) above 1. PT depends on various clotting factors, several of which are vitamin K dependent. INR compares the blood coagulation status of an individual to that of the normal population. Thus, an INR greater than 1 indicates that coagulation is slower than in the control group (Table 104-1).
Management and Therapy
A single dose (1 mg) of intramuscular vitamin K after birth is effective in preventing classic HDN. Oral vitamin K prophylaxis has been shown to improve indices of coagulation status at 1 to 7 days but has not been tested in randomized trials. Because it takes approximately 2 hours for systemically administered vitamin K to increase levels of vitamin K–dependent factors, infants with bleeding secondary to vitamin K deficiency should also be treated with plasma.
Future Directions
Randomized, controlled trials are needed to define the safe lower limit for platelet counts in sick newborns and provide evidence that platelet transfusion improves neonatal outcomes. With regard to HDN, a trial comparing multiple oral doses of vitamin K with a single intramuscular dose could potentially provide a cost-effective and less invasive alternative to vitamin K injection at birth.
Leukocyte Disorders
Neutropenia
Etiology and Pathogenesis
When evaluating neonates with neutropenia, it is important to first determine whether the cytopenia results from a defect in cellular production or peripheral destruction or is of mixed etiology (Box 104-1). The most common causes of neonatal neutropenia are maternal PIH, sepsis, and congenital viral infections (e.g., CMV, parvovirus, HIV, hepatitis B, rubella). Infants with severe and prolonged neutropenia should be evaluated for immune-mediated conditions and inherited genetic mutations.
Clinical Presentation and Differential Diagnosis
The neutropenia of maternal pregnancy-induced hypertension and sepsis is generally transient, rarely persisting for more than 72 hours. In contrast, infants with immune-mediated and inherited disorders show evidence of severe neutropenia for many weeks to months and can have recurrent bacterial infections. Clinical signs of neutropenia in this population include ulcerations of the oral mucosa or gingival inflammation. Otitis media, skin infections (cellulitis, pustules, abscesses), adenitis, pneumonia, and bacterial sepsis can also occur. The most common offending organisms are Staphylococcus aureus and gram-negative bacteria derived from the child’s skin or bowel flora.
Diagnostic Approach
Normal values for the absolute neutrophil count (ANC) vary by age, particularly during the first weeks after birth. The lower limit of normal is 6000/µL during the first 24 hours after birth, 5000/µL for the first week, 1500/µL during the second week, and 1000/µL between 2 weeks and 1 year of age. Severe neutropenia is defined as an ANC of less than 500/µL.
The initial evaluation of an infant with severe neutropenia should include a thorough history and physical examination. It is critical to know whether there is a family history of neutropenia or associated congenital anomalies suggestive of an inherited syndrome. If additional evaluation is warranted, antineutrophil antibody titers (elevated in immune-mediated neutropenias) and immunoglobulin quantification (decreased levels in underlying immunodeficiency) may be performed. Neonates with severe and prolonged neutropenia should be referred to a hematologist for bone marrow examination and possible genetic testing.
Management and Therapy
Infants with fever and severe neutropenia should be hospitalized and started on parenteral broad-spectrum antibiotics. Specific recommendations for antibiotic coverage depend on the prevalence of organisms in each community or hospital and their susceptibility patterns.
If recovery from neutropenia is not expected, as in inherited syndromes, granulocyte colony-stimulating factor (G-CSF) administration or stem cell transplantation may be necessary. G-CSF has been shown to mobilize preformed neutrophils from the bone marrow, promote neutrophil precursor proliferation, and enhance phagocytic bactericidal function. However, there is currently insufficient evidence to support the use of G-CSF in neutropenic neonates with systemic infection or as prophylaxis to prevent systemic infection in high-risk neonates.
Future Directions
Although children with inherited bone marrow failure syndromes often respond to treatment with G-CSF, many physicians remain concerned that it may increase the risk of malignant transformation. Further research is needed to help elucidate the mechanisms by which such transformations occur and to monitor long-term clinical outcomes.
Aher S, Malwatkar K, Kadam S. Neonatal anemia. Semin Fetal Neonatal Med. 2008;13(4):239-247.
Alcock GS, Liley H: Immunoglobulin infusion for isoimmune haemolytic jaundice in neonates. Cochrane Database Syst Rev (3):CD003313, 2002.
Sarkar S, Rosenkrantz TS. Neonatal polycythemia and hyperviscosity. Semin Fetal Neonatal Med. 2008;13(4):248-255.
Soll R, Schimmel MS, Ozek E: Partial exchange transfusion to prevent neurodevelopmental disability in infants with polycythemia (protocol). Cochrane Database Syst Rev (1):CD005089, 2009.
Steiner LA, Gallagher PG. Erythrocyte disorders in the perinatal period. Semin Perinatol. 2007;31(4):254-261.
Platelet Disorders and Coagulopathies
Puckett RM, Offringa M: Prophylactic vitamin K for vitamin K deficiency bleeding in neonates. Cochrane Database Syst Rev (4):CD002776, 2000.
Roberts I, Stanworth S, Murray NA. Thrombocytopenia in the neonate. Blood Rev. 2008;22(4):173-186.
van den Akker ES, Oepkes D, Lopriore E, et al. Noninvasive antenatal management of fetal and neonatal alloimmune thrombocytopenia: safe and effective. BJOG. 2007;114(4):469-473.
Carr R, Modi N, Doré C: G-CSF and GM-CSF for treating or preventing neonatal infections. Cochrane Database Syst Rev (3):CD003066, 2003.
Rivers A, Slayton WB. Congenital cytopenias and bone marrow failure syndromes. Semin Perinatol. 2009;33(1):20-28.
Segel GB, Halterman JS. Neutropenia in pediatric practice. Pediatr Rev. 2008;29(1):12-23.