Epilepsy
EPILEPSY
Overview and Definition
Epilepsy comes from the Greek word meaning possession. The Greek people believed that seizures were caused by demons. Stigma and prejudice surrounding epilepsy continue, and people living with epilepsy often are reluctant to admit it or to seek treatment.2
A fundamental difference exists between seizure and epilepsy. Epilepsy is defined as a chronic disorder of various causes characterized by recurrent seizures—sudden, usually unprovoked attacks of subjective experiential phenomena, altered awareness, involuntary movements, or convulsions. Although a diagnosis of epilepsy requires the presence of seizures, not all seizures imply epilepsy. Epilepsy in children is usually considered constitutional, whereas in the older person, it is probably related to a provoking cause that develops over time.29
A seizure is a finite event; it has a beginning and an end. Seizures are a result of paroxysmal excessive discharge of cerebral neurons resulting in transient impairment or loss of consciousness. Seizures can be induced in any normal human brain by a variety of different electrical or chemical stimuli. The cerebral cortex in particular contains within its anatomic and physiologic structure a mechanism that is inherently unstable and capable of producing a seizure. Seizures are a relatively common symptom of brain dysfunction, and they may occur in many acute medical or neurologic illnesses in which brain function is temporarily disrupted. These seizures are most often self-limited and do not persist after the underlying disorder has resolved. Seizures also can occur as a reaction of the brain to physiologic stress, sleep deprivation, fever, and alcohol or sedative drug withdrawal. Isolated seizures also occur sometimes for no discernible reason as unprovoked events in presumably healthy people. None of these kinds of seizures represents epilepsy.6
Incidence
Epilepsy affects about 45 million people worldwide. Incidence is highest among young children and the elderly, and men are affected slightly more often than women (1.5: 1). In the United States figures range from 31 to 57 per 100,000 population.
The incidence in the older population, once thought to be low, has increased to approximately 77 per 100,000. In individuals over age 80, this incidence rises to 159 per 100,000. Epilepsy is the third most common serious neurologic disease of old age after dementia and stroke.26,30
Although seizure activity in children is rare, at less than 1%, seizures are the most common symptoms requiring medical attention in the infant. It has been estimated that 75% of cases of epilepsy have their onset before 20 years of age.
Etiologic and Risk Factors
In many individuals, several predisposing factors coexist, and the development of epilepsy reflects the interaction of acquired brain pathology and genetic predisposition. Although genetic diseases account for only about 1% of cases, heritable factors are important in a much higher percentage, especially in children. Forms that are demonstrably more heritable than others are termed idiopathic or primary epilepsies. Common features include a variable family history, generalized spike-wave abnormality on electroencephalogram (EEG), and onset in childhood or adolescence.
Although mutations in single genes account for a few rare epileptic syndromes, in most cases of epilepsy, data are most consistent with complex, polygenic influences.19 One gene for juvenile myoclonic epilepsy has been located on chromosome 6. Mutations of the EFHC1 gene may underlie different syndromes. The function of EFHC1 is unclear; roles in apoptosis and ciliary function have been proposed.3 This genetic predisposition to seizure activity may explain why one individual with brain damage develops epilepsy while another with similar damage does not develop seizure activity.22
Adult-Onset Seizure Disorder.: The causes of symptomatic epilepsy are multiple, and the symptoms may be transient, resolving after treatment of the primary disorder. Box 36-1 describes some of the causes of symptomatic epilepsy. Epilepsy can be caused by virtually any major category of serious disease or human disorder. It can result from congenital malformations, infections, tumors, vascular disease, degenerative diseases, or injury. In people over age 50, cerebrovascular disease is the most common cause of seizures, which often accompany or follow a stroke, and increases the risk of seizure by 17%.15,28 It is believed that perhaps even in the absence of stroke, cerebrovascular disease may predispose an individual to seizure activity. Seizures may be the first symptom of an intracranial mass, and it is suggested that 10% to 15% of older adult-onset epilepsy is a result of neoplasm. When the seizure is because of a permanent lesion or scar, the seizure activity may persist. Head trauma is the most common preventable cause of epilepsy. More than 3000 cases of epilepsy are added every year in the U.S. population because of head injury.27 Subdural hematoma can cause seizure activity and is common in older adults, often after a fall that may have seemed trivial.25 Pneumonia, which carries the possibility of hypoxia, especially in the aged, can cause seizures that can become recurrent.27 Alcohol abuse often leads to uncontrolled seizure activity.
Hormonal changes with increase in the levels of estrogen that occur during ovulation and menstruation can be a trigger for seizure in some women. Fertility appears to be reduced by seizure activity when it is triggered by hormonal changes. Disruptions of reproductive function in women include anovulatory cycles that may increase the risk for infertility, migraine, emotional disorders, and reproductive cancers. Moreover, both epilepsy itself and use of medications have been implicated as causal or contributory factors that can alter reproductive hormone levels and promote the development of reproductive endocrine disorders, especially polycystic ovarian syndrome. Menopause also tends to occur earlier in women with epilepsy, and this early menopause is associated with a history of high seizure frequency.9
Infantile Seizures.: The age-dependent appearance of spontaneous seizures in the primary epilepsies appears to depend on a critical period in cerebral maturation when the genetically determined defect is expressed clinically as a manifest change in behavior. Changes known as microdysgenesis occur in the brains of these infants. These changes include an increase in cell density, abnormal arrangements of cortical neurons, and an increase in white matter neurons.7
Seizures may occur in the neonatal period, within the first 24 to 72 hours, and they are usually of focal cerebral origin. The seizure type relates to the level of development of the brain. The seizures tend to be unilateral because of the immaturity of the forebrain and corpus callosum, limiting movement from one hemisphere to the other. Seizure activity represents the relative development of the limbic system, diencephalon, and brainstem. Hypoxic-ischemic brain insult is the most common cause of neonatal convulsions and is because of compromise of oxygen to the brain during or before delivery. Seizures resulting from cerebral contusion are often a result of prolonged or traumatic labor.
The other important cause of seizures arising in the early neonatal period is hypoglycemia, most often seen in babies who are small for gestational age. The neurologic symptoms of hypoglycemia consist of irritability, drowsiness, hypotonia, and apnea. Approximately half of the babies with hypoglycemia develop neurologic symptoms, and about one fourth of these go on to develop seizures.
Hypocalcemia (serum level below 7 mg/100 ml) may occur in the first 2 to 3 days of life either in low-birth-weight infants or in association with the complications of birth asphyxia. It may then contribute to seizures but is rarely the primary cause. Hypercalcemia occurring at age 6 to 8 days is usually the primary cause of the clinical features of neonatal tetany, including jitteriness and jaw, knee, and ankle clonus. Hypocalcemia is rare in infants who are breastfed or fed formula created to simulate human milk. Hypernatremia, resulting from alteration in sodium level, may also lead to neonatal seizures. Hypernatremia is associated with dehydrating illnesses.
Pathogenesis
All seizure activity is a result of chaotic electrical discharge in the central nervous system (CNS). Epileptic seizures result from the sudden, excessive electrical discharges of large aggregates of neurons. Because there are no consistent, demonstrable pathologic changes in the brains of individuals with idiopathic generalized epilepsy, susceptibility to these seizures most likely results from inherited biochemical, membrane, or neurotransmitter defects that result in abnormal excitability within the involved circuits.
The main inhibitory neurotransmitter of the CNS is γ-aminobutyric acid (GABA). When stimulated, GABA receptors modulate chloride ion flux, inhibiting membrane depolarization. GABA antagonists or functional depletion of GABA increases membrane depolarization and may result in seizures. GABA agonists (direct or indirect) therefore play a vital role in seizure termination. Loss of GABA-mediated inhibition results in seizures.31
Activation of the N-methyl-D-aspartate (NMDA) type of glutamate receptors potentiates cellular excitability and leads to sustained neuronal depolarization and calcium influx. Extracellular potassium and intracellular calcium concentrations increase and contribute to the overall excitability of the epileptic neuronal aggregate. During the seizure itself, neurons are tonically depolarized and fire continuously in a sustained, high-frequency discharge (tonic phase). The seizure ends as phasic repolarizations interrupt the continuous firing pattern (clonic phase) and gradually restore membrane potentials to normal or to a temporary hyperpolarized state (postictal depression). Prolonged NMDA receptor activation and excessive accumulation of intracellular calcium also result in neuronal toxicity and may lead to cell death. In some areas of the cortex such as the hippocampus, subsets of neurons that normally fire in bursts may serve as pacemaker cells for other groups of neurons during epileptogenic activities. In the focal epilepsies, abnormal neuronal behavior originates in and may remain confined to a restricted area of the cortex.19
The thalamus plays a role in generating generalized seizures and the generalized spike-wave EEG patterns that accompany them. The substantia nigra also is crucial to the expression of generalized convulsions, especially the tonic phase; GABA-ergic inhibitory transmission in the substantia nigra plays a regulatory role in the propagation of primary and secondarily generalized seizure discharges. Generalized seizures and the rhythmic spike-wave discharges are dependent on ionic conductance of the neurons in the thalamic nucleus reticularis, allowing them to function as pacemaker control cells.
About 70% to 80% of complex partial seizures arise from the temporal lobe, and more than 65% of these originate in mesial temporal lobe structures, especially the hippocampus, amygdala, and parahippocampal gyrus. Remaining cases of complex partial seizures arise mainly from the frontal lobe, with smaller percentages originating in the parietal and occipital lobes. Hippocampal or mesial temporal sclerosis, or scarring, is characterized by variable degrees of pyramidal cell loss and gliosis in the hippocampal subfields and dentate gyrus. This condition represents the most common pathologic substrate of partial epilepsy in adolescents and adults.11 Fig. 36-1 shows mesial sclerosis identified through coronal magnetic resonance imaging (MRI).
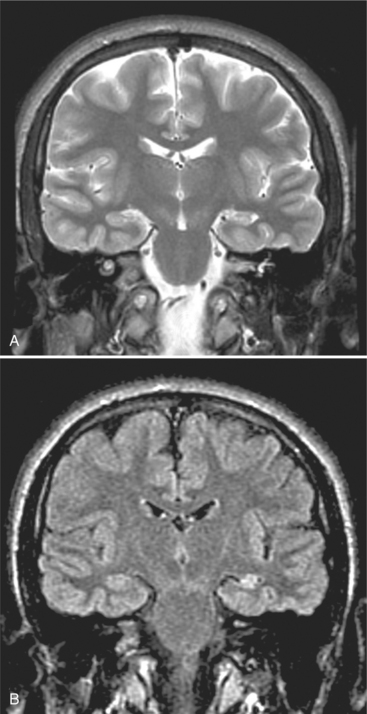
Figure 36-1 Temporal mesial sclerosis—coronal magnetic resonance image (MRI). The coronal projection is essential to reveal hippocampal abnormalities. Fluid-attenuated inversion recovery (FLAIR) MRI is superior to T2 weighting to show signal abnormalities, because the saturation nullifies the signal from the cerebrospinal fluid. A, A T2-weighted scan shows volume reduction of the left hippocampus. B, A FLAIR sequence shows the abnormal high signal (arrow), not seen on the T2 scan. (From Adam A, Dixon AK, Grainger RG, et al, eds: Grainger and Allison’s diagnostic radiology: a textbook of medical imaging, ed 4, Philadelphia, 2001, Churchill Livingstone.)
The hippocampus plays a critical role in both seizure activity and mood disorders. This suggests that pathology in this area of the brain might provide a link between epilepsy and depression. Remodeling of the hippocampal spine synapses may play a significant role in the neurobiology of depression and the effects of antidepressant therapy. Because the effects of estrogens on hippocampus parallel those of antidepressants, loss of estrogen appears to be a critical contributor to the etiology of depressive disorders. The increased incidence of depression observed in women with epilepsy might therefore reflect a hormonal deficiency state, although it is probably not the only factor that contributes to depression.8
Shifts in levels of brain-derived neurotrophic factor (BDNF) in the brain may represent another common link between the distinctive patterns of epilepsy and depression seen in women. Seizure incidence varies across the reproductive cycle, peaking in the periovulatory period. Dramatic fluctuations in estrogen levels in women may explain their greater vulnerability to depression, even as estrogen-related surges in BDNF expression may lead to an increased propensity for seizures.
Another potential explanation of the facilitation of seizures is kindling. Kindling refers to the processes that mediate long-lasting changes in brain function in response to repeated, gradually augmented stimulation of the brain resulting in epileptiform activity. Kindling also refers to sensitization of neuronal tissue by the addition of a drug or electrical stimulus that renders it susceptible to subsequent seizure activity. This may explain why exposures to localized, repetitive, low-intensity electrical stimuli induce increasingly pathologic responses. This is a possible mechanism that exists between the occurrence of a brain insult and the later onset of epilepsy. The kindling model is currently used in the study of partial epilepsy, particularly that involving the mesial temporal structures.6
Chronic focal epileptogenic lesions can cause distant areas to become capable of generating abnormal electrical discharges and seizures. This focus continues to function independently even after ablation of the primary lesion. This has important implications for the surgical treatment of epilepsy.
Clinical Manifestations
Signs and Symptoms.: In most individuals, seizures occur unpredictably at any time and without any relationship to posture or ongoing activities.10,14 In some individuals, seizures are provoked by specific stimuli such as flashing lights or a flickering television. See Box 36-2 for the typical triggers to seizure. The presence of focal signs after the seizure suggests that the seizure may have a focal origin. Prodromal symptoms (premonitory symptoms that indicate an impending seizure) may include headache, mood alterations, lethargy, and myoclonic jerking.
In the tonic phase of a tonic-clonic seizure, the body becomes rigid and the person is at risk of falling. A cry may be uttered, and the person may become cyanotic. In general the jaw is fixed and the hands are clenched. This phase usually lasts for 30 to 60 seconds. The clonic phase begins with rhythmic, jerky contractions and relaxation of all body muscles, especially in the extremities. Fig. 36-2 demonstrates the tonic and clonic phases of seizure. Biting of the tongue, lips, or inside of the mouth may occur. Saliva is blown from the mouth, with a froth appearing on the lips.
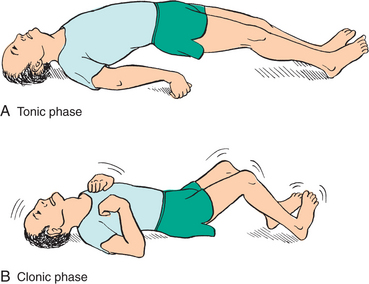
Figure 36-2 Tonic and clonic phases of seizure activity. (From Black JM: Medical-surgical nursing: clinical management for positive outcomes, ed 7, Philadelphia, 2004, Saunders.)
Minor motor seizures are reflected by involuntary jerking of the major muscles (myoclonus), momentary loss of muscle movement (akinesia), or total loss of muscle tone, which causes the person to fall to the floor (atonia).
Nonconvulsive status epilepticus is difficult to define, but it may be described as a syndrome in which the most fundamental feature is a change in the individual’s behavior. A degree of clouding of mental processes occurs, ranging from drowsiness and confusion to disorientation and dysphagia.
MEDICAL MANAGEMENT
The history obtained from the client and the observations of bystanders are of importance in establishing a diagnosis and classifying the seizure disorder correctly. To determine seizure type, a series of questions must be answered regarding the location of the seizure activity, the level of consciousness, the level of generalized motor activity, and the preceding level of seizure activity. The state of the client after the episode, including the level of confusion, sleepiness, or headache, must be determined. These data may be gathered from the paramedic or emergency personnel in the case of an individual who has been found on the floor.
It is important to recognize events that may mimic a seizure but are not related to the diagnosis of epilepsy. Early morning confusion and headache could be related to hypoglycemia in individuals on hypoglycemic medications. If the event is a transient ischemic attack, a loss of consciousness usually does not occur, and the neurologic insult is in the form of weakness or numbness. With a seizure, however, a twitching and tingling is evident. Behavioral disturbances as seen in dementia usually have a predictable pattern, such as occurring at a certain time of day.12 Syncope, or a transient loss of consciousness, is related to an acute change in cerebral perfusion. The related transient cerebral anoxia may itself cause seizure activity. Recurrent cardiac arrhythmia can be confused with recurrent seizures.17
On the other hand, epileptic events may not be recognized. Because the temporal lobe is the terminus of the vestibular pathways, the symptoms of activity may be nonspecific dizziness, or episodic vertigo. Fluctuations in mental status may be attributed to other causes or regarded as a dementing process.
Electroencephalography.
The EEG has a central role in the diagnosis of epilepsy. The EEG records the integrated electrical activity generated by synaptic potentials in neurons in the superficial layers of a localized area of cortex. In the epileptic focus, neurons in a small area of the cortex are activated for a brief period in a synchronized pattern and then inhibited. Interictal (between-seizure) activity on the EEG provides strong presumptive evidence that the event was a seizure. The best way to diagnose the presence of seizures and to classify the seizure is to observe, simultaneously, the seizure and the associated EEG recording. A normal reading does not rule out the diagnosis, and in the older individual, it may be more difficult to distinguish normal from abnormal.30
Prolonged EEG monitoring with simultaneous closed-circuit video recording is reserved for complicated cases of protracted and unresponsive seizures. It provides an invaluable method for recording ictal seizure events that are rarely captured during routine EEG studies. This technique is extremely helpful in the classification of seizures because it can accurately determine the location and frequency of seizure discharges while recording alterations in the level of consciousness and the presence of clinical signs.14
Metabolic studies are ideally performed at the time of the seizure occurrence, when values are most likely to be abnormal. Lumbar puncture should be considered for children with repeated seizures and other evidence of neurodevelopmental disability. It may be useful for detecting low cerebrospinal fluid glucose in glucose transporter disorder; alterations in amino acids, neurotransmitters, or cofactors in metabolic disorders; or evidence of chronic infection.
Aside from glucose determination, laboratory testing such as serum electrolyte levels and toxicology screening should be ordered based on individual clinical circumstances, such as evidence of dehydration. An EEG is not warranted after a simple febrile seizure but may be useful for evaluating individuals with an atypical feature or with other risk factors for later epilepsy. Similarly, neuroimaging is also not useful for children with simple febrile convulsions but may be considered for children with atypical features, including focal neurologic signs or preexisting neurologic deficits.14
Specialized neuropsychologic testing is often needed in evaluating clients with seizures. These tests not only help determine general intelligence and state of brain functioning but also often help to localize lesions.
A radiographic examination of the brain is indicated to rule out mass effect or vascular disease. MRI is the method of choice, since the lesions that cause epilepsy are often subtle.
TREATMENT.
Control of seizures is paramount in the treatment of epilepsy, but it is only part of the treatment. Support and education provided by health care professionals is critical to amelioration of the behavioral, social, and economic consequences of uncontrolled seizures. Reassurance should be given that for most individuals, epilepsy does not indicate serious brain damage. The risk-to-benefit ratio for antiepileptic treatments is one of the biggest challenges in seizure management. Because antiepileptic drugs have various adverse effects, which may interfere with normal developmental processes and affect cognitive functions, physicians must weigh the adverse effects of more aggressive treatments against the benefits of complete seizure control.4
Antiepileptic drugs carry risks of side effects that are particularly important in children. The decision as to whether or not to treat children and adolescents who have experienced a first unprovoked seizure must be based on a risk-benefit assessment that weighs the risk of having another seizure against the risks of long-term antiepileptic drug therapy. There appears to be no benefit of treatment with regard to the prognosis for long-term seizure remission.13 Suppressing interictal discharges can improve behavior in children with epilepsy and behavioral problems, particularly partial epilepsy. Focal discharges may be involved in the underlying mechanisms of behavioral problems in children with epilepsy.23
Antiepileptic medications are used according to the type of seizure. In most people with seizures of a single type, satisfactory control can be achieved with a single anticonvulsant drug. Monitoring of plasma drug levels has increased the ability to maintain the drug at the maximal tolerated dose. Generally, drug testing should be performed during the course of treatment and again when good seizure control has been established. Subsequent tests may be performed with changes in control or if side effects occur. Dose adjustments are carefully monitored, and noncompliance can be identified. It sometimes takes a trial of several different medications at different doses to find the best fit. In people who do not comply with the drug regimen, no drug may be better than an inconsistent dosage.1,3
Older individuals more often have difficulty with the neurologic side effects of dizziness, imbalance, drowsiness, and tremors. Cognitive deficits may worsen with topiramate, and mood disorders may worsen with phenobarbital, topiramate, and benzodiazepines.
Drugs and other therapies related to seizure management are listed here:
• Gabapentin (Neurontin) is an anticonvulsant is used as an add-on drug for individuals with refractory complex partial and secondarily generalized tonic-clonic seizures. The mechanism of action is binding of the drug to neuronal membranes (glutamate synapses) and increased brain GABA turnover. It has no drug interactions, and so can be used for most individuals.
• Lamotrigine (Lamictal) is a phenyltriazine compound used as an add-on drug for the management of complex partial and generalized tonic-clonic seizures. Lamotrigine is effective as monotherapy for some children with the Lennox-Gastaut syndrome and generalized absence seizures. Pharmacologic studies suggest that the drug acts at voltage-sensitive sodium channels to stabilize neuronal membranes and inhibit neuronal release, particularly of glutama-te.14
• Topiramate (Topamax) produces anticonvulsant action by blocking voltage-dependent sodium channels. This drug is used as adjunctive therapy for refractory complex seizures with or without secondary generalization. It can be used in the individual newly diagnosed with epilepsy.14
• Tiagabine (Gabitril) inhibits seizure activity by blocking reuptake of the neuroinhibitory transmitter GABA into neuronal and glial cells and is effective in the management of complex partial seizures as an add-on drug.
• Levetriacetam (Keppra) acts by an unknown mechanism and is indicated for use as an adjunctive treatment for partial seizures.
• Retigabine is structurally and functionally unrelated to any antiepileptic drugs in use today and was discovered though screening. The drug acts on the potassium channels and through an ancillary mechanism of potentiating GABA-evoked currents. Since the discovery that mutations in the genes that encode KCNQ2 and KCNQ3 are associated with benign familial neonatal convulsions, more attention has been paid to retigabine. The drug is particularly effective against kindled seizures. Retigabine is also effective against neuropathic pain in animals.21
• Divalproex sodium (Depakote) and sodium valproate (Valproate) are from the chemical compound valproic acid and are broad-spectrum anticonvulsants. They act by blocking voltage-dependent sodium channels and increase calcium-dependent potassium conductance. These drugs are useful for the management of many seizure types, including generalized tonic-clonic, absence, atypical absence, and myoclonic seizures. They rarely induce behavioral changes but are associated with mild gastrointestinal disturbances, alopecia, tremor, and hyperphagia. Two rare but serious side effects of valproate are a Reye-like syndrome and irreversible hepatotoxicity.14
• Benzodiazepines exert anticonvulsant activity by binding to a specific GABA site. The drugs diazepam and lorazepam delivered intravenously are used for initial management of status epilepticus. Clobazam may increase the serum drug levels of carbamazepine, phenytoin, phenobarbital, and valproic acid when used concomitantly.
• Carbamazepine is effective for the management of generalized tonic-clonic and partial seizures. It acts similarly to phenytoin by decreasing the sustained repetitive firing of neurons by blocking sodium-dependent channels and by decreasing depolarization-dependent calcium uptake.
• Ethosuximide provides its anticonvulsant action by blocking calcium channels associated with thalamocortical circuitry. Ethosuximide is an effective drug for the management of typical absence epilepsy.
• Phenobarbital and primidone are relatively safe anticonvulsants that are particularly useful for generalized tonic-clonic seizures. However, there is a chance of severe behavioral changes when these drugs are taken. Neurologically abnormal children are at greater risk. Furthermore, there is evidence that phenobarbital may adversely affect the cognitive performance of children treated on a long-term basis.
• Phenytoin acts by decreasing the sustained repetitive firing of single neurons by blocking sodium-dependent channels and decreasing depolarization-dependent calcium uptake. Phenytoin is used for primary and secondary generalized tonic-clonic seizures, partial seizures, and status epilepticus.14
• Vigabatrin acts by binding to the degradative enzyme GABA transaminase receptor, causing an increase in GABA levels and inhibition of neurotransmission. It is effective in infantile spasms, particularly in children with tuberous sclerosis. Fig. 36-3 diagrams the pharmacologic effects of some antiepileptic drugs at the GABA class A (GABAA) receptor.
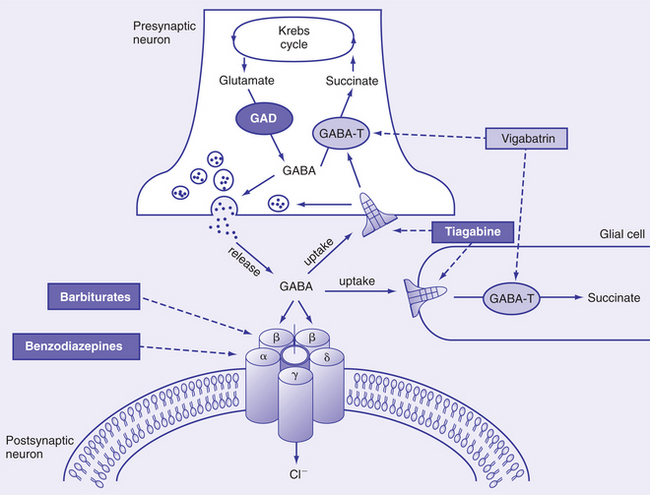
Figure 36-3 Pharmacologic effects of antiepileptic drugs at the γ-aminobutyric acid class A (GABAA) receptor. Barbiturates bind to a β-subunit of the GABAA receptor to potentiate the action of the endogenous agonist GABA and prolong the opening time of the chloride ion channel. Benzodiazepines bind to an α-subunit of GABAA to potentiate the action of GABA and increase the frequency of opening of the chloride ion channel. Vigabatrin irreversibly binds to GABA transaminase (GABA-T) to inhibit degradation of the inhibitory neurotransmitter GABA. Tiagabine blocks the uptake of synaptically released GABA into both presynaptic neurons and glial cells, allowing GABA to remain at the site of action for longer periods. GAD, Glutamic acid decarboxylase. (From Leach JP, Brodie MJ: Tiagabine, Lancet 351:203, 1998.)
• Adrenocorticotropic hormone (ACTH) is the preferred drug for the management of infantile spasms, although the dose and duration of therapy are not uniform. Prednisone is equally effective. ACTH and prednisone are equally effective for the treatment of cryptogenic and symptomatic seizures, and control can be expected in approximately 70% of individuals. There is no relationship between the ease or degree of seizure control and ultimate neurologic and cognitive outcome. The response to medication is usually apparent within a few weeks of therapy, but one third of individuals who respond suffer relapse when the ACTH or prednisone is discontinued.
• Ketogenic diet is a valuable therapeutic approach for epilepsy, used mostly with children. It is a diet high in fats, with enough protein for normal growth and energy and a low threshold for carbohydrates. Although the mechanism by which the diet protects against seizures is unknown, there is evidence that it causes effects on intermediary metabolism that influence the dynamics of the major inhibitory and excitatory neurotransmitter systems in brain. During consumption of the ketogenic diet, marked alterations in brain energy metabolism occur, with ketone bodies partly replacing glucose as fuel. Whether these metabolic changes contribute to acute seizure protection is unclear; however, the ketone body acetone has anticonvulsant activity and could play a role in the seizure protection afforded by the diet. In addition to acute seizure protection, the ketogenic diet provides protection against the development of spontaneous recurrent seizures in models of chronic epilepsy. The use of valproic acid is contraindicated in association with the ketogenic diet, because the risk of hepatotoxicity is enhanced.10
Anticonvulsive drugs are safe, but side effects do occur, especially at the start of drug therapy. Side effects of the medication may be ataxia, dysarthria, dizziness and blurring, or double vision. Fatigue is a common complaint.2 When phenytoin is taken, osteomalacia may occur as a result of increased metabolism of vitamin D. Hyponatremia can occur at low doses of carbamazepine and should be of concern when combined with a diuretic or in individuals with cardiac failure.24 Newer medications, such as lamotrigine, gabapentin, vigabatrin, and topiramate, are shown to be effective as add-on drugs demonstrating low interaction with other drugs.9 These drugs currently are more expensive, and studies have not shown them to be more effective when used as monotherapy compared with the current standard regimen. Allergic reactions are often in the form of a rash, and in these cases a change to another medication should be made.
When drug therapy does not control the seizures or drugs become toxic at effective dosages, then surgical treatment is indicated. Lobectomies, cortical resections, and sectioning of the corpus callosum are the types of surgeries most often performed. Hemispherectomies, the removal of one of the hemispheres, can be effective for the severe, uncontrollable seizures usually found in children.
Vagal nerve stimulation can be provided through an implantable pulse generator. By stimulating the left vagal nucleus, an inhibitory projection influences the entire cerebral cortex. A 50% reduction in seizure with vagal nerve stimulation has been reported. Recent studies indicate that it may be a safe adjunctive therapy for individuals with seizure disorders refractory to other therapies.
Among the complications associated with treatment with antiepileptic drugs is the reduction of efficacy of oral contraceptives, most of which involve hormonal intervention in the reproductive cycle to prevent ovulation and to inhibit fertilization and implantation of the ovum. A variety of pituitary hormone abnormalities in women with epilepsy are known to be caused by antiepileptic drugs, including elevated prolactin levels, disrupted luteinizing hormone and follicle-stimulating hormone levels, and abnormal concentrations of sex hormones. Seizure control must be the primary goal of therapy, with birth control a secondary concern. Understanding the continuum of estrogenic modulation of hippocampal BDNF levels may be relevant to the understanding, treatment, and even prevention of these disorders in women, possibly by the use of novel neutrophin-and estrogen-based therapies for their management.9
Many people with epilepsy experience a higher frequency of seizures with large doses of caffeine. Amphetamines and other stimulants should be avoided, and some asthma drugs can increase the incidence of seizure activity.
Some evidence shows that biofeedback and conditioning techniques are effective in helping control epilepsy in some people.7
PROGNOSIS.
Persons with epilepsy have increased mortality rates compared with the general population. Much of this increased risk occurs in individuals with symptomatic epilepsy in whom mortality relates to the underlying condition.
Death from asphyxia is the greatest concern in instances when the individual has a seizure during eating or when breathing passages are compromised during the seizure or in the postepileptic phase. Drowning during a seizure has been documented and remains a serious consequence of swimming or bathing alone. There is twentyfold increased risk of sudden unexplained death, presumably secondary to cardiac arrhythmia, pulmonary edema, or myocardial infarction. Sudden unexplained death is the most common cause of seizure-related mortality in persons with severe, chronic epilepsy.19 With status epilepticus, 3% of children and 10% of adults die during an attack.22
Duration of epilepsy is a composite factor that probably reflects the influence of several factors acting in combination. The longer an individual suffers from intractable epilepsy, the higher the possibility of exposure to factors that result in seizure-induced neuronal damage, the more prolonged the exposure to antiepilepsy drugs, and the greater the risk of seizure-related accidents, including closed head injuries.18
The correlation between depression and epilepsy is strong, and attempted suicide is higher than the norm in those with epilepsy. People with epilepsy are more likely to be hospitalized for depression. Young people are at particular risk for development of depression and other psychiatric disorders and are less likely to be treated. Despite evidence that two thirds of children with epilepsy have a diagnosable psychiatric disorder, only about one third receive psychiatric treatment.9
In people with epilepsy of no known cause and for whom a diagnosis is made before age 10, a 75% remission rate (defined as five seizure-free years) is seen. A child with epilepsy who has been free of seizures for more than 4 years while taking antiepileptic drugs has about a 70% chance of remaining in permanent remission when the drugs are withdrawn.
Chronic epilepsy is more likely when associated neurologic impairment is present at birth, when the seizures begin before 2 years of age, and when complex partial seizures are predominant.16
Classification of Seizures
An essential distinction in the modern system of classification is between partial and generalized seizures. Some classifications include a third group, partial seizures progressing to generalized seizures. Fig. 36-4 shows the seizure types, the typical area of the brain in which the seizure begins, and where it may travel. The following seizure types have been classified by the International League Against Epilepsy (Box 36-3). The initial events of a seizure, described either by the individual or by an observer, are usually the most reliable indication to determine whether a seizure begins focally.5 Fig. 36-5 charts the proportion of seizure types as a function of age.
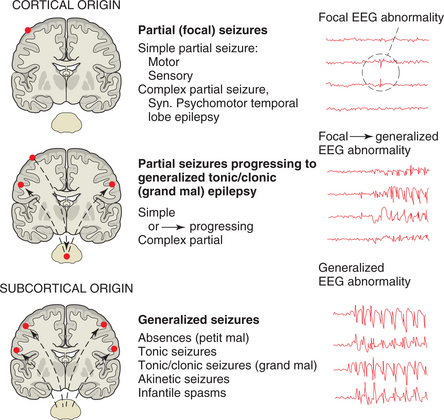
Figure 36-4 The nature of the attack is used to classify seizure types. (From Lindsay KW, Bone I, Callander R: Neurology and neurosurgery illustrated, New York, 1986, Churchill Livingstone.)
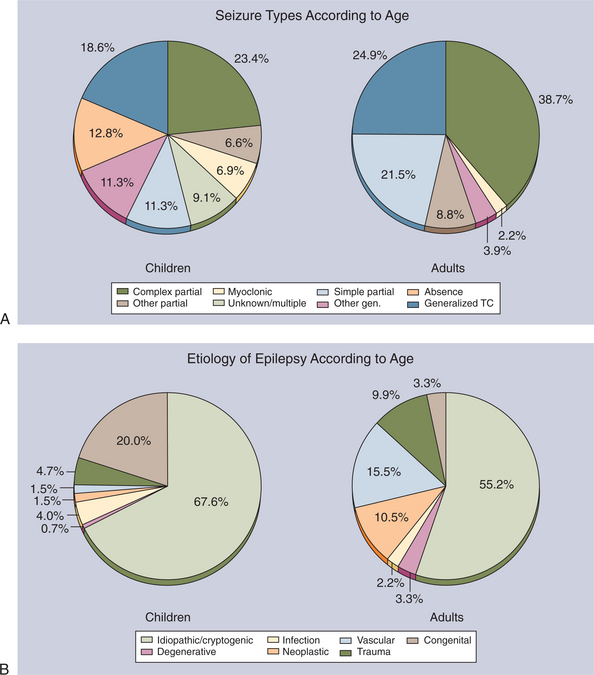
Figure 36-5 A, Proportion of seizure types as a function of age in newly diagnosed cases of epilepsy in Rochester, Minnesota, 1935 to 1984. B, Etiology of epilepsy in all newly diagnosed cases in Rochester, Minnesota, 1935 to 1984. TC, Tonic-clonic. (Modified from Hauser WA, Annegers JF, Kurland LT: Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935-1984, Epilepsia 34:453-468, 1993.)
Partial Seizures
Partial seizures have clinical or EEG evidence of a local onset. The abnormal discharge usually arises in a portion of one hemisphere and may spread to other parts of the brain during a seizure. The syndrome is characterized by the locus of onset of the attacks. Consciousness is not depressed, and individuals can interact normally with their environment except for limitations imposed by the seizure on specific localized brain functions. The seizures are identified as temporal, frontal, parietal, or occipital. Sensory symptoms such as localized paresthesias, numbness, vertigo, auditory hallucinations, and unformed visual hallucinations occur with seizures beginning in the corresponding primary sensory areas. Psychoillusory symptoms arise from ictal discharges in limbic and association cortex and include dysmnesic symptoms, such as feelings of familiarity (déj vu) and unfamiliarity (jamais vu); dreamy states, such as feelings of unreality and depersonalization; time distortion; emotional symptoms, such as fear or depression; visual illusions, such as multiple images (polyopsia) or distortions of size (micropsia and macropsia); and hallucinatory phenomena, such as unpleasant smells, stereotyped visions, or familiar voices. Autonomic symptoms reflect ictal involvement of limbic structures that lie in the mesial temporal or frontal lobe and project to the hypothalamus and brainstem.19 This group encompasses the most frequent and severe epilepsy problems of adults. Partial seizures can be subdivided into three groups: simple partial seizures, complex partial seizures, and partial seizures becoming secondarily generalized.
Many symptoms or phenomena can be the expressions of simple partial seizures. Subjective sensory and psychoillusory phenomena collectively are termed auras and affect about 60% of individuals with focal epilepsy. Examples of autonomic phenomena include an epigastric rising sensation (especially common with seizures beginning in the mesial temporal lobe), nausea, lightheadedness, pallor or flushing, pupillary dilation, piloerection, salivation, and urinary incontinence.
Simple Partial Seizures.: Simple partial seizures (formally known as focal seizures) are associated with preservation of consciousness and unilateral hemispheric involvement. Simple partial seizures result when the ictal discharge occurs in, and remains limited to, a circumscribed area of cortex. This site often is termed the epileptogenic focus. They may be manifested by focal motor symptoms (jerking) or somatosensory symptoms (paresthesias or tingling) that spread to different parts of the body. Because of their large cortical representation, muscles of the face and hand often are involved. Ictal discharges often involve supplementary or other secondary motor areas of the frontal lobe and produce contralateral flexion and elevation of the arm, contralateral turning of the head and eyes, and tonic extension of the ipsilateral arm (the so-called fencer’s posture). Light flashes, buzzing, or abnormal sensations of taste and smell represent involvement of the areas of the brain that control visual, auditory, or olfactory and gustatory responses. Sometimes autonomic symptoms create nausea, pallor, flushing, or pupillary dilation. Cognitive and affective changes can occur. Psychotic responses to seizure activity include illusions or hallucinations, and a sudden sense of fear is common. Focal weakness may follow a simple partial motor seizure; numbness, a sensory seizure; and blindness or amblyopia, an occipital lobe seizure. These reversible neurologic deficits collectively are called Todd’s paralysis and rarely last for more than 48 hours.
Complex Partial Seizures.: Complex partial seizures are associated with alteration or loss of consciousness and bilateral hemispheric involvement.
About 70% to 80% of complex partial seizures arise from the temporal lobe, hippocampus, amygdala, or parahippocampal gyrus. Remaining cases of complex partial seizures arise mainly from the frontal lobe, with smaller percentages originating in the parietal and occipital lobes. Many complex partial seizures evolve from simple partial seizures; consciousness becomes impaired as the seizure progresses. Complex partial seizures preceded by an olfactory aura are called uncinate fits because of their origin in or near the uncus of the medial temporal lobe. Uncinate fits may have a higher association with brain tumors than other types of complex partial seizures. In temporal lobe seizures, loss of consciousness results when the ictal discharge spreads bilaterally to involve areas of the hippocampus and amygdala; the parahippocampal gyri; and, to some extent, the entorhinal cortex and frontal regions.19
The person appears dazed and confused with random walking, mumbling, head turning, or pulling at clothing. Automatic behaviors, without conscious control, may manifest but cannot be recalled by the person. There may be clumsy perseveration of ongoing motor tasks, such as eating, drawing, walking, or washing dishes. Complex partial seizures usually last 45 to 90 seconds and are followed by confusion and disorientation lasting several more minutes. Characteristically, individuals are amnesic for details of the seizure that occurred after the aura. There may be transient postictal aphasia when the seizure involves the dominant temporal lobe.
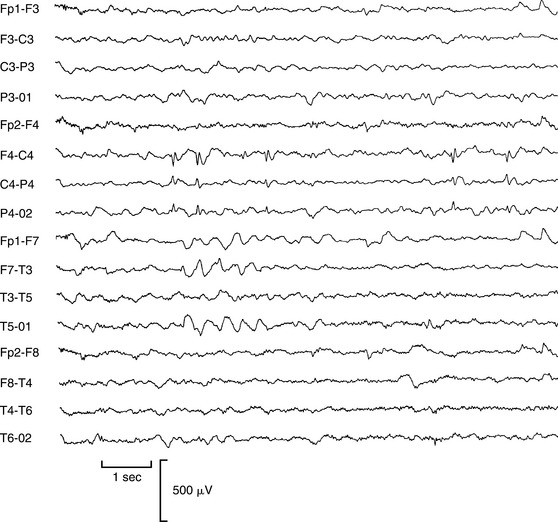
Complex partial seizures of frontal lobe origin are atypical and often differ dramatically from seizures originating in the temporal lobe. Although there are many variations, frontal lobe complex partial seizures tend to begin and end abruptly; have few, if any, postictal symptoms; and involve often bizarre motor manifestations, such as asynchronous thrashing or flailing of arms and legs, pelvic thrusting, pedaling leg movements, and loud vocalizations that may appear as psychiatric disorders.19 Fig. 36-6 presents the EEG of a woman with complex partial seizures, showing focal spike discharges in the right central region.
Generalized Seizures
In a generalized seizure, localized onset is not evident and the brain shows diffuse EEG abnormalities.
Absence Seizures.: Generalized absence seizures, formerly called petit mal seizures, consist of the sudden cessation of ongoing conscious activity with only minor convulsive muscular activity or loss of postural control. Generalized seizures begin diffusely and involve both cerebral hemispheres simultaneously from the outset. The person often stares into space. Onset and termination of attacks are abrupt. Absences are not preceded by an aura and are followed by normal activity. If attacks occur during conversation, the person may miss a few words or may break off in midsentence for a few seconds. The person is unaware of the loss of conscious control. Often, these seizures occur in children and frequently disappear by adolescence. Atypical absence seizures are similar to absence seizures but coexist with other forms of generalized seizure.
Myoclonic Seizures.: Myoclonic seizures are sudden, brief, single or repetitive muscle contractions involving one body part or the entire body. The myoclonic jerks range from small movements of the face or hands to massive bilateral spasms that simultaneously affect the head, limbs, and trunk. Repeated myoclonic seizures may seem to crescendo and terminate in a generalized tonic-clonic convulsion. Although they can occur at any time, myoclonic seizures often cluster shortly after waking or while falling asleep.19
Atonic Seizures.: Atonic seizures, also known as “drop attacks,” are brief losses of consciousness and postural tone not associated with tonic muscular contractions. Atonic seizures occur most often in children with diffuse encephalopathies and are characterized by sudden loss of muscle tone that may result in falls with self-injury. Sometimes the loss of muscle tone is limited or fragmentary, producing only a head drop.
Tonic-Clonic Seizure
Formerly called grand mal seizure, tonic-clonic seizure is the archetypal seizure, which means total loss of control. The seizure begins with a sudden loss of consciousness, and falls are common. The generalized rigidity (tonic) phase is followed by very rapid generalized jerking movements (the clonic stage). In adults, incontinence of bowel and bladder occur. Generalized tonic-clonic seizures result in many striking but transient physiologic changes, including hypoxia, lactic acidosis, elevated plasma catecholamine levels, and increased concentrations of serum creatine kinase, prolactin, corticotropin, cortisol, β-endorphin, and growth hormone. In the tonic phase respiration can cease briefly. Recovery may be swift after a short seizure, but a prolonged seizure may induce a deep sleep. Altered speech and transient paralysis or ataxia may follow, as well as headache, disorientation, or muscle soreness. Another seizure may follow without recovery of consciousness or, after recovery of consciousness, the person may experience seizure again.22 As recovery progresses, many individuals complain of headache, muscle soreness, mental dulling, lack of energy, or mood changes lasting 24 hours. Complications include oral trauma, vertebral compression fractures, shoulder dislocation, aspiration pneumonia, and sudden death, which may be related to acute pulmonary edema, cardiac arrhythmia, or suffocation. Although this is the type of epilepsy that most people associate with the disorder, it is less common than partial seizures.
Status Epilepticus
Status epilepticus is a condition in which seizures are so prolonged or so repeated that recovery does not occur between attacks. Convulsive status epilepticus occurs when the person has generalized tonic-clonic seizures and no return to consciousness occurs between seizures. It is a medical emergency. The molecular events that cause death can occur with the first few seizures. Tonic-clonic status epilepticus is more common in people whose seizures have a known cause. Often it is the result of tumor, CNS infection, or drug abuse. Febrile seizures are a common cause of status epilepticus in children under the age of 3 years.
Epilepsies of Infancy and Childhood
Infantile spasms represent a nonspecific reaction on the part of the brain to a wide variety of insults. It is likely that the condition is more age specific than disease specific. It is generally agreed that infants who develop spasms demonstrate a cessation of normal psychologic development and often show developmental deterioration that relates to the frequency of the spasms.
Infantile spasms are typically classified into two groups: cryptogenic and symptomatic. A child with cryptogenic infantile spasms has an uneventful pregnancy and birth history as well as normal achievement of developmental milestones before the onset of seizures. The neurologic examination and the computed tomographic (CT) and MRI scans of the head are normal, and there are no associated risk factors. Approximately 10% to 20% of infantile spasms are classified as cryptogenic, and the remainder are classified as symptomatic.
Symptomatic infantile spasms are related directly to several prenatal, perinatal, and postnatal factors. Prenatal and perinatal factors include hypoxia-ischemia, congenital infections, inborn errors of metabolism, tuberous sclerosis, and prematurity. Postnatal conditions include CNS infections, head trauma (especially subdural hematoma and intraventricular hemorrhage), and hypoxic-ischemic encephalopathy. The fact that infantile spasms and immunizations often occur simultaneously around 6 months of age is a coincidence of timing rather than a cause-and-effect relationship with any immunization antigen.
Infants with cryptogenic infantile spasms have a good prognosis, whereas those with the symptomatic type have an 80% to 90% risk of mental retardation. The underlying CNS disorder has a major role in the neurologic outcome. Several theories have been advanced with regard to the pathogenesis of infantile spasms, including dysfunction of the monoaminergic neurotransmitter system in the brainstem, derangement of neuronal structures in the brainstem, and an abnormality of the immune system. Another possibility is that specified stresses or injury to an infant during a critical period of neurodevelopment causes corticotropin-releasing hormone (CRH) overproduction, resulting in neuronal hyperexcitability and seizures. The number of CRH receptors reaches a maximum in the infant brain, followed by spontaneous reduction with age, which perhaps accounts for the eventual resolution of infantile spasms even without therapy. Exogenous ACTH and glucocorticoids suppress CRH synthesis, which may account for their effectiveness in treating infantile spasms.
Severe Myoclonic Epilepsy of Infancy
Severe myoclonic epilepsy of infancy is a syndrome of early normal development followed by treatmentresistant seizures of various types and by psychomotor retardation, usually beginning around 5 to 6 months of age. These attacks are often long and may include status epilepticus. Mental retardation is noted in all cases.
Benign Myoclonic Epilepsy of Infancy
Benign myoclonus begins during infancy and consists of clusters of myoclonic movements confined to the neck, trunk, and extremities. The myoclonic activity may be confused with infantile spasms; however, the EEG is normal in individuals with benign myoclonus. The prognosis is good, with normal development and the cessation of myoclonus by 2 years of age. An anticonvulsant is not indicated.
Lennox-Gastaut Syndrome
Lennox-Gastaut syndrome usually begins between 1 and 6 years of age. The most common seizures are atonic-akinetic, resulting in loss of postural tone. Violent falls occur suddenly followed by immediate recovery and resumption of activity, with the attack lasting less than 1 second. Injuries to the head and face are common. Tonic attacks consist of sudden flexion of the head and trunk. Clusters of attacks are common, followed by automatic behavior. Consciousness is usually clouded rather than completely lost. Neurologic abnormalities such as spasticity are common in severely affected patients.16
Acquired Epileptic Aphasia (Landau-Kleffner Syndrome)
Acquired epileptic aphasia (Landau-Kleffner syndrome) is characterized by an acquired aphasia in the absence of other neurologic abnormalities. Often epileptic seizures and psychomotor disturbances develop at the same time or shortly afterward. This disorder often begins between ages 3 and 9. The age of onset is critical for the long-term loss of language. It is postulated that recently acquired skills are particularly vulnerable to any disturbance of brain function. If the formation of intracerebral connections is prevented by abnormal neuronal firings of epilepsy during optimal periods of language learning in children, then such connections may never become functional and the ability to develop language is lost.11
Benign Childhood Epilepsy with Centrotemporal Spikes
Benign childhood epilepsy with centrotemporal spikes typically occurs between the ages of 3 and 13 and is characterized by brief, simple partial hemifacial motor seizures. Childhood epilepsy with occipital paroxysms is a syndrome similar to benign childhood epilepsy with centrotemporal spikes, but it includes visual symptoms at onset, and some children have associated migraine headache.
Childhood Absence Epilepsy
Childhood absence epilepsy begins between ages 4 and 10 years. The attacks may occur many times a day. Absence attacks often can be precipitated by hyperventilation. The attacks were previously described as petit mal and are characterized by a blank stare with unresponsiveness, rhythmic blinking, and, sometimes, a few small clonic jerks of arms or hands. Behavior and awareness return immediately to normal. There is no postictal period and usually no recollection that a seizure has occurred. The attacks last for between 10 and 45 seconds. Longer absence attacks are accompanied by automatisms, usually of a perseverative type, in about 70% of cases. Absence seizures commonly coexist with generalized tonic-clonic or myoclonic seizures. Untreated, absence seizures can occur hundreds of times each day, a condition referred to as pyknolepsy. Fig. 36-7 shows the EEG of a child with absence epilepsy.
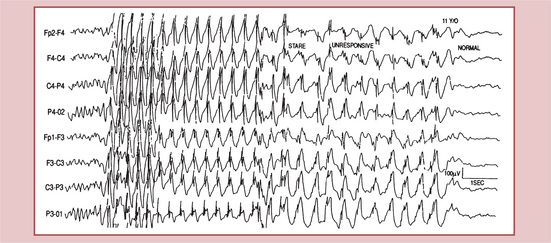
Figure 36-7 Childhood absence epilepsy. Electroencephalogram shows the typical pattern of generalized 3-Hz spike-wave complexes associated with a clinical absence seizure. (From Goldman L: Cecil textbook of medicine, ed 22, Philadelphia, 2004, Saunders.)
Lapses of awareness that have a more gradual onset, do not resolve as abruptly, and are accompanied by autonomic features or loss of muscle tone are called atypical absence seizures. These events occur most often in children with mental retardation, and they do not respond as well to antiepileptic drug treatment. Typical and atypical absence seizures also must be distinguished from complex partial seizures manifested only by brief lapses of consciousness, because cause, treatment, and prognosis differ among these three seizure types.19
Juvenile Myoclonic Epilepsy (Janz Syndrome)
Juvenile myoclonic epilepsy (Janz syndrome) has its onset in early adolescence, between ages 8 and 20 years, in otherwise healthy individuals with normal intelligence and a family history of similar seizures. Consciousness is unimpaired, and mental retardation is not evident. It begins with early morning jerks of the head, neck, and upper limbs, making hair combing and toothbrushing difficult. As the myoclonus tends to abate later in the morning, most individuals do not seek medical advice at this stage and some deny the episodes. A few years later, early morning generalized tonic-clonic seizures develop. A gene locus has been identified on chromosome band 6p21.
Febrile Convulsions
Febrile convulsions, the most common seizure disorder during childhood, generally have an excellent prognosis but may also signify a serious underlying acute infectious disease such as sepsis or bacterial meningitis. They are rare before 6 months and after age 5 and are a result of fever. The typical febrile convulsion is brief, generalized, and tonic-clonic in sequence, and the body temperature is high. The seizures occur more often when the child is asleep. The convulsion is often the first indication that the child is ill, because 90% of all seizures occur in the first 24 hours of fever. The rise in temperature, which may be a result of increased oxygen demands on cerebral oxidative mechanisms, may be the most important factor. Severe febrile seizures are most often unilateral, and a possibility exists for permanent brain damage, with the development of epilepsy, if the seizure lasts more than 30 minutes.16
Although most affected children have no long-term consequences, febrile seizures increase the risk of future epilepsy. This risk is low for most children, but increases when there is a family history of afebrile seizures or if there were neurologic abnormalities before the first febrile seizure. Febrile seizures neither are associated with nor do they cause mental retardation, below-average IQ, poor school performance, or behavior problems. Prophylactic treatment generally is not indicated because of the benign prognosis. Routine treatment of a normal infant with simple febrile convulsions includes a careful search for the cause of the fever; active measures to control the fever, including the use of antipyretics; and reassurance of the parents. Prolonged anticonvulsant prophylaxis for preventing recurrent febrile convulsions is controversial and no longer recommended. Antiepileptics such as phenytoin and carbamazepine have no effect on febrile seizures. Phenobarbital can be effective in preventing recurrent febrile seizures but may also decrease cognitive function in treated children compared with untreated children. Sodium valproate is also effective in the management of febrile seizures, but the potential risks of the drug do not justify its use in a disorder with an excellent prognosis regardless of treatment. The incidence of fatal valproate-induced hepatotoxicity is highest in children younger than 2 years of age. Oral diazepam is an effective and safe method of reducing the risk of recurrence of febrile seizures.14
Posttraumatic Epilepsy
After penetrating wounds and other severe head injuries, about one third of individuals have seizures within 1 year. Although most individuals experience seizures within 1 to 2 years of injury, new-onset seizures still may appear 5 or more years later. Two thirds of individuals with posttraumatic epilepsy have partial or secondarily generalized seizures. Mild head injuries (e.g., uncomplicated brief loss of consciousness, no skull fracture, absence of focal neurologic signs, no contusion or hematoma) do not increase the risk of seizures to a clinically significant degree.
Impact seizures (a generalized convulsion occurring at the time of, or immediately after, the injury) and early seizures (seizures occurring within the first 1 to 2 weeks) represent acute reactions of the brain to the trauma. Seizures beginning after 10 to 14 days reflect an increased risk of posttraumatic epilepsy development.
Early seizures should be treated with phenytoin. To minimize complications from seizures occurring during acute management, phenytoin also should be given prophylactically for 1 to 2 weeks to individuals who have had severe head injuries. In the absence of overt attacks, phenytoin use should be discontinued after 2 weeks, because no data indicate that antiepileptic drugs prevent the development of later epilepsy.19
Epilepsia Partialis Continua
Epilepsia partialis continua is characterized by continuous focal seizures that can involve part or all of one side of the body. In adults, epilepsia partialis continua occurs with severe strokes, primary or metastatic brain tumors, metabolic encephalopathies, encephalitis, and subacute or rare chronic inflammatory diseases of the brain. Antiepileptic drugs are usually ineffective, as are corticosteroids and antiviral agents. Seizures remit spontaneously in some cases. Rasmussen encephalitis is one cause of epilepsia partialis continua. The onset is usually before age 10. Sequelae include hemiplegia, hemianopia, and aphasia. The disease is progressive and potentially lethal but more often becomes self-limited with significant neurologic deficits. The disease may be due to autoantibodies that bind to and stimulate the glutamate receptors. Studies have identified cytomegalovirus in several surgical specimens of individuals with Rasmussen encephalitis.14
References
1. Aminoff, MJ. Nervous system. In: Tierney LM, McPhee SJ, Papadakis MA, eds. Current medical diagnosis and treatment. Norwalk, CT: Appleton & Lange, 1994.
2. Brown, SW. The treatment of epilepsy: a patient’s viewpoint. London: British Epilepsy Association, 1996.
3. Berkovic, SF. December 12 highlight and commentary: epilepsy genetics: complexity unmasked, conundrums revealed. Neurology. 2006;67(11):1907.
4. Cole, AJ. Guidelines for new epilepsy drugs. N Engl J Med. 2004;10(7):6–7.
5. Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised clinical and electroencephalographic classifications of epileptic seizures. Epilepsia. 1999;22:489–501.
6. Foldvary-Schaefer, N, Wyllie, E. Epilepsy. In Goetz CG, ed.: Textbook of clinical neurology, ed 2, Philadelphia: Saunders, 2003.
7. Gumnit, RJ. The epilepsy handbook: the practical management of seizures, ed 2. New York: Raven Press, 1995.
8. Hajszan, T. Neurologic links between epilepsy and depression in women: is hippocampal neuroplasticity the key? Neurology. 2006;66(6 suppl 3):S13–S22.
9. Harden, CL. Adolescent female with epilepsy: mood, menstruation, and birth control. Neurology. 2006;66(6 suppl 3):S3–S4.
10. Hartman, AL. The neuropharmacology of the ketogenic diet. Pediatr Neurol. 2007;36(5):281–292.
11. Haut, SR. Susceptibility of immature and adult brains to seizure effects. Lancet Neurol. 2004;3(10):608–617.
12. Hesdorffer, DC, et al. Dementia and adult-onset unprovoked seizures. Neurology. 1996;46:727–730.
13. Hirtz, D. Practice parameter: treatment of the child with a first unprovoked seizure: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2003;60(2):166–175.
14. Johnston, MV. Seizures in childhood. In Behrman RE, Kliegman M, Jenson HB, eds.: Nelson Textbook of pediatrics, ed 17, Philadelphia: Saunders, 2004. [chap 586].
15. Kilpatrick, CJ, et al. Epileptic seizures in acute stroke. Arch Neurol. 1990;47:157–160.
16. O’Donohoe, NV. Epilepsies of Childhood, ed 3. Oxford: Butterworth-Heinemann, 1994.
17. Overstall, P. Drop attacks. In: Kenny RA, ed. Syncope in the older patient: causes, investigations and consequences of syncope and falls. London: Chapman & Hall, 1996.
18. Oyegbile, TO. The nature and course of neuropsychological morbidity in chronic temporal lobe epilepsy. Neurology. 2004;62(10):1736–1742.
19. Pedley, TA. The epilepsies. In Goldman L, ed.: Cecil textbook of medicine, ed 22, Philadelphia: Saunders, 2004. [chap 434].
20. Perucca, E. The new generation of antiepileptic drugs: advantages and disadvantages. Br J Clin Pharmacol. 1996;42:531–543.
21. Pollard, JR. Antiepileptic drugs in development. Lancet Neurol. 2006;5(12):1064–1067.
22. Porter, RJ, Theodore, WH. Epilepsy: 100 elementary principles. Philadelphia: Saunders, 1995.
23. Pressler, RM. Treatment of interictal epileptiform discharges can improve behavior in children with behavioral problems and epilepsy. J Pediatr. 2005;146(1):112–117.
24. Rimmer, EM, Richens, A. Clinical pharmacology and medical treatment. In Laidlaw J, Richens A, Oxley J, eds.: A textbook of epilepsy, ed 3, Edinburgh: Churchill Livingstone, 1988.
25. Rowan, AJ, Ramsay, GR. Seizures and epilepsy in the elderly. Boston: Butterworth-Heinemann, 1997.
26. Sander, JW, Hart, YM, Johnson, AL, et al. National General Practice Study of Epilepsy: newly diagnosed epileptic seizures in a general population. Lancet. 1990;336:1267–1270.
27. Schnell, SS. Nursing care of clients with cerebral disorders. In Black JM, Matassarin-Jacobs E, eds.: Luckmann and Sorensen’s medical-surgical nursing, ed 4, Philadelphia: Saunders, 1993.
28. So, EL, et al. Population-based study of seizure disorders after cerebral infarctions. Neurology. 1996;46:350–355.
29. Tallis, RC. Epilepsy in elderly people. London: Martin Dunitz, 1995.
30. Tallis, RC. Epilepsy. In: Tallis RC, ed. Brockelhurst’s textbook of geriatric medicine and gerontology/neurology. Edinburgh: Churchill Livingstone, 1998.
31. Wills, B. Chemically induced seizures. Clin Lab Med. 2006;26(1):185–209. [ix].