RECEPTORS AND HORMONE ACTION
Introduction
All hormones act by binding to receptors in their target cells and by doing so bring about an intracellular response. It is the presence of receptors, which are highly specific binding proteins, that defines the target cells for a hormone: target cells of a hormone are those cells that have receptors for the hormone. The location of these receptors in each cell depends to some extent on the chemical nature of the hormone. Peptide hormones act on receptors located in the cell membrane, while steroid hormones act on intracellular receptors. There are many forms of receptor and several different ways in which the action of a hormone binding to a receptor can cause a change in intracellular activity. In this chapter we shall explore the many different forms of hormone action.
General characteristics of receptors
Receptor agonists and antagonists
A receptor is a specialized protein, located in the cell membrane, cytoplasm or nucleus of a target cell, which acts to pass on a chemical message. Receptors have binding sites to receive the message and the effect of this interaction is to bring about changes in the receptor which result in the message being passed on to initiate a cellular response. Although the receptor has a ‘specific binding site’ for the physiological chemical message, receptors will usually bind any compound which is structurally similar to the message. Any compound which binds to a receptor is called a ‘ligand’ for that receptor. So a hormone is a naturally occurring ligand for its target cell receptor.
However, not all ligands will bring about the normal conformational changes in the receptor. Ligands which bind to the receptor but do not initiate the cellular response are called ‘antagonists’ because they block the normal function of the receptor. Ligands which do initiate the cellular response are called ‘agonists’, so hormones are agonists at their receptors. Both receptor agonists and antagonists are often used pharmacologically to mimic or to block the effects of hormones.
Let’s think of some examples. Cortisol (a naturally occurring glucocorticoid hormone), methylprednisolone (a synthetic glucocorticoid) and mifepristone (an anti-glucocorticoid drug) all bind to the glucocorticoid receptor. Both cortisol and methylprednisolone are agonists and induce a cellular response by activating the glucocorticoid receptor. Mifepristone is an antagonist which binds to the glucocorticoid receptor but does not pass on the normal message and so can be used to block the effects of circulating glucocorticoids.
Dose–response effects
A feature of a receptor-mediated effect is that is has dose–response characteristics (Fig. 2.1): the response increases with increasing amounts of the hormone, until a plateau is reached. At the plateau the receptor system is saturated. The dose–response curves are used to investigate the effects of agonists and antagonists at the receptor. In the presence of some antagonists, a higher concentration of the hormone is required to elicit the effect.
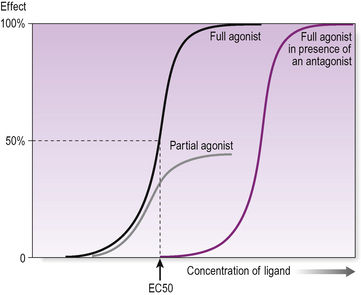
Figure 2.1 Receptor binding: a dose–response curve showing a typical sigmoid shape of increasing effect with increasing ligand concentration. A partial agonist never produces the same magnitude of effect, even at maximal concentrations. The presence of an antagonist has the effect of moving the dose–response curve to the right so that a higher concentration of agonist is required to produce the same effect, but the same maximal effect is still achievable.
A drug or hormone that is a partial agonist at a receptor elicits a lesser maximal response than a full agonist at that receptor (Fig. 2.1).
Receptor binding properties
Receptors have two important binding characteristics, affinity and specificity. Binding affinity relates to how tightly the hormone binds the receptor while specificity refers to whether the receptor binds just one hormone or whether it might bind other closely related molecules. This is particularly relevant when we are looking at steroid receptors where the steroid hormones are structurally very similar.
A receptor needs to have a high affinity for the hormone in order to bind to it. However, hormone binding to a receptor is a reversible process and in an equilibrium the hormone and receptor constantly associate and dissociate. Because of the law of mass action:

The dissociation constant, KD, for the receptor is defined as:
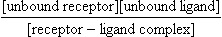
This means that KD is the concentration of ligand when half the receptors are occupied, as [unbound receptor] and [receptor-ligand complex] will then be the same and cancel each other out. So KD is measured by the concentration of hormone required to produce 50% receptor occupancy; and affinity is defined as 1/KD (Fig. 2.2) and high affinity means a low KD.
The usual analogy for receptors and ligands is a lock and a big bunch of keys. Several keys (ligands) might fit into the lock (receptor) but only one or two keys (agonists) are likely to open it. Keys which fit but do not open the lock have the effect of blocking the receptor (antagonists).
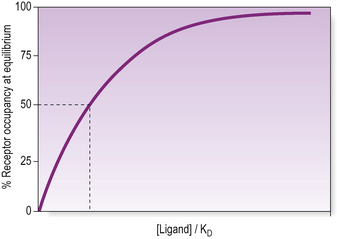
Figure 2.2 Dissociation constant. The dissociation constant is the concentration of ligand at which half the receptor sites are occupied when the reaction is at equilibrium. This is derived experimentally by incubating a sample containing receptors with increasing quantities of radiolabelled ligand. At equilibrium the bound ligand is separated from the free ligand and a saturation curve of increasing receptor occupancy such as this is obtained.
Of course, this rather simplistic analogy breaks down for partial agonists. These compounds are particularly fascinating because they bind to the receptor and activate it, but the cellular response is less than that produced by a ‘full agonist’. In the presence of a full agonist, a partial agonist will compete for binding sites and reduce the normal cellular response, in other words it can also have antagonist effects. You will come across several examples of partial agonists in this book.
To add another level of complexity, conventional pharmacology assumes that each receptor is only linked to one system which passes on the message. However, membrane receptors, which can move around in a fluid membrane, may associate with more than one second messenger or other signalling system. So different ligands binding to the same receptor can activate different signalling pathways within the cell. This situation, where different cellular responses triggered by a receptor are ligand-dependent is known as ‘functional selectivity’. Going back to our lock and key analogy, it is as if one lock could open any of several different doors, depending on which key you use!
Hormones circulate in very low concentrations indeed. The lower the concentration of a hormone, the higher the receptor affinity needs to be in order to elicit a response. Most receptor’s KD for its hormone is therefore very low, typically in the picomolar range.
Ligand properties
Two properties of ligands that are particularly important in pharmacology are efficacy and potency. Efficacy is a measure of the amount of bound ligand required to produce a given response. A ligand with a high efficacy will produce a large response even when it has occupied only a small number of binding sites. In contrast, a ligand with low efficacy will need to occupy a high proportion of binding sites to elicit the same response, and a partial agonist, even with all the binding sites occupied, will elicit a sub-maximal response.
Potency is a measure of the concentration of ligand required to produce a given response. A ligand with high potency will produce a large response even at low concentrations. The measure of potency is called the EC50, the concentration of a ligand required to produce a half-maximal response (Fig. 2.1). The more potent the ligand, the lower the EC50.
As you will have noted, efficacy and potency are related to each other via affinity.
Types of hormone receptors
There are three major classes of hormone receptor: G-protein coupled receptors; kinase-linked receptors (both located in the plasma membrane of the cell); and intracellular receptors. Table 2.1 shows the major classes of receptors and the hormones which interact with each type. We shall consider each of these receptor types in turn, looking first at the two types of cell-membrane receptor.

Cell-membrane receptors
Peptides, glycoproteins and catecholamines are either too large or too hydrophilic to enter the cell and so these hormones bind to receptors located in the plasma membrane, with their ligand binding domain (hormone binding site) on the extracellular surface. There are two broad classes of cell surface receptor: those which are G-protein coupled and act through the generation of a second messenger and those that directly activate a protein kinase.
G-protein coupled receptors (GPCRs) and second messengers
This group of receptors is probably the most widespread through the endocrine system. Receptors are linked via G-protein activation to second messenger production or ion channel opening (Fig. 2.3).
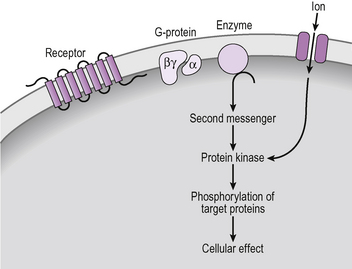
Figure 2.3 G-protein coupled receptor signalling by generation of a second messenger or opening of an ion channel.
The receptor protein spans the cell membrane with seven helices, an external N-terminal and an intracellular C-terminal. These receptors are also called seven transmembrane domain receptors. The receptor, when occupied, can interact with a protein on the intracellular face of the cell membrane, called a G-protein (Fig. 2.4). A G-protein consists of three distinct subunits, termed alpha, beta and gamma. The beta and gamma subunits are constant but many different forms of the alpha subunit exist and these different forms interact with different targets, either enzymes or ion channels. A summary of the different forms of alpha subunit and their effects is shown in Table 2.2.

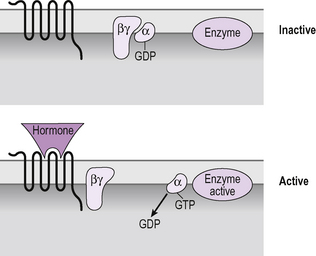
Figure 2.4 G-protein interactions with a seven transmembrane domain receptor. In the absence of hormone binding the G-protein subunits are associated and the α subunit binds GDP. When the hormone binds to the receptor a conformational change causes the α subunit to bind GTP instead of GDP and to move away from the βγ units. The α subunit is then able to exert an effect on an enzyme or ion channel. Inbuilt hydrolase activity converts the GTP to GDP which inactivates the α subunit causing it to return to the basal state.
In the resting state, the three G-protein subunits are physically close to each other and the alpha subunit binds guanosine diphosphate (GDP). It is the guanosine binding capability that gives G-proteins their name. When the hormone binds to the seven transmembrane domain receptors it causes the receptor to change shape, known as inducing a conformational change in the protein (Fig. 2.4). That shape change causes the G-protein to drop the GDP and bind a GTP molecule instead. The alpha subunit, with its bound GTP, separates from the beta and gamma subunits and moves through the membrane to interact with its target protein. When it comes adjacent to the target, such as the enzyme adenylyl cyclase, it causes a conformational change in the enzyme, which activates it. In this way the G-protein is the signal transduction mechanism which passes the signal from the receptor to stimulate production of a second messenger.
Once it has been activated, how is the enzyme switched off? The signal from the G-protein is stopped when the alpha subunit is no longer bound to GTP. The alpha subunit possesses GTP-ase activity which breaks down the GTP into GDP and it is this which returns the alpha subunit to its inactive form, so that it diffuses back through the membrane to associate with the beta and gamma subunits. In the absence of the activated alpha subunit the enzyme returns to its inactive form and stops producing second messengers.
Second messenger systems
A range of compounds are employed by cells to act as ‘second messengers’. These are chemical signals that relay the hormonal signal (the first messenger) within the cell. The common feature of second messengers is that they all have a very short half-life within the cell and are deactivated very rapidly by either chemical degradation or by re-uptake.
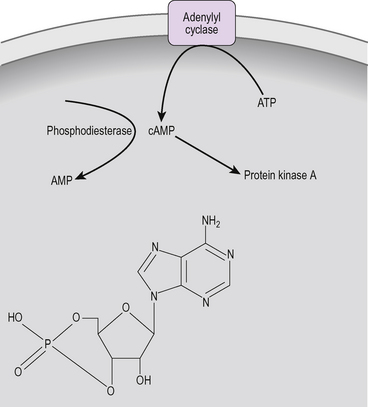
Cyclic AMP
The commonest second messenger is cyclic adenosine monophosphate (cAMP, Fig. 2.5). This is produced by the dephosphorylation of ATP by the enzyme adenylyl cyclase which is located in the cell membrane. Some GPCRs activate adenylyl cyclase while others, acting through alpha i, inhibit cAMP production (Table 2.2). Within the cell, cAMP acts by binding to protein kinase A (see Fig. 2.8). Intracellular cAMP is rapidly broken down by phosphodiesterase.
Phosphatidylinositol bisphosphate
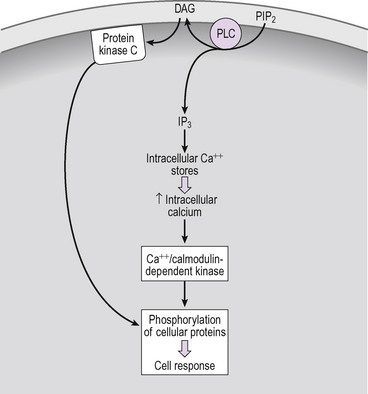
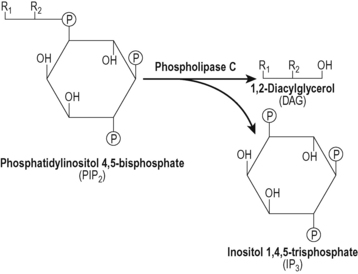
Another second messenger system is activated when the GPCR is coupled to activation of phospholipase C (Fig. 2.6). This enzyme cleaves a membrane phospho-lipid, phosphatidylinositol bisphosphate (PIP2) to give two second messengers, inositol trisphosphate (IP3) and diacylglycerol (DAG) (Fig. 2.7). The DAG remains in the cell membrane where it attracts and activates protein kinase C. The IP3 is an important component of calcium signalling.
Calcium signalling
There are two elements to calcium signalling: first is the G-protein-dependent opening of ligand gated ion channels in the cell membrane, allowing an influx of calcium into the cell. The second is release of intracellular calcium stores, mediated by release of IP3 by the actions of phospholipase C (above). The IP3 remains within the cytoplasm and acts to open calcium channels in the endoplasmic reticulum causing the release of calcium into the cytoplasm (Fig. 2.6). Together these two mechanisms cause increases in the cytoplasmic calcium concentration. Calcium acts as a second messenger, activating calcium/calmodulin dependent protein kinase.
Protein kinases and phosphatases
You will have noted, reading the section above on second messengers, that second messengers commonly act through protein kinases to influence intracellular events. Protein kinases are regulatory proteins which bind the second messenger and then phosphorylate target proteins in the cell, usually enzymes, either activating or inactivating them by phosphorylation.
Protein kinase A, also known as cAMP-dependent protein kinase is perhaps the best characterized of all these. It consists of two regulatory subunits and two catalytic subunits (Fig. 2.8). When cAMP binds to the regulatory subunits they undergo a conformational change and separate from the catalytic subunits. This physical separation causes activation of the two catalytic subunits which then phosphorylate serine and threonine residues on specific cellular proteins. When the bound cAMP dissociates from the binding sites on the regulatory subunits the kinase reassembles itself and the catalytic activity stops.
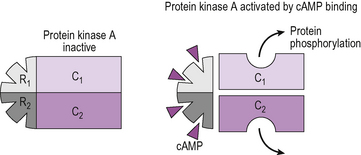
Figure 2.8 Signalling through cAMP: interaction with protein kinase A (PKA). The PKA consists of four subunits, two regulatory (R1 and R2) and two catalytic (C1 and C2). In the absence of cAMP these are closely associated. When cAMP binds to the sites on the regulatory subunits, the four units dissociate, which activates the catalytic subunits.
Phosphorylation of these cellular proteins, usually enzymes, causes either activation or deactivation of the enzyme. This is reversed by the action of cellular phosphatases, which dephosphorylate the protein, returning the cellular activity to its basal state. An example of a cellular protein which is activated in response to phosphorylation by protein kinase A is cholesterol ester hydrolase (a hormone-sensitive lipase), which acts to liberate cholesterol in steroidogenic cells in preparation for steroid biosynthesis.
Receptor desensitization and downregulation: GPKs and beta arrestin
When a hormone binds to a G-protein coupled receptor the receptor is usually quickly de-sensitized and eventually internalized within the cell for subsequent breakdown or recycling. The first step of this process involves two families of intracellular proteins which, like the G-proteins, are able to interact with all members of this receptor family. These are the beta arrestins and the G-protein coupled receptor kinases (GPK) (Fig. 2.9). Hormone binding to a receptor causes interaction of the receptor with G-proteins as we have seen above. It also attracts an enzyme of the GPK family which phosphorylates the receptor. The phosphorylated receptor has a lower binding affinity for the hormone but also attracts beta arrestin binding, which physically blocks interaction of the receptor with G-proteins and so prevents further signalling through second messengers.
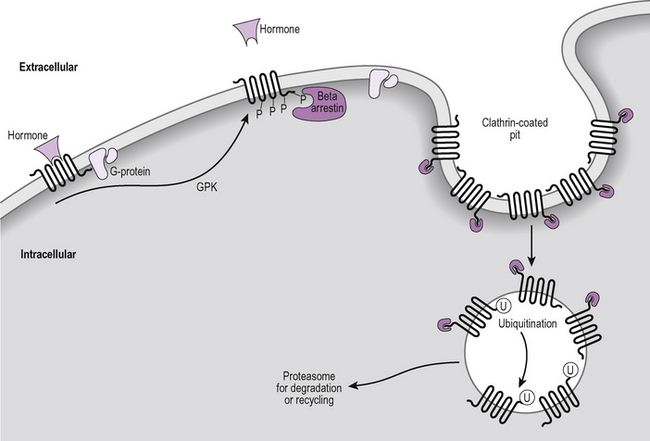
Figure 2.9 Receptor desensitization and internalization. Binding of a hormone to a G-protein coupled receptor attracts a kinase, GPK, which phosphorylates the receptor (P). The phosphorylated receptor attracts beta-arrestin which binds, preventing the receptor from activating G-proteins. Receptors gather in clathrin-coated pits and are internalized, ubiquinated (U) and processed in proteasomes.
The desensitized receptors then move through the plasma membrane to accumulate in clathrin-coated pits (specialized areas of the cell membrane involved in endocytosis) which become internalized within the cell. The beta arrestin has a further role in recruiting proteins which bring about the ubiquitination of both the receptor and the beta arrestin. Ubiquitination is the process of attaching the protein, ubiquitin (so named because it is found in every cell and so is said to be ubiquitous), to the receptor. This has the effect of tagging the receptor for transport to a proteasome where receptors undergo degradation.
Receptors which directly activate a protein kinase
The first group of cell-membrane receptors we looked at all act by generating a second messenger which then activates a protein kinase. The other group of cell-surface hormone receptors takes a short-cut by activating a kinase without going through a second messenger. One group of these receptors actually has a protein kinase within the structure of the receptor: this group includes the insulin and growth factor receptors. The second group acts by attracting kinases to the activated receptor: this group includes the growth hormone receptor and cytokine receptors. In both of these receptor groups, dimerization of the receptor is usually an important feature of their activation.
The insulin and growth factor receptor family: receptors with inherent tyrosine kinase activity
This receptor family includes insulin, insulin-like growth factor and a large number of other growth factor receptors (Fig. 2.10). These receptors have a single transmembrane domain, an extracellular ligand binding site and an intracellular tyrosine kinase domain. When the hormone or growth factor binds to the receptor, the conformational change causes two receptors to associate together. This is called receptor dimerization and is an important step in the signalling process for most of these receptors, although the epidermal growth factor receptor (EGF-R) does not appear to form dimers in order to become activated. Dimerization results in activation of the receptor’s intrinsic tyrosine kinase activity and phosphorylation of tyrosine residues on target proteins, which include the receptor itself. The phosphorylated receptor attracts a number of accessory proteins which each have the capacity to activate different signalling pathways. All the accessory proteins have a common area called an SH2 domain, which is an area of sequence homology with the src proto-oncogene. This appears to be important for the interaction with the phosphorylated receptor. There is, frankly, a bewildering array of these accessory proteins, with names such as ‘son of sevenless’ which is known as SOS. The study of these intracellular signalling proteins is worth a book in itself.
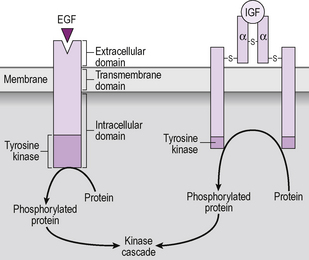
Figure 2.10 Insulin and growth factor receptors. Hormone action through a growth factor receptor, with intrinsic tyrosine kinase activity. The receptor exists in the plasma membrane either as a single transmembrane unit such as the epidermal growth factor (EGF) receptor, or a homodimer such as the insulin or insulin-like growth factor (IGF) receptor. The insulin receptor consists of a dimer with each part having an α and β subunit. The whole receptor is held together by disulphide bridges, indicated as –s– in the diagram. When the hormone binds to the receptor it causes a conformational change within the receptor which activates the protein tyrosine kinase, resulting in the direct phosphorylation of intracellular proteins, and initiation of a kinase cascade.
The insulin receptor is thought to signal by phosphorylating target proteins called insulin receptor substrate (IRS) proteins. There is a whole family of these proteins which then activate kinase cascades. A large number of proteins are involved in the coordinated cellular response to insulin and growth factor receptor activation.
The growth hormone and cytokine receptors: receptors which attract kinases
These are single-transmembrane domain receptors that do not have inherent kinase activity. After binding of the hormone to one receptor, the receptor dimerizes, apparently via the binding of the same hormone molecule to a second receptor (see Fig. 2.11). The dimerized receptor complex attracts and activates the Janus-associated kinase-signal transducer and activator of transcription (JAK-STAT) pathway (Fig. 2.11). The JAK phosphorylates STAT proteins which then dimerize, move into the nucleus, bind to specific regulatory elements on gene promoters and so regulate transcription. JAK also phosphorylates the receptor and causes other accessory proteins to associate with the receptor, in a similar manner to the growth factor receptors (above). Again, this results in the activation of a kinase cascade. There are several examples of these, for example the family of mitogen-activated protein kinases (MAPK) which are involved in cell division (Fig. 2.12).
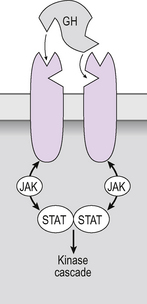
Figure 2.11 Growth hormone and cytokine receptors: Hormone action through a cytokine receptor (or growth hormone, GH receptor). Binding of the hormone to the first receptor causes a conformational change which allows the receptor to dimerize and the hormone to bind to the second receptor subunit; in this case the conformational change in the receptor causes the Janus-associated kinase (JAK) to migrate to the receptor, become activated and phosphorylate signal transducer and activator of transcription (STAT) proteins. JAK was originally called ‘Just Another Kinase’ but now has the more prosaic name of Janus-associated kinase.
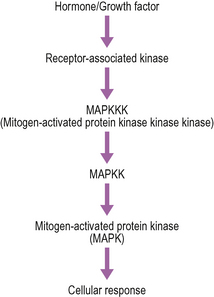
Figure 2.12 The mitogen-activated protein kinase cascade. Each of the kinases in this cascade refers to a family of proteins, which is associated with different aspects of cellular function. For example, the MAPK family includes the kinase P38 which has a role in apoptosis, and ERK1 and ERK2 which have roles in cell growth.
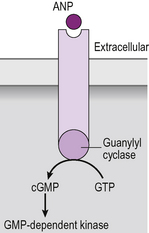
There is one small family of receptors which falls somewhere between the G-protein coupled receptors and those that directly activate a kinase. The atrial natriuretic peptide family of receptors signals through the generation of cyclic guanosine monophosphate (cGMP). Cyclic GMP is a second messenger like cAMP, produced by the action of an enzyme called guanylyl cyclase. However, the activation of guanylyl cyclase is not mediated by a G-protein. Instead, the receptor itself is a single trans-membrane domain protein which possesses intrinsic guanylyl cyclase activity, so cGMP is generated by the receptor itself (Fig. 2.13).
Hormonal regulation of transcription
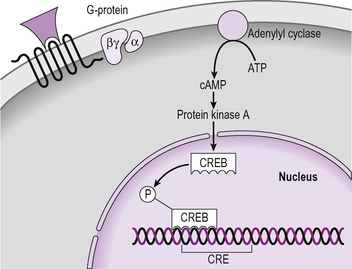
The effects of all hormones result, at some point, in changes in gene transcription. Hormones that act through kinases all eventually exert effects on the nucleus. In order to achieve this there are regions in the promoter of every hormone-regulated gene that contain ‘response elements’. These response elements are consensus sequences of DNA that act as binding sites for specific transcription factors. There is, for example, a cyclic AMP response element (CRE, Fig. 2.14). The protein kinase A, activated by cAMP, translocates to the nucleus and activates a protein called CREB protein which binds to the cAMP response element on the DNA. This attracts CREB binding protein which also binds, activating transcription. Just to complicate matters, there is also a cyclic AMP response element modulator (CREM) which modifies the actions of CREB protein. Proteins such as CREB, which bind to DNA and alter transcription, are called ‘transcription factors’. There are many transcription factors in human cells and they regulate the transcription of all genes. From the endocrine point of view, the most important transcription factors are the intracellular hormone receptors.
Intracellular receptors
Steroids, thyroid hormones and calcitriol (active vitamin D), being small and lipophilic, are thought to pass readily across the cell membrane and bind to intracellular receptors, located in the cytoplasm or the nucleus. There is some evidence that these hormones, particularly thyroid hormones, may also be actively transported into the cell. The hormone–receptor complex binds to DNA, to specific response elements in the promoter region of specific genes, and stimulates gene transcription. In this way, steroid and thyroid hormones increase the production of specific proteins and thereby alter cellular function (Fig. 2.15).
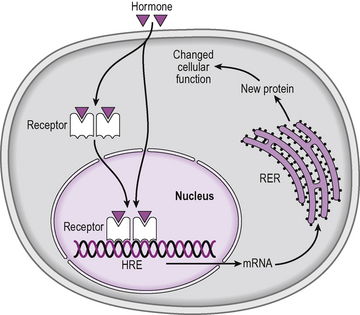
Figure 2.15 Receptors for steroid and thyroid hormones are found inside the cell, in either the cytoplasm or the nucleus. The hormone enters the cell and binds to the receptor. The hormone–receptor complex forms a dimer and binds to hormone response elements (HRE) in the promoter region of certain genes. This can activate or repress transcription of that gene, causing changes in mRNA and therefore new protein formation in the cell. RER is rough endoplasmic reticulum.
The intracellular receptors activated by steroid and other hormones may usefully be considered to be members of the family of transcription factors. Together they are called the nuclear receptor superfamily, comprising 48 members in total. The receptors are structurally closely related although their ligands are diverse. The basic structure of these receptors is shown in Figure 2.16. Each receptor comprises five regions, A, B, C, D and E. AB is a region which is not well-conserved and there is little homology between family members. Region C is the DNA binding domain and is highly conserved. It contains two structures called ‘zinc fingers’ which enable the receptor to bind to specific sites on the DNA. Zinc fingers are given this name because for each ‘finger’ there is a zinc atom linked to four cysteine residues, which gives this region a stable and characteristic shape (Fig. 2.17). In between the zinc fingers is an area called the P-box which determines the specificity of the receptor for the particular region of DNA. Region E is the hormone binding domain.
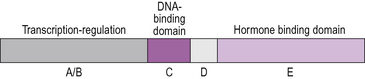
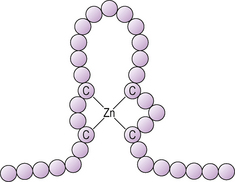
Figure 2.17 A zinc finger motif in a steroid hormone receptor. Each circle represents an amino acid. The ‘finger’ is stabilized by a zinc atom (Zn) held between cysteine or histidine residues.
The nuclear hormone receptors which we shall be looking at all form dimers when they bind to DNA, although not all transcription factors behave in this way. The dimerized hormone–receptor complex attracts other proteins, co-activators and co-repressors, which together act to regulate gene transcription (Fig. 2.18).
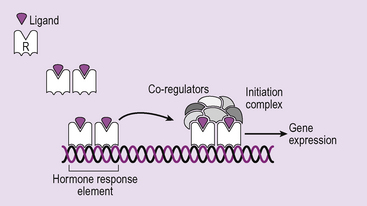
Figure 2.18 Nuclear receptor dimerization and interaction with DNA. In response to ligand binding the receptors dimerize. This can be between two receptors of the same type (homo-dimer) or between two different types of nuclear receptor (hetero-dimer). In both cases the dimer binds to the hormone response element DNA sequence in the gene promoter. The complex attracts co-regulators which can be either co-activators (which permit transcription) or co-repressors (which inhibit transcription). The observation that the same hormone can have different effects in different cells can be explained by the fact that different co-regulators are found in different cells.
Many drugs act through these nuclear receptors, including the contraceptive pill and anti-diabetic drugs. An understanding of these receptors and how they work is important in understanding drug actions. Mutations in the genes encoding these receptors also gives rise to a number of more rare endocrine disorders, particularly syndromes of hormone resistance: thyroid hormone, vitamin D and androgen resistance. Interestingly, mutations in a nuclear receptor also cause severe insulin insensitivity even though insulin acts through a cell-membrane receptor.
There are two classes of nuclear receptors, class I and class II. We will consider each in turn.
Class I receptors
This receptor subfamily includes all the steroid hormone receptors, but not the calcitriol receptor. There are five types of steroid hormone receptor, corresponding to the five classes of steroid hormone: receptors for glucocorticoid (GR), mineralocorticoid (MR), progesterone (PR), oestrogen (ER) and androgen (AR). These receptors may be located in either the cytoplasm or the nucleus of the target cell and in their resting state, when not bound to a hormone, they are bound to heat shock proteins (HSP), also through the hormone binding domain (Fig. 2.19). In class I receptors, the DNA binding domain of the receptor also contains a region which permits dimerization of the receptors in the presence of DNA. These receptors form only homodimers, which means two receptors of the same type forming a dimer. The receptor only binds to DNA when it is bound to a hormone and it binds to highly specific palindromic (which means reading the same backwards as forwards) regions of the DNA which are called hormone response elements.
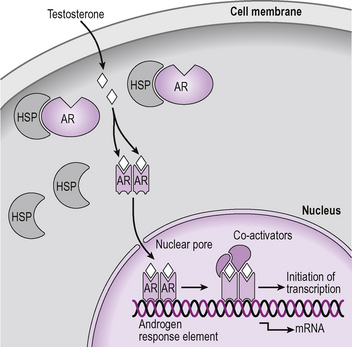
Figure 2.19 Cellular actions of androgens. Androgen receptors are intracellular and in the absence of testosterone they are bound to heat shock protein (HSP) and located in the cytoplasm. In the presence of androgen the HSP dissociates from the receptor allowing the hormone receptor complex to move into the nucleus where it dimerizes with another androgen receptor–hormone complex and binds to the androgen response element on a gene promoter. Various co-activators are attracted to the complex and gene transcription occurs.
Class II receptors
This subfamily of nuclear receptors includes the thyroid hormone receptor and the vitamin D (calcitriol) receptor (Fig. 2.20). The retinoic acid receptor (RXR) and the orphan nuclear receptor called peroxisome proliferator-activated receptor (PPAR gamma) are also included in this group and are significant in the mechanism of action of hormone receptors. The receptors are only located in the nucleus and never in the cytoplasm. Class II receptors are able to bind to DNA even in the absence of ligand, and generally bind as heterodimers (two different types of receptors dimerizing: often the hormone receptor dimerizes with RXR). In the absence of ligand, these receptors recruit co-repressors and act to block transcription. When hormone binds to the receptor there is a conformational change in the ligand binding domain so that the co-repressors dissociate and co-activators are recruited. These cause local histone acetylation and transcriptional activation.
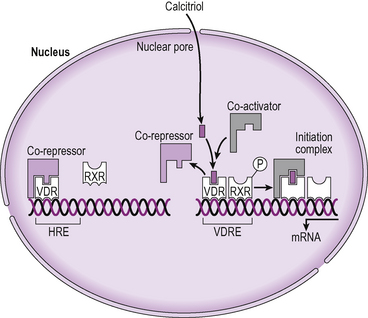
Figure 2.20 Molecular action of calcitriol. Calcitriol binds to vitamin D receptors (VDR) which are located in the nucleus of target cells. Binding of the calcitriol causes the VDR to become phosphorylated which allows it to recruit the retinoic acid receptor (RXR) to form a dimer which binds to the vitamin D response element (VDRE) in a gene promoter. The dimer attracts co-activators to form an initiation complex and permit gene transcription to proceed.
Disorders of receptor function
There are some well-characterized clinical conditions which arise from mutations of the genes encoding different receptors: Laron syndrome, for example, where there is a defect in the growth hormone receptor. In general, receptor gene mutations cause varying degrees of loss of function of the receptor. Where there is a significant loss of function in a developmentally significant receptor this usually causes spontaneous abortion of the foetus. In some cases we have learned a great deal about the normal functioning of a receptor system from examples of gene mutations causing changes in function. Several clinical conditions resulting from receptor defects are described in the following chapters, but these generally do not involve G-protein coupled receptors (GPCR). Despite the complexity of the GPCR system, receptor defects are very rare. The most commonly cited examples of G protein receptor mutations are those which result in constitutive activity of the receptor, which means that the receptor is able to activate signalling pathways even when no ligand is present. Spontaneous mutations of the thyroid stimulating hormone (TSH) receptor produce a constitutively active receptor leading to the development of highly active (‘hot’) thyroid nodules which secrete excess thyroid hormone. There is also an hereditary disorder of a constitutively active luteinizing hormone (LH) receptor which causes precocious puberty in males.