Chapter 15 Pharmacological treatments in rheumatic diseases
KEY POINTS
 The pathogenesis of rheumatic diseases is diverse and often involves a complex mix of immunological and biomechanical mechanisms. Prior to prescribing pharmacological therapy, the needs of the patients, alternative non-pharmacological remedies and drug interactions (the pharmaco-dynamics and pharmaco-kinetics) need to be considered. Symptomatic management of pain involves analgesics (e.g. paracetamol, NSAIDs, opioids) and therapy is titrated according to severity of pain with such therapies tailored to individual needs and co-morbidities. For people with osteoarthritis, current therapies are aimed at symptom control. However for people with inflammatory arthritis, treatment of the underlying inflammatory process is required. This will involve anti-inflammatory drugs, glucocorticosteroids, and disease modifying anti-rheumatic drugs (DMARDs). There is evidence that DMARDs are able to modulate the disease process in inflammatory arthritis and especially with the newer biologic drugs, prevent progression of joint damage and even induce remission. While increased understanding and knowledge of the pathogenesis of rheumatic diseases has resulted in increased therapeutic options, all pharmacological therapies are associated with adverse events. For optimal outcomes consumer and clinician education are essential, together with a multi-disciplinary approach for complex problems.
The pathogenesis of rheumatic diseases is diverse and often involves a complex mix of immunological and biomechanical mechanisms. Prior to prescribing pharmacological therapy, the needs of the patients, alternative non-pharmacological remedies and drug interactions (the pharmaco-dynamics and pharmaco-kinetics) need to be considered. Symptomatic management of pain involves analgesics (e.g. paracetamol, NSAIDs, opioids) and therapy is titrated according to severity of pain with such therapies tailored to individual needs and co-morbidities. For people with osteoarthritis, current therapies are aimed at symptom control. However for people with inflammatory arthritis, treatment of the underlying inflammatory process is required. This will involve anti-inflammatory drugs, glucocorticosteroids, and disease modifying anti-rheumatic drugs (DMARDs). There is evidence that DMARDs are able to modulate the disease process in inflammatory arthritis and especially with the newer biologic drugs, prevent progression of joint damage and even induce remission. While increased understanding and knowledge of the pathogenesis of rheumatic diseases has resulted in increased therapeutic options, all pharmacological therapies are associated with adverse events. For optimal outcomes consumer and clinician education are essential, together with a multi-disciplinary approach for complex problems.INTRODUCTION
In rheumatic diseases, pharmacotherapies are largely used for control of inflammatory processes or for pain control; such therapies are usually used in conjunction with non-pharmacological therapies such as muscle strengthening. Over the past decade, a greater understanding of the underlying mechanism involved in inflammatory arthritides has facilitated the development of new drugs and raised therapeutic goals. The aims of modern therapy are early diagnosis and prompt initiation of therapy to control inflammation, thereby preventing pain and subsequent joint damage with associated impaired function.
The decision regarding initiation and selection of drug therapy is not only based on the diagnosis, but on a person’s age, past medical history (e.g. previous peptic ulcer), the presence of co-morbidities (e.g. renal impairment) and the presence of concomitant medications. Compliance with medication and patient expectations are other important concepts to be taken into consideration when prescribing. It is worth briefly reviewing the underlying processes for which pharmacotherapies are employed.
PAIN
The word pain comes from Latin ‘poena’ meaning fine or penalty. Pain is a complex phenomenon involving biopsychosocial interactions, and detailed mechanisms are beyond the scope of this chapter. In brief, pain perception peripherally is mediated by the terminal endings of finely myelinated A delta and of non-myelinated C fibres. Chemicals produced locally as a result of injury produce pain by direct stimulation or by sensitizing the nerve endings. A-delta fibres are responsible for the acute pain sensation, which may be followed by a slower onset, more diffuse pain mediated by the slower conducting C fibres. Most sensory input enters the spinal cord via the dorsal spinal roots. When the signal reaches the spinal cord, a signal is immediately sent back along motor nerves to the original site of the pain, triggering the muscles to contract (Kidd et al 2007). The pain signal is also sent to the brain. Only when the brain processes the signal and interprets it as pain do people become conscious of the sensation. Pain receptors and their nerve pathways differ in different parts of the body and the type of pain felt depends on the stimuli. These stimuli may be mechanical, thermal or chemical.
It is worth noting that the anatomical source of pain in many arthritic conditions is not always clear, and may often be multifactorial in nature. In early rheumatoid arthritis (RA) where synovitis is the primary pathology, the pain is presumably derived from inflammatory mediators in the synovium. In osteoarthritis (OA), the sources of pain are more controversial and may arise from the synovium or subchondral bone. The source of pain in lateral epicondylitis may be different again and relate to specific entheseal pathology. Musculoskeletal pain may be present without the classical signs of inflammation.
INFLAMMATION
Inflammation results in heat, redness, swelling, pain and loss of function. Acute inflammation involves: increased blood supply in the region of injury; an increase in local capillary permeability; exudation of vascular fluid; migration of inflammatory cells out of the blood vessels and into the surrounding tissue; and the release of mediators of inflammation. Acute inflammation may resolve or can lead to chronic inflammation, tissue death (necrosis), scarring or fibrosis.
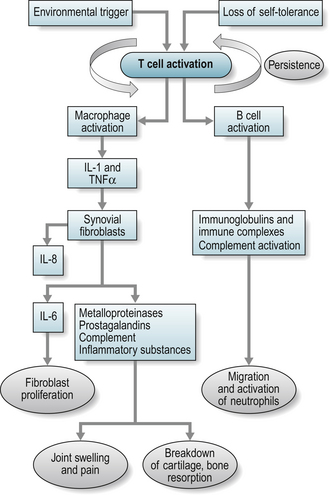
However, the triggering event for inflammation in many inflammatory arthritides is unclear and both environmental and genetic triggers have been considered i.e. exposure to an environmental antigen in a genetically predisposed individual. It is known that a foreign antigen activates T-cells resulting in an inflammatory response. This process is paramount in the body’s normal defence against infection, when the process is controlled and subject to inhibitory mechanisms. Uncontrolled and autonomous, this process can result in diseases such as rheumatoid arthritis (RA). In RA, activated T-cells secrete inflammatory cytokines that lead to chronic activation of B cells and immunoglobulin synthesis (see Fig. 15.1). The pro-inflammatory cytokines include interleukin (IL)-1 and tumour necrosis factor α (TNF) that in turn stimulate the production of more cytokines such as IL-6 and IL-8 (Jovanovic et al 1998). The result is pain, chronic synovitis and eventually local tissue and bone destruction.
PRINCIPLES OF CLINICAL PHARMACOLOGY
Before prescribing a therapeutic agent, two main questions must be answered:
Pharmadynamics is the study of the biochemical and physiological effects of drugs and the mechanism of drug action, and the relationship between drug concentration and effect.
Pharmacokinetics is a branch of pharmacology dedicated to the determination of the fate of substances administered externally to a living organism. In practice, this discipline is applied mainly to drug substances, though in principle it concerns itself with all manner of compounds ingested or otherwise delivered externally to an organism, such as nutrients, metabolites, hormones, toxins, etc. Pharmacokinetics is often divided into several areas including, but not limited to, the extent and rate of absorption, distribution, metabolism and excretion.
Absorption is the process of a substance entering the body. Distribution is the dispersion or dissemination of substances throughout the fluids and tissues of the body. Metabolism is the transformation of the substance and its daughter metabolites. Excretion is the elimination of the substances from the body. In rare cases, some drugs irreversibly accumulate in a tissue in the body.
When calculating dosage, it is important to know the half-life of the drug. This refers to the time required for the amount in the body to fall to 50% and is influenced by drug clearance and the volume of distribution. Drug clearance is defined as the volume of plasma that would contain the amount of drug excreted per minute. Drug clearance is a measure of the ability of the body to eliminate a drug from the circulation. It determines daily dosage. Drugs may be cleared by the renal system (mostly excreted, e.g. penicillin) or by the hepatic system (mostly metabolised e.g. paracetomol). Some drugs undergo substantial removal from the portal circulation by the liver after oral administration (e.g. Buprenorphine). This ‘first-pass’ effect can significantly reduce the amount of active drug that reaches the systemic circulation. The volume of distribution for a drug is determined by its degree of water or lipid solubility, the extent of plasma- and tissue-protein binding, and the perfusion of tissues. It gives an indication of initial or loading dose e.g. hydroxychloroquine (see later).
DRUGS USED IN RHEUMATIC DISEASES
ANALGESICS
Paracetamol
Paracetamol, also known as acetaminophen in the USA, is one of the most commonly used non-narcotic analgesics; it is also an anti-pyretic agent (lowers temperature). It is recommended as the first line analgesic in OA by the American College of Rheumatology (ACR) and European League against Rheumatism (EULAR). It is usually taken orally but may be used per rectum. Orally it is well absorbed and peak plasma concentrations are reached in 30–60 minutes. A proportion is bound to plasma proteins and the drug is inactivated in the liver. The plasma half-life of paracetamol is 2–4 hours. It is a weak inhibitor of cyclo-oxygenase (COX-1) (Warner et al 1999) at therapeutic doses.
The usual dose of paracetamol in adults is in the range of 1 gram three or four times daily. The incidence of side effects at therapeutic levels is low so routine monitoring is not required. Toxic doses (two to three times the maximum therapeutic dose) can cause fatal renal and liver failure. This effect is exacerbated by concomitant alcohol ingestion and in patients with active liver disease (Clissold 1986).
OPIOIDS
This group of drugs are used to relieve moderate to severe pain. The term opioid applies to substance that produces morphine-like effects. Opium is an extract of the juice of the poppy ‘papaver somniferum’ which has been used for social and medicinal purposes for thousands of years to induce euphoria, analgesia and sleep, and prevent diarrhoea. These effects are reflected in the potential toxicity of opioids in clinical use: nausea, constipation and confusion. Repeated administration can result in mild tolerance and (much more uncommonly) cause dependence. The most severe and serious adverse reaction associated with opioid use is respiratory depression, the mechanism behind fatal overdose.
Opioids are available in a variety of forms, and are usually used parenterally for acute pain, while oral and transdermal preparations have been developed for chronic ambulatory use. Some oral agents are also available in slow release preparations. A number of common drugs are now highlighted.
Morphine
Morphine can be administered parenterally (intramuscularly, subcutaneously, intravenously) and orally. The former route is preferred as the drug undergoes substantial first pass effect after oral dosing. Morphine has a half-life of 2–3 hours. Morphine commonly causes nausea and vomiting (and therefore is often given with an anti-emetic), constipation and drowsiness. It may also lead to respiratory depression and hypotension.
Pethidine
Unlike morphine, pethidine is a synthetic opioid, but has the same mechanism of action. It produces prompt but short lasting analgesia and is not recommended for chronic use because of a high risk of tolerance and dependency. Like morphine, it is absorbed well orally but undergoes extensive first pass metabolism and so is only used parenterally. The side-effect profile is similar to morphine but pethidine can also cause convulsions and other central nervous system disorders.
Codeine and codeine/paracetomol preparations
Codeine is an opium derivative pro-drug; once metabolised about 5–10% of codeine will be converted to morphine. It is usually prescribed orally as it is not as extensively metabolised on first pass through the liver. Codeine is effective for mild to moderate pain but may be too constipating for long-term use. The combination of codeine with paracetomol (generic name, co-codomol) is also effective for patients with mild to moderate pain, though evidence for superior efficacy of the combination is minimal.
Tramadol
Tramadol is a centrally acting analgesic, which possesses opioid agonist properties and activates monoaminergic spinal inhibition of pain. It is metabolised in the liver and excreted via the kidneys. It may be administered orally, rectally, intravenously or intramuscularly for patients with moderate to severe pain. Tramadol is about 10% as potent as morphine and consequently may have fewer of the typical opioid side effects. Tramadol is well tolerated in short term use with dizziness, nausea, sedation, dry mouth and sweating being the principal adverse effects.
Buprenorphine
Buprenorphine is a thebaine derivative, and its analgesic effect is due to partial agonist activity at μ-opioid receptors, i.e. when the molecule binds to a receptor, it is only partially activated in contrast to a full agonist such as morphine. Buprenorphine also has very high binding affinity for the μ receptor such that opioid receptor antagonists (e.g. naloxone) only partially reverse its effects. Buprenorphine is administered via a transdermal route in 35, 52.5 and 70 mcg/hour patches that deliver the dose over 96 hours. It is not administered orally, due to very high first-pass metabolism. Buprenorphine is metabolised by the liver and the metabolites are eliminated mainly through excretion into bile. The elimination half-life of buprenorphine is 20–73 hours (mean 37). Common adverse drug reactions associated with the use of buprenorphine are similar to those of other opioids and include: nausea and vomiting, drowsiness, dizziness, headache, itch, dry mouth, miosis, orthostatic hypotension, male ejaculatory difficulty, decreased libido and urinary retention. Constipation and central nervous system (CNS) effects are seen less frequently than with morphine. Hepatic necrosis and hepatitis with jaundice have been reported with the use of buprenorphine, especially after intravenous injection of crushed tablets.
Fentanyl
Fentanyl transdermal patches work by releasing fentanyl into body fats, which then slowly release the drug into the bloodstream over 72 hours, allowing for long lasting relief from pain. Fentanyl patches are manufactured in five patch sizes: 12.5 micrograms/h, 25 μg/h, 50 μg/h, 75 μg/h, and 100 μg/h. Rate of absorption is dependent on a number of factors and body temperature, skin type and placement of the patch can have major effects. Fentanyl is metabolised by the liver and excreted renally. The side effect profile is similar to buprenorphine.
ANTI-INFLAMMATORY DRUGS
Anti-inflammatory drugs are used extremely commonly across the world for acute and chronic musculoskeletal pain. The drugs may be classified into three categories:
NSAIDs
This very commonly used group of drugs is effective in reducing the immediate signs and symptoms of inflammation, although much of their use is as analgesics in conditions where the underlying inflammatory nature of the condition is not always clear (e.g. commonly used over-the-counter for headaches and lower back pain). NSAIDs are commonly used in patients suffering from all types of inflammatory arthritis and are also recommended for patients with OA if paracetamol is ineffective.
There are many different NSAIDs currently available and a list of common NSAIDs with their usual daily doses are shown in Table 15.1. The dose used in inflammatory arthritis is often higher than that used in osteoarthritis or soft tissue problems. The usual rule in dosing is to use the lowest effective dose for the shortest possible period of time.
Table 15.1 Common non-steroidal anti-inflammatory drugs (NSAIDs) with usual oral daily dosage
| NSAID | DAILY DOSE (MG) |
|---|---|
| Meloxicam | 7.5–15 |
| Ibuprofen | 1200–2400 |
| Diclofenac | 75–150 |
| Naproxen | 500–1000 |
| Indometacin | 50–150 |
| Celecoxib | 100–400 |
| Etoricoxib | 60–120 |
In general the NSAID mechanism of action is by inhibition of cyclooxygenase and thus inhibition of the production of prostaglandins and thromboxanes. There are two cyclooxygenase enzymes, COX-1 and COX-2. COX-1 is important in producing prostaglandins with physiological functions such as protection of the gastrointestinal mucosa and vascular homeostasis; its levels are reasonably constant. On the other hand, COX-2 is up-regulated at sites of inflammation and it is responsible for the production of inflammatory mediators. All NSAIDs vary in their degree of COX-1 and COX-2 selectivity.
NSAIDs can be given orally, topically, intramuscularly or per rectum. The drugs tend to be well absorbed in the gastrointestinal tract and are highly plasma bound. They only have a small first pass effect. There is no consistent evidence that one NSAID is superior to another in terms of analgesic or anti-inflammatory efficacy. However they do have different pharmacokinetic properties according to half life. Drugs such as indometacin, diclofenac and ibuprofen have short half lives ( < 6 hours), whilst naproxen and meloxicam have long half lives ( > 10 hours). In addition, many of these agents come in slow-release or modified release preparations to extend their half-life. This difference in half life is important for daily dosing regimens, and it is worth considering what time of day the person’s symptoms are greatest.
Topical NSAIDs may be useful for knee and hand OA (Moore et al 1998). They generally require multiple daily applications. Their advantages are fewer serious side effects because they do not achieve high plasma levels. The most common problems are rash and pruritis (itching) at the application site.
As a class, oral NSAIDs are associated with a large number of side effects, as presented in Table 15.2. The gastrointestinal side effects are the most frequent, and risk factors for gastrointestinal side effects include: age over 65 years, history of peptic ulcer disease, use of oral corticosteroids or anticoagulants. Given the large numbers of people who use NSAIDs, they are associated with a significant mortality and morbidity. There are also important clinical pharmacodynamic interactions associated with NSAID use: they can antagonise antihypertensive medication leading to high potassium levels.
Table 15.2 Common side-effects of NSAIDs
| Gastrointestinal | Nausea, diarrhoea Dyspepsia, bleeding, ulceration |
| Hypersensitivity | Rashes, angio-oedema, bronchospasm |
| Cardiovascular | Potential increased risk of ischaemic heart disease and stroke, hypertension |
| Renal | Fluid retention, renal impairment |
| Central nervous system | Headache, dizziness, depression |
NSAIDs and cardiovascular risk
In order to overcome the considerable gastrointestinal (GI) toxicity (see Table 15.2) and mortality associated with NSAIDs, selective COX-2 inhibitors were developed (e.g. celecoxib, etoricoxib), and initial studies suggested some benefit from these agents in reducing serious gastrointestinal complications. However the withdrawal from the market of one of these agents, rofecoxib, due to an increased risk of thrombotic vascular complications, led to a re-evaluation and understanding that both COX-2 and traditional NSAIDs have a pro-thrombotic tendency. All COX-2 agents and NSAIDs should be used with caution (lowest dose for shortest period of time) or where possible avoided completely in people with cardiovascular risk factors or who are taking aspirin (as aspirin is used in people with increased cardiovascular risk, but also because of concerns about an interaction between NSAIDs and aspirin that may reduce the benefits of aspirin) (Graham 2006). Another strategy for reducing GI side effects is co-prescription of gastroprotective agents such as proton-pump inhibitors (PPI eg omeprazole); in the UK, the National Institute for Health and Clinical Excellence (NICE) have recommended that all people with osteoarthritis requiring NSAIDs for moderate to long term use should have a PPI co-prescribed, based on a cost-effectiveness analysis (Conaghan et al 2008).
CORTICOSTEROIDS
Oral corticosteroids (also known as glucocorticosteroids) have been used in patients with RA and other inflammatory joint conditions for many years. Corticosteroids are effective in rapidly reducing inflammation. Some patients require low dose maintenance doses of corticosteroid ( < 10 mg/day) but long-term usage and higher doses are associated with increased side effects. Patients with severe RA, vasculitis or active systemic lupus erythematosus (SLE) may require intravenous, high-dose corticosteroids.
Corticosteroids are powerful immunosuppressants. The corticosteroid receptor is a transcription factor, and after binding the receptor –steroid complex enters the cell nucleus and binds to specific DNA sequences modifying DNA transcriptions. The result is inhibition of various leucocytes, especially lymphocytes and macrophages. Corticosteroids can reduce prostaglandin and leukotriene production and antagonise the effect of some pro-inflammatory cytokines.
The pharmacokinetics of corticosteroids depends on the preparation. The most widely used, prednisolone, has a half-life of 2–3 hours. Other preparations include methylprednisolone, hydrocortisone and triamcinolone. Corticosteroid therapy may be associated with adverse effects (see Table 15.3), which are generally related to the cumulative dose of drug. Of particular concern is the ability of corticosteroids to induce bone loss and osteoporosis. All patients requiring long-term steroid therapy (e.g. prednisolone > 7.5 mg/day for 3 months) require bone protection therapy with a calcium supplement and probably a bisphosphonate (e.g. alendronate). Long-term oral corticosteroid therapy leads to suppression of the body’s own hypothalamic pituitary – adrenal axis. Abrupt cessation of the drug may induce a withdrawal syndrome involving fatigue, myalgia, anorexia, weight loss and sometimes collapse with hypotension and electrolyte imbalance. To prevent this all patients should be educated, wear medi-alert bracelets and undergo gradual reduction of steroid dose prior to cessation.
Table 15.3 The adverse effects of corticosteroid therapy
| Musculoskeletal | Osteoporosis Myopathy Avascular necrosis of the femoral head |
| Immunological | Increased susceptibility to infection |
| Endocrine | Truncal obesity, moon-like face Hyperglycaemia or frank diabetes Acne Hirsuitism Salt and water retention |
| Dermatological | Thinning of skin Increased fragility of skin |
| Cardiovascular | Hypertension Exacerbation of congestive heart failure |
| Gastrointestinal | Peptic ulceration Reduced rate of ulcer healing Pancreatitis |
| Neurological | Cataracts Psychosis Change in mood (especially high doses) |
Intra-articular corticosteroid
Intra-articular corticosteroids are important in the management of mono-articular inflammatory arthritis and therapy-resistant joints in polyarthritis. They are also used for their short-term (up to approximately 4 weeks) analgesic efficacy in common musculoskeletal disorders such as OA knee, subacromial impingement syndrome, tennis elbow and carpal tunnel syndrome. Corticosteroids are also used in caudal epidurals for patients suffering from radicular-pattern leg pain.
Most commonly used corticosteroid preparations for intra articular joint injections are hydrocortisone, methylprednisolone and triamcinolone. The dose used depends largely on the joint to be injected, although there is little consensus across clinicians on the type or dose of steroid recommended. For example, a knee may require 80 mg of methylprednisolone and an elbow may require 40 mg. 4 mg of methylprednisolone or triamcinolone and 20 mg of hydrocortisone are equivalent to 5 mg prednisolone.
The long-term side-effects of intra-articular corticosteroids are unclear with no good human studies and animal model evidence for both beneficial (related to effects on synovitis) and detrimental effects on cartilage. However, the detrimental consequences of persistent synovitis versus the side effects of the steroid need to be considered. As a general rule, an individual joint should not be injected more than 3–4 times a year. Following an intra-articular corticosteroid patients are advised to rest the relevant joint for 24 hours and monitor for signs and symptoms of infection. Serious side-effects are highly unlikely, with the most important being septic arthritis. Occasionally patients notice a flare in their joint pain within the first 24 hours after an injection. This usually settles on its own within a couple of days. Occasionally with intra-articular and peri-articular injections some thinning (loss of subcutaneous fat) or change in the colour of the skin may occur at the injection site, so care needs to be taken in areas where appearance may be important. The risk of side-effects, particularly thinning of the skin, is greatest with the stronger corticosteroid preparations.
DISEASE MODIFYING ANTI-RHEUMATIC DRUGS
These drugs modify the inflammatory process in patients with RA and other inflammatory arthritides. In modern management of RA, disease modifying anti-rheumatic drugs (DMARDs) are initiated at time of diagnosis to control inflammation, prevent joint damage and preserve function and quality of life. These drugs are slow to work and it often takes weeks before any clinical effect is noticeable. There is no consensus on the order of usage if a drug is stopped due to lack of efficacy or toxicity, and there is increasing use of combination therapies with evidence-based therapeutic synergy. The more commonly prescribed DMARDs are now briefly reviewed.
Methotrexate
Methotrexate (MTX) is an established treatment for inflammatory joint conditions and is commonly the first DMARD used for RA. The use of MTX has escalated since the 1980s with good evidence now available for its long term effectiveness as a single agent and part of combination therapy with other DMARDs and biologic agents. The safety profile has been carefully studied and documented with guidelines for its use and monitoring (Pincus et al 2003).
Methotrexate acts as a folate antagonist by inhibiting the enzyme dihydrofolate reductase, which reduces intracellular folate. This folate is required for the synthesis of purines, important in cell replication. Methotrexate is commonly prescribed orally with a bioavailability of about 65%. It can also be given subcutaneously if the oral route is not tolerated. The half-life is 5–6 hours and about 50% is excreted via the renal route.
Most patients require a dose of methotrexate of about 15–20 mg given once per week (maximum dose 25 mg per week). Patients are prescribed folic acid supplements to reduce the common, mild adverse effects of MTX such as nausea or mouth ulcers. Other serious side-effects include bone marrow suppression and abnormal liver function tests. The incidence of serious adverse events is reduced by regular monitoring of full blood count (FBC) and liver function tests (LFT). It must be used with caution in patients with renal impairment as the risk of adverse events is increased. Methotrexate is potentially teratogenic (causing birth defects) so pregnancy is contraindicated whilst the patient is taking the drug and for 3 months after stopping. The consumption of alcohol is also contraindicated in patients on MTX.
Sulphasalazine
Sulphasalazine (SAS) is a combination of sulphapyridine, an antibacterial agent, and 5 aminosalicylic acid, which has anti-inflammatory properties. It is commonly used for the treatment of RA, peripheral joint disease in ankylosing spondylitis (AS) and psoriatic arthritis (PsA) and inflammatory bowel conditions such as ulcerative colitis (UC).
SAS is administered orally at doses of 1–1.5 grams twice a day. The dose is gradually increased as this improves tolerability. Side effects include nausea, upper abdominal pain and rash. Rarely a leucopenia (low white blood cells) is observed. Regular blood monitoring (FBC) will allow early detection of haematological abnormalities. There have been no accounts of teratogenicity (abnormalities to the developing foetus) but it can induce a reversible reduction in sperm count.
Antimalarials, e.g. hydroxychloroquine (HCQ)
Hydroxychloroquine is probably the weakest acting of the DMARDs. It tends to be used in combination with other drugs in patients with RA and in the arthritis of SLE. The exact mechanism of action is unknown but it is thought to have an effect on the signaltransduction pathway, inhibiting pro-inflammatory gene expression. The bioavailability is variable and ranges from 20–100% but is constant within an individual. It has a long half-life of 40 days and hence may take 3 months to achieve its full effect. As a consequence, a loading dose of 200 mg twice a day is given for 1 month before reducing to a standard dose of 200 mg once a day.
The anti-malarials are the least toxic of all DMARDs. Rarely, they can cause myopathy, abnormal skin pigmentation and peripheral neuropathy. Of most concern, although rare, is irreversible retinopathy resulting in permanent visual loss. It is recommended that baseline ophthalmology screening (visual acuity and assessment for blurred vision) is performed if patients have pre-existing ocular pathology or visual disturbance, impaired renal function or are over the age of 60.
Leflunomide
Leflunomide is a competitive inhibitor of pyrimidine synthesis. It is primarily used in RA and prevents the multiplication of pro-inflammatory lymphocytes. After oral administration it undergoes rapid chemical conversion to its active metabolite. This metabolite has a long half-life of 15 to 18 days. Therefore, like HCQ, leflunomide requires a loading dose to achieve steady state concentrations. The loading dose of 100 mg once a day for 3 days is however often omitted in clinical practise because of poor tolerability. The usual maintenance dose is 10–20 mg per day. The most important adverse reactions to leflunomide are hepatic. Other side effects include diarrhoea, nausea, weight loss, rashes, hair loss, teratogenicity and hypertension. Regular monitoring of blood pressure, full blood count (FBC), urea and electrolytes (U&E’s) and lung function tests (LFTs) are required.
TUMOUR NECROSIS FACTOR ANTAGONIST AGENTS
Tumour necrosis factor (TNF α) is a pivotal pro inflammatory cytokine that is released by activated monocytes, macrophages and T lymphocytes (see Fig. 15.1). It promotes inflammatory responses that are important in the pathogenesis of RA (Bazzoni & Beutler 1996, Taylor et al 2000) TNF binds to 2 receptors, the type 1 receptor (p55) and the type 2 receptor (p75). The advent of TNF antagonist therapies (etanercept, adalimubab, infliximab) has markedly improved control of inflammation in RA and reduced radiographic damage progression (Klareskog et al 2004, Maini et al 1999, Weinblatt et al 2006). They have also successfully been used in AS (Baraliakos et al 2005, Marzo-Ortega et al 2005), PsA (Antoni et al 2005, Mease et al 2006) and inflammatory bowel disease. Superior efficacy is observed when TNF antagonists are used in combination with MTX. Currently in the UK, the use of these therapies is limited to patients that have failed traditional DMARDs and have high levels of disease activity.
Etanercept is a soluble fusion protein composed of two dimers, each with an extra cellular, ligand-binding portion of the higher affinity type 2 TNF receptor (p75) linked to the Fc portion of human IgG1, thereby preventing each from binding to its respective receptors (Olsen & Stein 2004). It is administered subcutaneously (50 mg once a week or 25 mg twice a week) and takes 50 hours to reach peak concentrations. The half-life is around 4 days. It can be prescribed as mono-therapy but data from clinical trials show superior efficacy when taken with methotrexate (Klareskog et al 2004).
Adalimubab is a recombinant human IgG1 monoclonal antibody that binds to human TNF α with high affinity. Therefore it impairs both cytokine binding to its receptors and lysing cells that express TNF α on their surface. It is administered subcutaneously reaching peak concentrations after 130 hours. Adalimubab may be prescribed as mono-therapy but like etanercept superior efficacy is seen when combined with methotrexate.
Infliximab is a chimeric IgG1 anti-TNF α antibody containing the antigen binding region of a mouse antibody and the constant region of a human antibody. It binds to soluble and membrane bound TNF α with high affinity, impairing the biding of TNF α to its receptor. In pharmacokinetic terms, there appears to be marked variations among patients, with trough levels varying by a factor of 100 following the standard dose of 3 mg per kilogram every 8 weeks given intravenously. However when used as mono-therapy patients often experienced a loss of response over time due to the development of anti-infliximab antibodies (human anti-chimeric antibodies). Combination therapy with methotrexate reduces the tendency to form antibodies.
Adverse effects of TNF antagonist therapy
Similar adverse events have been documented in all 3 TNF antagonist agents (see Table 15.4). They form the basis of the screening (including TB screening) and monitoring procedures for all patients prior to starting and whilst receiving TNF antagonist therapy.
Table 15.4 The adverse events of TNF antagonist agents
| Infection | Serious bacterial and opportunistic infections (e.g. aspergillosis) have been reported Increased risk of tuberculosis (TB) |
| Malignant disease | TNF antagonists do not confer an increased risk of malignancy above that of RA with the possible exception of skin malignancy |
| Heart disease | May exacerbate heart failure |
| Injection site/infusion reactions | Redness and itching at injection site Infusion reactions include mild headache or nausea or in 2% hypersensitivity reaction |
| Autoimmune response | Induction of anti-nuclear antibody (ANA), double stranded DNA and anti-nuclear cytoplasmic antibody. However, clinical manifestations of drug induced lupus or vasculitis are surprisingly rare |
| Lung disease | Exacerbation of existing lung fibrosis |
OTHER BIOLOGIC AGENTS
Rituximab
Rituximab is a chimeric anti-CD20 monoclonal antibody that causes a selective depletion of CD20+ B cells. B cells can produce autoantibodies that, in some diseases, are directly pathogenic (Dass et al 2006). This drug was originally developed for the treatment of refractory CD20+ B cell non Hodgkin’s lymphoma. In 2006, Rituximab in conjunction with methotrexate was licensed in the US and the EU for use in patients with active, moderate to severe RA that is refractory to treatment with TNF antagonist agents. The results from two major trials (Cohen et al 2006, Emery et al 2006) highlight successful clinical outcomes in terms of laboratory, patient-reported and radiographic measures.
Rituximab is given as two infusions of 1 g on days 1 and 15 with infusions of methylprednisolone. The mean half-life is 20 days. The main adverse events are minor infections and there have been no reported cases of TB. Most adverse infusion reactions occur during or immediately after the infusion. Up to 30% of patients experience pruritis, urticaria (allergic reaction in which red round wheals develop on the skin), pyrexia, throat irritation and hypo- and hypertension. The incidence of infusion reactions is reduced by concomitant intravenous corticosteroid. Small studies have also shown rituximab to be beneficial in connective tissue disease such as SLE (Eisenberg 2006).
Abatacept
The pathogenesis of RA is dependent on T cell activation. This requires two signals: firstly presentation of the antigen to the T cell receptor and secondly various co-stimulatory molecules. Abatacept is a soluble fusion protein that targets co-stimulation. It has demonstrated good results in patients with RA that are non responsive to MTX or failed previous TNF antagonist therapy (Genovese et al 2005, Kremer 2005). Abatacept (10 mg per kg) is given as an infusion every month and has a favourable safety profile, with no increase in malignancy or TB.
CONCLUSION
The therapeutic options for rheumatic diseases continue to expand and modern biologic therapies mean that at least RA patients are reaching levels of disease control previously thought impossible; these drugs are expensive and place a huge burden on health systems. For the vast majority of musculoskeletal patients with OA or mechanical joint pain, our use of pharmacological analgesia remains largely empiric with few good trials of how to optimally use individual drugs or combination therapies. Despite increasing therapeutic options, a multi disciplinary approach remains key to the management of patients with rheumatic diseases.
Identify all patient information leaflets that would be applicable to a patient presenting to a therapist with difficulties with pain control. Consider five main messages on pharmacological approaches that you can identify from the information in these leaflets. List these and keep them for future reference.References and further reading
Antoni C.E., Kavanaugh A., Kirkham B., et al. Sustained benefits of infliximab therapy for dermatologic and articular manifestations of psoriatic arthritis: results from the infliximab multinational psoriatic arthritis controlled trial (IMPACT). Arthrit. Rheum.. 2005;52(4):1227-1236.
Baraliakos X., Brandt J., Listing J., et al. Outcome of patients with active ankylosing spondylitis after two years of therapy with etanercept: clinical and magnetic resonance imaging data. Arthrit. Rheum.. 2005;53(6):856-863.
Bazzoni F., Beutler B. The tumor necrosis factor ligand and receptor families. N. Engl. J. Med.. 1996;334(26):1717-1725.
Clissold S.P. Paracetamol and phenacetin. Drugs. 1986;32(Suppl. 4):46-59.
Cohen S.B., Emery P., Greenwald M.W., et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthrit. Rheum.. 2006;54(9):2793-2806.
Conaghan P.G., Dickson J., Grant R.L. Care and management of osteoarthritis in adults: summary of NICE guidance. Br. Med. J.. 2008;336:502-503.
Dass S., Vital E.M., Emery P., et al. Rituximab: novel B-cell depletion therapy for the treatment of rheumatoid arthritis. Expert Opin. Pharmaco.. 2006;7(18):2559-2570.
Eisenberg R. Targeting B cells in SLE: the experience with rituximab treatment (anti-CD20). Endocr. Metab. Immune Disord. Drug Targets. 2006;6(4):345-350.
Emery P., Fleischmann R., Filipowicz-Sosnowska A., et al. The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: results of a phase IIB randomized, double-blind, placebo-controlled, dose-ranging trial. Arthrit. Rheum.. 2006;54(5):1390-1400.
Genovese M.C., Becker J.C., Schiff M., et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. N. Engl. J. Med.. 2005;353(11):1114-1123.
Graham D.J. COX-2 Inhibitors, other NSAIDs and cardiovascular risk; the seduction of common sense. J. Am. Med. Assoc.. 2006;296(13):1653-1656.
Jovanovic D.V., Di Battista J.A., Martel-Pelletier J., et al. Il-7 stimulates the production and expression of pro-inflammatory cytokines, IL-beta and TNF-alpha by human macropgages. J. Immunol.. 1998;160:3513-3521.
Kidd B.L., Langford R.M., Wodehouse T. Arthritis and pain. Current approaches in the treatment of arthritic pain. Arth. Res. Ther.. 2007;9(3):214.
Klareskog L., van der Heijde D., de Jager J.P., et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. The Lancet. 2004;363(9410):675-681.
Kremer J.M. Selective costimulation modulators: a novel approach for the treatment of rheumatoid arthritis. J. Clin. Rheumatol.. 2005;11(Suppl. 3):S55-S862.
Maini R., St Clair E.W., Breedveld F., et al. Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. ATTRACT study group. The Lancet. 1999;354(9194):1932-1939.
Marzo-Ortega H., McGonagle D., Jarrett S., et al. Infliximab in combination with methotrexate in active ankylosing spondylitis: a clinical and imaging study. Ann. Rheum. Dis.. 2005;64(11):1568-1575.
Mease P.J., Kivitz A.J., Burch F.X., et al. Continued inhibition of radiographic progression in patients with psoriatic arthritis following 2 years of treatment with etanercept. J. Rheumatol.. 2006;33(4):712-721.
Moore R.A., Tramèr M.R., Carroll D., et al. Quantitative systematic review of topically applied non-steroidal anti-inflammatory drugs. Br. Med. J.. 1998;316(7128):333-338.
Olsen N.J., Stein C.M. New drugs for rheumatoid arthritis. N. Engl. J. Med.. 2004;350(21):2167-2179.
Pincus T., Yazici Y., Sokka T., et al. Methotrexate as the “anchor drug” for the treatment of early rheumatoid arthritis. Clin. Exp. Rheumatol.. 2003;21(5 Suppl. 31):S179-S185.
Taylor P.C., Peters A.M., Paleolog E., et al. Reduction of chemokine levels and leukocyte traffic to joints by tumor necrosis factor alpha blockade in patients with rheumatoid arthritis. Arthrit. Rheum.. 2000;43(1):38-47.
Warner T.D., Giuliano F., Vojnovic I., et al. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc. Natl. Acad. Sci. USA. 1999;96(13):7563-7568.
Weinblatt M.E., Keystone E.C., Furst D.E., et al. Long-term efficacy and safety of adalimu-mab plus methotrexate in patients with rheumatoid arthritis: ARMADA 4 year extended study. Ann. Rheum. Dis.. 2006;65(6):753-759.