CHAPTER 17 Paediatric tumors
Clinical aspects
Incidence of paediatric tumors and histologic types
Paediatric tumors show a distinctive incidence, histology, and biologic behavior from those in adults. In addition, fetal and neonatal malignancies tend to differentiate or regress spontaneously, leading to high survival and curability rates.1 In the United States, only 2% of patients with a malignancy are in the paediatric age group, with almost 9000 children under the age of 15 presenting with a malignancy each year.2 Even with improved overall 5-year survival from 27% in 1960 to over 70% in the 1990s, cancer still remains a leading cause of childhood mortality. The type of malignancy varies considerably within age groups. In children younger than 5 years, acute lymphoblastic leukemia is the most frequent and most lethal cancer.3 The most common solid tumours of childhood are primary posterior fossa brain tumors and teratoma, followed by neuroblastoma and soft tissue sarcomas. Osteosarcoma is the most common primary bone tumor followed by Ewing’s sarcoma.
Diagnostic accuracy of FNA cytology
Fine needle aspiration biopsy (FNAB) has many advantages in the diagnosis of paediatric tumors, especially the ease of performance and repeatability with no essentially morbidity or risk of tumor upstaging.4-6 Centers experienced in performing paediatric FNAB have shown excellent results with sensitivity and specificity rates approaching 93% and 100%, respectively, comparable to those in the adult population.5-21 The availability and applicability of diagnostic ancillary techniques such as histochemistry, immunocytochemistry, electron microscopy, flow cytometry, cytogenetics, and molecular analysis to the cytology smears often enable the pathologist to give a definitive diagnosis.
FNAB of paediatric mass lesions has been slow in gaining popularity compared with its utilization in adult patients.5-21 Many pathologists and clinicians are hesitant to employ fine needle aspiration (FNA) as a diagnostic method in children for a variety of reasons, including rarity of paediatric tumors with different morphology from those of adults, lack of experience of general pathologist with such tumors, morphological overlap between different tumor types, lack of specific immunocytochemical markers and unrepresentative samples as a result of degenerative changes or tumors with heterologous components such as hepatoblastoma or Wilms’ tumor. These lead to difficulties in interpretation of FNA of paediatric tumors.1,22 There is a recent decline in the number of aspirates from patients with a history of cancer as a result of recent advances of image techniques to document recurrence and relapses.23
Obtaining and handling of specimens
The procurement and preparation of specimens from paediatric FNA is no different from that in adults. However, children may not be as cooperative as adults and require sedation in radiographically guided FNA of deep-seated masses. For superficial masses, aspiration without local anesthesia is usually well tolerated with proper immobilization of the child. Multiple passes can be obtained in most cases. To avoid repeating the FNA, it is important to immediately assess the sample sufficiency. An on-site pathologist, preferably performing the aspiration procedure, can evaluate each pass for adequacy by use of a Romanowsky staining method (Wright-Giemsa or Diff-Quik®), and for triage of material for appropriate ancillary tests such as immunocytochemistry (cell-block preparation), cytogenetics, and electron microscopy.
Some centers, such as ours, have been using FNA as the first diagnostic modality in the work-up of mass lesions (superficial and deep seated) in children for providing rapid diagnostic results, thereby aiding in proper triage and management of the patient.20,24-26 Intraoperative cytology (IOC) can be useful in cases with limited tissue available, leading to a rapid assessment of the nature of the lesion, thereby, optimizing triage of tissue for the most appropriate ancillary studies (cultures, cell blocks, cytogenetics, flow cytometry, etc.). This allows for preservation of tissue for permanent section examination, especially in HIV-positive patients (avoiding cryostat contamination), or tissue prone to difficulties in cutting at the time of frozen section such as bony fragments and fatty specimens.
We use both the Papanicolaou and Romanowsky stains since the two are complimentary. Nuclear features such as chromatin distribution and granularity are better evaluated on the Papanicolaou stain, while the Romanowsky stain is superior for evaluation of lymphoreticular lesions (benign and malignant). Lesions with stromal and matrix components are also more easily discernible by Romanowsky staining, such as the bright magenta color of neuropil in the case of neuroblastoma. For immunocytochemistry, a cell block preparation works best. However, direct smears and cytospins can also be used.26
Radiographic imaging
Image-guided FNA biopsy of deep-seated masses is a well established diagnostic modality in adults and is now used more widely in the paediatric population. The modality of choice is dependent on the site of the lesion as well as the clinical impression. For most abdominal and pelvic masses, ultrasound is the method of choice for real-time FNA guidance. Computed tomography (CT) is more often used to define and localize lesions in the lung and mediastinum. Lesions involving the head and neck region can usually be visualized under ultrasound guidance. However, deep-seated lesions may require the use of contrast enhanced CT to demonstrate the exact location, especially in the case of lymphomas. Correlation of the imaging characteristics and location of the mass lesion with the patient’s age and clinical findings is necessary for an accurate diagnosis.
Ancillary techniques
Nowhere has the use of immunohistochemistry and molecular genetic studies impacted the rendering of histogenetic-specific diagnoses more than in the childhood soft tissue sarcomas, particularly small round cell tumor (SRCTs).21,27 As most paediatric patients are enrolled in histogenetic-specific protocols (e.g. Paediatric Oncology Group) following a diagnosis of sarcoma, most authors agree that although the gold standard for diagnosis remains the light microscopic evaluation, ancillary diagnostic procedures are helpful to render the specific subtyping of SRCTs.27 FNAB permits a rapid diagnosis with the availability of performing ancillary techniques such as immunocytochemistry, flow cytometry, cytogenetics, and electron microscopy (EM). It is for these reasons that FNA has been incorporated by many institutions into the diagnostic algorithm of paediatric tumors. Cell block preparations are the ideal for performance of immunocytochemical studies as they allow the performance of a panel of IHC markers. Gurley et al. reviewed the diagnostic contribution of ancillary studies performed on aspirated material in the work-up of paediatric biopsies.28 Ancillary studies were performed in 40% of their cases. Immunohistochemistry helped to narrow the differential diagnosis or classify the disease process in 42%, confirm the cytologic impression in 47%, and gave contradictory results in 10% of cases. Seventy-four percent of cases had adequate material for electron microscopy which was diagnostic or helped to classify the lesions in one-third of cases, helped exclude diagnostic consideration in 21% and was noncontributory in only 14% of cases.28
The utility of FNA material in the molecular characterization has been shown in several studies, and the majority of aspirates can yield sufficient material to perform the necessary molecular studies.22,28-30 In a study by Kilpatrick et al., among 27 patients clinically eligible for histogenetic-specific protocols, an accurate diagnosis was rendered by FNA in 25 (92%) cases.21 Kilpatrick and colleagues also showed that cytogenetic analysis can be accurately performed using FNA biopsy material to confirm the t(11;22) translocation in Ewing’s sarcoma and the t(x;18) translocation in synovial sarcomas, supporting the FNA cytologic impression (Table 17.1).21
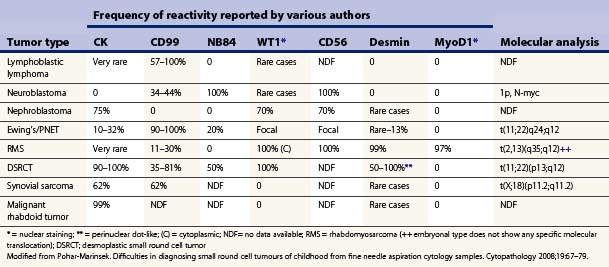
Diagnostic approach
As with FNA material from other sites, a multidisciplinary approach is critical when evaluating aspirated material from children. Specifically, besides patient age, tumor size, mobility, anatomic location of the mass, and clinical presentation (rapid versus slow growth), the radiographic findings need to be correlated with the FNA cytologic features to narrow down the differential diagnoses and avoid misdiagnoses. Most authors agree that a definitive cytologic diagnosis must be based on a combination of the cytologic findings (i.e. adequate specimen, cytomorphology) correlated with results of ancillary studies (immunohistochemistry, flow cytometry, cytogenetic analysis, electron microscopy), clinical and/or radiographic data. Close interaction between the clinician and cytopathologist is therefore an essential component to the success of paediatric FNA.
An approach to the cytologic work-up of paediatric FNA is to divide lesions conceptually into two broad groups based on the size of the malignant cells, uniformity of cell appearance, and patterns of cell arrangement.24 The majority of paediatric malignancies can be classified into either the small or large cell categories, although there are occasional tumors such as rhabdomyosarcoma that have cytologic features that bridge both groups.20 This is especially important since many of the paediatric malignancies fall into the category of small round cell (blue cell) tumors (SRCTs) of childhood.2,20,31 These neoplasms consist of a uniform cell populations having diameters up to approximately three times that of a small mature lymphocyte and typically possess only a single hyperchromatic nucleus with finely granular, evenly distributed chromatin. The cytoplasm is generally scanty, resulting in very high nuclear to cytoplasmic ratios. The much less frequent ‘large cell’ category of malignancies is composed of pleomorphic cell populations with more abundant cytoplasm and may include multinucleated cells.20 Characteristically, coarsely clumped chromatin with irregular distribution and prominent nucleoli are present. However, for both categories, particularly SRCTs, definitive diagnosis often requires ancillary studies such as immunocytochemistry, EM, cytogenetic and molecular studies.21,23,25,28,32
This chapter will focus on the FNA cytologic features of neoplasms that are seen predominantly in the paediatric age group. Lesions that occur in both the adult and paediatric population are discussed elsewhere in this book.
Cytological findings
Small round cell tumors (SRCTs)
Definitive cell typing of SRCT is mandatory for enrollment of patients in specific therapeutic protocols, which has led to a significant increase in the disease-free survival rates. Immediate cytologic assessment is a critical step that helps to establish the initial diagnostic impression and points to the need for additional tissue material for pertinent ancillary studies.25,29,33 Aktar et al.26 and Layfield15 have expressed the importance of a complete history, physical examination, and radiological and laboratory evaluations in arriving at a definitive diagnosis of SRCTs.
Neoplasms which are conventionally considered in the SRCT category include the prototypical neuroblastoma along with rhabdomyosarcoma, Ewing’s sarcoma/primitive neuroectodermal tumor (EWS/PNET), intra-abdominal desmoplastic small round cell tumor, leukemia and malignant lymphoma.10,34 Other childhood malignancies that are in the differential diagnosis include small cell osteosarcoma, undifferentiated (small cell) hepatoblastoma, blastemal Wilms’ tumor, rhabdoid tumor of soft tissue, synovial sarcoma and granulocytic sarcoma.35,36
All SRCTs are uniformly characterized by the presence of sheets of monomorphic cells with subtle architectural and cytomorphologic features that serve as clues to the correct diagnosis.22 Morphological similarity and lack of immunocytochemical specificity in SRCT are reasons for difficulties in specific diagnosis of SRCT on cytology.22 Some SRCTs are so poorly differentiated that they lack specific antigens. In addition, cross-reactivity exists for many antigens among certain SRCTs.22 However, correct cell typing is possible and achievable in 92% of FNAB of SRCTs with the judicious use of various ancillary studies.4,18,21 A variety of cytologic variables including nuclear, cytoplasmic, and architectural features were analyzed as to their association with the final histologic typing to determine the most predictive characteristics for each sarcoma. Using the previous cytologic features, Layfield et al. performed logistic regression analysis of 59 cases with diagnoses of Ewing’s sarcoma/PNET, rhabdomyosarcoma, neuroblastoma, Wilms’ tumor and lymphoma.34 Among the entire group, Ewing’s sarcoma/PNET was the most difficult to specifically diagnose by cytomorphology alone. In most cases, it was the ‘lack’ of diagnostic criteria for the other round cell sarcomas that was used to make a diagnosis of Ewing’s sarcoma.34
Neuroblastoma is considered the ‘prototypical’ small round cell tumor of childhood.25 It is the most common solid nonlymphoreticular malignancy in the paediatric age group, with the majority of patients diagnosed before 5 years of age and 50% before 2 years of age. It is also the most common malignancy in the neonate.20,37,38
Neuroblastoma can be found in any site which harbors sympathetic neural tissue. The most common sites of occurrence, comprising two-thirds of cases, are the adrenal gland and retroperitoneal sympathetic ganglia.18 Neuroblastoma also occurs in other neural crest sites such as the thoracopulmonary, mediastinal, cervical and, less commonly, pelvic regions.2,20,30 Approximately one-third of children with neuroblastoma present with metastatic disease to regional lymph nodes, bone marrow, liver, bone and/or lung at the time of diagnosis.37,38 The prognosis of patients with neuroblastoma correlates with age, site of involvement and stage. Adrenal neuroblastoma has a less favorable prognosis than its extra-adrenal counterpart.14 In addition, the presence of a stroma-rich matrix, low mitotic–karyorrhectic cell index, and increased numbers of differentiating tumor cells are correlated with favorable prognosis.35,39-41
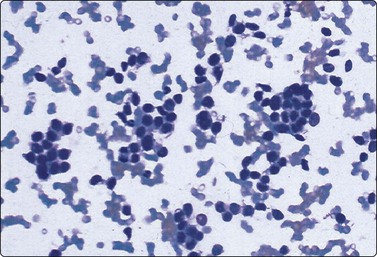
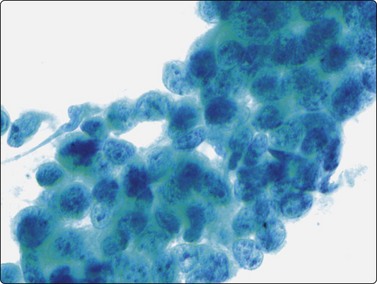
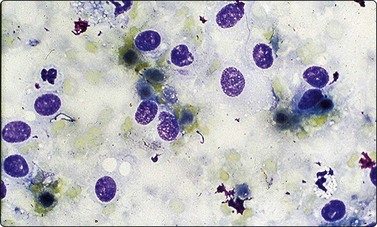
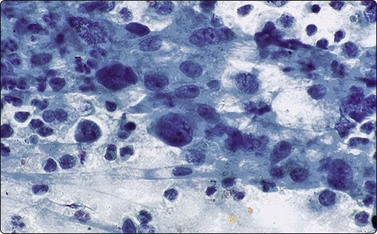
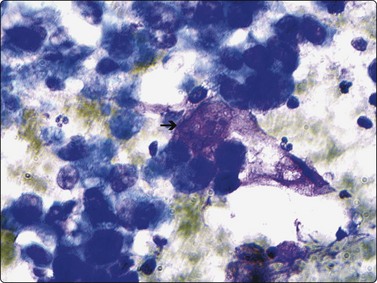
Neuroblastoma usually forms a well-defined solid tumor <10 cm in size with hemorrhage and extentive necrosis. The FNA usually yields hypercellular smears with predominant individually scattered small anaplastic cells, showing prominent nuclear molding.27,41,42 The prototypical neuroblastic cells have high nuclear to cytoplasmic ratios with single nuclei that are oval to slightly irregular in shape containing evenly dispersed granular chromatin (salt and pepper) and small to inconspicuous nucleoli (Fig. 17.1). Small round cells are arranged in moderately or well-formed Homer-Wright rosettes surrounding centrally located neuropil, which stains pink or blue–gray in Giemsa-stained smears. The presence of Homer-Wright rosettes is diagnostic but not present in all cases (Fig. 17.2). Neuropil, either associated with the rosettes or present in the smear background, is the most helpful cytologic feature for rendering a definitive cytologic diagnosis of neuroblastoma.34 Neuropil consists of a fibrillary tangle of neuritic processes with or without associated neuroblastic cells (Fig. 17.3). Mitotic–karyorrhectic cells and calcifications can occasionally be recognized in aspirate smears. Larger differentiating neuroblasts with moderate amounts of cytoplasm and binucleated to multinucleated ganglion cells can also be present in the smear. Some neuroblastomas may undergo different grades of maturation, forming ganglioneuroblastoma or ganglioneuroma. In ganglioneuroblastoma, the smear is pleomorphic with prominent anisonucleosis and abundant neuropil background but without ganglion cells, while ganglioneuroma demonstrates characteristic ganglion cells (Fig. 17.4).27,42
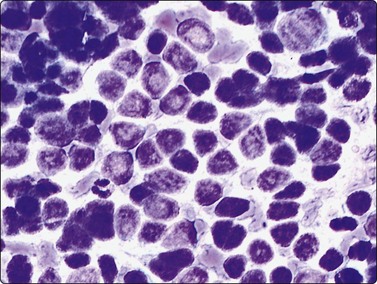
Hypercellular smears demonstrate numerous singly scattered cells with high nuclear to cytoplasmic ratios, round to oval irregular nuclei with fine granular chromatin and inconspicuous nucleoli (Diff-Quik, ×400).
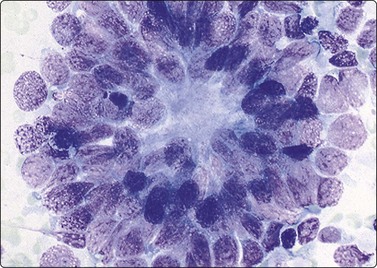
Cluster of cells arranged in a Homer-Wright rosette containing central neuropil. The presence of neuropil is the most helpful cytologic feature for rendering a definitive diagnosis of neuroblastoma (Diff-Quik, ×400).
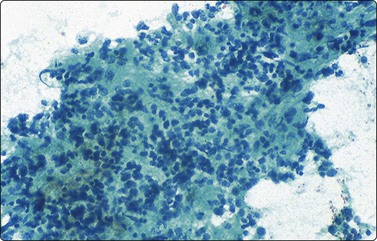
Medium power of aspirate smear containing neuroblastic cells associated with fibrillary to granular background neuropil (Pap, ×200).
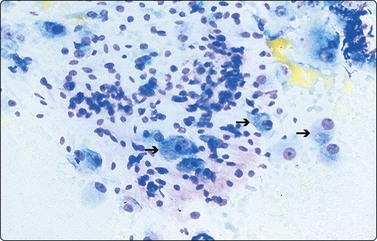
Compared with neuroblastic cells, neoplastic ganglion cells (arrows) demonstrate larger nuclei with prominent nucleoli, and moderate amounts of coarsely granular cytoplasm. These cells are seen in the two related lesions, ganglioneuroblastoma and ganglioneuroma (Diff-Quik, ×400).
The differential diagnosis of neuroblastoma includes other members of SRCTs group.42 The presence of apoptotic nuclei, nuclear molding, paranuclear ‘blue bodies’, necrotic background, with the absence of lymphoglandular bodies will help to distinguish neuroblastoma from lymphomas.24,43 Neuroblastoma composed of dissociated primitive cells without rosettes formation, is morphologically undistinguishable from blastema cells of a Wilms’ tumor or EWS/PNET. The immunocytochemical profile supportive of neuroblastoma includes positive staining for neuron-specific enolase (NSE), microtubule-associated proteins (MAPs) and/or neurofilament protein.28 Positive staining for S-100 and/or glial fibrillary acidic protein (GFAP) has occasionally been observed. CD56 can be positive in both neuroblastoma cells and EWS/PNET. However, CD56 was reported uniformly and strongly positive in all neuroblastomas and only focally in EWS/PNET.28,43 In addition, EWS/PNET is usually positive for CD99, while neuroblastoma CD99 is usually negative. Conversely, Wilms tumor can be WT1 negative in 30% and CD56 positive.43 In this setting, low molecular weight cytokeratin is helpful to differentiate neuroblastoma from Wilms’ tumor, since Wilms’ tumor is cytokeratin positive while neuroblastoma is negative, although blastema cells can occasionally be cytokeratin negative.22 Neuroblastomas are negative for desmin, myogenin, smooth muscle actin, muscle-specific actin, CD34, and CD45.43 Ultrastructural features of neuroblastoma include small dense core neurosecretory-type granules and abundant processes containing microtubules.43 Neuroblastoma usually shows a deletion or rearrangement of material of chromosome 1p and amplification of N-myc.44
This is the most frequent intraocular tumor in children, with similar cytomorphology to neuroblastoma. One-third of these tumors are bilateral and are usually seen in children younger than 2 years. Retinoblastomas appear as high-grade undifferentiated small round cell tumors. Smears show hypercellular smears formed by sheets of small round or oval hyperchromatic nuclei with numerous mitoses and inconspicuous nucleoli, often with necrotic background.1,5 Cells show molding and typically arranged in rosettes with a central lumen containing acid mucopolysaccharides resistant to hyaluronidase (Flexner-Wintersteiner rosettes), and less frequently in Homer-Wright rosettes, in which cells are arranged around neuropil matrix.4
Olfactory neuroblastoma (ONB) is a rare tumor of neuroectodermal origin that arises from epithelium that lines the upper aerodigestive tract (nasal septum, superior turbinates, cribriform plate) malignancies. Although ONB show similar cytomorphology to SRCTs, they are mainly seen in adult populations.
Nephroblastoma is the most frequent malignancy of the kidney in childhood and accounts for 6% of all paediatric cancers.20,29 Wilms tumor (WT) is the most frequent paediatric cancer in children less than 3 years old. Approximately 90% of all intra-abdominal malignancies in childhood will be either Wilms’ tumor or neuroblastoma.20 The preoperative diagnosis of Wilms’ tumor by FNA biopsy has become increasingly important since neoadjuvant chemotherapy has become the standard of care.45,46
Wilms’ tumors usually present as a single tumor, but it is sometimes multicentric and bilateral. Most cases are >10 cm in size and has a soft consistency, even surface, and well-delimited border with frequent areas of necrosis and hemorrhage.1 FNA biopsy for the primary diagnosis and management of Wilms’ tumor is contraindicated in children with a resectable renal mass.47 However, patients with unresectable Wilms’ tumor requiring neoadjuvant chemotherapy, or children with metastatic disease at the time of presentation may benefit from FNAB technique.48,49 According to the current guidelines,48 FNA through a posterior approach does not upstage the tumors, while an anterior approach technique, will upstage the tumor to stage III because of the possibility of generalized peritoneal contamination.
FNAB of conventional Wilms’ tumor shows a triphasic pattern consisting of complex tubules (epithelial component), mesenchymal differentiation and a primitive blastemal component.20,35 The blastemal cells are characterized by a relatively small size with high nuclear to cytoplasmic ratios and only a scant rim of extremely fragile cytoplasm (Figs 17.5 and 17.6). Nuclear molding can also be seen. The larger cells of the epithelial component may be arranged in tubules which range from simple tubular outlines to complex, branching luminal patterns (Fig. 17.7). These tubules have a well-defined apical border surrounding an empty luminal space with palisading of the epithelial nuclei. They can potentially be confused with the Homer-Wright rosettes of neuroblastoma.42-51 However, Homer-Wright rosettes are less complex in architecture and consist of cells arranged around central fibrillary neuropil. The mesenchymal component of Wilms’ tumor consists of fibroconnective tissue fragments containing short spindle-shaped cells with bland nuclear features, which can occasionally predominate and demonstrate a myxoid or collagenous appearance in the smears. Occasional cases can show skeletal muscle differentiation. Histologically, anaplasia in Wilms’ tumor is defined as the combination of enlarged nuclei equal to or greater than three times the size of the nuclei of adjacent cells along with nuclear hyperchromasia and abnormal multipolar mitotic figures.45 Recognition of anaplasia is crucial since it is associated with aggressive tumor behavior and decreased survival, thereby necessitating more aggressive therapy. An overdiagnosis of anaplasia in the aspirated smears, however, can be due to a number of artifacts such as calcification and stain precipitate which can simulate enlarged hyperchromatic nuclei. DNA-smearing artifact and basophilic extracellular mucinous material can also simulate anaplastic nuclei. Conversely, sampling errors can lead to an underdiagnosis of anaplasia.45
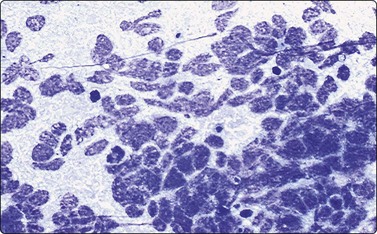
Fig. 17.5 Wilms’ tumor (nephroblastoma)
Blastemal cells form the majority of the cellular component in aspirates, present either singly or in small clusters. Blastemal cells are small with finely granular nuclear chromatin, inconspicuous nucleoli, and scant, extremely fragile cytoplasm rendering large numbers of stripped nuclei on the smears (Diff-Quik, ×400).
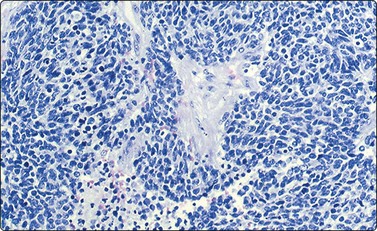
Fig. 17.6 Wilms’ tumor (nephroblastoma)
Blastemal cells can surround and/or be incorporated into stromal fragments (Diff-Quik, ×200).
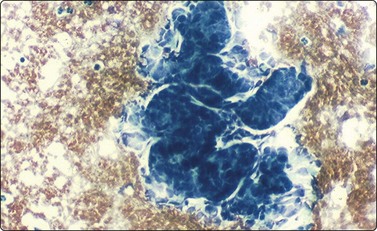
Fig. 17.7 Wilms’ tumor (nephroblastoma)
Aspirate smear demonstrating the epithelial component with glandular arrangements of primitive cells that are arranged in complex branching patterns (Diff-Quik ×400).
Separation of WT from other SRCTs can be extremely difficult, particularly if the undifferentiated blastemal component is the predominant or sole component in the smears.45,51 A careful search for epithelial and mesenchymal components favors WT, whereas a fibrillary background confirms the diagnosis of intrarenal neuroblastoma.50 Ancillary studies become crucial in rendering a specific diagnosis and separating this lesion from other SRCTs. In WT, blastemal cells stain positively for vimentin, cytokeratin (AE1/AE3), epithelial membrane antigen (EMA) and CD99 negative.30,50,52 Neuroblastomas are positive for neuron-specific enolase (NSE) but negative for cytokeratin and EMA. Lymphomas can occur in the kidney and are characterized by leukocyte common antigen (LCA, CD45) positive. The background typically demonstrates abundant diagnostic lymphoglandular bodies.
Ewing’s sarcoma/primitive neuroectodermal tumor (EWS/PNET)
Ewing’s sarcoma/primitive neuroectodermal tumor is the second most frequent malignant paediatric primary bone tumor, accounts for approximately 20% of soft tissue sarcomas in the first 2 decades of life and is the most common thoracic neoplasm.20,21,53 Around 25% of patients have metastasis in the lungs and bones at the time of diagnosis. Both PNET and Ewing’s sarcoma stain positively with monoclonal antibodies FLI-1 and CD99 (MIC-2 oncogene) and share a common chromosomal abnormality (11;22 translocation).27 The demographic and biologic behavior of both lesions are similar enough to consider these lesions as a continuous spectrum of the same tumor.54,55
Smears are cellular, composed of small cohesive groups of undifferentiated small round cells without nuclear molding (Fig 17.8). Although single dispersed cells are seen in the smear background, EWS/PNET can yield the most cohesive pattern of the SRCTs group. The cells have indistinct borders and may form pseudorosettes. A dimorphic population of lighter and darker cells may be present in Diff-Quik-stained smears, mimicking the histologic findings. Lighter-staining cells are large (approximately the size of histiocytes) and have round-to-oval, slightly pleomorphic nuclei, smooth nuclear membranes with fine chromatin and one or two small nucleoli. Cells have a moderate amount of finely vacuolated-to-clear cytoplasm that contains abundant, periodic acid–Schiff-positive, diastase-digestible glycogen granules.56 Darker-staining or lymphocytoid cells have a small, irregularly contoured nucleus with dense chromatin and a narrow rim of cytoplasm, changes that are likely a manifestation of apoptosis. The two cell types are usually intermingled in no discernible pattern, with lighter-staining cells predominating. Binucleation, multinucleation, and stromal matrix formation are not features of EWS/PNET. Homer-Wright rosettes are not usually seen. In the Diff-Quik smear, peripheral cytoplasmic vacuolization and membranous cytoplasmic blebs can be noted in occasional cases and considered as characteristic features.56,57
Highly cellular aspirate smears demonstrate individually scattered and small loose clusters of uniform small cells with round nuclei, evenly dispersed finely granular chromatin, distinctly absent nucleoli, and only a scant rim of cytoplasm (Diff-Quik, ×200).
In rare cases of EWS/PNET, the majority of the cell population has slightly oval nuclei, mimicking other SRCTs, particularly monophasic synovial sarcoma. The so-called large cell variant of EWS/PNET may be confused with large cell lymphoma, but immunophenotyping settles this diagnostic issue in most cases.55,56
Although positive staining for CD99 is quite helpful in the diagnosis of PNET/Ewing’s sarcoma, it is not specific.22,30 CD99 can be demonstrated in various percentages in other SRCTs. CD99 was reported in 57–100% of lymphoblastic lymphomas, 34–44% of neuroblastomas, 11–30% of rhabdomyosarcoma, 35–81% of DSRCT and 62% of synovial sarcomas.22,30 Ewing/PNET tumors can be negative for CD99 (8.6%). EWS/PNET can be occasionally positive for cytokeratin and desmin (about 10%). Molecular analysis plays an important diagnostic role in doubtful cases, whereas 90% show t(11;22)(q24;q12) translocation, while 10% show t(21;22)(q22;q12) translocation.22,30,54,55
Rhabdomyosarcoma (RMS) is the most frequent soft tissue sarcoma in children with two age peaks, with the first peak occurring at 4 to 5 years of age and the second in late adolescence.21 It arises often in the head and neck, followed by genitourinary tract, extremities, and trunk of children younger than 5 years old. In the paediatric population, there are two major histologic forms: the alveolar and embryonal subtypes. Embryonal subtype is the most common form in early age. In contrast, the alveolar subtype is more frequently seen in adolescents and originates most often in the extremities, paranasal sinuses and retroperitoneum.58
Although rhabdomyosarcoma is generally considered one of the SRCTs of childhood, it is associated with a relatively broad spectrum of histologic appearances.58,59 FNA demonstrates a wide range of morphology and the degree of variability greater than that seen in the other SRCTs.4 Aspiration biopsies of both embryonal and alveolar rhabdomyosarcomas are characterized by moderately to highly cellular samples which include both numerous individually dispersed tumor cells (rhabdomyoblasts) and densely packed aggregates with prominent overlapping of the cells.45,60 The smear background may show characteristic loose myxoid material which may have a metachromatic appearance with the Romanowsky stains. In other instances, the smear background is simply clean, collagenous or bubbly (tigroid) in appearance, resembling that seen in aspirates from germinomas.4,45,54,59 Some investigators have attempted to differentiate embryonal from alveolar subtype in FNAC smears and were successful in 80% of cases. The embryonal subtype may be composed almost exclusively of small primitive cellular elements with round or polygonal contours, extremely high N : C ratios, solitary nuclei and minute nucleoli. Some cells may show a higher level of differentiation with increasing volumes of eosinophilic cytoplasm and eccentrically positioned nuclei, mimicking rhabdoid tumors (Fig. 17.9).4,59 Smears of alveolar rhabdomyosarcoma are more cellular with more mature rhabdomyoblasts than embryonal rhabdomyosarcoma.59,60 Rhabdomyoblastic cells may be arranged in alveolar structures. Compared with the embryonal subtype, the malignant cells comprising the alveolar subtype of rhabdomyosarcoma are generally larger and more uniform in appearance with rounded contours, solitary hyperchromatic nuclei, prominent nucleoli and high N : C ratios (Fig. 17.10). A helpful diagnostic feature of alveolar subtype is the presence of multinucleated tumor giant cells with nuclei arranged in a wreathlike manner (Fig. 17.10). Characteristic strap cells with a solitary tapered cytoplasmic tail may be occasionally seen.58,61,62 Pohar-Marinsek and Bracko observed that the alveolar rhabdomyosarcoma exhibited two major architectural patterns: one characterized by completely dissociated cells and the other one containing many clustered formations.60 Presence of binucleate cells was an important criterion for the diagnosis of alveolar rhabdomyosarcoma.60
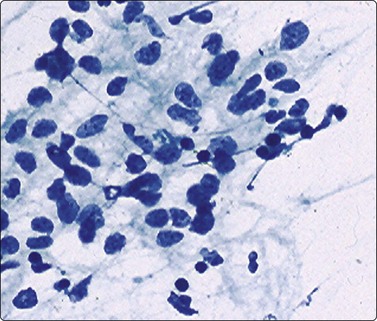
Fig. 17.9 Rhabdomyosarcoma, embryonal subtype
The cellular appearance in this subtype is highly variable with more undifferentiated cells (shown here) admixed with cells showing higher degrees of myogenic differentiation. The cells vary from round to spindled, with frequent eccentrically placed nuclei and tapering cytoplasmic tails (Pap, ×400).
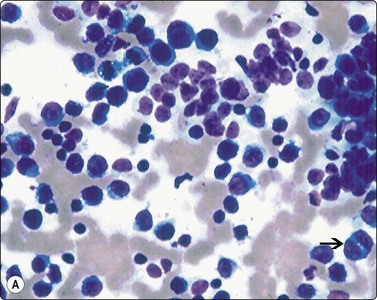
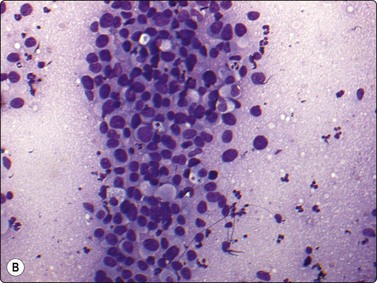
Fig. 17.10 Rhabdomyosarcoma, alveolar subtype
(A) Highly cellular smears contain large uniform cells with round nuclei, prominent nucleoli, and high nuclear : cytoplasmic ratios. A helpful diagnostic feature of the alveolar subtype is the presence of multinucleated tumor cells (arrow) (Diff-Quik, ×400); (B) Highly cellular smear shows large uniform cells with bubbly (tigroid) in appearance, resembling that seen in aspirates from germinomas (Diff-Quik, ×400).
It was believed for a long time that desmin was a specific marker for RMS.28 However, desmin positivity has been seen in other members of SRCTs, while rhabdomyosarcoma can be positive for CD99.30,45,62 WT1 shows a strong cytoplasmic staining in rhabdomyosarcoma and a nuclear one in DSRCT, while is only focal in EWS/PNET. Unlike EWS/PNET positivity for cytokeratin, rhabdomyosarcoma is negative or show only a rare positive individual cell. Rare cases have been shown to express lymphoid markers including CD10, CD19 and CD20. Recently, more specific skeletal muscle markers have become available, including myogenin and MyoD1, and are highly sensitive in the diagnosis of rhabdomyosarcoma.59,63 These markers are expressed by positive nuclear staining. In challenging cases, molecular analysis shows a specific chromosomal aberration, t(2;13)(q35;q14) chromosomal translocation and/or the gene fusion transcripts PAX-FKHR in 80% alveolar subtype, but no specific translocation has been identified in the embryonal subtype.22,61,63
Rhabdoid tumors are easily confused with rhabdomyosarcoma. The presence of perinuclear cytoplasmic globular inclusions is usually more characteristic of rhabdoid tumors and eccentrically placed nuclei with a prominent central nucleoli. Hypercalcemia, and cytokeratin positive/desmin negative immunostaining, help in accurate tumor typing,19 although some cases can be desmin positive.21,22,61,64 Aspiration biopsies yield uniform-appearing, moderately sized neoplastic cells which are characterized by rounded contours, and solitary eccentric nuclei and uniform, intensely eosinophilic cytoplasm.61 The nuclei have thick nuclear membranes and a massive nucleolus. In contrast to rhabdomyosarcomas, these smears do not contain a spectrum of cellular appearances presenting different maturation stages of rhabdomyoblasts.
Intra-abdominal desmoplastic small round cell tumor of childhood
Intra-abdominal desmoplastic small round cell tumor (DSRCT) is a very rare malignancy predominantly seen in male patients between 16 and 18 years of age.20,65,66 Although a variety of sites have been reported, the tumor most commonly involves the omentum or peritoneum with secondary invasion of the bowel wall. FNA cytologic findings include the presence of groups of undifferentiated malignant cells showing nuclear molding, associated with desmoplastic stroma (Fig. 17.11).14,67,68 DSRCT demonstrates multidirectional differentiation based on expression of both epithelial and mesenchymal immunocytochemical markers including cytokeratin (AE1/AE3), EMA, desmin and NSE.66,68 The majority of cases are also positive for vimentin and WT1.68 A specific chromosomal abnormality can be demonstrated [t(11;22)(p13;q12)], representing the fusion of the EWS and WT1 genes.67,69,70
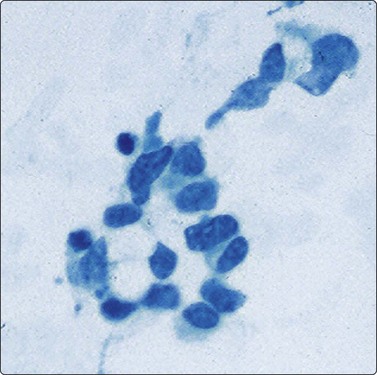
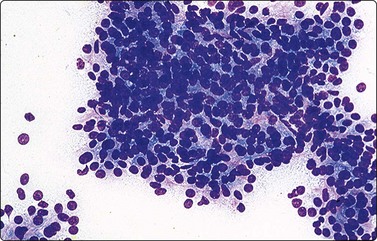
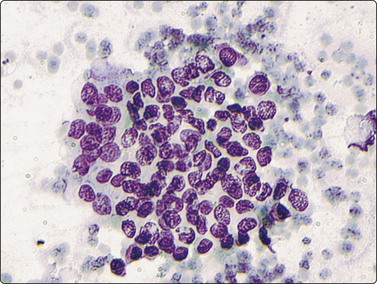
Over 90% of non-Hodgkin lymphomas in children are high-grade lymphomas.14 The most common types are T and B lymphoblastic, Burkitt and anaplastic large cell lymphoma while diffuse large B-cell lymphoma is less common.20 Lymphoblastic, Burkitt and diffuse large cell lymphomas in children can be mistaken for any other SRCTs on the basis of morphology alone. Within the paediatric population, each of these lymphomas has distinct clinical features which are important to recognize.71 Lymphoblastic lymphoma typically affects young teenage boys, and is a common cause of a mediastinal mass as well as peripheral, usually supradiaphragmatic, lymphadenopathy. Most of these are T-cell immunophenotype and positive for terminal deoxynucleotidyl transferase (TdT). Aspiration smears are highly cellular with a singly dispersed monotonous population of lymphoblasts with no evidence of true intercellular cohesiveness. The cells are twice the size of normal lymphocytes. Nuclei demonstrate finely granular chromatin with inconspicuous nucleoli, and only a thin rim of delicate cytoplasm (Fig. 17.12). Abundant lymphoglandular bodies are present in the background and serve as an important diagnostic clue of the lymphoid lesion. Neuroblastoma enters into the differential diagnosis as it can present as a primary mediastinal lesion. However, intercellular cohesion, nuclear molding, typical Homer-Wright rosettes and absence of lymphoglandular bodies favor the diagnosis of neuroblastoma.
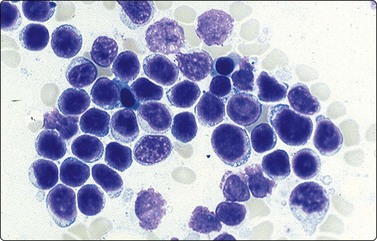
Fig. 17.12 Lymphoblastic lymphoma
Highly cellular smears contain a monomorphic population of lymphoblasts with no true intracellular cohesiveness. The cells demonstrate uniform round nuclei with finely dispersed chromatin, inconspicuous nucleoli, and a thin rim of cytoplasm. Numerous lymphoglandular bodies are readily identifiable in the background.
The small non-cleaved cell lymphomas such as Burkitt and non-Burkitt types, usually presents in the nonendemic form with enlargement of abdominal lymph nodes and/or visceral organs.4,45,71 Ovarian involvement may be seen in females. Aspiration smears are highly cellular and demonstrate monotonous population of singly dispersed cells with round nuclei, moderate to high nuclear to cytoplasmic ratios, coarse clumping of chromatin, and prominent nucleoli (Fig. 17.13). Multiple nucleoli are seen in Burkitt-type and single nucleoli are noted in the non-Burkitt type. Nuclear irregularities and polylobated nuclei are often seen. On Romanowsky-stained smears, numerous cytoplasmic vacuoles are seen in Burkitt lymphoma (Fig. 17.14). The vacuoles are due to the accumulation of neutral lipids, which can be stained with oil-red-O. Abundant mitotic figures and apoptosis are seen and are indicative of a tumor with extremely rapid turnover. Phagocytic histiocytes are often seen in the background.
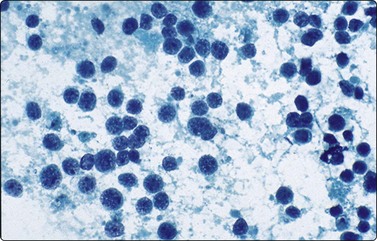
Aspirates are extremely cellular, containing intermediate-sized, predominantly round nuclei with coarsely clumped chromatin and one or more prominent nucleoli (Papanicolaou, ×200).
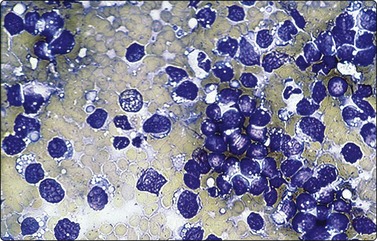
Romanowsky-stained smears demonstrate cytoplasmic lipid vacuoles which are lost with alcohol fixation. Nuclei can be polylobated and irregular (Diff-Quik×200).
Immunocytochemistry and flow cytometry have proved to be an invaluable ancillary technique in diagnosing challenging cases of lymphomas. However, some lymphoblastic lymphomas in bones may be negative for CD45, CD3 and CD20, while CD99 and cytokeratin can be positive, erroneously excluding a lymphoma diagnosis.72,73 Burkitt and non-Burkitt lymphomas demonstrate a characteristic chromosomal translocation t(8;14) and in some cases a t(2;8) chromosomal translocation may be present.74
Langerhans cell histiocytosis (LCH) encompasses a group of histologically similar lesions including eosinophilic granuloma, Hand-Schüller-Christian disease, and Letterer-Siwe disease. LCH occurs at any age but there is a predilection in children under 5 years of age, males being more commonly affected. The etiology is unknown, but viruses such as EBV, herpes, and adenovirus are believed to be involved in its pathogenesis.73 The skeleton is most commonly affected, especially the craniofacial region such as frontal, temporal and zygomatic regions, followed by the femur, pelvis, ribs, and spine.4,75 Radiograpically, these are well-demarcated intramedullary osteolytic lesions which commonly show cortical destruction. FNA usually yields cellular smears with singly scattered Langerhans cell histiocytes with characteristic reniform, convoluted nuclei with distinct longitudinal nuclear grooves. Bi- and multinucleated cells are commonly seen (Figs 17.15 and 17.16). A variable number of lymphocytes, eosinophils, and neutrophils are present and may be the predominant cellular component in some cases (Fig. 17.17).4,75 LCH can be misclassified as SRCTs on FNAB.24 This inaccurate interpretation is usually associated with paucicellular smears, crushing artifacts, and the impression of cohesion.75 Immunohistochemically, the cells are S-100 positive, and most express CD1a, CD68, and vimentin and negative for the majority of B- and T-cell markers, myeloperoxidase, CD34, EMA, and the follicular dendritic cell lineage markers CD21 and CD35. Ultrastructural examination reveals histiocytic/dendritic cells with classic Birbeck granules.75
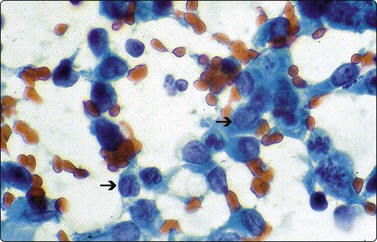
Fig. 17.15 Langerhans cell histiocytosis
Small, singly scattered uniform Langerhans histiocytes are shown in this smear and are characterized by oval nuclei, finely reticulated evenly dispersed chromatin and classic longitudinal nuclear grooves (arrows) (Pap, ×400).
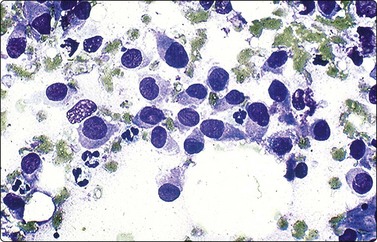
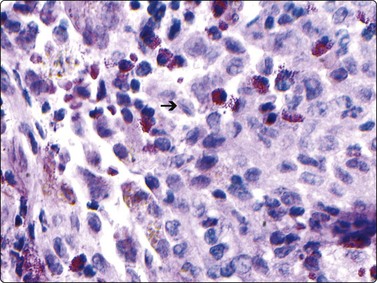
Leukemias are the most common form of cancer in children in the United States, accounting for 30–35% of all new paediatric malignancies.5 Most are acute leukemias and occur in children under 5 years of age. The diagnosis of leukemia is made by bone marrow aspirate and biopsy in the majority of cases. The diagnostic role of cytology is usually in the identification of relapses of the CNS with examination of CSF fluid specimens. Rarely, acute leukemias can form a mass lesion, including lymphadenopathy, mostly in cases of unexpected relapses. The role of FNA is very limited aside from rare case reports of the application of FNA to examine testicular enlargement in males with acute lymphoblastic leukemias.76
Hepatoblastoma is the most common primary hepatic malignancy in children and the third most common intra-abdominal tumors after neuroplastoma and Wilm’s tumor. Almost 90% of hepatoblastomas affect children younger than 5 years.20,77,78 There is a relatively constant annual incidence in Western countries of 0.5–1.5 per million children.75 Hepatoblastoma has an incidence of approximately 10% of Wilms’ tumour and 5% of leukemia. It accounts for a little less than half of all primary liver neoplasms of children with a male-to-female ratio of approximately 1 : 2.5.2,20 Serum alpha fetoprotein levels are elevated in 84–91% of patients, usually with very high titres.79 The dramatic improvement in the survival of children with hepatoblastoma, with the current cure rates of approximately 75%, is a reflection of both contemporary chemotherapeutic protocols and surgical resection.79 Children with extrahepatic tumor extension, multifocal tumor, vascular invasion, and distant metastases have a poor prognosis. Histologic subtype has not been shown to be of prognostic significance.79
Hepatoblastoma usually causes a single mass involving more often the right side of the liver.68 The neoplasms range in size from 3 cm to 20 cm and is generally not associated with cirrhosis. Typically, most children present with abdominal enlargement and occasional hemihypertrophy, precocious puberty, hypoglycaemia, Wilms’ tumour or the Beckwith-Wiedemann syndrome.14 Hepatoblastoma can be classified as either purely epithelial or mixed epithelial–mesenchymal type.45,78 Distinct subtypes of the epithelial cell component include fetal, embryonal, and anaplastic (small cell).44 Fetal tumor cells resemble normal fetal hepatocytes and can be associated with foci of extramedullary hematopoiesis.77 In the embryonal subtype, cells are small and spindled, with hyperchromatic nuclei and poorly defined plasma membranes. Approximately one-third of hepatoblastomas are of the mixed type. Mesenchymal elements consist of either primitive mesenchyme in the form of immature spindle cells, osteoid, cartilage and/or skeletal muscle.20
Aspirates are hypercellular and composed of a uniform population of small to intermediate round to oval cells arranged in trabeculae and cords along with individually scattered cells (Figs 17.18 and 17.19).77,78 The trabeculae and cords are often covered by sinusoidal lining cells. Occasionally, pseudoacinar formation can be seen. Intact cells usually have spherical and slightly eccentrically placed nuclei with several small to inconspicuous nucleoli (Fig. 17.20). A scant to moderate amount of surrounding cytoplasm is seen. Occasional small cytoplasmic vacuoles may be present but bile is rarely seen.80
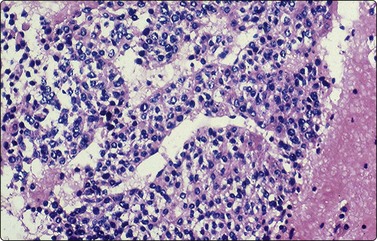
Cell block preparation demonstrating small to medium-sized polygonal fetal-type cells with moderate amounts of granular cytoplasm and arranged in thick trabeculae (H&E, ×200).
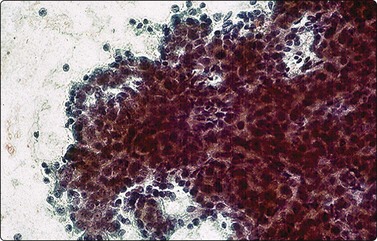
Aspirate smear with cells arranged in cohesive clusters and scattered individually. Neoplastic cells possess prominent central nucleoli and scant cytoplasm, representing the more primitive embryonal cells (Pap, ×200).
In this Diff-Quik-stained smear, neoplastic cells demonstrate moderate amounts of granular cytoplasm consistent with fetal cell type. The cells are arranged in loose clusters and dispersed individually in the background (Diff-Quik, ×400).
The differential diagnosis of hepatoblastoma includes well-differentiated hepatocellular carcinoma, which is the second most common primary malignant liver tumor in children.20 Most hepatocellular carcinomas are diagnosed in children older than 5 years of age (mean age between 12 and 14 years old) and, importantly, occur in already diseased livers with manifestation of hepatitis, cirrhosis, or storage diseases.78-80 Cytological features that support a diagnosis of hepatocellular carcinoma include the presence of cells larger than those seen in hepatoblastoma with a greater degree of pleomorphism. Anisonucleosis, macronucleoli, tumor giant cells, and intranuclear cytoplasmic inclusions, including bile, are readily seen.20 Characteristically, the cells are arranged in anastomosing trabeculae which are broader than those seen in hepatoblastoma.
FNAB of small cell undifferentiated (anaplastic) hepatoblastoma may show features that can be confused with other metastatic SRCTs.2,20,80 Cancers that commonly metastasize to the liver in children include neuroblastoma, followed by Wilms’ tumor, Ewing’s sarcoma/PNET, embryonal and alveolar rhabdomyosarcoma, synovial sarcoma and retinoblastoma.43 In contrast to most cases of hepatoblastoma, multiple discrete nodules are typically present. Hepatoblastomas are positive for low molecular weight cytokeratin and alpha fetoprotein and negative for vimentin, high molecular weight cytokeratin, epithelial membrane antigen (EMA) and carcinoembryonic antigen (CEA).80 Electron microscopic (EM) examination demonstrates ultrastructural features of immature hepatocytes including variably prominent rough endoplasmic reticulum, abundant mitochondria, well-developed microvilli with focal canaliculus formation and intercellular junctions.20
Undifferentiated (embryonal) sarcoma of the liver
This typically occurs in the first decade of life and is the most common type of primary stromal malignancy of childhood.45,80 The cytologic findings include the presence of small to medium-sized, round, spindled and stellate-shaped cells set in a myxoid background often mixed with mono- and multinucleated tumor giant cells. Periodic acid–Schiff (PAS)-positive intracytoplasmic globules are present in occasional cases. The tumor is positive for desmin, myoglobin, vimentin, α1-antitrypsin and α1-antichymotrypsin.80 Another very rare hepatic malignancy that has been described in the cytology literature is primary yolk sac tumor of the liver.81 Cytologic examination reveals loosely cohesive groups of cells having nuclei with small nucleoli and surrounding cytoplasm showing vacuolization. Individually scattered cells are also present as well as extracellular metachromatically staining amorphous material of basement membrane origin in the air-dried Romanowsky-stained smears.81
Synovial sarcomas account for 2–10% of soft tissue sarcomas in children and more frequent in adolescents. The tumor frequently locates in soft tissues of the lower extremities, close to joints cavities. Synovial sarcomas can show either a biphasic or monophasic histologic pattern.82 FNAB of monophasic type revealed a hypercellular specimen consisting of numerous microtissue fragments as well as individually scattered single cells. The malignant cells have high nuclear-to-cytoplasmic ratios with round to oval nuclei. Occasional tumor cells show a slight tendency to spindle, with wispy cytoplasmic processes. Smears may show predominantly dissociated tumor cells, which vary in size and shape from round, ovoid to plasmacytoid in cytology smears, mimicking morphologically other SRCTs.83 Small cell variant smears show numerous, small, round cells with very high nuclear-to-cytoplasmic ratios, can potentially be confused with other paediatric SRCTs.84 FNA of the biphasic type show two cell population: polygonal or columnar epithelioid cells arranged in solid nests, pseudoglands and/or papillae, and atypical spindle cells.85 Tumor cells demonstrate positive staining for cytokeratin (CAM 5.2), CK7, epithelial membrane antigen (EMA), and CD99 and negative staining for S-100 and vimentin. Cytogenetic studies have shown a very specific chromosomal translocation t(X;18)(p11.2;q11.2), with an estimated prevalence of 85% in synovial sarcoma.83-85
Ameloblastoma is a locally aggressive maxillary neoplasm of the odontogenic epithelia that originates from residual epithelial components of the developing teeth. There are two histologic variants: follicular and plexiform. The plexiform is the most frequent type and is formed by nests and cords of epithelial cells surrounded by a fibrous connective stroma; peripheral cells of nests are arranged in palisades and show inverse polarization (nuclei shifted to the apical pole of cells).86 Fine needle aspirates show aggregates and/or compacted cords of epithelial cells without atypia with inverse polarization of peripheral cells, sometimes a few fragments of fibrous stroma, and a clean background.1,86
Fine needle aspiration cytology has successfully been used to document both primary and metastatic malignant rhabdoid tumor of the kidney.87 This aggressive renal tumor occurs most commonly in infants and very young children with a median age of 13 months and has a very high mortality.87 It is rare, representing only 2% of paediatric renal neoplasms.44 It is often single, gray, soft mass, and without a capsule. Cytologic smears are hypercellular with clusters and individually scattered cells demonstrating enlarged eccentric nuclei, vesicular chromatin, and prominent nucleoli.45,50,87 There is abundant eosinophilic cytoplasm with characteristic large intracytoplasmic hyaline eosinophilic globular inclusions. Rhabdoid tumor is immunoreactive for vimentin, pankeratin, and CD99.
Clear cell sarcoma of the kidney is also known as bone-metastasizing sarcoma of the kidney. Aspirates consist of stellate, spindled and polygonal-shaped cells having pale cytoplasm and round to oval uniform nuclei scattered within mucoid material in the background best appreciated on Romanowsky-stained smears.35,50
Mesoblastic nephroma is an infrequent renal congenital tumor that is generally benign. It presents as a well-defined single unilateral abdominal tumor. FNAB shows isolated spindle cells, similar to smooth muscle cells without atypia or mitoses, sometimes forming small bundles with a clean background. The cellular variant has significant cellular atypia and numerous mitoses, mimicking SRCTs, but with clean background.49 These tumors are immunoreactive for CD34.45,52 Cystic nephroma yields atypical cells forming papillary clusters which potentially could be misdiagnosed as a renal cell carcinoma.45,50,88 The cytologic features helpful in making the diagnosis of cystic nephroma include hypocellular smears with relatively few spindle cells without necrosis in an aspirate from a child with a cystic renal mass.44 Drut also identified benign-appearing epithelial cells arranged individually and in uniform sheets.88
Neuroblastoma and malignant lymphoma are the most common secondary malignancies to involve the kidneys. Cytologic features of malignant lymphoma and neuroblastoma are discussed elsewhere in this chapter.
Paediatric pancreatic malignancies are rare. Islet cell tumors are the most common pancreatic tumor in children, in contrast to the dominance of ductal adenocarcinomas in the adult population.89 The nonendocrine neoplasms in the paediatric age group include the very rare conventional ductal adenocarcinoma, papillary cystic tumor, acinar cell carcinoma and pancreatoblastoma.89
Islet cell tumors are capable of producing a variety of peptide hormones leading to a host of clinical syndromes. However, many remain hormonally silent, presenting only as a mass lesion. Aspirates are moderately to highly cellular and are composed of a uniform discohesive population of small to medium-sized cells (Fig. 17.21). Most cells are arranged singly but loose clusters can be seen.90,91 A notable diagnostic feature is the characteristic plasmacytoid appearance of the cells, showing eccentrically placed nuclei and moderate amounts of granular basophilic cytoplasm. This is best appreciated in Romanowsky-stained smears (Fig. 17.22). The characteristic ‘salt and pepper’ chromatin pattern with the lack of prominent nucleoli is seen best in the alcohol-fixed, Papanicolaou-stained smears (Fig. 17.23). Due to the fragility of the cells, naked nuclei and a granular background smear pattern will be present along with occasional delicate blood vessels lined by tumor cells.90,91 Most cases do not require immunocytochemical confirmation, although occasionally the smear patterns will almost exclusively consist of clusters of cells that can mimic nonendocrine lesions. Neuroendocrine markers such as NSE, CD56, chromogranin and synaptophysin and/or EM examination can be helpful. Positive staining for specific markers such as insulin, gastrin and somatostatin can be helpful in selected cases.91
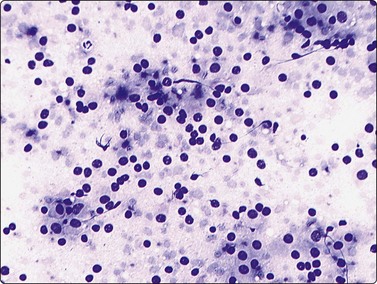
This aspirate is dominated by numerous stripped neoplastic nuclei that are singly dispersed. Some of the neoplastic cells have a more abundant granular cytoplasm and eccentrically placed nuclei (Diff-Quik, ×200).
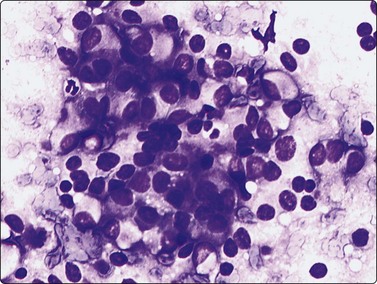
A characteristic diagnostic cytologic feature is shown in this aspirate consisting of clusters of neoplastic cells with a plasmacytoid appearance. The nuclei are eccentrically placed with finely granular chromatin and abundant eosinophilic cytoplasm (Diff-Quik, ×400).
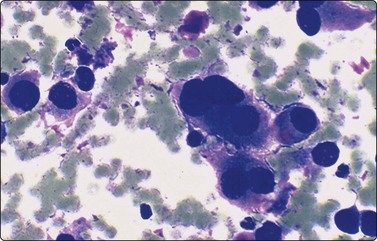
Pap-stained smear highlighting the salt and pepper chromatin pattern of neoplastic nuclei (Pap, ×600).
Pancreatoblastoma is an extremely rare embryonal tumor of childhood with only 60 cases reported in the literature since the tumor was first described.92 They occur in children between 15 months to 13 years of age. Pancreatoblastomas are more frequent in girls less than 5 years old, and approximately one-third of them produce metastasis. These tumors are usually large and can occur in any portion of the pancreas. The tumors arise from multi-potential stem cells.89,92 The overall prognosis is favorable. FNA cytology demonstrates cellular smears formed by uniform epithelial cells arranged in organoid structures, often with nests of squamoid cells and keratinization foci. Cells show a moderate amount of granular cytoplasm. Glandular structures of columnar to cuboidal cells with round to oval vesicular nuclei, occasional nucleoli and a moderate amount of granular to foamy cytoplasm are seen in the smears. Spindle-shaped, elongated and triangular epithelial cells, admixed with occasional smaller cells having high nuclear-to-cytoplasmic ratios and denser cytoplasm may be seen (Fig. 17.24).92,93 Abundant fragments of cellular stroma, some with a peripheral layer of neoplastic epithelial cells, can serve as an important diagnostic clue (Fig. 17.25).93 Immunocytochemical studies demonstrate positive staining of the epithelial cells for cytokeratin (AE1/3), carcinoembryonic antigen (CEA), α1-antitrypsin, as well as neuroendocrine markers.20,92 EM is helpful and reveals cells containing abundant large electron-dense zymogen-like granules measuring 400–600 nm as well as smaller neuroendocrine granules measuring 100–200 nm.92 Most tumor cells will contain only one type of granule, although occasional cells demonstrate both types.
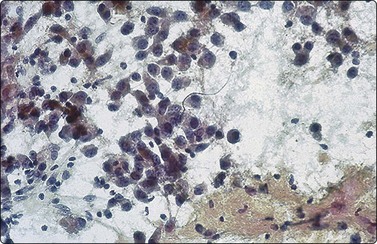
Aspirate smear showing small clusters and individually scattered cells with irregular nuclear contours and moderate amounts of granular cytoplasm (Diff-Quik, ×400).
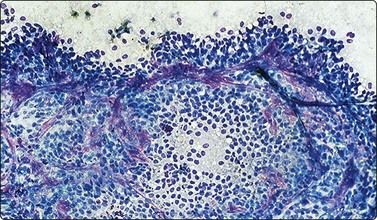
This aspirate highlights the variability of cellular contours with round, oval, triangular, and spindled cells with vesicular nuclei and prominent nucleoli (Pap, ×200).
Papillary-cystic tumor of the pancreas is an uncommon pancreatic neoplasm that occurs almost exclusively in adolescent and young adult females.94 This low-grade pancreatic malignancy can be cured with surgical resection in most cases, although metastatic disease has been reported.80 The characteristic cytologic features of papillary-cystic tumor are the presence of papillary fronds that consist of fibrovascular stalks lined by a uniform population of small cells along with individually scattered and small clusters of similar-appearing cells (Figs 17.26 and 17.27). The neoplastic cells possess oval nuclei with finely granular chromatin, characteristic longitudinal nuclear grooves, inconspicuous to small nucleoli and moderate amounts of cytoplasm.94,95 Occasional cases may demonstrate reddish-pink hyaline globules in the Romanowsky stains.
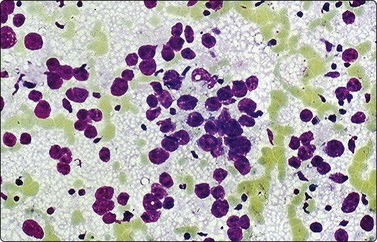
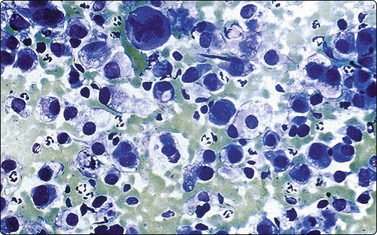
Epithelial tumors of the ovary account for only 15–20% of all ovarian childhood neoplasms, with germ cell tumors involving the ovary in up to one-third of paediatric cases.95 Benign cystic teratomas account for 30–90% of germ cell tumors followed by a variety of other germ cell tumors.16 Aspirates of benign cystic teratoma are of variable cellularity containing benign-appearing epithelial and mesenchymal components derived from the three germinal layers.96 As would be expected, squamous cells, choroid and glial tissue, cartilage and glandular cells can be present.67 In immature teratoma, primitive neuroblastic small cells resembling distinct nests of SRCTs are also present.97 Yolk sac tumors are rare and may be a challenge to the cytopathologist on FNA. Yolk sac tumors can have a variety of patterns including groups of malignant cells that are arranged in papillary, glandular or large pleomorphic tumor balls.81 The cytoplasm of the neoplastic cells can contain small or large vacuoles. A helpful diagnostic feature is the presence of eosinophilic inclusions within the cytoplasm of some of the tumor cells as well as basement membrane material in the background that is best seen in the Diff-Quik stain.96,98 Well-defined cytoplasmic vacuoles corresponding to intracytoplasmic deposits of glycogen, as well as nuclear vacuoles within the malignant cells have been identified.98 Positive immunohistochemical studies for alpha fetoprotein, cytokeratins AE1/AE3, and CAM 5.2 are useful in supporting the diagnosis.81
Seminomas and dysgerminomas yield highly cellular aspirates composed of both loose clusters and individually scattered round tumor cells with round nuclei containing finely granular chromatin and a prominent single nucleolus (Fig. 17.28).96 A moderate to abundant amount of cytoplasm can be present and PAS-positive cytoplasmic vacuoles are often seen. The smear background contains chronic inflammatory cells with occasional granulomas. Due to the spillage of the glycogen-rich cytoplasm, a characteristic ‘tigroid’ background can be appreciated and serves as a useful diagnostic clue (Fig. 17.28). In contrast, embryonal carcinoma consists of large three-dimensional clusters of tumor cells with indistinct cell boundaries. Papillary and glandular clusters may contain delicate branching blood vessels. The neoplastic cells show pleomorphism with coarsely granular chromatin and multiple prominent nucleoli (Fig. 17.29). As previously mentioned, FNA biopsy of the testicle has also been used to document relapse of acute lymphoblastic leukemia in children and could be considered as an alternative procedure to surgical biopsy.71,76,99
Fine needle aspiration of the lung has been used in the work-up of acute pneumonias in children.25 However, based on our experience, FNA of the lung is most often performed to document metastatic malignancies and/or lung abscesses.24 The most common benign lung masses in the paediatric age group are inflammatory pseudotumor, lung sequestration, and hamartoma.20,100 Primary lung malignancies are exceedingly uncommon in childhood.20 Paediatric pulmonary malignancies that can present as primary intrathoracic tumors include primary neuroblastoma, teratoma, malignant lymphoma and adenocarcinoma of fetal type.99
Inflammatory pseudotumor (inflammatory myofibroblastic tumor) is the most frequent pulmonary tumor of children older than 5 years. It can represent as a single, round well-defined lesion measuring between 3 and 10 cm. FNA smears are slightly cellular, showing spindle cells with a fibroblastic proliferation, placed in an isolated fashion and/or small bands. Fragments of collagen and inflammatory cells can appear, especially lymphocytes, plasmocytes, and macrophages.1,45 The background is often clean.1
Pulmonary blastoma is rare and most frequent in children older than 12 years. It can be associated with medulloblastoma, ovarian teratoma, leukemias, Hodgkin lymphoma, and germ cell malignancies. Pulmonary blastoma metastasizes in 25% of cases, especially to the brain, spinal cord, and bones. There have also been rare FNA case reports of pulmonary blastoma and pleuropulmonary blastoma.18 Both of these neoplasms demonstrate the presence of undifferentiated cells, but pleuropulmonary blastoma also has an admixture of mesenchymal cells. Due to the rarity of many of these malignancies, tissue confirmation is often needed to make a definitive diagnosis.
Aspiration biopsies of benign and malignant disorders involving the lymph nodes are presented in detail elsewhere in this book and will be only briefly covered in this section. In children, lymph nodes, especially in the head and neck region, serve as one of the sites most commonly aspirated by FNA. The majority of lymph node enlargements are benign due to hyperplasia. However, it is important to emphasize that specific malignant lymphomas show a marked predilection in the paediatric population and must always remain in the differential diagnosis of lymphadenopathy. Aspirate smears from reactive lymph nodes generally show a dissociated cell population and moderate to high smear cellularity in the majority of cases. However, follicular center cell fragments as well as granulomatous inflammation can yield predominant cell clusters. Identification of a predominant cell type and a monomorphic cell population can be helpful in separating benign from malignant lesions. A heterogeneous cell population with predominance of small mature lymphoid cells is typically seen in benign processes. Lymphoglandular bodies which represent cytoplasmic fragments are a consistent background element in aspirates from lymph nodes and serve as an important diagnostic clue in identifying the lymphoid origin of lesions that can be either benign or malignant. These are best appreciated on Romanowsky-stained smears and stain blue to blue–gray, but can also be appreciated in the Papanicolaou preparation.
The more common reactive lesions include acute lymphadenitis, granulomatous lymphadenitis, infectious mononucleosis and reactive lymph node hyperplasia.43 With infectious processes, either a single lymph node or a group of nodes (i.e. cervical chain) may be involved. Smears that demonstrate sheets of neutrophils with associated bacteria and/or necrosis signify an acute suppurative lymphadenitis (Fig. 17.30). Occasionally, smears can demonstrate fungal organisms; therefore, histochemical stains for fungal, mycobacterial, and bacterial organisms should be performed and material should be obtained with additional passes for microbiological cultures. In some cases of mycobacterial infection, Romanowsky-stained smears demonstrate the presence of ‘negative images’ of the mycobacteria which do not stain, and this can serve as an important diagnostic clue (Fig. 17.31).
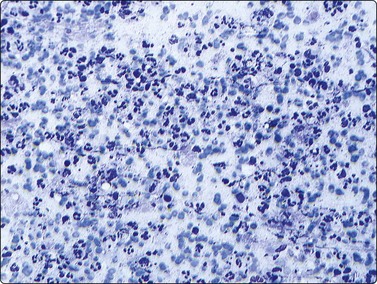
Fig. 17.30 Acute suppurative lymphadenitis
Cellular smear demonstrating numerous neutrophils with associated necrotic debris in the background (Diff-Quik, ×200).
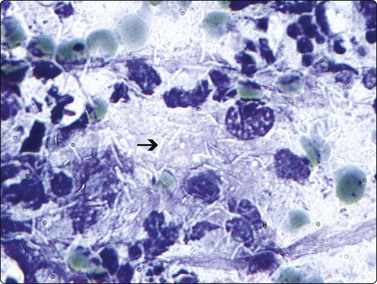
Fig. 17.31 Tuberculous lymphadenitis
Romanowsky-stained smears demonstrate abundant ‘negative images’ of mycobacterial organisms (arrow) (Diff-Quik, ×600).
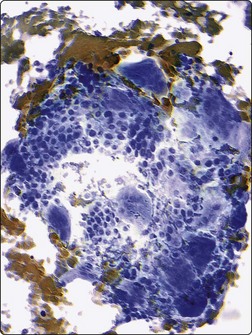
The head and neck is the most common site of involvement for granulomatous lymphadenitis. Granulomas can be either necrotizing or non-necrotizing. Smears demonstrate variable cellularity with tightly clustered groups of epithelioid histiocytes containing characteristic bent or ‘boomerang-shaped’ nuclei (Fig. 17.32). Associated necrosis and acute inflammation can be seen in necrotizing granulomatous lymphadenitis for which the etiological agents include cat scratch disease, mycobacteria, tularemia, brucellosis, and fungi, among others. Infectious mononucleosis is caused by Epstein-Barr virus (EBV) and is a self-limited disorder predominantly affecting adolescents. Enlarged cervical lymph nodes are most often aspirated and the smears are highly cellular, containing a heterogeneous lymphoid cell population with the presence of numerous immunoblasts (Figs 17.33 and 17.34).45 However, confirmatory serologic testing is needed, with most patients demonstrating positive antibody titers for various antibodies specific for EBV.
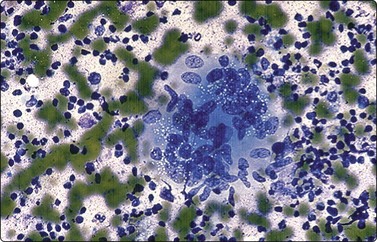
Fig. 17.32 Granulomatous lymphadenitis
This smear shows a well-formed epithelioid granuloma. The epithelioid histiocytes are round to elongated with curved or bent ‘boomerang-shaped’ nuclei that contain finely reticulated cytoplasm. The background contains abundant neutrophils (Diff-Quik, ×400).
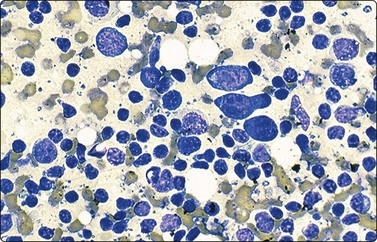
Fig. 17.33 Infectious mononucleosis
This highly cellular smear contains a heterogeneous lymphoid population with prominent immunoblasts and plasmacytoid cells (Diff-Quik, ×400).
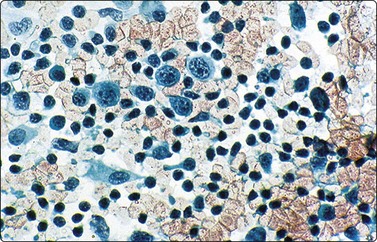
Fig. 17.34 Infectious mononucleosis
In Papanicolaou-stained smears, the cellular elements appear more uniform in size but are still dominated by increased numbers of immunoblasts which contain irregularly clumped chromatin, prominent nucleoli and moderate amounts of granular cytoplasm (Pap, ×200).
Rosai-Dorfman disease (sinus histiocytosis) presents with lymphadenopathy involving the cervical lymph nodes of young children, and has a predilection for blacks. The etiology is unknown. Unlike the infection-related lymphadenopathies, node enlargement is often massive, bilateral, and painless. Aspirate smears reveal features of reactive hyperplasia with a heterogeneous cell population. Increased numbers of accompanying histiocytes demonstrating emperipolesis (engulfment of red blood cells and lymphocytes) are seen throughout the smears and serve as an important differentiating feature to separate Rosai-Dorfman disease from other reactive hyperplasias (Fig. 17.35).45 These histiocytes also demonstrate positive staining for S-100.
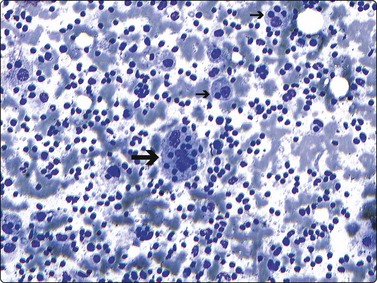
Fig. 17.35 Rosai-Dorfman disease
Highly cellular smears contain a mixture of small and large lymphocytes. The diagnostic feature of abundant histiocytes demonstrating emperiopolesis is shown here (arrows) with the cytoplasm of histiocytes containing phagocytosed lymphocytes (Diff-Quik, ×200).
Hodgkin lymphoma is relatively common in older children and adolescents and rare before the age of 5 years. Nodular sclerosis is the most common subtype. The cellularity of smears varies from low to intermediate in most cases, depending on the degree of sclerosis present. Smears demonstrate a picture similar to reactive hyperplasias; therefore, a diligent search for Reed-Sternberg cells and its mononuclear variants is necessary for an accurate diagnosis (Figs 17.36 and 17.37).45 Immunostaining can be helpful as the Reed-Sternberg cells express both CD15 and CD30 and are negative for CD45 and CD20, while anaplastic large cell lymphoma shows positive staining for CD30, CD45 and ALK, along with negative staining for CD15.45,74
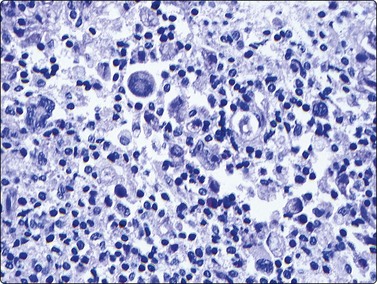
An extremely cellular aspirate is seen here with a polymorphic background lymphoid population with lymphocytes, eosinophils, plasma cells and histiocytes, amongst which are scattered larger atypical Reed-Sternberg cell variants (Diff-Quik, ×200).
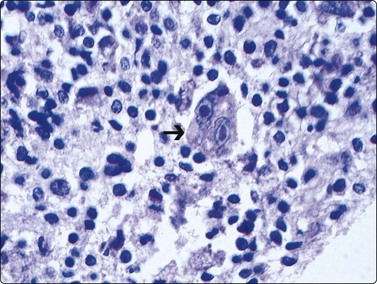
A classic Reed-Sternberg cell is shown here (arrow) with two relatively uniform nuclei containing finely reticulated chromatin and massive nucleoli. A moderate amount of cytoplasm is present (Diff-Quik, ×400).
The small noncleaved malignant lymphomas (Burkitt and non-Burkitt type) and lymphoblastic lymphomas have been discussed previously in this chapter.
There are a great variety of soft tissue tumors in children, such as hamartomas, inflammatory tumors, reactive proliferative tumors, and benign or malignant neoplasms. Infantile soft tissue tumors differ from those of adults in frequency, location, histologic type, and prognosis; the majority are benign, and only 7–10% are malignant. Rhabdomyosarcoma, malignant fibrohistiocytomas, PNET and undifferentiated sarcomas account for >80% of malignant soft tissue tumors.1,100
The separation of benign from malignant lesions of soft tissue and bone by FNA can often be accomplished with high diagnostic accuracy, thereby facilitating expeditious and proper patient triage and therapeutic management. FNA is increasingly being used as the diagnostic modality of choice for a first-time diagnosis of lesions from both soft tissues and bone. FNA in conjunction with ancillary techniques can afford a diagnostic accuracy of 70–90%, comparable to open and core biopsy techniques. The reported false-positive and false-negative rates range from less than 1% to 4% for adequate specimens.27 As emphasized throughout this chapter, a multidisciplinary approach and the presence of an on-site pathologist for immediate evaluation and appropriate triage for ancillary studies at the time of biopsy are crucial for rendering an accurate diagnosis. Otherwise, core needle biopsy and/or open biopsy may be preferable. For bone lesions to be accessible by FNA, soft tissue extension and/or cortical bone destruction needs to be present. Sclerotic tumors such as osteoma, osteoid osteoma, osteoblastoma, low-grade osteosarcoma, fibrous dysplasia, and low-grade chondrosarcoma are not adequately sampled by FNA under most circumstances.21
Some benign soft tissue spindle cell lesions are unique to childhood and include fibrous hamartoma of infancy, fibromatosis colli, and infantile digital fibromatosis. Fibromatosis colli, also called sternocleidomastoid tumor of infancy, occurs at all ages in the region of the sternocleidomastoid muscle. Myofibroma/myofibromatosis (congenital fibromatosis) is rare but seen most commonly in infancy and children less than 2 years of age. Clinically, this lesion is rapidly growing and the differential diagnosis includes nodular fascitis and fibrosarcoma. FNA of benign soft tissue spindle cell lesions shows paucicellular smears with bland loosely cohesive spindle cells and isolated bare oval nuclei. All of these lesions are fibroblastic/myofibroblastic in origin; therefore, ancillary studies such as immunohistochemistry and EM are of little aid to establish the diagnosis. However, both fasciitis and fibrosarcoma usually produce relatively cellular specimens.100-102
An extremely uncommon benign tumor, the fetal rhabdomyoma, almost always occurs in children 3 years of age or younger as a mass lesion in the head and neck region.45 These are slow-growing lesions. Fine needle aspirates show immature muscle fibers with multiple nuclei and transverse striation and arranged in lobulated bundles. The presence of cellular atypia and hypercellularity may suggest a rhabdomyosarcoma.
Osteosarcoma is the most common primary bone malignancy in children, peaking in adolescence (80% of cases) with a slight male predominance. The majority are sporadic, and about 20% of patients develop bone or lung metastasis. Osteosarcoma can have various subtypes which are all linked by the production of osteoid/bone by the malignant cells. Conventional osteosarcoma represents the vast majority of all osteosarcoma (80–90%).21,103 The metaphyseal regions of the long bones are most commonly affected, followed in decreasing order by: distal femur, proximal tibia, and proximal humerus. Radiographically, this is a permeative lesion associated with a lytic/sclerotic reaction, and soft tissue extension is commonly seen. Therefore, most osteosarcomas are amenable to FNA with an accuracy rate for establishing a diagnosis exceeding 90%.103 Smears demonstrate moderate to high cellularity with small aggregates and singly scattered polygonal cells with a plasmacytoid appearance, one or more nucleoli, and abundant cytoplasm (Figs 17.38 and 17.39). On Romanowsky-stained smears, metachromatic matrix material compatible with osteoid is present, associated with the malignant cells (Fig. 17.40).103 Small cell osteosarcoma is composed of mostly discohesive, oval to round cells with high nuclear to cytoplasmic ratios, and inconspicuous to prominent nucleoli, mimicking EWS/PNET. However, osteosarcoma shows more cells variation than Ewing’s sarcoma, with osteoid formation. There is no reliable immunohistochemical pattern of staining to help in the diagnosis of osteosarcoma, including small cell variant. Occasional staining with CD99 has been observed in osteosarcoma, adding to the confusion with EWS/PNET. Therefore, the diagnosis rests primarily on the cytomorphologic findings and the identification of osteoid/bone production by the malignant cells.104
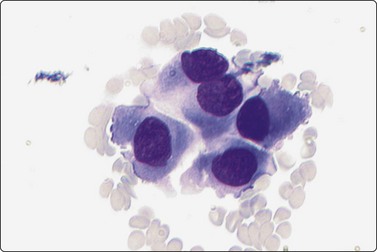
This smear demonstrates a small aggregate of polygonal cells with a plasmacytoid appearance and abundant granular cytoplasm (Diff-Quik, ×400).
These are infrequent in children (<5% of primary bone tumors), often located in the distal femur and proximal region of the tibia (>50% of cases). X-rays show an osteolytic subchondral lesion. Microscopically, it is formed by mononuclear cells with oval or spindled nuclei grouped in solid masses and numerous osteoclast-like cells arranged in a diffuse fashion. Giant cells have numerous nuclei that look like those of mononuclear cells. There are few mitoses, high vascularization, hemorrhagic and necrotic foci, and macrophages with hemosiderin and/or foamy histiocytes.105 FNA smears show two cell types: disperse histiocytoid mononuclear and/or three-dimensional aggregates and giant multinucleated osteoclast-like cells. Hemosiderin-containing macrophages can often be observed.1,104
Chondroblastomas are seen most often in the first and second decade of life, with a male predominance. They occur predominantly in the epiphyseal and epimetaphyseal regions of long bones, the proximal and distal femur, tibia and proximal humerus. FNA smears are generally hypercellular with sheets and individually scattered uniform round to polygonal cells with nuclei displaying prominent longitudinal grooves and inconspicuous nucleoli similar to those seen in Langerhans cell histiocytosis (Fig. 17.41).105 Chondroid matrix material is arranged in lobules and plates recapitulating mature cartilage and is best seen on Romanowsky-stained smears. Associated multinucleated giant cells can be seen (Fig. 17.42). The cells are positive for S-100 and vimentin and occasionally positive for cytokeratin.104 Simple curettage is the treatment of choice.
Aspirates demonstrate a monomorphic population of singly dispersed and loosely cohesive cell clusters. The chondroblasts contain solitary ovoid nuclei with finely granular chromatin, inconspicuous nucleoli, and nuclear grooves (Diff-Quik, ×400).
Scattered amongst the chondroblasts are variable numbers of osteoclast-like giant cells which contain nuclei resembling those seen in the mononuclear chondroblastic cells (Diff-Quik, ×200).
As in adults, metastases to bone are far more common than primary malignant bone tumors. Lymphoreticular metastases and those from neuroblastoma are most common in children.
Pilomatrixoma is the most frequent benign tumor of the pilosebaceous unit, often affecting children younger than 10 years, forming a hard and well-defined nodule in the skin of the head and neck.106 The diagnosis of pilomatrixoma by FNAC can frequently pose a diagnostic dilemma to cytopathologists. Classically, the presence of cohesive epithelial clusters with basaloid features and ghost cells are diagnostic; however, both of these components may not always be apparent in the aspirate smears.106 SRCTs can also enter into the differential diagnosis when ghost cells are not appreciated. When pilomatrixomas occur in the salivary gland region, other basaloid neoplasms enter into the differential diagnosis, such as adenoid cystic carcinoma, monomorphic adenoma, and pleomorphic adenoma.106 However, knowledge of the superficial location and other cytologic clues such as multinucleated giant cells, calcification, and keratin debris as well as the lack of stroma can aid in the diagnosis. Cell block preparations aid in the diagnosis of pilomatrixoma, as ghost cells become more apparent on cell block sections.106
Fine needle aspiration (FNA) of the thyroid is seldom performed in the paediatric population. Therefore, the clinical utility of thyroid FNA in this patient group has not been adequately addressed. Thyroid nodules are relatively common in adolescents, with prevalence ranging from 0.2% to 1.8%.107 Although most of these nodules are benign, malignant tumors, particularly papillary carcinomas, are known to occur in this age group.108 FNA biopsy of thyroid nodules in young patients has similar high sensitivity and specificity as the procedure in the adult population.107,108
1 Schalper JA. Paediatric Tumours. In: Bibbo M, Wilbur D, editors. Comprehensive Cytopathology. 3rd ed. Saunders Elsvier Publisher; 2008:915-950. Chapter 29
2 Grovas A, Fremgen A, Rauck A, et al. The National Cancer Data Base Report on Patterns of Childhood Cancers in the United States. Cancer. 1997;80:2321-2332.
3 Buchino JJ. Cytopathology in Paediatrics. In: Wied GL, editor. Monographs in Clinical Cytology. Basel: Karger; 1991:1-7. 13
4 Dave B, Shet T, Ramadwar M, et al. Cytologic evaluation of head and neck tumours in children – A pattern analysis. Diagn Cytopathol. 2006;34:434-446.
5 Cohen MB, Bottles K, Ablin AR, et al. The use of fine-needle aspiration biopsy in children. West J Med. 1989;150:665-667.
6 Rajwanshi A, Rao KL, Marwaha RK, et al. Role of fine-needle aspiration cytology in childhood malignancies. Diagn Cytopathol. 1989;5:378-382.
7 Taylor SR, Nunez C. Fine-needle aspiration biopsy in a population. Report of 64 consecutive cases. Cancer. 1984;54:1449-1453.
8 Wakely PE, Kardos TF, Frable WJ. Application of fine needle aspiration biopsy to paediatrics. Human Pathol. 1988;19:1383-1386.
9 Silverman JF, Gurley AM, Holbrook CT, et al. Paediatric fine-needle aspiration biopsy. Am J Clin Pathol. 1991;95:653-659.
10 McGahey BE, Moriarty AT, Nelson WA, et al. Fine needle aspiration biopsy of small round blue cell tumours of childhood. Cancer. 1992;69:1067-1073.
11 Gorczyca W, Bedner E, Juszkiewicz P, et al. Aspiration cytology in the diagnosis of malignant tumours in children. Am J Pediatr Hematol Oncol. 1992;14:129-135.
12 Akhtar M, Ali MA, Sabbah R, et al. Fine-needle aspiration biopsy diagnosis of round cell malignant tumours of childhood. A combined light and electron microscopic approach. Cancer. 1985;55:1805-1817.
13 Diament MJ, Stanley P, Taylor SR. Percutaneous fine needle biopsy in paediatrics. Pediatr Radiol. 1985;15:409-411.
14 Verdeguer A, Castel V, Torres V, et al. Fine-needle aspiration biopsy in children: experience in 70 cases. Med Pediatr Oncol. 1988;16:98-100.
15 Layfield LJ, Reichman A. Fine needle aspiration cytology: utilization in paediatric pathology. Dis Markers. 1990;8:301-315.
16 Layfield LJ, Glasgow B, Ostrzega N, et al. Fine-needle aspiration cytology and the diagnosis of neoplasms in the paediatric age group. Diagn Cytopathol. 1991;7:451-461.
17 Obers VJ, Phillips JI. Fine needle aspiration of paediatric abdominal masses. Cytologic and electron microscopic diagnosis. Acta Cytol. 1991;35:165-170.
18 Valkov I, Boijkin B. Fine-needle aspiration biopsy of abdominal and retroperitoneal tumours in infants and children. Diagn Cytopathol. 1987;3:129-133.
19 Shakoor KA. Fine needle aspiration cytology in advanced paediatric tumours. Pediatr Pathol. 1989;9:713-718.
20 Geisinger KR, Silverman JF, Wakely PEJr. Paediatric cytopathology. Chicago: American Society of Clinical Pathologists; 1994.
21 Kilpatrick SE, Ward WG, Chauvenet AR, et al. The role of fine-needle aspiration biopsy in the initial diagnosis of paediatric bone and soft tissue tumours: an institutional experience. Mod Pathol. 1998;11:923-928.
22 Pohar-Marinsek Z. Difficulties in diagnosing small round cell tumours of childhood from fine needle aspiration cytology samples. Review Article. Cytopathology. 2008;19:67-79.
23 Howell LP. Changing role of fine-needle aspiration in the evaluation of paediatric masses. Diagn Cytopathol. 2001;24:65-70.
24 Eisenhut CC, King DE, Nelson WA, et al. Fine-needle biopsy of paediatric lesions: a three-year study in an outpatient biopsy clinic. Diagn Cytopathol. 1996;14:43-50.
25 Layfield LJ. Fine-needle aspiration biopsy in the diagnosis of paediatric tumours. West J Med. 1991;154:90-91.
26 Akhtar M, Iqbal MA, Mourad W, et al. Fine-needle aspiration biopsy diagnosis of small round cell tumours of childhood: A comprehensive approach. Diagn Cytopathol. 1999;21:81-91.
27 Singh HK, Kilpatrick SE, Silverman JF. Fine needle aspiration biopsy of soft tissue sarcomas: utility and diagnostic challenges. Adv Anat Pathol. 2004;11:24-37.
28 Silverman JF, Joshi VV. FNA biopsy of small round cell tumours of childhood: cytomorphologic features and the role of ancillary studies. Diagn Cytopathol. 1994;10:245-255.
29 Gautam U, Srinivasan R, Rajwanshi A, et al. Comparative evaluation of flow-cytometric immunophenotyping and immunocytochemistry in the categorization of malignant small round cell tumours in fine-needle aspiration cytologic specimens. Cancer (Cytopathology). 2008;114:494-503.
30 Gurley AM, Silverman JF, Lassaletta MM, et al. The utility of ancillary studies in paediatric FNA cytology. Diagn Cytopathol. 1992;8:137-146.
31 Plasschaert SLA, Willem AK, Vellenga E, et al. Prognosis in childhood and adult lymphoblastic leukemia: a question of maturation? Cancer Treat Rev. 2004;30:37-51.
32 Brahim U, Srinivasan R, Komal HS, et al. Comparative analysis of electron microscopy and immunocytochemistry in the cytologic diagnosis of malignant small round cell tumours. Acta Cytol. 2003;47:443-449.
33 Leon ME, How JS, Galindo LM, et al. Fine needle aspiration of adult small-round-cell tumours studies with flow cytometry. Diagn Cytopathol. 2004;31:147-154.
34 Layfield LJ, Liu K, Dodge RK. Logistic regression analysis of small round cell neoplasms: a cytologic study. Diagn Cytopathol. 1999;20:271-277.
35 Joshi VV, Silverman JF, Alshuler G, et al. Systematization of primary histopathologic and fine-needle aspiration cytologic features and description of unusual histopathologic features of neuroblastic tumours: a report from the Paediatric Oncology Group. Human Pathol. 1993;24:493-504.
36 Stephenson CF, Bridge JA, Sandberg AA. Cytogenetic and pathologic aspects of Ewing’s sarcoma and neuroectodermal tumours. Human Pathol. 1992;23:1270-1277.
37 Triche TJ, Askin FB. Neuroblastoma and the differential diagnosis of small-, round- blue-cell tumours. Human Pathol. 1983;14:569-595.
38 Triche TJ, Askin FB, Kissane JM. Neuroblastoma, Ewing’s sarcoma, and the differential diagnosis of small-, round-, blue-cell tumours. In: Finegold M, editor. Pathology of neoplasms in children and adolescents. Philadelphia: Saunders; 1986:145-195.
39 Shimada H, Chatten J, Newton WAJr, et al. Histopathologic prognostic factors in neuroblastic tumours: definition of subtypes of ganglioneuroblastoma and age-linked classification of neuroblastomas. J Natl Cancer Inst. 1984;73:405.
40 Frostad B, Martinsson T, Tani E, et al. The use of fine-needle aspiration cytology in the molecular characterization of neuroblastoma in children. Cancer. 1999;87:60-68.
41 Thiesse P, Hany MA, Combaret V, et al. Assessment of percutaneous fine needle aspiration cytology as a technique to provide diagnostic and prognostic information in neuroblastoma. Eur J Cancer. 2000;36:1544-1551.
42 Silverman JF, Dabbs DJ, Ganick DH, et al. Fine needle aspiration cytology of neuroblastoma, including peripheral neuroectodermal tumour, with immunohistochemical and ultrastructural confirmation. Acta Cytol. 1988;32:367-376.
43 Sebire NJ, Gibson S, Rampling D, et al. Immunohistochemical findings in embryonal small round cell tumours with molecular diagnostic confirmation. Appl Immunohistochem Mol Morphol. 2005;13:1-5.
44 Barroca H, Carvalho JL, da Costa MJ, et al. Detection of N-myc amplification in neuroblastomas using Southern blotting on fine needle aspirates. Acta Cytol. 2001;45:169-172.
45 Geisinger KR, Stanley MW, Raab SS, et al. Liver, Kidney, Lymph node and spleen. Modern cytopathology. Philadelphia: Churchill Livingstone; 2004. p. 505–42, 579–618, 643–687
46 Barroca H. Nephroblastoma is a success of paediatric oncologic therapy. How further can we go? Results of a cyt-histologic correlation study. Diagn Cytopathology. 2009.
47 Bray GL, Pendergrass TW, Schaller RTJ, et al. Preoperative chemotherapy in the treatment of Wilms’ tumour diagnosed with the aid of fine needle aspiration biopsy. Am J Pediatr Hematol Oncol. 1986;8:75-78.
48 Geisinger KR, Wakely PEJr, Wofford MM. Unresectable stage IV nephroblastoma. A potential indication for fine-needle aspiration biopsy in children. Diagn Cytopathol. 1993;9:197-201.
49 Sarinen UM, Wilkstrom S, Korkimies O, et al. Percutaneous needle biopsy preceeding preoperative chemotherapy; the management of massive renal tumours in children. J Clin Oncol. 1991;9:406-415.
50 Ravindra S, Kini S. Cytomorphology and morphometry of small round-cell tumours in the region of the kidney. Diagn Cytopathol. 2005;32:211-216.
51 Serrano R, Rodriguez-Peralto JL, De Orbe GG, et al. Intrarenal neuroblastoma diagnosed by fine-needle aspiration: a report of two cases. Diagn Cytopathol. 2002;27:294-297.
52 Schmidt D, Beckwith JB. Histopathology of childhood renal tumours. Hematol Oncol Clin North Am. 1995;9:1179-2000.
53 Silverman JF, Berns LA, Holbrook CT, et al. Fine needle aspiration cytology of primitive neuroectodermal tumours. A report of three cases. Acta Cytol. 1992;36:541-550.
54 Kilpatrick SE, Renner J. Small round cell tumours of bone and soft tissues. In: Kilpatrick SE, editor. Diagnostic musculoskeletal surgical pathology with clinicopathologic and cytologic correlations. Philadelphia: Saunders; 2003:37-70.
55 Sanati S, Lu DW, Schmidt E, et al. Cytologic diagnosis of Ewing sarcoma/peripheral neuroectodermal tumour with paired prospective molecular genetic analysis. Cancer (Cytopathology). 2007;111:192-199.
56 Folpe AL, Goldblum JR, Rubin BP, et al. Morphologic and immunophenotypic diversity in Ewing family tumours: a study of 66 genetically confirmed cases. Am J Surg Pathol. 2005;29:1025-1033.
57 Frostad B, Tani E, Brosjo O, et al. Fine needle aspiration cytology in the diagnosis and management of children and adolescents with Ewing sarcoma and peripheral primitive neuroectodermal tumour. Med Pediatr Oncol. 2002;38:33-40.
58 Newton WAJ, Gehan EA, Webber BL, et al. Classification of rhabdomyosarcomas and related sarcomas, pathologic aspects and proposal for a new classification – an Intergroup Rhabdomyosarcoma Study. Cancer. 1995;76:1073-1085.
59 Wang NP, Marx J, McNutt MA, et al. Expression of myogenic regulatory proteins (myogenin and MyoD1) in small blue cell tumours of childhood. Am J Pathol. 1995;147:1799-1810.
60 Pohar-Marinsek Z, Bracko M. Rhabdomyosarcoma: cytomorphology, subtyping and differential diagnostic dilemmas. Acta Cytol. 2000;44:524-532.
61 Klijanienko J, Caillaud JM, Orbach D, et al. Cyto-histologic correlations in primary, recurrent and metastatic rhabdomyosarcoma: The Institute Curie’s Experience. Diagn Cytopathol. 2007;35:482-487.
62 Atahan S, Aksu O, Ekinci C. Cytologic diagnosis and subtyping of rhabdomyosarcoma. Cytopathology. 1998;9:389-397.
63 Morotti RA, Nicol KK, Parham DM, et al. Children’s Oncology Group. An Immunohistochemical algorithm to facilitate diagnosis and subtyping of rhabdomyosarcoma: the Children’s Oncology Group Experience. Am J Surg Pathol. 2006;30:962-968.
64 Kilpatrick SE, Renner J. Epithelioid/polygonal cell tumours. In: Kilpatrick SE, editor. Diagnostic musculoskeletal surgical pathology with clinicopathologic and cytologic correlations. Philadelphia: Saunders; 2003:71-96.
65 Layfield LJ, Lenarsky C. Desmoplastic small cell tumours of the peritoneum coexpressing mesenchymal and epithelial markers. Am J Clin Pathol. 1991;96:536-543.
66 Ali SZ, Nicol TL, Port J, et al. Intraabdominal desmoplastic small round cell tumour: cytopathologic findings in two cases. Diagn Cytopathol. 1998;18:449-452.
67 Ordi J, de Alava E, Torne A, et al. Intraabdominal desmoplastic small round cell tumour with EWS/ERG fusion transcript. Am J Surg Pathol. 1998;22:1026-1032.
68 Granja NM, Brgnami MD, Bortolan J, et al. Desmoplastic small round cell tumour: Cytological and immunocytochemical features. Case report. CytoJournal. 2005;2:1-6.
69 Roberts P, Burchill SA, Beddow RA, et al. A combined cytogenetic and molecular approach to diagnosis in a case of desmoplastic small round cell tumour with a complex translocation (11;22;21). Cancer Genet Cytogenet. 1999;108:19-25.
70 Ferlicot S, Coue O, Gilbert E, et al. Intraabdominal desmoplastic small round cell tumour: report of a case with fine needle aspiration, cytologic diagnosis and molecular confirmation. Acta Cytol. 2001;45:617-621.
71 Mora J, Fillipa DA, Qin J, et al. Lymphoblastic lymphoma of childhood and the LSA2-L2 Protocol; The 30 year experience at Memorial Sloan-Kettering Cancer Center. Cancer. 2003;98:1283-1291.
72 Hameed M. Small round cell tumours of bone. Arch Pathol Lab med. 2007;131:192-204.
73 Zhao XF, Young KH, Frank D, et al. Paediatric primary bone lymphoma-diffuse large B-cell lymphoma: morphologic and immunohistochemical characteristics of 10 cases. Am J Clin Pathol. 2007;127:47-54.
74 McManus AP, Gusterson BA, Pinkenton R, et al. The molecular pathology of small round cell tumours – relevance to diagnosis, prognosis, and classification. J Pathol. 1996;178:116-121.
75 Rapkiewicz A, Le BT, Simsir A, et al. Spectrum of head and neck lesions diagnosed by fine needle aspiration cytology in the paediatric population. Cancer (Cytopathology). 2007;111:242-251.
76 Akhtar M, Ali MA, Burgess A, et al. Fine-needle aspiration biopsy (FNAB) diagnosis of testicular involvement in acute lymphoblastic leukemia in children. Diagn Cytopathol. 1991;7:504-507.
77 Perez JS, Perez-Guillermo M, Bernal AB, et al. Hepatoblastoma: An attempt to apply histologic classification to aspirates obtained by fine needle aspiration cytology. Acta Cytol. 1994;38:175-182.
78 Douglass EC. Hepatic malignancies in childhood and adolescence (hepatoblastoma, hepatocellular carcinoma, and embryonal sarcoma). Cancer Treat Res. 1997;92:201-212.
79 Schnater JM, Kohler SE, Lamers WH, et al. Where do we stand with hepatoblastoma? Cancer. 2003;98:668-678.
80 Wakely PEJ, Silverman JF, Geisinger KR, et al. Fine needle aspiration biopsy cytology of hepatoblastoma. Mod Pathol. 1990;3:688-693.
81 Gilbert KL, Bergman S, Dodd LG, et al. Cytomorphology of yolk sac tumour of the liver in fine-needle aspiration: A paediatric case. Diagn Cytopathol. 2006;34:421-423.
82 Klijanienko J, Caillaud JM, Lagace R, Vielh P. Cytohistologic correlations in 56 synovial sarcomas in 36 patients. The Institut Curie experience. Diagn Cytopathol. 2002;27:96-102.
83 Akerman M, Willen H, Carlen B, et al. Fine needle aspiration (FNA) of synovial sarcoma-comparative histological-cytological study of 15 cases, including immunohistochemical, electron microscopic and cytogenetic examination and DNA-ploidy analysis. Cytopathology. 1996;7:187-200.
84 Silverman JF, Landreneau RJ, Sturgis CD, et al. Small-cell variant of synovial sarcoma: Fine needle aspiration with ancillary features and potential diagnostic pitfalls. Diagn Cytopathol. 2000;23:118-123.
85 Ackerman M, Ryd W, Skytting B. Scandinavian sarcoma Group. Fine-needle aspiration of synovial sarcoma: criteria for diagnosis: retrospective reexamination of 37 cases, including ancillary diagnostics. A Scandinavian Sarcoma Group Stidy. Diagn Cytopathol. 2003;28:232-238.
86 Gardner DG, Heikinheimo K, Shear M, et al. Ameloblastomas. In: Barnes L, Eveson J, Reichard P, et al, editors. World health Organization Classification of Tumours. Pathology and Genetics of Head and Neck Tumours. Lyon: IARC Press, 2005.
87 Barroca HM, Costa MJ, Carvalho JL. Cytologic profile of rhabdoid tumour of the kidney. A report of 3 cases. Acta Cytol. 2003;47:1055-1058.
88 Drut R. Cystic nephroma. Cytologic findings in fine needle aspiration biopsy. Diagn Cytopathol. 1992;18:593-595.
89 Shorter NA, Glick RD, Klimstra DS, et al. Malignant pancreatic tumours in childhood and adolescence: The Memorial Sloan-Kettering experience, 1967 to present. J Pediatr Surg. 2002;37:887-892.
90 Al-Kaisi N, Weaver MG, Abdul-Karim FW, et al. Fine needle aspiration cytology of neuroendocrine tumours of the pancreas. A cytologic, immunohistochemical, and electron microscopic study. Acta Cytol. 1992;36:655-660.
91 Shaw JA, Vance RP, Geisinger KR, et al. Islet cell neoplasms. A fine-needle aspiration cytology study with immunocytochemical correlations. Am J Clin Pathol. 1990;94:142-149.
92 Silverman JF, Holbrook CT, Pories WJ, et al. Fine needle aspiration cytology of pancreatoblastoma with immunocytochemical and ultrastructural studies. Acta Cytol. 1990;34:632-640.
93 Kerr NJ, Chun YH, Yun K, et al. Pancreatoblastoma is associated with chromosome 11p loss of heterozygosity and IGF2 overexpression. Med Pediatr Oncol. 2002;39:52-54.
94 Pettinato G, Di Vizio D, Manivel JC, et al. Solid-pseudopapillary tumour of the pancreas: a neoplasm with distinct and highly characteristic cytological features. Diagn Cytopathol. 2002;27:325-334.
95 Salla C, Chatzipantelis P, Konstantinou P, et al. Endoscopic ultrasound-guided fine-needle aspiration cytology diagnosis of solid pseudopapillary tumour of the pancreas: A case report and literature review. World J Gastroenterol. 2007;13:5158-5163.
96 Akhtar M, Dayel FA. Is it feasible to diagnose germ cell tumours by fine needle aspiration biopsy? Diagn Cytopathol. 1997;16:72-77.
97 Balco MT, Burroughs FH, Ali SZ. Cytopathologic findings in an immature cystic teratoma: Report of an unusual case. Diagn Cytopathol. 2007;35:120-122.
98 Allias F, Chanoz J, Blache G, et al. Value of ultrasound guided fine-needle aspiration in the management of ovarian and parovarian cysts. Diagn Cytopathol. 2000;22:70-80.
99 De Almeida MM, Chagas M, de Sousa JV, et al. Fine needle aspiration cytology as a tool for the early detection of testicular relapse of acute lymphoblastic leukemia in children. Diagn Cytopathol. 1994;10:44-46.
100 Layfield LJ. Cytopathology of Bone and Soft Tissue Tumours. New York: Oxford University Press; 2002.
101 Dey P, Mallik MK, Gupta SK, et al. Role of fine needle aspiration cytology in the diagnosis of soft tissue tumours and tumour-like lesions. Cytopathology. 2004;15:32-37.
102 Kurtycz DF, Logrono R, Hoerl HD, et al. Diagnosis of fibromatosis colli by fine needle aspiration. Diagn Cytopathol. 2000;23:338-342.
103 Kilpatrick SE, Ward WG, Bos GD, et al. The role of fine needle aspiration biopsy in the diagnosis and management of osteosarcoma. Pediatr Pathol Mol Med. 2001;20:175-187.
104 Gupta K, Dey P, Goldsmith R, et al. Comparison of cytologic features of giant-cell tumour and giant-cell tumour of tend sheath. Diagn Cytopathol. 2004;30:14-18.
105 Kilpatrick SE, Renner J. Chondroid tumours of bone and soft tissues. In: Kilpatrick SE, editor. Diagnostic musculoskeletal surgical pathology with clinicopathologic and cytologic correlations. Philadelphia: Saunders; 2003:323-328.
106 Wang J, Cobb CJ, Martin SE, et al. Pilomatrixoma: clinicopathologic study of 51 cases with emphasis on cytologic features. Diagn Cytopathol. 2002;27:167-172.
107 Hosler GA, Clark I, Zakowski MF, et al. Cytopathologic analysis of thyroid lesions in the paediatric population. Diagn Cytopathol. 2006;34:101-105.
108 Amrikachi M, Ponder TB, Wheeler TM, et al. Thyroid fine-needle aspiration biopsy in children and adolescents: Experience with 218 aspirates. Diagn Cytopathiol. 2005;32:189-192.