CHAPTER 77 Adrenocortical Hormones
The adrenal gland is composed of two distinct parts: (1) an inner adrenal medulla, which is functionally related to the sympathetic nervous system and secretes mainly epinephrine but some norepinephrine and (2) an outer adrenal cortex, which forms the bulk of the gland and secretes corticosteroids. The primary corticosteroids secreted by the adrenal cortex are as follows:
The secretion of mineralocorticoids and glucocorticoids is essential to life. Only small amounts of sex hormones are normally secreted by the adrenal cortex, and they have little effect on reproductive function.
Chemistry of Adrenocortical Secretion (p. 921)
The Adrenal Cortex Is Composed of Three Distinct Layers or Cell Types: Zona Glomerulosa, Zona Fasciculata, and Zona Reticularis
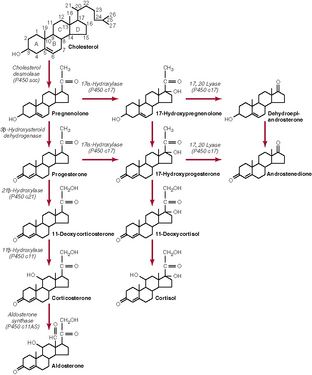
Adrenocortical Hormones Are Synthesized from Cholesterol
Most of the cholesterol in adrenocortical cells is taken up from the circulation and then esterified and stored in lipid droplets. The rate-limiting step in the synthesis of adrenocortical hormones is the side-chain cleavage of cholesterol to form pregnenolone (see Fig. 77–1). This step includes the delivery of cholesterol to the inner mitochondrial membrane and the enzymatic cleavage (through cholesterol desmolase) of a six-carbon unit from cholesterol to yield pregnenolone. In all three zones of the adrenal cortex, this initial step in steroid biosynthesis is stimulated by the controllers of the major hormone products (aldosterone and cortisol). The conversion of cholesterol to pregnenolone and all the subsequent steps in the synthesis of adrenocortical hormones occur either in the endoplasmic reticulum or mitochondria. Not all of the compounds shown in Figure 77–1 are produced in all three zones of the adrenal cortex.
Adrenocortical Hormones Are Bound to Plasma Proteins
Approximately 90% to 95% of the cortisol in the plasma is bound to plasma proteins, especially transcortin or corticosteroid-binding globulin (CBG). As a result of this high degree of binding to plasma proteins, cortisol has a long half-life (about 60 to 90 minutes). Corticosterone is bound to plasma proteins to a lesser degree than cortisol and has a half-life of approximately 50 minutes. Even smaller amounts of aldosterone are bound to plasma proteins; consequently, aldosterone has a half-life of only approximately 20 minutes.
Adrenocortical Hormones Are Metabolized in the Liver
Cortisol and aldosterone are metabolized to various compounds in the liver and then conjugated to glucuronic acid. These inactive conjugates are freely soluble in plasma and are not bound to plasma proteins. Once released into the circulation, they are readily excreted in urine. The rate of inactivation of adrenocortical hormones is depressed in liver disease.
Functions of the Mineralocorticoids—Aldosterone (p. 924)
Aldosterone Is the Primary Mineralocorticoid Secreted by the Adrenal Cortex
Aldosterone accounts for approximately 90% of the mineralocorticoid activity of adrenocortical hormones. Most of the remainder of the mineralocorticoid activity can be attributed to (1) deoxycorticosterone, which has approximately 3% of the mineralocorticoid activity of aldosterone and is secreted at a comparable rate, and (2) cortisol, a glucocorticoid with weak mineralocorticoid activity that is normally present at plasma concentrations of more than 1000 times that of aldosterone. In vitro studies have shown that cortisol binds with high affinity to mineralocorticoid receptors. Because the kidneys have the enzyme 11β-hydroxysteroid dehydrogenase type 2, cortisol is converted to cortisone, which does not avidly bind mineralocorticoid receptors. Consequently, cortisol does not normally exert significant mineralocorticoid effects in vivo. Under conditions in which 11β-hydroxysteroid dehydrogenase is either congenitally absent or inhibited (e.g., during excessive licorice ingestion), cortisol may have substantial mineralocorticoid effects.
Aldosterone Increases Sodium Reabsorption and Potassium Secretion
Aldosterone and other mineralocorticoids act on the distal nephron, especially the principal cells of the collecting duct, to increase sodium reabsorption and potassium secretion. These effects occur after the binding of aldosterone to intracellular receptors and the subsequent synthesis of proteins, including Na,K-ATPase in the basolateral membrane and sodium and potassium channel proteins in the apical membrane. As a result of increased Na,K-ATPase activity, sodium is pumped out of the tubular cells into the blood and exchanged for potassium. Potassium then diffuses into the tubular urine. As sodium is reabsorbed under the influence of aldosterone, there is enhanced tubular secretion of potassium ions. Aldosterone also causes secretion of hydrogen ions in exchange for sodium in the intercalated cells of the cortical collecting tubules. Because protein synthesis is required to mediate the tubular actions of aldosterone, a lag time of about 60 minutes occurs between exposure to aldosterone and its onset of action.
Aldosterone Affects Electrolyte Transport in Organs Other Than the Kidneys
Aldosterone binds to mineralocorticoid receptors in epithelial cells other than those of the kidney. Aldosterone increases sodium reabsorption from the colon and promotes potassium excretion in the feces. Similarly, aldosterone has an effect on sweat and salivary glands, decreasing the sodium/potassium ratio in their respective secretions.
Controllers of Aldosterone Secretion—Angiotensin II and Potassium
Angiotensin II Stimulates Aldosterone Secretion
Angiotensin II directly stimulates the cells of the zona glomerulosa to secrete aldosterone. This effect of angiotensin II is mediated via increments in intracellular levels of calcium and the phosphatidylinositol products, diacylglycerol and inosital triphosphate. These second messengers activate protein kinase C, which in turn stimulates both early (cholesterol desmolase) and late (aldosterone synthase) steps in the biosynthesis of aldosterone.
The control of aldosterone secretion by angiotensin II is closely linked to the regulation of extracellular fluid volume and arterial pressure (see Chapter 29). The renin-angiotensin system is activated in the presence of hypovolemia and hypotension; and high plasma levels of angiotensin II stimulate aldosterone secretion. In turn, aldosterone increases sodium reabsorption in the distal nephron; as fluid retention returns body fluid volumes and arterial pressure to normal levels, the stimulus for activation of the renin-angiotensin system wanes, and aldosterone secretion falls to basal levels. Accordingly, the activity of the renin-angiotensin system is inversely related to dietary sodium intake.
Potassium Stimulates Aldosterone Secretion
The cells of the zona glomerulosa are sensitive to small changes in the plasma potassium concentration. Increments in plasma potassium concentration increase aldosterone secretion by depolarizing the cell membrane, opening calcium channels, thereby increasing the intracellular calcium concentration. In response to these events, aldosterone secretion increases as a result of stimulation of the same early and late biosynthetic steps affected by angiotensin II (see previous discussion).
Aldosterone plays a critical role in eliminating ingested potassium and in feedback regulation of the plasma potassium concentration (see Chapter 29). Increments in plasma potassium concentration increase aldosterone secretion, which in turn stimulates tubular secretion of potassium. As plasma potassium concentrations fall to normal levels, the stimulus for aldosterone secretion is removed. The opposite sequence of events occurs when plasma potassium concentration decreases. Increases in plasma potassium concentration depolarize the cell membrane, activating voltage-dependent calcium channels. The rise in cytoplasmic calcium stimulates aldosterone secretion by the mechanism described above for angiotensin II.
ACTH Plays a Permissive Role in the Regulation of Aldosterone Secretion
So long as normal plasma levels of ACTH are present, the responsiveness of the zona glomerulosa to its major controllers, angiotensin II and potassium, is maintained. In contrast, if ACTH is chronically deficient, the aldosterone response to angiotensin II and potassium is diminished. High plasma levels of ACTH, which occur acutely during stress, stimulate aldosterone secretion; but in states of chronic ACTH excess (e.g., with Cushing’s disease), hyperaldosteronism is not sustained.
Functions of the Glucocorticoids (p. 928)
Cortisol Is the Primary Glucocorticoid Secreted by the Adrenal Cortex
More than 95% of glucocorticoid activity exerted by the adrenocortical hormones can be attributed to cortisol; most of the remaining glucocorticoid activity is due to corticosterone. Cortisol mediates most of its effects by binding with intracellular receptors in target tissues and inducing or repressing gene transcription; this results in alterations in the synthesis of enzymes that alter cell function.
Cortisol Has Widespread Effects on Metabolism
There are pronounced disturbances in carbohydrate, fat, and protein metabolism in adrenal insufficiency. Some of the metabolic effects of cortisol are permissive in that cortisol does not initiate the changes, but its presence at normal plasma levels permits certain metabolic processes. Cortisol exerts the following effects on metabolism:
Increased Cortisol Secretion Is Important for Resistance to Stress
Physical or mental stress increases ACTH secretion, which in turn stimulates the adrenal cortex to secrete cortisol. Although it is not clear how hypercortisolism mediates this response, the large rise in cortisol secretion in response to many stressors is essential to survival. Patients with adrenal dysfunction who are administered maintenance doses of steroids require extra glucocorticoid under stressful conditions.
Pharmacological Doses of Glucocorticoids Have Anti-Inflammatory and Antiallergic Effects and Suppress Immune Responses
Large doses of glucocorticoids decrease the inflammatory response to tissue trauma, foreign proteins, or infections through several effects, including the following:
Controller of Cortisol Secretion—ACTH (p. 931)
ACTH Stimulates Cortisol Secretion
The secretion of cortisol is under the control of the hypothalamic-pituitary, corticotropin-releasing hormone (CRH)-ACTH axis. The release of ACTH (corticotropin) from the pituitary is dependent on the hypophysiotropic hormone CRH. Once ACTH is secreted into the blood, it has a rapid effect on the inner two zones of the adrenal cortex, especially the zona fasciculata, to increase the secretion of cortisol. This effect of ACTH is achieved by increasing the conversion of cholesterol to pregnenolone and is mediated via the second messenger cyclic AMP. Chronic stimulation of the adrenal cortex by ACTH causes hypertrophy and hyperplasia of the zona fasciculata and zona reticularis and increased synthesis of several enzymes that convert cholesterol into the final product cortisol. Under conditions of chronic ACTH excess, such as with Cushing’s syndrome, there are sustained increases in the secretion of cortisol and adrenal androgens.
Blood levels of free (unbound) cortisol are controlled in a negative feedback fashion. Increased plasma levels of cortisol decrease ACTH secretion through a direct effect on the pituitary as well as indirect inhibition of CRH release from the hypothalamus. The secretion of cortisol is highest in the early morning and reaches its lowest in the late evening because there is a diurnal or circadian rhythm in ACTH secretion as a result of changes in the frequency and duration of CRH bursts from the hypothalamus. Because of the cyclic changes in cortisol secretion, plasma levels of cortisol are meaningful only when expressed in terms of the time of day when blood sampling occurred.
Stress Increases ACTH Secretion
Several physical and mental stressors stimulate the neuroendocrine cells of the hypothalamus to secrete CRH; as a result, there is increased ACTH secretion, which stimulates release of cortisol. Under conditions of stress, the inhibitory effect of cortisol on ACTH secretion is insufficient to counteract the extra neural input to the neuroendocrine cells secreting CRH. Consequently, plasma levels of ACTH are increased.
Adrenal Androgens (p. 934)
The adrenal androgens DHEA and androstenedione are secreted in appreciable amounts, but they have only weak androgenic effects. Consequently, the normal plasma concentrations of these hormones exert little effect on secondary sex characteristics, especially in males, in whom large amounts of testosterone, the most potent androgen, are secreted by the testes. In females, adrenal androgens are responsible for pubic and axillary hair. Most of the androgenic activity of adrenal hormones may be due to the conversion of adrenal androgens to testosterone in peripheral tissues. In contrast to the normal state, when adrenal androgens are secreted in excessive amounts, as with Cushing’s syndrome, appreciable masculinization may be produced in both males and females. The secretion of adrenal androgens is stimulated by ACTH.
Abnormalities of Adrenocortical Secretion (p. 934)
Increased Plasma Levels of Glucocorticoids (Cortisol) Produce Cushing’s Syndrome
Excess cortisol secretion can be caused by an adrenal tumor, a pituitary tumor that is secreting large amounts of ACTH and causing bilateral adrenal hyperplasia (Cushing’s disease), or a tumor of the lungs or other tissues (ectopic tumors) that is secreting large amounts of ACTH and causing bilateral adrenal hyperplasia. Cushing’s syndrome may also be produced by the administration of large amounts of exogenous glucocorticoids.
Symptoms of Cushing’s syndrome include the following:
Conn’s syndrome (primary aldosteronism) is caused by a tumor in the zona glomerulosa. When a tumor is present in the zona glomerulosa that produces large amounts of aldosterone, the most notable features are hypertension and hypokalemia; usually hypertension is relatively mild because there is only a small increase in extracellular fluid volume resulting from “sodium escape” (see Chapter 29). The hypertension and hypokalemia are exacerbated by increased sodium intake. Because of expansion of the extracellular fluid volume and the rise in arterial pressure, plasma renin activity is suppressed. The potassium depletion in Conn’s syndrome decreases the concentrating ability of the kidneys, leading to polyuria, and causes muscle weakness and metabolic alkalosis.
Impaired secretion of adrenocortical hormones occurs in Addison’s disease. Destruction of the adrenal cortex can result from autoimmune disease, tuberculosis, or cancer. These processes usually are gradual, leading to a progressive reduction in glucocorticoid and mineralocorticoid function. As a result of the decreased cortisol secretion, there is a compensatory increase in ACTH secretion, which produces hyperpigmentation. Symptoms of Addison’s disease include the following.