29.14 Cyanide
Introduction and epidemiology
Cyanide is used in a variety of commercial processes including metal extraction and recovery, metal hardening and in the production of agricultural and horticultural pest control compounds. Exposure can also occur to hydrogen cyanide (HCN) gas, produced when inorganic cyanide comes in contact with mineral acids as in electroplating, or accidentally when cyanide solutions are poured into acid waste containers. Cyanide off-gassing in house fires is well documented with significant blood levels being reported in 59% of smoke inhalation victims.1
Death from cyanide poisoning is one of the most rapid and dramatic seen in medicine, and antidotal therapy must be given early to alter outcome. A dose of 200 mg of ingested cyanide, or 3 min exposure to HCN gas, is potentially lethal.2
Fortunately, serious acute cyanide poisoning is rare. Of 2 424 180 human poison exposures reported to the American Association of Poison Control Centers during 2005, only 214 involved cyanide poisoning. Of the 118 cases in which clinical outcome is known, only six were reported to develop major symptoms, and only six patients died.3 However, the incidence of cyanide poisoning may be significantly underestimated. Blood cyanide concentrations greater than 40 μmol/L were found in 74% of victims found dead at the scenes of fires.1
Toxicokinetics and pathophysiology
The uptake of cyanide into cells is rapid and follows a first-order kinetic simple diffusion process. The half-life of cyanide is from 2 to 3 h.
While the precise in vivo action of cyanide is yet to be determined, it is thought that its major effect is due to binding with the ferric ion (Fe3+) of cytochrome oxidase, the last cytochrome in the respiratory chain. This results in inhibition of oxidative phosphorylation, leading to a net accumulation of hydrogen ions, a change in the NAD:NADH ratio and greatly increased lactic acid production. Other enzymatic processes, involving antioxidant enzymes, catalase, superoxide dismutase and glutathione, may contribute to toxicity..4 Cyanide is also a potent stimulator of neurotransmitter release, in both the central and the peripheral nervous systems.5
Humans detoxify cyanide by transferring sulphane sulphur, R–Sx–SH, to cyanide to form thiocyanate (SCN). The availability of R–Sx–SH is the rate-limiting step. This reaction is thought to be catalyzed by the liver enzyme rhodanese. However, other enzymes, such as β-mercaptopyruvate sulphur transferase, may be important. Other routes of biotransformation include oxidative detoxification.4
Clinical features
Cyanide toxicity is characterized by effects on the central nervous system (CNS), respiratory and cardiovascular systems, and by metabolic acidosis.2
CNS manifestations, in order of increasing severity of cyanide exposure, are headache, anxiety, disorientation, lethargy, seizures, respiratory depression, CNS depression and cerebral death. An initial tachypnoea gives way to respiratory depression as CNS depression develops.
Cardiovascular manifestations include hypertension followed by hypotension, tachycardia followed by bradycardia, arrhythmias, atrioventricular block and cardiovascular collapse. The classic finding of bright red skin and blood is not observed if significant myocardial, respiratory or CNS depression has already occurred; in these situations the patient may appear cyanotic. Other cardiovascular parameters of interest include decreased systemic vascular resistance, increased cardiac output and decreased arterio-venous oxygen gradient.
Clinical investigation
Arterial blood gas analysis and serum lactate measurements reveal metabolic acidosis with a raised lactate. Concentration decay curves suggest that serum lactate concentration is closely related to blood cyanide concentration. In smoke-inhalation victims without severe burns, plasma lactate concentrations above 10 mmol/L correlate with blood cyanide concentrations above 40 μmol/L, with a sensitivity of 87%, a specificity of 94% and a positive predictive value of 95%.6
Cyanide is concentrated ten-fold by erythrocytes and whole-blood cyanide concentrations are used as the benchmark when comparing levels. A level of 40 μmol/L is considered toxic, and a level of 100 μmol/L potentially lethal. Symptomatic intoxication starts at levels of about 20 μmol/L.7
Treatment
Attention to airway, breathing, circulation and other resuscitative measures must be instituted immediately.
In the case of cyanide poisoning from smoke inhalation or self-poisoning with clinical signs and associated lactic acidosis and where cyanide poisoning is suspected to be the cause of coma or cardiovascular instability, antidote administration is indicated. A number of antidotes are available (see discussion below) but the regime below is recommended if available:
Cyanide antidotes
Dicobalt edetate (Kelocyanor®)
This inorganic cobalt salt was introduced as a cyanide antidote in the late 1950s. It complexes with cyanide to form cobalt cyanide, thus removing cyanide from the circulation and reducing toxicity. However, unless cyanide is forced into the extracellular fluid, tissue levels are minimally affected.
Adverse effects are considerable and may be life-threatening.7 Severe hypotension, cardiac arrhythmias, convulsions and gross oedema are reported.8 These effects are exacerbated when drug is administered to an individual who is not cyanide poisoned. In life-threatening situations where cyanide poisoning is suspected the antidote is a must. The treating physician therefore faces a significant dilemma when presented with a critically ill patient in whom the history of exposure is unclear. A semiquantitative bedside test for cyanide in blood is available and may be helpful in determining the need for antidotal therapy when time permits.9 The recommended initial dose of dicobalt edetate is 300 mg i.v‥ Further doses may be required. Therapeutic endpoints are improvement in conscious state, haemodynamic stability and improvement in metabolic acidosis.
Hydroxocobalamin
Hydroxocobalamin (vitamin B12A) is the cyanide antidote most widely used in Europe. It complexes with cyanide, on a mole-for-mole ratio, to form cyanocobalamin. Antidotal doses of hydroxocobalamin are approximately 5000 times the physiological dose.
Hydroxocobalamin and cyanocobalamin are excreted by the kidney. The half-life of hydroxocobalamin in cyanide-exposed patients is 26.2 h.10 As the half-life of cyanide in smoke inhalation victims is calculated to be between 1.2 and 3.0 h, it is suggested that hydroxocobalamin can be satisfactorily used as single-dose therapy. The amount of cyanocobalamin formed after a dose of 5 g hydroxocobalamin correlates linearly until a blood cyanide level of 40 μmol/L is reached. At higher blood cyanide concentrations there is little further rise in plasma cyanocobalamin, and it is suggested that the rate-limiting step in the formation of cyanocobalamin is the availability of antidote, not the absence of cyanide ions.11
Extensive research has demonstrated the safety of this drug.12 In healthy adult smokers, 5 g of i.v. hydroxocobalamin is associated with a transient reddish discolouration of the skin, mucous membranes and urine, and a mean elevation in systolic blood pressure of 13.6%, with a concomitant 16.3% decrease in heart rate. No other clinical adverse effects are noted.13 Allergic reactions are rare.14 There is substantial experimental evidence to support the efficacy of hydroxocobalamin at lower levels of toxicity.10,12 Hydroxocobalamin has been shown to be safe and efficacious in mild-to-moderate cyanide poisonings with levels up to 150 μmol/L and has been given successfully to patients with severe cyanide toxicity.15 In cases of ingestion of cyanide with suicidal intent (where blood cyanide levels may be >150 μmol/L or plasma lactate concentrations >20 mmol/L), the usual dose of 5–10 g may be insufficient.
There are no data comparing the efficacy of hydroxocobalamin with dicobalt edetate so it is not possible to make any definitive conclusion about which antidote is best. However, in the emergency situation hydroxocobalamin appears to offer a greater margin of safety.
Limited volunteer studies suggest a synergistic effect of hydroxocobalamin and thiosulphate. Thiosulphate used on its own is limited by a slow onset of action and thus cannot be used alone as a first-line antidote. Case reports document successful outcomes in patients with extremely high levels of cyanide (494 μmol/L) with combination therapy.15
Hydroxocobalamin has been recommended as the treatment of choice for mass casualty chemical disasters where cyanide poisoning is suspected.16
Eli lilly cyanide kit
Administration of sodium nitrite followed by sodium thiosulphate is a long-accepted antidote for cyanide poisoning. The current Eli Lilly Cyanide kit was devised in 1970 and contains:
The kit is based upon the premise that humans can tolerate up to 30% methaemoglobinaemia.17,18 Conversion of haemoglobin to methaemoglobin promotes the movement of cyanide out of the cytochrome system; 4 mg/kg of sodium nitrite takes 30 min to achieve 7–10.5% methaemoglobin.15 The formation of sodium thiocyanate allows for the reformation of Hb2+, restoring the oxygen-carrying capacity of haemoglobin. Cellular respiration can continue as normal with cyanide removed from the respiratory chain. The observation that dramatic improvements in symptoms have occurred well before methaemoglobin levels have peaked has led many authors to suggest different mechanisms of action, such as vasodilatation and extracellular redistribution of cyanide.7,14 In smoke inhalation victims with suspected combined carbon monoxide and cyanide poisoning, the availability of an antidote that will not exacerbate any oxygen carriage or delivery problem, or cause toxicity by its own action, is highly desirable. The combination of 10% methaemoglobin with carboxyhaemoglobin has synergistic detrimental effects on the oxyhaemoglobin dissociation curve.
1 Baud FJ, Barriot P, Toffis V, et al. Elevated blood cyanide levels in victims of smoke inhalation. New England Journal of Medicine. 1991;325:1761-1766.
2 Gonzales J, Sabatini S. Cyanide poisoning: pathophysiology and current approaches to therapy. International Journal of Artificial Organs. 1989;12(6):347-355.
3 Lai MW, Klein-Schwartz, Rodgers GC, et al. Annual report of the American Association of Poison control Centers’ National Poisoning and Exposure Database. Clinical Toxicology. 2006;44:803-932.
4 Curry SC. Hydrogen cyanide and inorganic salts. In: Sullivan JB, Krieger GR, editors. Hazardous materials toxicology. Philadelphia Williams and Wilkins: Clinical principles of environmental health; 1992:698. 670
5 Isom GE, Borowitz JL. Modification of cyanide toxicodynamics mechanistic based antidote development. Toxicology Letters. 1995;82/83:795-799.
6 Baud FJ, Borron SW, Bavoux E, et al. Relationship between plasma lactate and blood cyanide concentrations in acute poisoning. British Medical Journal. 1996;312:26-27.
7 Marrs TC. Antidotal treatment of acute cyanide poisoning. Advances in drug reaction. Acute Poisoning Review. 1988;4:179-206.
8 Dodds C, McKnight C. Cyanide toxicity after immersion and the hazards of dicobalt edetate. British Medical Journal. 1985;291:785-786.
9 Fligner CL, Luthi R, Linkaityle-Weiss F, et al. Paper strip screening method for detection of cyanide in blood using the CYANOTESTMO test paper. American Journal of Forensic Medical Pathology. 1992;13(1):81-84.
10 Houeto P, Borron SW, Sandauk P, et al. Pharmacokinetics of hydroxocobalamin in smoke inhalation victims. Clinical Toxicology. 1996;34(4):397-404.
11 Houeto P, Hoffman JR, Imbert M, et al. Relation of blood cyanide to plasma cyanocobalamin concentration after a fixed dose of hydroxocobalamin in cyanide poisoning. Lancet. 1995;346:605-608.
12 Riou B, Baud FJ, Borron SW, et al. In vitro demonstration of the antidotal efficacy of hydroxocobalamin in cyanide poisoning. Journal of Neurosurgical Anaesthetics. 1990;2(4):296-304.
13 Forsyth JC, Mueller PD, Becker CE, et al. Hydroxocobalamin as a cyanide antidote: safety, efficacy and pharamacokinetics in heavily smoking normal volunteers. Journal of Toxicology and Clinical Toxicology. 1993;31:277-294.
14 Borron SW, Baud FJ. Acute cyanide poisoning: clinical spectrum, diagnosis and treatment. Arhiv za Higijenu Rada i Toksikologiju (Zagreb). 1996;47:307-322.
15 Tassan H, Joyon D, Richard T, et al. Potassium cyanide poisoning treated with hydroxocobalamin. Annales Françaises d’Anesthésie et Réanimation. 1990;4:383-385.
16 Sauer SW, Keim ME. Hydroxocobalamin: improved public health readiness for cyanide disasters. Annals of Emergency Medicine. 2001;37:635-641.
17 Kirk MA, Gerace R, Kulig KW. Cyanide and methaemoglobin kinetics in smoke inhalation victims treated with the cyanide antidote kit. Annals of Emergency Medicine. 1993;22:1413-1418.
18 Kiese M, Weger N. Formation of ferrihaemoglobin with aminophenols in the human for the treatment of cyanide poisoning. European Journal of Pharmacology. 1969;7:97-105.
19 Hart GB, Strauss MB, Lennon PA, et al. Treatment of smoke inhalation by hyperbaric oxygen. Journal of Emergency Medicine. 1985;3:111.
29.15 Corrosive ingestion
Introduction
Corrosives cause injury by an acid–base reaction with tissues. Strong solutions, capable of causing significant injury, are those with a pH of less than 2 or greater than 12 (Table 29.15.1). The pH of a solution is dependent on the concentration and dissociation constant (pKa) of the chemical. Strong acids have a pKa ≤0 and strong alkalis have a pKa ≥14 (Table 29.15.2). The extent of injury also depends on the volume ingested, contact time and viscosity.
Table 29.15.1 Approximate pH of some common solutions
| Solution | pH |
|---|---|
| Battery acid (1% solution) | 1.4 |
| Domestic toilet cleaner (1%) | 2.0 |
| Bleach (1% solution) | 9.5–10.2 |
| Automatic dishwasher detergents | 10.4–13 |
| Laundry detergents | 11.6–12.6 |
| Domestic ammonium cleaners | 11.9–12.4 |
| Drain cleaner (containing NaOH, KOH) | 13.3–14 |
Table 29.15.2 pKa of some common corrosives
| Chemical | pKa | Highly corrosive? |
|---|---|---|
| Hydrochloric acid | −3 | Yes |
| Bromic acid | <1 | Yes |
| Nitric acid | <1 | Yes |
| Sulphuric acid | 1.9 | |
| Arsenic acid | 2.3 | |
| Nitrous acid | 3.3 | |
| Hydrofluoric acid | 3.4 | |
| Ammonia | 9.3 | |
| Ammonium hydroxide | 9.3 | |
| Magnesium hydroxide | 10 | |
| Zinc hydroxide | 11 | |
| Calcium hydroxide | 11.6 | |
| Lithium hydroxide | >14 | Yes |
| Potassium hydroxide | >14 | Yes |
| Sodium hydroxide | >14 | Yes |
| Calcium oxide | >14 | Yes |
| Sodium carbonate | >14 | Yes |
| Potassium carbonate | >14 | Yes |
| Sodium hypochlorite | >14 | Yes |
Domestic hypochlorite bleaches and ammonia products are the commonest substances ingested, but severe injury generally does not occur unless large amounts are swallowed.1 Death results mainly from the ingestion of drain or toilet cleaners. Powdered automatic dishwasher detergents are also capable of causing severe injuries.2,3
Pathophysiology
Acid–base reactions cause injury by disrupting organic macromolecules. Heat generation may cause thermal burns. Highly exothermic reactions occur between strong acids and bases, or between light metallic compounds and water. Chemical reactions may also result in the production of other compounds that can cause additional injury to the gastrointestinal (GI) tract and lungs (Table 29.15.3).
Table 29.15.3 Chemical reactions resulting in the production of further toxic chemicals
| Chemical | Plus | Produces |
|---|---|---|
| Chlorine | Water | Hydrochloric acid Hypochlorous acid Oxygen radicals Heat |
| Ammonia | Water | Ammonium hydroxide Heat |
| Nitrogen dioxide | Water | Nitric acid Nitrous acid |
| Ammonia | Hypochlorite | Chloramine gas (NH2Cl and NHCl2) |
| Hypochlorite | Acid | Chlorine gas Hydrogen Sulphide |
| Sulphur compounds (e.g. plaster casts) | Acid | Sulphur oxide |
Alkalis cause ‘liquefactive’ necrosis, a process that involves saponification of fats, dissolution of proteins and emulsification of lipid membranes. Disruption of cellular membranes enhances penetration of alkali through the tissues.
Acids cause ‘coagulative’ necrosis, a process that involves denaturation of protein. The denatured protein forms a hard eschar that may limit further penetration of the acid.
In both settings, tissue injury progresses rapidly and can continue for several hours following ingestion. Granulation tissue develops after 3–4 days, but collagen deposition may not begin until the second week, making the healing tissue extremely fragile during this period. Complete repair of the epithelium may take weeks. From the third week newly deposited collagen begins to contract and may produce strictures of the oesophagus, stomach and affected bowel.
Hydrocarbon compounds can produce injury by dissolving lipids and coagulating proteins. Other chemicals can injure tissues by redox reactions and alkylation.
Following corrosive ingestion tissue inflammation, necrosis and infection can result in hypovolaemia, acidosis and organ failure.
Narrowings in the GI tract are most at risk from corrosive ingestion: the cricopharyngeal area, the diaphragmatic oesophagus, antrum and pylorus.4 Up to 80% of patients have injuries at multiple sites.5 Alkalis are more likely to produce oesophageal injury than are acids, which typically injure the stomach.4,6-11 Solid corrosives are more likely to affect the mouth, pharynx and upper oesophagus, and to cause deeper burns.
The main acute complications of corrosive ingestion are haemorrhage, perforation and fistula formation. These result from severe burns causing full-thickness necrosis.
Full-thickness necrosis of the stomach may be associated with injury to the transverse colon, pancreas, spleen, small bowel, liver and kidneys. Perforation of the upper anterior oesophagus may lead to the formation of a tracheoesophageal fistula. Formation of a tracheoesophago-aortic fistula is a rapidly lethal complication.
Clinical features
Symptoms and signs associated with significant alkali ingestion include mouth and throat pain, drooling, pain on swallowing, vomiting, abdominal pain and haematemesis.7 If the larynx is involved, local oedema may produce respiratory distress, stridor and a hoarse voice.9,12,13
Extensive tissue injury may be associated with fever, tachycardia, hypotension and tachypnoea.
Inspection of the oropharynx may reveal areas of mucosal burn. The absence of visible burns to the lips, mouth or throat does not imply an absence of significant burns to the oesophagus.3,5,7,9-11,14-17
Symptoms and signs associated with the life-threatening complications of oesophageal perforation and mediastinitis include chest pain, dyspnoea, fever, subcutaneous emphysema of the chest or neck and a pleural rub. Perforation of the abdominal oesophagus or stomach is associated with the clinical features of chemical peritonitis, including abdominal pain, fever and ileus.5,6,10,18 Septic shock, multiorgan failure and death may develop rapidly if perforation is not recognized.
The systemic effects of large acid ingestion include hypotension, metabolic acidosis, haemolysis, haemoglobinuria, nephrotoxicity, pulmonary oedema and hypotension. Features of systemic toxicity can result from the ingestion of arsenic, cyanide and other heavy metal salts, fluoride, ammonia, hydrazine, hydrochloric acid, nitrates, sulphuric acid and phosphoric acid. Ingestion of ammonia can cause coma, hypotension, acidosis, pulmonary oedema, liver dysfunction and coagulopathy.19 Systemic effects of phenol and related compounds include haemolysis and renal failure.20
Long-term complications
The major late complication of corrosive ingestion is the development of an oesophageal stricture. All patients with full-thickness necrosis of the oesophageal wall develop strictures, as do 70% of those with deep ulceration.5,10 Symptoms of oesophageal narrowing (principally dysphagia) may develop within 2 weeks; 80% occur within the first 2 months. Early onset of symptoms is associated with a more rapidly progressive and severe obstruction. Strictures do not develop in areas of superficial mucosal ulceration.6,11,18,21-25 Strictures can also affect the mouth, pharynx and stomach. Only 40% of gastric outlet strictures become symptomatic.5,10,11 A very late complication of alkali ingestion is the development of oesophageal carcinoma, reported to develop 22–81 years after exposure.26
Clinical investigation
Initial investigations in symptomatic patients should include an ECG, arterial blood gas, blood count, type and cross-match, coagulation profile, serum electrolytes, blood glucose and liver and renal function.
Chest and upright abdominal X-rays should be assessed for evidence of mediastinal widening, pleural effusions, pneumomediastinum, pneumothorax and subphrenic gas.
All patients who are symptomatic or have visible oropharyngeal burns should undergo upper GI endoscopy within 24 hours. Endoscopy should also be considered in any patient who has intentionally ingested a strong acid or alkali. Endoscopy is the only way to fully assess the extent of injury to the GI tract, and the findings are the best guide to prognosis and subsequent management. The entire upper GI tract may be safely examined with a small-diameter flexible endoscope, provided it is not retroflexed or forced through areas of narrowing.4,5,27 It is not necessary to terminate the examination at the first circumferential or full-thickness lesion. The cricopharynx should be assessed initially to identify any laryngeal burns. If laryngeal oedema or ulceration is encountered, endotracheal intubation may be necessary before continuing with endoscopy.
Oesophageal burns can be graded according to the depth of ulceration and the presence of necrosis, as determined at endoscopy (Table 29.15.4). Some parallel grading systems are used for thermal skin burns; others differentiate several levels of ulceration and necrosis. Injuries can be divided into three main groups:
Table 29.15.4 Classification of gastrointestinal corrosive burns
| Grade I | First-degree |
| Mucosal inflammation | Mucosal inflammation, oedema or superficial sloughing |
| Grade IIA | Second-degree |
| Haemorrhages, erosions and superficial ulceration | Damage extends to all layers of, but not through, the oesophagus |
| Grade IIB | |
| Isolated discrete or circumferential superficial ulceration | |
| Grade IIIA | Third-degree |
| Small scattered areas of necrosis | Ulceration through to perioesophageal tissues |
| Grade IIIB | |
| Extensive necrosis involving the whole oesophagus |
Contrast oesophagography with a water-soluble contrast agent is useful for the detection of perforation, but is less sensitive than endoscopy in evaluating ulceration.28
Treatment
Patients should initially be assessed for the presence of any symptomatic airway burns or respiratory distress. The need for urgent intubation should be considered in any patient with stridor or hypoxia.
Efforts at decontamination must not induce vomiting, as this may exacerbate the oesophageal injury. The mouth should be rinsed thoroughly with water. Dilution of an ingested solid chemical by drinking 250 mL of water or milk is recommended. The value of administering oral fluids following ingestion of a liquid corrosive is controversial.8,29 Patients should otherwise be given nothing by mouth. Neutralization, aspiration and administration of activated charcoal are all contraindicated.
Patients with persistent symptoms should be admitted for observation and undergo endoscopy 12–24 h later. Further management is dictated by the findings at endoscopy.
Patients with endoscopic evidence of superficial injury can be managed on a general medical ward with supportive care only. Complete healing can be expected. Patients with deep discrete ulceration, circumferential ulceration or isolated areas of necrosis should be admitted to high-dependency or the intensive care unit and kept nil by mouth. Intravenous fluid replacement, accurate fluid and electrolyte balance and symptom control are the mainstays of therapy. These patients may require prolonged i.v. access, and parenteral feeding and central venous access should be considered.
If perforation or penetration is suspected clinically or documented by endoscopy or contrast radiography, urgent laparotomy with or without thoracotomy must be considered. Early excision of areas with extensive full-thickness necrosis has been proposed, but this needs to be weighed against mortality rates of 40–50% for patients undergoing such emergency surgery.
Prophylactic broad-spectrum antibiotics are only indicated where there is evidence of GI tract perforation.
Strictures are dilated by endoscopy 3–4 weeks after ingestion. Reconstructive surgery may be required if the oesophageal lumen becomes completely obstructed, or if perforation occurs.
Disposition
Asymptomatic patients can be discharged after observation. They should be instructed to return if they develop pain, respiratory symptoms or difficulty swallowing. Symptomatic patients should be admitted for endoscopy with subsequent disposition dependent on the findings, as detailed above.
1 Litovitz TL, Klein-Schwartz W, White S, et al. 2000 annual report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. American Journal of Emergency Medicine. 2001;19:337-395.
2 Clausen JO, Nielsen TL, Fogh A. Admission to Danish hospitals after suspected ingestion of corrosives. A nationwide survey (1984–1988) comprising children aged 0–14 years. Danish Medical Bulletin. 1994;41:234-237.
3 Kynaston JA, Patrick MK, Shepherd RW, et al. The hazards of automatic-dishwasher detergent. Medical Journal of Australia. 1989;151:5-7.
4 Sugawa C, Lucas CE. Caustic injury of the upper gastrointestinal tract in adults: a clinical and endoscopic study. Surgery. 1989;106:802-806.
5 Zargar SA, Kochhar R, Mehta S, et al. The role of fiberoptic endoscopy in the management of corrosive ingestion and modified endoscopic classification of burns. Gastrointestinal Endoscopy. 1991;37:165-169.
6 Estrera A, Taylor W, Mills LJ. Corrosive burns of the esophagus and stomach: a recommendation for an aggressive surgical approach. Annals of Thoracic Surgery. 1986;41:276-283.
7 Gorman RL, Khin-Maung-Gyi MT, Klein-Schwartz W, et al. Initial symptoms as predictors of esophageal injury in alkaline corrosive ingestions. American Journal of Emergency Medicine. 1992;10:189-194.
8 Penner GE. Acid ingestion: toxicology and treatment. Annals of Emergency Medicine. 1980;9:374-379.
9 Vergauwen P, Moulin D, Buts JP, et al. Caustic burns of the upper digestive and respiratory tracts. European Journal of Pediatrics. 1991;150:700-703.
10 Zargar SA, Kochhar R, Nagi B, et al. Ingestion of strong corrosive alkalis: spectrum of injury to upper gastrointestinal tract and natural history. American Journal of Gastroenterology. 1992;87:337-341.
11 Zargar SA, Kochhar R, Nagi B, et al. Ingestion of corrosive acids: spectrum of injury to upper gastrointestinal tract and natural history. Gastroenterology. 1989;97:702-707.
12 Moulin D, Bertrand JM, Buts JP, et al. Upper airway lesions in children after accidental ingestion of caustic substances. Journal of Pediatrics. 1985;106:408-410.
13 Scott JC, Jones B, Eisele DW, et al. Caustic ingestion injuries of the upper aerodigestive tract. Laryngoscope. 1992;102:1-8.
14 Crain EF, Gershel JC, Mezey AP. Caustic ingestions: symptoms as predictors of esophageal injury. American Journal of Diseases of Childhood. 1984;138:863-865.
15 Gaudreault P, Parent M, McGuigan MA, et al. Predictability of esophageal injury from symptoms and signs: a study of caustic ingestion in 378 children. Pediatrics. 1983;71:767-770.
16 Christesen HB. Prediction of complications following unintentional caustic ingestion in children. Is endoscopy always necessary? Acta Pediatrica. 1995;84(10):1177-1182.
17 Muhlendahl KE, Oberdisse U, Krienke EG. Local injuries by accidental ingestions of corrosive substances by children. Archives of Toxicology. 1978;39:299-314.
18 Ray JR, Meyers W, Lawton BR. The natural history of liquid lye ingestion: rationale for aggressive surgical approach. Archives of Surgery. 1974;109:436-439.
19 Zitnik RS, Burchell HB, Shepherd JT. Hemodynamic effects of inhalation of ammonia in man. American Journal of Cardiology. 1969;24:187-190.
20 Lin CH, Yang JY. Chemical burn with cresol intoxication and multiple organ failure. Burns. 1992;18:162-166.
21 Middlekamp JN, Ferguson TB, Roper CL, et al. The management and problems of caustic burns in children. Journal of Thoracic and Cardiovascular Surgery. 1969;57:341-347.
22 Hawkins DB, Demeter MJ, Barness TE. Caustic ingestions: controversies in management – a review of 214 cases. Laryngoscope. 1980;90:98-109.
23 Anderson KD, Rouse TM, Randolph JG. A controlled trial of corticosteroids in children with corrosive injury of the esophagus. New England Journal of Medicine. 1990;323:637-640.
24 Webb WR, Koutras P, Eckker RR, et al. An evaluation of steroids and antibiotics in caustic burns of the esophagus. Annals of Thoracic Surgery. 1970;9:95-102.
25 Cannon S, Chandler JR. Corrosive burns of the esophagus: analysis of 100 patients. Eye Ear Nose Throat Monthly. 1963;42:35-44.
26 Isolauri J, Markkula H. Lye ingestion and carcinoma of the esophagus. Acta Chirurgica Scandinavica. 1989;155:269-271.
27 Chung RS, DenBesten L. Fibreoptic endoscopy in the treatment of corrosive injury of the stomach. Archives of Surgery. 1975;110:725-728.
28 Mansson I. Diagnosis of acute corrosive lesions of the esophagus. Journal of Laryngology and Otology. 1978;92:499-503.
29 Rumack BH, Burrington JD. Caustic ingestions: a rational look at diluents. Clinical Toxicology. 1977;11:27-34.
30 Howell JM, Dalsey WC, Hartsell FW, et al. Steroids for the treatment of corrosive esophageal injury: a statistical analysis of past studies. American Journal of Emergency Medicine. 1992;10:421-425.
31 Oakes DD, Sherck JP, Mark JB. Lye ingestion: clinical patterns and therapeutic implications. Journal of Thoracic and Cardiovascular Surgery. 1982;83:194-204.
29.16 Hydrofluoric acid
Introduction
The inorganic acid of fluoride, hydrofluoric acid (HF), is a moderately corrosive chemical widely used in industry for the etching of glass, metal and stone, and in the preparation of silicon computer chips. HF is also a common constituent of rust and scale removers, car wheel cleaners, brick cleaners and solder flux mixtures. These products may be for either commercial or home use and are often found in containers with inadequate labelling in regard to the potential toxicity. Concentrations of commercially available HF may vary from 50 to 100%. Products containing HF for domestic use generally have a concentration of less than 10% but higher concentration products may be obtained illicitly for home use.
The most common route of accidental exposure to HF is topical.1-3 This may occur when high-concentration HF leaks through damaged gloves in the industrial setting or when HF products are used in the home without gloves. Massive topical HF exposure and inhalational exposure to HF may also occur in the industrial setting.4-7 Finally, ingestion of HF products may occur accidentally in the home in the paediatric age group or as a result of deliberate self-harm in adults.8,9
Pathophysiology
HF is a relatively weak acid with less corrosive effects than other stronger acids such as hydrochloric or sulphuric. In particular, low concentrations of HF (<20%) may result in little or no perceptible corrosive injury to the skin immediately following exposure. This is due to the relatively small dissociation constant (pKa = 3.8), which limits the concentration of free hydrogen ions on the skin surface.2,3,8 As HF tends to remain in an undissociated, neutral state its ability to penetrate through the skin into deeper tissues is enhanced. Gradual dissociation of HF producing free fluoride ions in the tissues leads to local tissue injury characterized by liquefactive necrosis rather than the coagulative necrosis more commonly associated with acid burns.2,3 As the concentration of HF increases so does the potential for corrosive injury.3,10 Nevertheless, chemical burns may result from exposures to dilute (<5%) solutions of HF, and fatal systemic poisoning has resulted from relatively small (<5% total body surface area (BSA)) burns caused by more concentrated solutions.6,11-14
The primary mechanism of tissue damage resulting from exposure to HF is related to fluoride toxicity following on from dissociation of the acid in exposed tissues.2,3 A number of pathological mechanisms may be involved in both local and systemic fluoride poisoning. Fluoride binds divalent cations, especially calcium and magnesium, to form insoluble fluoride salts. The resulting hypocalcaemia and hypomagnesaemia may have profound local and systemic effects on cellular and organ functions. Fluoride is a cellular poison. It inhibits both aerobic and anaerobic metabolic enzyme systems and interferes with cellular respiration.6,7 Fluoride also interferes with Na+/K+ ATPase activity and opens calcium-dependent potassium channels in cell membranes, resulting in the leak of potassium into the extracellular space with the potential for systemic hyperkalaemia.1,15 Precipitation of calcium may also interfere with calcium-dependent clotting factors, resulting in coagulopathy.16 Finally, exposure to HF may produce direct corrosive injury.
Fatal systemic fluoride poisonings have also been reported following inhalational and gastrointestinal (GI) exposures.5,6 Once absorbed, fluoride ions are distributed to virtually every tissue and organ resulting in widespread disruption of organ function. Fluoride is slowly eliminated in the urine and elevations of urinary fluoride excretion can be detected following exposure to HF although these do not correlate with clinical toxicity.3,17
Clinical features
Exposure to HF in the industrial setting is usually recognized as such and patients will often have been decontaminated and had topical therapy applied prior to arrival at hospital. They are also likely to be in possession of appropriate information in the form of material safety data sheets. Acute dermal exposures in the domestic setting may present a more difficult diagnostic dilemma. Domestic product labels may be incomplete and offer no advice regarding the use of protective apparel such as gloves.18 Additionally, the onset of the signs and symptoms of HF injury may be delayed after exposure to low concentration domestic products and the patient may not recognize that the symptoms are related to chemical exposure.3,18
Highly concentrated (>70%) HF contains enough free hydrogen ions to produce a burning sensation on the skin providing some degree of warning of an acute exposure with symptom onset within 1–2 h.1,10 However, low concentrations of HF (<10%), found in products such as over-the-counter rust removers, often produce no symptoms at the time of contact and patients can present with gradually increasing pain from 6 to 12 h following exposure.1,19
The primary presenting complaint of acute topical HF injury is pain out of all proportion to any physical signs. HF exposure should always be considered in this situation. The pain is usually described as a tingling sensation that progresses to a burning pain and, then to a typical deep, throbbing and severe pain.1,10,19-21
Visible evidence of HF burns also follows a fairly common pattern. Initially, the burn site is erythematous and may be oedematous. As tissue injury progresses, the site becomes pale and blanched, progressing to a classical silvery grey appearance.2,22 Local vesiculation and frank tissue necrosis may ensue. This process can progress over several days in untreated patients, resulting in the development of deep ulceration and extensive tissue loss.
Dermal exposure to HF commonly occurs on the hands or feet with relatively small areas of the skin being exposed. Systemic fluoride poisoning is rarely a problem under these circumstances. The risk of systemic toxicity increases with the percentage of BSA exposed to HF and the concentration of HF.3 In general, if more than 3–5% BSA has been exposed to HF, there is a risk of hypocalcaemia.3,23 Systemic fluoride toxicity is more likely following large dermal exposures, ingestion or inhalation of HF.3,23,24
Systemic fluoride toxicity is manifest by various effects, the most lethal of which are the severe electrolyte abnormalities produced by direct interaction with fluoride or effects on cell membranes and cellular enzyme systems.3,6,7,15 Hypocalcaemia is due to the complexing of calcium by fluoride ions. Hypomagnesaemia may also occur. However, the primary cause of the lethal arrhythmias (refractory ventricular tachycardia, ventricular fibrillation and pulseless idioventricular rhythm) is the development of hyperkalaemia.8,15 Patients with systemic fluoride poisoning also develop a significant metabolic acidosis.3,8 This is the result of fluoride interference with intracellular metabolism. The systemic manifestations of significant hypocalcaemia include carpopedal spasm, hyperreflexia, tetany and coagulopathy.16 Headache, paraesthesiae and visual complaints may be noted. In severe cases coma, seizures, shock and dysrhythmias often precede death.
Fluoride inhalation is associated with pulmonary injury, including the development of non-cardiogenic pulmonary oedema, adult respiratory distress syndrome (ARDS) and the potential for systemic fluoride toxicity.3,5,25
Clinical investigation
No investigations are necessary following dermal exposures to dilute domestic preparations involving less than 5% of BSA. Following significant dermal exposures to HF or any ingestion or inhalational exposure, serum electrolytes including magnesium and calcium, baseline coagulation studies and a 12-lead ECG (looking for evidence of hypocalcaemia or hyperkalaemia) are indicated. A chest radiograph should be performed in any patient with respiratory symptoms or severe systemic toxicity.
Treatment
All patients with significant dermal HF exposures (>5% BSA, exposure to concentrated industrial preparations) or any ingestion or inhalation exposures should have continuous cardiac monitoring and intravenous access established on arrival.
The initial management of an acute topical HF exposure is thorough skin decontamination with a water flush. This ideally should be performed as a first-aid measure as soon as possible following the exposure as the delayed presentation of most patients makes it unlikely that significant amounts of HF still remain on the surface of the skin. Pre-hospital use of hexafluorine preparations offers no benefit in terms of local burn minimization or prevention of systemic toxicity when compared to water irrigation of exposed surfaces.26-28 Other first-aid measures in the work place for known or suspected HF burns include topical treatments such as calcium gluconate gel (2.5–10%) or soaks with quaternary ammonium salts such as benzalkonium or benzethonium chloride. Topical therapies are intended to form insoluble complexes with any surface fluoride ion thus preventing tissue penetration and minimizing deeper injury. Topical therapy is probably of little value once fluoride ions penetrate to deeper tissues but should be initiated on presentation to the emergency department (ED). Experimental evidence suggests that improved calcium penetration and burn control may occur with iontophoretic enhancement of divalent cation delivery; however, these observations have not yet been validated in the clinical setting.29 Calcium gluconate gel can be applied to the hand in a rubber glove. It may provide relief to some patients with low concentration HF exposures to the digits. In most cases, topical therapy is a temporizing measure until more invasive methods of calcium administration can be employed. If calcium gluconate gel is not readily available, a 4.8% preparation can be rapidly prepared by mixing one ampoule of calcium gluconate injection BP in 10 g of KY jelly.
The definitive treatment of dermal HF burns involves the administration of calcium gluconate into the tissues affected by the exposure.1-3,10,20,30-32 This may be achieved by a number of methods; direct tissue infiltration, regional intravenous infusion using a Bier’s block technique and intra-arterial infusion.1-3,10,20,30-32 The choice of method depends upon the site and concentration of HF involved.
Direct injection of approximately 0.5 mL per square centimetre of 10% calcium gluconate solution at the burn site can be considered in areas with little skin tension such as the trunk, forearms and legs. A small needle (25 gauge) should be used to minimize discomfort and care should be taken to infiltrate into, around and beneath the burn area as completely as possible. Only calcium gluconate should be used for local infiltration as calcium chloride is more concentrated and produces direct injury when injected into tissues.3,32
HF burns to the hands are relatively common. In view of the lack of loose tissues in the digits, direct dermal injection may be extremely painful and only small amounts of calcium gluconate may be injected. Additionally, the introduction of hyperosmolar calcium solutions to these limited tissues spaces may exacerbate oedema and result in vascular compromise.1,2 HF may also penetrate beneath the fingernails. In the past, removal of fingernails was advocated to allow for injection of calcium gluconate into the nail bed.32,33 Fortunately, the advent of focused, parenteral calcium administration techniques to the affected limb have meant that nail removal is rarely, if ever indicated.
Two techniques are available for direct injection of calcium gluconate into digital HF burns. The first is intra-arterial infusion of calcium gluconate. This technique involves inserting an arterial canula into the radial (for burns of the thumb, index and middle fingers) or brachial artery (for more extensive hand involvement) and slowly infusing a dilute solution of calcium gluconate utilizing an infusion pump. This allows the calcium to be delivered to the affected tissues through the vascular supply and avoids the pain and tissue distension associated with direct injection.20,33,34 A typical dose is 10–20 mL of 10% calcium gluconate in 50–100 mL of dextrose, 5% in water infused over 4 h and repeated as necessary. The endpoint for therapy is the absence of pain. The number of intra-arterial infusions required for pain relief may vary from one to four or five, and depends on the concentration of HF to which exposure occurred. There is case report level evidence for the use of continuous infusions.19,35
Regional intravenous calcium gluconate infusion using a Bier’s block technique has also been employed in the treatment of HF burns to the limbs.10,30,36 Success has been observed for digital, hand and forearm exposures as well as for exposures to the leg.10,30,36 The technique is similar to that described by Bier for regional limb anaesthesia and has the advantages of relative simplicity and of not requiring arterial cannulation.37 An intravenous cannula is inserted in the dorsum of the hand of the affected limb and the arm is raised to exsanguinate the superficial venous system. A pneumatic tourniquet is applied to the upper arm and inflated to a pressure 100 mmHg above systolic blood pressure. Ten to 15 mL of 10% calcium gluconate is diluted to a total volume of 50 mL with normal saline and injected via the cannula into the ischaemic arm. The tourniquet is sequentially released after 20 to 25 min.10 Pain relief is usually apparent within 30 min of tourniquet release.
There have been no controlled studies comparing any of these techniques in the treatment of HF burns. However, intra-arterial calcium infusion appears to be a better technique for distal digital exposures, particularly in cases where exposure has been to high concentrations (>20%) or where multiple digits are involved.10 Intra-arterial infusion of calcium has the advantages of more focal provision of calcium to the site of digital exposures and the potential for multiple infusions in patients with ongoing pain. If the intravenous route is selected as the primary therapy and is unsuccessful following one treatment, intra-arterial calcium infusion should then be used. The use of intra-arterial magnesium sulphate in place of calcium gluconate has resulted in tissue necrosis requiring surgical debridement in a small case series and cannot be recommended.38
It is sometimes difficult to determine whether ongoing pain at the exposure site is due to continued tissue destruction from fluoride still present in the tissues, or to established tissue damage. This is particularly the case with patients who have had digital exposures to high concentrations of HF and received multiple infusions of intra-arterial calcium which do not seem to produce further pain relief. It also applies to patients who present more than 24 h post-exposure with ongoing pain despite calcium therapy. In both instances, failure to achieve pain relief with repeated infusions of calcium gluconate suggests that pain may be related to established tissue damage rather than ongoing tissue destruction.
Ocular HF exposures can result in serious consequences if left untreated. Patients should be treated as for other chemical exposures to the eye with copious saline irrigation, and local anaesthetic drops for pain relief. Calcium gluconate (10–20 mL/L) may be added to saline irrigation fluid although animal studies suggest that calcium gluconate eye drops are no better than copious irrigation with normal saline and may, in fact, result in delayed corneal healing.39 Clinical case reports of calcium gluconate eye drop use suggest that this treatment is not harmful but controlled studies are lacking.40,41
Systemic fluoride poisoning resulting from HF ingestion, inhalation or significant dermal exposures is potentially life-threatening. Patients with HF ingestion should receive rapid GI decontamination. Aspiration of HF through a small bore nasogastric tube may limit absorption if the patient presents within an hour of ingestion. Calcium or magnesium-containing antacids can complex intragastric HF and prevent some systemic absorption of fluoride ions, although any benefit is likely to be marginal at best.42 Endoscopy should be performed following HF ingestion as soon as the patient is clinically stable to assess the extent of any upper GI corrosive injury.1 Nebulized calcium gluconate has been administered acutely to patients following HF inhalation.5,43 Serum calcium, magnesium and potassium levels should be closely monitored. Intravenous calcium and magnesium replacement should be commenced prior to any fall in serum Ca2+ or Mg2+ concentrations and replacement doses may be guided by the calculated dose of fluoride ingested. Large amounts of calcium (200–300 mmol) have been used in severe cases of systemic HF poisoning with hypocalcaemia.3,8,13 Hyperkalaemia may be recognized on the 12-lead ECG, but close monitoring of serum potassium levels is warranted. Hyperkalaemia in systemic fluoride poisoning is resistant to standard measures of potassium reduction, such as insulin, glucose and bicarbonate infusions. Ventricular arrhythmias associated with systemic fluoride poisoning are refractory to cardioversion and defibrillation, and may not respond to antiarrhythmic agents.15 Haemodialysis is indicated for severe or refractory hypocalcaemia, hyperkalaemia or clinical toxicity (e.g. arrhythmias) and may be useful for the removal of fluoride ions.3,7 Calcium and magnesium monitoring and replacement should continue during this procedure.
Disposition
Patients with minor dermal exposures in whom ED treatment produces complete resolution of symptoms may be discharged home with follow-up arranged within 24 h or should pain return. Those patients in whom tissue damage is evident require referral to a plastic or hand surgeon.
Patients at risk of systemic fluoride poisoning (exposure to high concentration solutions, greater than 5% BSA burns, inhalations and ingestions) require admission to an intensive care unit for ongoing monitoring and management of the electrolyte disturbances and other complications of systemic toxicity.
All patients with eye exposures require early ophthalmological referral.
1 Salzman M, O’Malley RN. Updates on the evaluation and management of caustic exposures. Emergency Medical Clinics of North America. 2007;25:459-476.
2 Burd A. Hydrofluoric acid-revisited. Burns. 2004;30:720-722.
3 Dunser MW, Ohlbauer M, Rieder J, et al. Critical care management of major hydrofluoric acid burns: a case report, review of the literature, and recommendations for therapy. Burns. 2004;30:391-398.
4 Blodgett DW, Suruda AJ, Crouch BI. Fatal unintentional occupational poisonings by hydrofluoric acid in the U.S. American Journal of Indian Medicine. 2001;40:215-220.
5 Kono K, Watanabe T, Dote T, et al. Successful treatments of lung injury and skin burn due to hydrofluoric acid exposure. International Archives of Occupational and Environmental Health. 2007;73(Suppl):S93-S97.
6 Caravati EM. Acute hydrofluoric acid exposure. American Journal of Emergency Medicine. 1988;6:143-150.
7 Bjornhagen V, Hojer J, Karlson-Stiber C, et al. Hydrofluoric acid induced burns and life threatening systemic poisoning – favourable outcome after haemodialysis. Journal of Toxicology – Clinical Toxicology. 2003;41(6):855-860.
8 Chan BS, Duggin GG. Survival after a massive hydrofluoric acid ingestion. Journal of Toxicology – Clinical Toxicology. 1997;35:307-309.
9 Holtstege C, Baer A, Brady WJ. The electrocardiographic toxidrome: the ECG presentation of hydrofluoric acid ingestion. American Journal of Emergency Medicine. 2004;23:171-176.
10 Graudins A, Burns MJ, Aaron CK. Regional intravenous infusion of calcium gluconate for hydrofluoric acid burns of the upper extremity. Annals of Emergency Medicine. 1997;30:604-607.
11 Chan KM, Svancarek WP, Creer M. Fatality due to acute hydrofluoric acid exposure. Journal of Toxicology – Clinical Toxicology. 1987;25:333-339.
12 Manoguerra AS, Neuman TS. Fatal poisoning from acute HF ingestion. American Journal of Emergency Medicine. 1988;4:362.
13 Mayer TG, Gross PL. Fatal systemic fluorosis due to hydrofluoric acid burns. Annals of Emergency Medicine. 1985;14:149-153.
14 Bordelon BM, Saffle JR, Morris SE. Systemic fluoride toxicity in a child with hydrofluoric acid burns: case report. Journal of Trauma. 1993;34:437-439.
15 Cummings CC, McIvor ME. Fluoride-induced hyperkalemia: the role of Ca++ dependent K+ channels. American Journal of Emergency Medicine. 1988;6:1.
16 Auguilera IM, Vaughan RS. Calcium and the anaesthetist. Anaesthesia. 2000;55:779-790.
17 Saady JJ, Rose CS. A case of non-fatal sodium fluoride ingestion. Journal of Analytical Toxicology. 1988;12:270-271.
18 Smith MA. A hand burn from unmarked hydrofluoric acid [letter]. Medical Journal of Australia. 1992;157:431.
19 Siegel DC, Heard JM. Intra-arterial calcium infusion for hydrofluoric acid burns. Aviation Space and Environmental Medicine. 1992;63:206-211.
20 Vance MV, Curry SC, Kunkel DB, et al. Digital hydrofluoric acid burns: treatment with intra arterial calcium infusion. Annals of Emergency Medicine. 1986;15:890-896.
21 Wilkes GJ. Intravenous regional calcium gluconate for hydrofluoric acid burns of the digits. Emergency Medicine. 1993;5:149-244.
22 Anderson WJ, Anderson JR. Hydrofluoric acid burns of the hand: mechanism of injury and treatment. Journal of Hand Surgery [Am]. 1988;13:52-57.
23 Mullett T, Zoeller T, Bingham H, et al. Fatal hydrofluoric acid cutaneous exposure with refractory ventricular fibrillation. Journal of Burn Care Rehabilitation. 1987;8:216-219.
24 Sadove R, Hainsworth D, Van Meter W. Total body immersion in hydrofluoric acid. Southern Medical Journal. 1990;83:698-700.
25 Watson AA, Oliver JS, Thorpe JW. Accidental death due to inhalation of hydrofluoric acid. Medicine, Science and the Law. 1973;13:277-279.
26 Hojer J, Personne M, Hulten P, Ludwigs U. Topical treatments for hydrofluoric acid burns: a blind controlled experimental study. Journal of Toxicology – Clinical Toxicology. 2002;40(7):861-866.
27 Hojer J, Personnes M, Hulten P, Ludwigs U. Existing evidence does not support the use of Hexafluorine (letter). Journal of Toxicology – Clinical Toxicology. 2003;41(7):1033-1034.
28 Hulten P, Hojer J, Ludwigs U, Janson A. Hexafluorine vs standard decontamination to reduce systemic toxicity after dermal exposure to hydrofluoric acid. Journal of Toxicology – Clinical Toxicology. 2004;42(4):355-361.
29 Yamashita M, Yamashita M, Suzuki M, et al. Iontophoretic delivery of calcium for experimental hydrofluoric acid burns. Critical Care Medicine. 2001;29(8):1575-1578.
30 Ryan JM, McCarthy GM, Plunkett PK. Regional intravenous calcium – an effective method of treating hydrofluoric acid burns to limb peripheries. Journal of Accident and Emergency Medicine. 1997;14:401-404.
31 Murao M. Studies on the treatment of hydrofluoric acid burn. Bulletin of the Osaka Medical College. 1989;35:39-48.
32 Bracken WM, Cuppage F, McLaury RL, et al. Comparative effectiveness of topical treatments for hydrofluoric acid burns. Journal of Occupational Medicine. 1985;27:733-739.
33 Trevino MA, Herrmann GH, Sprout WL. Treatment of severe hydrofluoric acid exposures. Journal of Occupational Medicine. 1983;25:861-863.
34 Kohnlein HE, Achinger R. A new method of treatment of HF burns of the extremities. Chirurgie Plastica. 1982;6:298.
35 Lin TM, Tsai CC, Lin SD, Lai CS. Continuous intra-arterial infusion therapy in hydrofluoric acid burns. Journal of Occupational and Environmental Medicine. 2000;42(9):892-897.
36 Henry JA, Hla KK. Intravenous regional calcium gluconate perfusion for hydrofluoric acid burns. Journal of Toxicology – Clinical Toxicology. 1992;30:203-207.
37 Bier A. Concerning a new method of local anaesthesia of the extremities. Archiv fur Klinische Chirurgia. 1908;86:123.
38 Vance M, Curry S, Gerkin R, et al. An update on the treatment of digital hydrofluoric acid burns with intra-arterial infusion techniques. Veterinary and Human Toxicology. 1986;28:486.
39 Beiran I, Miller B, Bentur Y. The efficacy of calcium gluconate in ocular hydrofluoric acid burns. Human and Experimental Toxicology. 1997;16:223-228.
40 Bentur Y, Tannenbaum S, Yaffe Y, Halpert M. The role of calcium gluconate in the treatment of hydrofluoric acid eye burn. Annals of Emergency Medicine. 1993;22:1488-1490.
41 Rubinfeld RS, Silbert DI, Arentsen JJ, Laibson PR. Ocular hydrofluoric acid burns. American Journal of Ophthalmology. 1992;114:420-423.
42 Heard K, Delgado J. Oral decontamination with calcium or magnesium salts does not improve survival following hydrofluoric acid ingestion. Journal of Toxicology – Clinical Toxicology. 2003;41(6):789-792.
43 Lee DC, Wiley JFD, Synder JWD. Treatment of inhalational exposure to hydrofluoric acid with nebulized calcium gluconate [letter]. Journal of Occupational Medicine. 1993;35:470.
29.17 Pesticides
Introduction
Pesticide poisoning occurs worldwide. Poisoning may occur due to either acute (intentional self-poisoning) or chronic (such as occupational) exposures. Acute poisoning is of more importance to the emergency physician and is the focus of this chapter.
A pesticide is any chemical used for the control of a plant or animal, which encompasses hundreds of chemicals. They can be sub-classified in terms of their intended target, the most common being insecticides, herbicides (selective or non-selective), fungicides, rodenticides and nematocides. Other methods for classification that have been used include mechanism of action and chemical structure.
Worldwide, pesticides are the most important cause of death from acute self-poisoning.1 As with pharmaceutical poisoning, the toxicity of pesticides varies between individual compounds but, in general, pesticides are intrinsically more toxic than pharmaceuticals.2,3 However, not all pesticide exposures lead to significant clinical toxicity. In Australasia, most acute pesticide exposures are accidental and the majority of patients do not require admission to hospital.
An accurate risk assessment is necessary for the proper management of patients with acute pesticide poisoning. This considers the dose ingested, time since ingestion, clinical features, patient factors and available medical facilities.4 If a patient presents to a facility that is unable to provide sufficient medical and nursing care or does not have ready access to necessary antidotes, then arrangements should be made to rapidly and safely transport the patient to a healthcare facility where this is available.5
Due to the low incidence, pesticide poisoning is not always considered in the differential diagnosis. A number of case reports from Australia have described a delay in the diagnosis of significant pesticide poisoning because it was not considered initially.6-8 These delays did not appear to adversely affect patient outcomes, but they highlight the importance for clinicians to be familiar with the clinical features of pesticide poisoning.6
This chapter primarily focuses on agrochemicals used in Australasia, in particular insecticides (organophosphorus pesticides (OPs) and carbamates) and herbicides (glyphosate and paraquat).
Aetiology, pathogenesis and pathology
Acute pesticide poisoning requiring admission to hospital and ongoing care usually occurs following acute intentional self-poisoning. Significant toxicity may also occur with accidental (e.g. storage of a pesticide in a milk carton) or criminal exposures.
The pathophysiology of acute pesticide poisoning, and therefore the clinical manifestations, vary widely between individual compounds. Many pesticides induce multisystem toxicity due to interactions with a number of physiological systems. The mechanism of toxicity in humans is discussed below for each pesticide; often it bears little relation to the mechanism of action in the target pest.
For many pesticides, the mechanism of toxicity is poorly described which usually means that less information is available to guide management of these exposures; however, it is an active area of research.9
It should be noted that proprietary pesticide products often contain other co-formulants, in particular hydrocarbon-based solvents. Herbicide products also contain surfactants to enhance herbicide penetration into the plant.10
Epidemiology
Acute pesticide poisoning is a major issue in developing countries of the Asia–Pacific region and OPs are considered the most important cause of death from acute poisoning worldwide.11 In developed countries, however, the incidence of severe pesticide poisoning is relatively low. In rural areas, the incidence of severe pesticide poisoning may be higher compared to urban regions due to easier access.
Prevention
Primary exposures
Restriction of the availability of pesticides by regulatory authorities may decrease the overall mortality.12 In Australia, for example, pesticides with a high case fatality such as paraquat, organochlorines and parathion are heavily regulated so poison exposures are increasingly rare. Proper storage, handling and use of pesticides can prevent accidental exposures.
Secondary exposures or nosocomial poisoning
There is much concern regarding the risk of nosocomial poisoning to staff and family members who are exposed to patients with acute pesticide poisoning, in particular OPs. Few cases of secondary poisoning, if any, have been confirmed by abnormal cholinesterase activities. While mild symptoms such as nausea, dizziness, weakness and headache have been reported in staff, these resolved after exposure to fresh air and were probably due to inhalation of the hydrocarbon solvent. Universal precautions using nitrile gloves are most likely to provide sufficient protection for staff members.13,14 Dermal decontamination is necessary. Wash pesticide spills from the patient with soap and water and remove and discard contaminated clothes, shoes and other leather materials.
Anticholinesterase pesticides
Anticholinesterase pesticides are among the most widely used types of pesticides and include organophosphorus (organophosphate, OP, OGP) and carbamate compounds. In Australasia, the most commonly encountered anticholinesterase compounds are chlorpyrifos, dimethoate, fenthion, malathion (maldison), diazinon and propoxur (carbamate).6,15
The relationship between exposure and clinical toxicity is poorly defined and therefore all exposures should be observed and treated as significant.6,15 Deliberate self-poisoning by ingestion is the scenario most likely to result in severe toxicity. Carbamates are generally less toxic and produce toxicity of a shorter duration than OPs. However, severe toxicity and death occur with some carbamates, in particular carbosulfan and carbofuran.16-19
Mechanism of toxicity
The effects of anticholinesterase compounds on human physiology are multiple, complex and incompletely described. Inhibition of acetylcholinesterase (AChE), thus preventing the hydrolysis of acetylcholine, is considered the most important mechanism.20,21 Accumulation of acetylcholine at cholinergic synapses interferes with systemic nervous function, producing a range of clinical manifestations which are known as the acute cholinergic crisis (Table 29.17.1).
Table 29.17.1 Clinical manifestations and treatment of acute anticholinesterase poisoning5,20,21,22,43,4,5
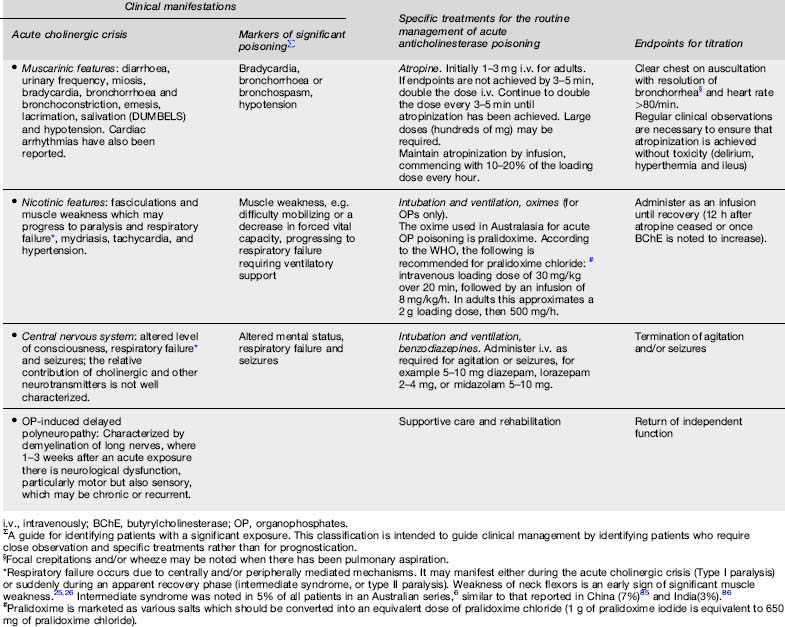
Inhibition of other esterases contributes to the clinical manifestations of acute poisoning. Inhibition of neuropathy target esterase leads to organophosphorus-induced delayed polyneuropathy (Table 29.17.1).22
Enzyme inhibition by an anticholinesterase compound is potentially reversible. In the case of OP-inhibited AChE, the enzyme can undergo spontaneous reactivation resulting in normal enzymatic function. But a proportion of inhibited AChE undergoes irreversible inhibition (‘ageing’) and enzyme resynthesis is required for restoration of nervous function. The rate of these competing reactions varies more than 10-fold between individual OPs.20,23 This influences the clinical manifestations and response to antidotes, which reactivate inhibited AChE. Due to structural differences between carbamates and OPs there is spontaneous reactivation of carbamate-inhibited AChE and ageing does not occur.23
Marked differences in the clinical manifestations of acute anticholinergic poisoning from different compounds are observed.20,21,24-26 This may reflect the variability in potency of enzyme inhibition,20,27 physiological adaptations following prolonged stimulation,20,23,26 pharmacokinetic factors,20,23,24 additional mechanisms of toxicity such as oxidative stress,28 inter-patient differences24,39 or a complex interplay of a number of these and other unknown factors.20,25,28
Clinical features
The initial manifestation of acute anticholinesterase poisoning is the acute cholinergic crisis (Table 29.17.1). The duration and manifestations of the acute cholinergic crisis vary between individual anticholinesterase compounds, as mentioned above.20,21,24-26
Gastrointestinal symptoms are most prevalent following oral exposures, probably a result of high pesticide concentrations in the gut prior to absorption, and the hydrocarbon solvent.6 Tachycardia does not always appear to correlate with hypotension or pneumonia;6 instead it may be secondary to catecholamine release from the adrenal medulla under nicotinic stimulation.29
Differential diagnosis
In situations where the history is not forthcoming, the differential diagnosis is broad and includes other toxins (clonidine, opioids, dopamine antagonists such as chlorpromazine or haloperidol), funnel web spider envenoming and pontine haemorrhage.
Clinical investigation
The diagnosis and management of acute anticholinesterase poisoning is primarily clinical but measurement of cholinesterase activity can assist. The reference ranges are wide due to the large inter-individual variability in baseline AChE and butyrylcholinesterase (BChE, plasma cholinesterase, pseudocholinesterase) activities.23,31 Cholinesterase inhibition is generally noted prior to clinical effects.23,32 AChE and BChE are generally depressed within 6 h, although enzyme inhibition may progress until 12–24 h post-ingestion.33 AChE or BChE activity, if <80% of the lower reference range, indicates significant anticholinesterase exposure.26,31,33 Patients in whom cholinesterase activity is higher might still have been exposed, but to a minimal degree only.
BChE inhibition is a sensitive biomarker of anticholinesterase exposure but has no relation to the severity of poisoning because the affinity of anticholinesterase compounds for BChE is highly variable and differs to that of AChE.23,30,32,34 Serial measurements of BChE may be useful for confirming systemic elimination of the anticholinesterase compounds. Once BChE activity starts to increase (the rate of this depends on hepatic function) it suggests that the plasma concentration of the anticholinesterase compound is negligible.5
Erythrocyte AChE is structurally similar to synaptic AChE and their activities change similarly in response to exogenous inhibition. The degree of AChE inhibition appears to correlate with severity of OP poisoning and it is considered the most useful biomarker of severity. In severe clinical toxicity, erythrocyte AChE activity is less than 20% of normal.21,23,35 Serial measurements of erythrocyte AChE activity can be useful for confirming the efficacy of oximes. If AChE activity normalizes following initiation of oximes, it suggests that ageing has not occurred and that the dose of oximes is sufficient.
Cholinesterase mixing tests have been used clinically in patients with acute OP poisoning to titrate the oxime regimen.36 In one method, the patient’s plasma is mixed with an equal volume of non-poisoned donor (control) plasma. If BChE activity in the mixed sample is less than the mean of the samples from the patient and control, it suggests that free anticholinesterase compounds are present.36 It has been suggested that a decrease in cholinesterase activity in mixing studies is an indication to increase the dose of oximes although the AChE response to oximes is probably a more useful parameter to guide oxime dosing. Despite being widely used, neither of these approaches to monitor therapy have been demonstrated to improve outcome.
Arterial blood gases and routine blood laboratory analyses are recommended for measuring metabolic and respiratory derangements; hypokalaemia secondary to vomiting and diarrhoea is not uncommon.
Criteria for diagnosis
Acute anticholinesterase poisoning is diagnosed on the basis of a history of exposure and development of characteristic clinical features (Table 29.17.1). Therefore, a high index of clinical suspicion is necessary. Since the correlation between intent, dose and severity of toxicity appears to be poor,6,15 and the clinical manifestations between individual compounds differ,24 each exposure requires a thorough review.
The onset of clinical toxicity is variable; however, the majority of patients who will develop severe toxicity are symptomatic within 6 h. Patients remaining asymptomatic for 12 h post-ingestion are unlikely to develop significant clinical toxicity.5,21,37 A possible exception is highly lipophilic compounds such as fenthion. These may produce only subtle cholinergic features initially but then go on to cause progressive muscle weakness over a number of days, including respiratory failure, requiring ventilatory support.24,26
Where there is doubt regarding the diagnosis or significance of an OP exposure, quantification of BChE or AChE activity is helpful if available. BChE is particularly useful because it is more widely available and a sensitive marker of exposure.23,27,32,38
Treatment
Resuscitation and early considerations
As with all acute poisonings, initial management begins with immediate assessment and management of disturbances in airway, breathing and circulation. Because suxamethonium is metabolized by BChE, this agent should not be used for intubation of patients with acute anticholinesterase poisoning because the duration of paralysis will be prolonged by many hours.39 Continuous clinical monitoring, including pulse oximetry, cardiac monitoring and blood pressure are required. Intravenous vasopressors should be used for hypotension not responding to fluid loading because hypotension is often due to a decrease in systemic vascular resistance.
Although the amount ingested as per history appears to be a poor predictor of the amount absorbed,32,40 all patients with intentional poisoning who are symptomatic should be managed in a centre with access to intensive care facilities. Oral decontamination with activated charcoal has been recommended for patients presenting within 1–2 h of ingestion;41 however, a recent randomized controlled study demonstrated that this was not beneficial.42
During the immediate assessment and resuscitation of the patient, all patients should undergo some degree of dermal decontamination. The removal and discarding of exposed clothing will reduce further anticholinesterase exposure.
Subsequent interventions depend on changes in clinical observations during continuous monitoring. Antidotal therapy should be administered rapidly, as outlined in Table 29.17.1. The acute cholinergic syndrome is potentially reversible with adequate doses of atropine. Oximes such as pralidoxime may reverse muscle weakness or paralysis if administered promptly. Established OP-induced delayed polyneuropathy (OPIDP) does not respond to antidotes; instead supportive care is the priority.5,43
Mild clinical toxicity and dermal exposures
Patients who present with a history of accidental poisoning who are asymptomatic or mildly symptomatic (limited to GI symptoms), often do not require hospital admission. Management priorities for these patients are rapid triage, a detailed risk assessment and consideration of forensic implications. If the exposure is trivial, the patient does not need medical review and can be observed at home or in the workplace. Other patients should be decontaminated and monitored clinically for a minimum of 6–12 h. If available, cholinesterase activity should be measured to confirm whether the exposure is significant. A normal cholinesterase activity at 6 h post-exposure may be sufficient to exclude a significant oral exposure, although this approach has not been sufficiently assessed.5
Patients with a single acute dermal exposure rarely develop significant clinical effects and probably do not require medical assessment. Volunteer studies document that the risk of significant clinical toxicity from a dermal exposure is far below that of an oral exposure. Although the rate of anticholinesterase absorption across the skin is slower than across the gut, patients who are asymptomatic at 12 h are unlikely to develop toxicity. Such patients should be given instructions to present for medical review if there is a significant worsening of signs and symptoms. If there is significant concern regarding a dermal exposure, testing for changes in cholinesterase activity is recommended.5
Moderate-to-severe clinical toxicity
Patients with moderate-to-severe anticholinesterase poisoning experience prolonged and complicated hospital admissions. Close observation is required to monitor for a rapid clinical deterioration, even if there is apparent recovery from the acute cholinergic crisis.25,44 Therefore, following resuscitation, these patients require ongoing management in an intensive care unit (ICU).6,43,44 Priorities post-admission to ICU include careful titration of antidotes and supportive care, including ventilation and inotropes/vasopressors.43
Antidotes
The three most widely used classes of antidotes are muscarinic antagonists (usually atropine), oximes (usually pralidoxime in Australasia) and benzodiazepines.5 The indications and dosing regimen of these specific antidotes are described in Table 29.17.1.
Antimuscarinic agents
Atropine is the most widely used antimuscarinic agent. It is carefully titrated to reverse muscarinic effects and has no effect on the neuromuscular junction and muscle weakness.43,45
Oximes
Oximes are used to reverse neuromuscular blockade by reactivating the inhibited AChE before ageing occurs and should therefore be administered as early as possible.20
Evidence supporting the efficacy of oximes and the dosing regimen is limited and their role in therapy is controversial.46 Much of the controversy relates to issues with study design and low dosing regimens. Recent meta-analyses point to the limited data supporting the use of oximes.47,48 However, the outcomes of these meta-analyses are questionable because they fail to account for the heterogeneity of the studies or the adequacy of their designs.46
More recently, a randomized controlled trial concluded that high doses of pralidoxime iodide are effective, although further studies were recommended.49 In this study, all patients received an initial bolus of 2 g pralidoxime iodide followed by either 24 g/day for 48 h, then 1 g every 4 h until recovery (high dose) or the 1 g every 4 h (lower dose) until recovery.
Because carbamate-inhibited AChE does not undergo ageing, the role for oximes appears limited. However, it remains controversial given that data have been presented to suggest that oximes may increase the reactivation of carbamate-inhibited AChE,50,51 although not consistently.52 Oximes appear to increase carbaryl toxicity for reasons that are not understood. Despite the lack of clinical data, it is not unreasonable for oximes to be administered to patients with an unknown exposure and evidence of AChE inhibition.5,53
On the basis of a few reasonably conducted studies and clinical experience, oximes are recommended for use in patients with significant OP poisoning.5,20
Benzodiazepines
Benzodiazepines are recommended for use in patients with agitation or seizures.45 It is proposed that early use of benzodiazepines may prevent cognitive deficits or improve the central control of respiration preventing the need for intubation; however, this has not yet been sufficiently studied.
Prognosis
The mortality in patients with anticholinesterase poisoning is variable, which may reflect differences in degree of exposure, reporting, resources, genetics or the types of compounds encountered.54-56 However, the mortality is generally high at greater than 10%,11 compared to a mortality of less than 0.5% for pharmaceuticals.57
Various tools are proposed to classify the severity of OP poisoning,54,58-60 but few have been widely adopted or validated. Generalized approaches to prognostication in OP poisoning are difficult given that individual compounds vary markedly in the onset, severity and manifestations of clinical toxicity.24 Further, they are not often useful for guiding management.5
In the case of dermal exposures, the potential for toxicity is poorly defined but prognosis appears favourable.
Paraquat (Bipyridyl Herbicides)
Paraquat is a non-selective contact herbicide and is considered one of the most toxic pesticides available. The mortality from acute poisoning is high, varying between 50 and 90%.61 Fortunately, cases of acute paraquat poisoning are increasingly rare in developed countries due to regulation of availability. However, paraquat continues to be an important cause of death in developing countries around Asia, where it is widely used in subsistence farming.
Diquat is another bipyridyl herbicide that is more widely available in Australasia.
Mechanism of toxicity
Oral exposures to paraquat are most likely to lead to poisoning. Because paraquat formulations are highly irritating (and potentially corrosive), GI toxicity occurs with all oral exposures.
Paraquat is rapidly absorbed and distributed to all tissues. Free oxygen radicals are generated and non-specifically damage the lipid membrane of cells, inducing cellular toxicity and death. The extent of dysfunction depends on the concentration of paraquat at the cellular level and the efficiency of protective mechanisms such as intracellular glutathione which is a free radical scavenger. Following an exposure of only 10–20 mL of the 20%w/v solution, these protective mechanisms are overwhelmed leading to multisystem toxicity and death within 24–48 h.
Paraquat displays specific toxicity in the lung and kidney due to active uptake in type II pneumocytes and renal tubular cells. Because paraquat concentrates in these cells they are more affected by oxidative damage than other cells. Therefore, in the event of a smaller exposure where multisystem toxicity does not occur, progressive renal and respiratory failure may occur. Pulmonary effects are characterized by an initial pneumonitis, followed by neutrophil infiltration with ongoing inflammation and progressive pulmonary fibrosis. Progressive renal impairment decreases the excretion of paraquat and prolongs toxicity. Death normally occurs due to pulmonary fibrosis a number of weeks post-ingestion.
The free oxygen radicals generated by paraquat require oxygen, and supplemental inspired oxygen may exacerbate pulmonary toxicity.
Diquat does not concentrate in the pneumocytes as readily as paraquat. Therefore, if the patient survives the acute phase of multiorgan dysfunction, delayed pulmonary fibrosis is less likely to occur.
Clinical features
Severe GI toxicity including vomiting and diarrhoea is the initial manifestation of acute paraquat poisoning. Necrosis of the oral mucosa is often noted about 12 h post-ingestion; it has been reported in patients who drink the paraquat solution without swallowing, despite the brief contact time.
Patients ingesting more than 20 mL are likely to develop severe poisoning with multisystem toxicity. This manifests as pneumonitis, hypotension, hepatitis, acute renal failure and severe diarrhoea. Oesophageal perforation with extensive subcutaneous emphysema may also occur due to the corrosive effects of the formulation. Death within 48 h of ingestion is expected.
Patients ingesting less than 20 mL are less likely to die so quickly. Instead, the clinical course of these exposures is characterized by increasing dyspnoea and hypoxia until death. Death from respiratory failure due to pulmonary fibrosis has been reported a number of months post-ingestion. This is associated with various degrees of hepatic and renal impairment.62
Differential diagnosis
Acute paraquat poisoning may resemble sepsis or poisoning with another cellular poison, such as phosphine, colchicine or iron.
Clinical investigation
A range of investigations have been proposed and tested in patients with acute paraquat poisoning. Because outcomes from paraquat poisoning are so poor, their principal role is to more accurately define prognosis.
Initial investigations aim to confirm significant exposure to paraquat and the easiest method to do this is by the dithionite urine test. This involves the addition of 1 mL of a 1% sodium dithionite solution to 10 mL of urine. A blue colour change indicates paraquat ingestion, the darker the blue, the higher the concentration. If the test is negative on urine passed 6 h after ingestion, a significant exposure is unlikely.
The concentration of paraquat can be quantified in plasma and graphed on a nomogram to determine the chance of survival or death. A number of similar nomograms have been developed.61 It is sometimes difficult to locate laboratories able to measure paraquat concentrations and the turn-around time may be too long for the test to be clinically useful.
Other investigations may be useful to determine the evolution of toxicity in other organ systems. Serial arterial blood gas measurements and chest X-rays demonstrate progression of the pneumonitis and pulmonary fibrosis.
Criteria for diagnosis
A diagnosis of paraquat poisoning is made on the basis of a history of exposure and the clinical symptoms, so a high index of clinical suspicion is required. The urinary dithionite test is a simple and quick method for confirming (or hopefully excluding) paraquat poisoning.
Treatment
Because death is reported following ingestion of as little as 10–20 mL, all exposures should be treated as significant and observed in hospital for 6 h post-ingestion so a dithionate urinary test can be conducted. Patients should be treated symptomatically, including intravenous fluids.
Systemic exposure to paraquat may be decreased by either reducing absorption or increasing clearance. Both Fuller’s earth and activated charcoal have been advocated to decrease absorption. There are no data to suggest which is more effective and neither has been demonstrated to improve outcomes.42,63
Methods for increasing clearance have also been disappointing. While haemoperfusion increases paraquat elimination and reduces lethality in dogs, it is ineffective if commenced more than a few hours post-ingestion due to paraquat’s rapid distribution.64
Treatment with antioxidants (e.g. vitamin C, vitamin E, acetylcysteine) has also been proposed but inadequately studied. There has been much research into the effect of immunosuppression (e.g. cyclophosphamide and corticosteroids). Initial studies were promising but not conclusive,61 and while a subsequent study reported benefit from this treatment,65 it was later criticized because of serious concerns with the study design.66 The role of immunosuppression and antioxidants in the treatment of acute paraquat poisoning requires more research.
Despite these fairly disappointing results it is obvious that, in the absence of treatment, the majority of patients will die. This has led some clinicians to treat patients with a number of these treatments concurrently (e.g. activated charcoal, acetylcysteine, vitamins C and E, immunosuppression and either haemodialysis or haemoperfusion) in the hope that there will be a favourable outcome. The choice of whether to commence this treatment is largely a personal one and requires discussion with the patient and family. Such a treatment regimen is probably reasonable in patients with a faintly positive dithionite urinary test. However, it seems unlikely to be of assistance to patients in whom this test is strongly positive, or those with evolving multi-organ dysfunction. Instead, palliation should be the priority, including oxygen for hypoxia and morphine for dyspnoea and oropharyngeal or abdominal pain.
Prognosis
Much research has focused on the evaluation of markers of prognosis in patients with acute paraquat poisoning. Two predictors of death are well established: the dose ingested or the plasma concentration of paraquat, relative to the time of poisoning.61 These require an accurate history and access to an appropriate laboratory. Direct markers of paraquat-induced organ toxicity that allow earlier prognostication might assist with determining which patients should be treated, palliated or discharged with confidence that harm will not occur.61
Alternative markers of prognosis have also been explored, although few have been validated. These include measurement of the respiratory index, temporal changes in haematological or biochemical measures such as creatinine, and pulmonary surfactants.61
Glyphosate
Glyphosate is a non-selective herbicide that acts by inhibiting the enzymatic synthesis of aromatic amino acids in plants. This target enzyme is not present in humans. Both ready-to-use (~1–5% ) and concentrated (~30–50%) formulations requiring dilution are available.
Glyphosate is absorbed from the GI tract and does not penetrate the skin to a significant extent. Respiratory, ocular and dermal symptoms may occur following occupational use but are usually of minor severity.67 Ingestion is the most significant route of exposure in clinical toxicology.
Mechanism of toxicity
The mechanism of toxicity of glyphosate-containing herbicides in humans has not been adequately described. Experimentally, there appears to be minimal (if any) mammalian toxicity from glyphosate itself.67,69 Toxicity has been largely attributed to surfactant co-formulants. Polyoxyethyleneamine (POEA; tallow amine) is the most common surfactant formulated in these products.
Poisoning is more severe following ingestion of concentrated formulations. This may reflect either the total dose ingested, or direct effects of the highly irritating compounds present in these products. Gastrointestinal corrosion is reported with high doses of the concentrated solutions.70,71
Patients with severe poisoning manifest multisystem effects. This suggests that glyphosate-containing herbicides may be non-specific in their action, or that they interfere with physiological functions that are common to a number of systems. Proposed mechanisms include disruption of cellular membranes and uncoupling of oxidative phosphorylation, although these may be inter-related.72,73
Clinical features
Abdominal pain with nausea, vomiting and/or diarrhoea, are the most common manifestations of acute poisoning. These may be mild and self-resolving, but in severe poisoning there may be inflammation, ulceration or infarction. Severe diarrhoea may also occur and vomiting may be recurrent, leading to dehydration.70-72,74-77
With more severe poisoning, hypotension, renal and hepatic dysfunction, pulmonary oedema or pneumonitis, altered level of consciousness and acidosis are reported. It is not understood which of these clinical features reflect primary or secondary toxic effects of the glyphosate-containing herbicides. These effects may be transient or severe, progressing over 12–72 h to shock and death.71,72,74-77 Some patients who subsequently died demonstrated minimal symptoms on admission.77
Differential diagnosis
The differential diagnoses are wide, including any poisoning or medical condition associated with GI symptomatology and progressive multisystem toxicity.
Clinical investigation
There are no specific clinical investigations to guide management.
Targeted laboratory and radiological investigations should be conducted in patients demonstrating anything more than mild GI symptoms. Serial blood gases may be useful for detection of metabolic disequilibria.
An assay for quantifying glyphosate is not available for clinical use. Since the relationship between blood concentration and clinical outcomes has not been determined, this does not appear to be a useful investigation.
Endoscopic investigations may be useful in assessing for erosions or ulceration, particularly following exposures to the concentrated formulation.70,71,74,75
Criteria for diagnosis
The principal criterion for diagnosis of acute poisoning with a glyphosate-containing herbicide is a history of exposure. Therefore, a high index of suspicion is necessary for diagnosis. A number of clinical criteria for the classification of severity have been suggested, but none have been validated.71,72,74,77
Treatment
All patients presenting with a history of acute ingestion should be observed for a minimum of 6 h. Patients reporting a history of intentional ingestion and GI symptoms should be observed for at least 24 h given that clinical toxicity may progress.
Treatment of acute poisoning with glyphosate-containing herbicides is empiric. All patients should receive prompt resuscitation and supportive care. Oral-activated charcoal may be given if the patient presents within 1–2 h of ingestion,41 although a recent randomized controlled trial did not support its efficacy for pesticides in general.42 Intravenous fluids should be administered to replace GI losses and haemodynamic monitoring is recommended. Biochemical and acid–base abnormalities should be corrected where possible.
No specific antidote has been proposed or tested for the treatment of acute poisoning with glyphosate-containing herbicides. This relates largely to the unknown mechanism of toxicity of these products.
Survival in two patients with severe poisoning was attributed to haemodialysis,78 however other patients have died despite this treatment.74,76,79 Early initiation of haemodialysis may be required to optimize outcomes, although the efficacy of haemodialysis for poison removal has not been determined.
Prognosis
All intentional exposures should be considered significant. Retrospective studies have suggested a correlation between increasing dose and severe poisoning and death.71,75,76
Mortality from acute poisoning with glyphosate-containing herbicides has been estimated at 10.1% (38/377 patients).72 This incidence was derived from pooled data in the literature, mostly from retrospective studies in tertiary centres, so there is the potential for a referral bias. Prospective studies in secondary hospitals suggest that the mortality is lower, around 3.5%.77
Tools for estimating prognosis in acute poisoning with glyphosate-containing herbicides have not been described in detail. Patients developing marked non-specific organ toxicity (e.g. renal failure, pulmonary oedema, sedation, arrhythmias) are more likely to die.76 Patients with more extensive erosions of the upper GI tract developed more severe systemic poisoning and required prolonged admission.70
1 Eddleston M, Phillips MR. Self poisoning with pesticides. British Medical Journal. 2004;328(7430):42-44.
2 Eddleston M, Sudarshan K, Senthilkumaran M, et al. Patterns of hospital transfer for self-poisoned patients in rural Sri Lanka: implications for estimating the incidence of self-poisoning in the developing world. Bulletin of the World Health Organization. 2006;84(4):276-282.
3 Eddleston M, Karalliedde L, Buckley N, et al. Pesticide poisoning in the developing world – a minimum pesticides list. Lancet. 2002;360:1163-1167.
4 Daly FFS, Little M, Murray L. A risk assessment based approach to the management of acute poisoning. Emergency Medical Journal. 2006;23:396-399.
5 Roberts DM, Aaron CK. Management of acute organophosphorus pesticide poisoning. British Medical Journal. 2007;334(7594):629-634.
6 Roberts DM, Fraser JF, Buckley NA, et al. Experiences of anticholinesterase pesticide poisonings in an Australian Tertiary Hospital. Anaesthesia .Intensive Care. 2005;33(4):469-476.
7 Hollis GJ. Organophosphate poisoning versus brainstem stroke. Medical Journal of Australia. 1999;170(12):596-597.
8 Teague B, Peter JV, O’Fathartaigh M, et al. An unusual cause for cardiac arrest. Critical Care Resuscitation. 1999;1(4):362-365.
9 Buckley NA, Karalliedde A, Dawson L. Where is the evidence for treatments used in pesticide poisoning? Is clinical toxicology fiddling while the developing world burns? Journal of Toxicology, Clinical Toxicology. 2004;42(1):113-116.
10 Tominack RL. Herbicide formulations. Journal of Toxicology – Clinical.Toxicology. 2000;8(2):129-135.
11 Eddleston M. Patterns and problems of deliberate self-poisoning in the developing world. Quarterly Journal of Medicine. 2000;93(11):715-731.
12 Roberts DM, Karunarathna A, Buckley NA, et al. Influence of pesticide regulation on acute poisoning deaths in Sri Lanka. Bulletin of the World Health Organization. 2003;81(11):789-798.
13 Roberts D, Senarathna L. Secondary contamination in organophosphate poisoning. Quarterly Journal of Medicine. 2004;97(10):697-698.
14 Little M, Murray L. Consensus statement: risk of nosocomial organophosphate poisoning in emergency departments. Emergency Medicine of Australasia. 2004;16(5–6):456-458.
15 Emerson GM, Gray NM, Jelinek GA, et al. Organophosphate poisoning in Perth, Western Australia, 1987–1996. Journal of Emergency Medicine. 1999;17(2):273-277.
16 Yang CC, Deng JF. Pattern of acute pesticide poisonings in Taiwan. Journal of Toxicology, Clinical Toxicology. 2003;41(4):523.
17 Ameno K, Lee S-K, In S-W, et al. Blood carbofuran concentrations in suicidal ingestion cases. Forensic Science International. 2001;116(1):59-61.
18 Paul N, Mannathukkaran TJ. Intermediate syndrome following carbamate poisoning. Clinical Toxicology. 2005;43(7):867-868.
19 Yang P-Y, Tsao TCY, Lin J-L, et al. Carbofuran-induced delayed neuropathy. Journal of Toxicology – Clinical Toxicology. 2000;38(1):43-46.
20 Eyer P. The role of oximes in the management of organophosphorus pesticide poisoning. Toxicology Review. 2003;22(3):165-190.
21 Namba T. Cholinesterase inhibition by organophosphorus compounds and its clinical effects. Bulletin of World Health Organisation. 1971;44:289-307.
22 Lotti M, Moretto A. Organophosphate-induced delayed polyneuropathy. Toxicology Review. 2005;24(1):37-49.
23 Lotti M. Cholinesterase inhibition: complexities in interpretation. Clinical Chemistry. 1995;41(12):1814-1818.
24 Eddleston M, Eyer P, Worek F, et al. Differences between organophosphorus insecticides in human self-poisoning: a prospective cohort study. Lancet. 2005;366:1452-1459.
25 Karalliedde L, Baker D, Marrs TC. Organophosphate-induced intermediate syndrome: aetiology and relationships with myopathy. Toxicology Review. 2006;25(1):1-14.
26 Eddleston M, Mohamed F, Davies JOJ, et al. Respiratory failure in acute organophosphorus pesticide self-poisoning. Quarterly Journal of Medicine. 2006;99(8):513-522.
27 Worek F, Diepold C, Eyer P. Dimethylphosphoryl-inhibited human cholinesterases: inhibition, reactivation, and aging kinetics. Archives of Toxicology. 1999;73(1):7-14.
28 Venkatesh S, Kavitha ML, Zachariah A. Progression of type I to type II paralysis in acute organophosphorus poisoning: is oxidative stress significant? Archives of Toxicology. 2006;80(6):354-361.
29 Petroianu G, Ruefer R. Poisoning with organophosphorous compounds. Emergency Medicine (Fremantle). 2001;13:258-260.
30 Carlock LL, Chen WL, Gordon EB, et al. Regulating and assessing risks of cholinesterase-inhibiting pesticides: divergent approaches and interpretations. Journal of Toxicology, Environmental and Health B Critical Review. 1999;2(2):105-160.
31 Cochran RC, Kishiyama J, Aldous C. Chlorpyrifos: Hazard assessment based on a review of the effects of short-term and long-term exposure in animals and humans. Food Chemical Toxicology. 1995;33(2):165-172.
32 Nolan RJ, Rick DL, Freshour NL. Chlorpyrifos: pharmacokinetics in human volunteers. Toxicology and Applied Pharmacology. 1984;73:8-15.
33 Solecki R, Davies L, Dellarco V, et al. Guidance on setting of acute reference dose (ARFD) for pesticides. Food Chemical Toxicology. 2005;43(11):1569-1593.
34 Nutenko I, Taitelman U. Affinity of organophosphate insecticides to acetylcholinesterase compared to plasma cholinesterase [abstract]. Journal of Toxicology – Clinical Toxicology. 1999;37(5):662.
35 Thiermann H, Szinicz L, Eyer P, et al. Correlation between red blood cell acetylcholinesterase activity and neuromuscular transmission in organophosphate poisoning. Chemico-Biological Interactions. 2005;157–158:345-347.
36 Dawson A, Buckley N, Whyte I. What target pralidoxime concentration? Journal of Toxicology – Clinical Toxicology. 1997;35(2):227-228.
37 Bardin PG, van Eeden SF, Moolman JA, et al. Organophosphate and carbamate poisoning. Archives of Internal Medicine. 1994;154(13):1433-1441.
38 Amitai G, Moorad D, Adani R, Doctor BP. Inhibition of acetylcholinesterase and butyrylcholinesterase by chlorpyrifos-oxon. Biochemical Pharmacology. 1998;56(3):293-299.
39 Sener EB, Ustun E, Kocamanoglu S, Tur A. Prolonged apnea following succinylcholine administration in undiagnosed acute organophosphate poisoning. Acta Anaesthesiology Scandinavica. 2002;46(8):1046-1048.
40 Eyer F, Meischner V, Kiderlen D, et al. Human parathion poisoning: a toxicokinetic analysis. Toxicological Review. 2003;22(3):143-163.
41 Chyka PA, Seger D, Krenzelok EP, Vale JA. American Academy of Clinical Toxicology, European Association of Poisons Centres and Clinical Toxicologists. Position paper: single-dose activated charcoal. Clinical Toxicology. 2005;43(2):61-87.
42 Eddleston M, Juszczak E, Buckley NA, et al. Randomised controlled trial multiple dose activated charcoal in acute self-poisoning. Lancet. 2008;371:579-587.
43 Eddleston M, Dawson A, Karalliedde L, et al. Early management after self-poisoning with an organophosphorus or carbamate pesticide – a treatment protocol for junior doctors. Critical Care. 2004;8:R391-R397.
44 Sungur M, Güven M. Intensive care management of organophosphate insecticide poisoning. Critical Care. 2001;5(4):211-215.
45 Eddleston M, Singh S, Buckley N. Organophosphorus poisoning (acute). Clinical Evidence. 2005;13:1744-1755..
46 Buckley NA, Eddleston M, Szinicz L. Oximes for acute organophosphate pesticide poisoning. The Cochrane Database Syst Rev. 25(1), 2005. CD005085
47 Peter JV, Moran JL, Graham P. Oxime therapy and outcomes in human organophosphate poisoning: an evaluation using meta-analytic techniques. Critical Care Medicine. 2006;34(2):502-510.
48 Rahimi R, Nikfar S, Abdollahi M. Increased morbidity and mortality in acute human organophosphate-poisoned patients treated by oximes: a meta-analysis of clinical trials. Human & Experimental Toxicology. 2006;25(3):157-162.
49 Pawar KS, Bhoite RR, Pillay CP, et al. Continuous pralidoxime infusion versus repeated bolus injection to treat organophosphorus pesticide poisoning: a randomised controlled trial. Lancet. 2006;368:2136-2141.
50 Dawson RM, Poretski M. Carbamylated acetylcholinesterase: acceleration of decarbamylation by bispyridinium oximes. Biochemical Pharmacology. 1985;34(24):4337-4340.
51 Harris LW, Talbot BG, Lennox WJ, Anderson DR. The relationship between oxime-induced reactivation of carbamylated acetylcholinesterase and antidotal efficacy against carbamate intoxication. Toxicology and Applied Pharmacology. 1989;98(1):128-133.
52 Dawson RM. Oxime effects on the rate constants of carbamylation and decarbamylation of acetylcholinesterase for pyridostigmine, physostigmine and insecticidal carbamates. Neurochemistry International. 1995;26(6):643-654.
53 Lifshitz M, Rotenberg M, Sofer S, et al. Carbamate poisoning and oxime treatment in children: a clinical and laboratory study. Pediatrics. 1994;93(4):652-655.
54 Karalliedde L, Feldman S, Henry J, Marrs T, editors. Organophosphates and health. London: Imperial College Press, 2001.
55 Costa LG, Cole TB, Furlong CE. Polymorphisms of paraoxonase (PON1) and their significance in clinical toxicology of organophosphates. Journal of Toxicology – Clinical Toxicology. 2003;41(1):37-45.
56 Allebrandt KV, Souza RL, Chautard-Freire-Maia EA. Variability of the paraoxonase gene (PON1) in Euro- and Afro-Brazilians. Toxicology and Applied Pharmacology. 2002;180(3):151-156.
57 Gunnell D, Ho D, Murray V. Medical management of deliberate drug overdose: a neglected area for suicide prevention? Journal of Emergency Medicine. 2004;21(1):35-38.
58 Bardin PG, van Eeden SF. Organophosphate poisoning: grading the severity and comparing treatment between atropine and glycopyrrolate. Critical Care Medicine. 1990;18(9):956-960.
59 Senanayake N, de Silva HJ, Karalliedde L. A scale to assess severity in organophosphorus intoxication: POP scale. Human & Experimental Toxicology. 1993;12:297-299.
60 Lee P, Tai DYH. Clinical features of patients with acute organophosphate poisoning requiring intensive care. Intensive Care Medicine. 2001;27:694-699.
61 Eddleston M, Wilks MF, Buckley NA. Prospects for treatment of paraquat-induced lung fibrosis with immunosuppressive drugs and the need for better prediction of outcome: a systematic review. Quarterly Journal of Medicine. 2003;96(11):809-824.
62 Bismuth C, Garnier R, Baud FJ, et al. Paraquat poisoning. An overview of the current status. Drug Safety. 1990;5(4):243-251.
63 Fountain JS. Gastrointestinal decontamination following paraquat ingestion. New Zealand Medical Journal. 2000;113(1118):406-407.
64 Pond SM, Rivory LP, Hampson EC, Roberts MS. Kinetics of toxic doses of paraquat and the effects of hemoperfusion in the dog. Journal of Toxicology – Clinical Toxicology. 1993;31(2):229-246.
65 Lin JL, Lin-Tan DT, Chen KH, Huang WH. Repeated pulse of methylprednisolone and cyclophosphamide with continuous dexamethasone therapy for patients with severe paraquat poisoning. Critical Care Medicine. 2006;34(2):368-373.
66 Gunawardena G, Roberts DM, Buckley NA. Randomized control trial of immunosuppression in paraquat poisoning. Critical Care Medicine. 2007;35(1):330-331.
67 Goldstein DA, Acquavella JF, Mannion RM, Farmer DR. An analysis of glyphosate data from the California Environmental Protection Agency Pesticide Illness Surveillance Program. Journal of Toxicology – Clinical Toxicology. 2002;40(7):885-892.
68 Williams GM, Kroes R, Munro IC. Safety evaluation and risk assessment of the herbicide Roundup and its active ingredient, glyphosate, for humans. Regulatory Toxicology and Pharmacology. 2000;31(2 Pt 1):117-165.
69 IPCS. Environmental health criteria 159 Glyphosate. Geneva: World Health Organization, 1994.
70 Chang C-Y, Peng Y-C, Hung D-Z, et al. Clinical impact of upper gastrointestinal tract injuries in glyphosate-surfactant oral intoxication. Human and Experimental Toxicology. 1999;18(8):475-478.
71 Tominack RL, Yang GY, Tsai WJ, et al. Taiwan National Poison Center survey of glyphosate-surfactant herbicide ingestions. Journal of Toxicology – Clinical Toxicology. 1991;29(1):91-109.
72 Bradberry SM, Proudfoot AT, Vale JA. Glyphosate poisoning. Toxicological Review. 2004;23(3):159-167.
73 Peixoto F. Comparative effects of the Roundup and glyphosate on mitochondrial oxidative phosphorylation. Chemosphere. 2005;61(8):1115-1122.
74 Talbot AR, Shiaw MH, Huang JS, et al. Acute poisoning with a glyphosate-surfactant herbicide (‘Roundup’): a review of 93 cases. Human and Experimental Toxicology. 1991;10(1):1-8.
75 Sawada Y, Nagai Y, Ueyama M, Yamamoto I. Probable toxicity of surface-active agent in commercial herbicide containing glyphosate. Lancet. 1988;1(8580):299.
76 Lee H-L, Chen K-W, Chi C-H, et al. Clinical presentations and prognostic factors of a glyphosate-surfactant herbicide intoxication: a review of 131 cases. Academic Emergency Medicine. 2000;7(8):906-910.
77 Roberts DM, Buckley NA. Acute intentional self-poisoning with glyphosate-containing herbicides. Clinical Toxicology. 2006;44(4):414.
78 Moon JM, Min YI, Chun BJ. Can early hemodialysis affect the outcome of the ingestion of glyphosate herbicide? Clinical Toxicology. 2006;44(3):329-332.
79 Stella J, Ryan M. Glyphosate herbicide formulation: a potentially lethal ingestion. Emergency Medicine Australasia. 2004;16(3):235-239.
80 Güven M, Sungur M, Eser B, et al. The effects of fresh frozen plasma on cholinesterase levels and outcomes in patients with organophosphate poisoning. Journal of Toxicology – Clinical Toxicology. 2004;42(5):617-623.
81 Li Y, Yu X, Wang Z, et al. Gastric lavage in acute organophosphorus pesticide poisoning (GLAOP) – a randomised controlled trial of multiple vs. single gastric lavage in unselected acute organophosphorus pesticide poisoning. BMC Emergency Medicine. 2006;6:10.
82 Roberts DM, Ai P, Kaiyuan Z, Buckley NA. Extracorporeal blood purification for acute organophosphorus pesticide poisoning. Journal of Intensive Care Medicine. 2007;22(2):124-126.
83 Pajoumand A, Shadnia S, Rezaie A, et al. Benefits of magnesium sulfate in the management of acute human poisoning by organophosphorus insecticides. Human and Experimental Toxicology. 2004;23(12):565-569.
84 Roberts D, Buckley NA. Alkalinisation for organophosphorus pesticide poisoning. Cochrane Database Systemic Review. (1):2005. CD004897
85 Fengsheng H, Haibing X, Fukuang Q. Intermediate myasthenia syndrome following acute organophosphate poisoning – an analysis of 21 cases. Human & Experimental Toxicology. 1998;17:40-45.
86 Samuel J, Thomas K, Jeyaseelan L, et al. Incidence of intermediate syndrome in organophosphorous poisoning. Journal of Association of .Physicians of India. 1995;43(5):321-323.
29.18 Ethanol and other alcohols
Introduction
Alcohols are hydrocarbons that contain a hydroxyl (OH) group. Ethanol, a two-carbon primary alcohol, is the most commonly used recreational drug in Australasia and elsewhere in the Western world. Ethanol misuse is a major cause of mortality and morbidity both directly and indirectly and EDs deal with the results on a daily basis. It was estimated that in 1997, 3290 Australians died from injury due to high risk drinking and there were 72 302 hospitalizations.1 In excess of 30% of all ED presentations are deemed to be ethanol related.
The complications of chronic alcohol consumption contribute to the development of a number of medical and surgical emergencies many of which are dealt with elsewhere in this text. This chapter confines its discussion to acute ethanol intoxication, ethanol withdrawal and two other important ethanol specific emergency presentations – Wernicke’s encephalopathy and alcoholic ketoacidosis.
A number of other alcohols, although far less frequently implicated in ED presentation than ethanol, are metabolized to form toxic organic acids and produce life-threatening clinical syndromes. These alcohols include methanol and ethylene glycol and are termed ‘toxic alcohols’. Early recognition and intervention can prevent significant morbidity and mortality.
Ethanol
Pharmacology
Ethanol is a small molecule that is rapidly and almost completely absorbed from the stomach and small intestine. Ethanol is both water and lipid soluble and rapidly crosses lipid membranes to distribute uniformly throughout the total body water. Ethanol is principally eliminated by hepatic metabolism with smaller amounts (5–10%) excreted unchanged by the kidneys, lungs and in sweat.
Ethanol is oxidized by cytosolic and microsomal cytochrome P450 (2EI and 1A2) alcohol dehydrogenases (ADH) to acetaldehyde, which in turn is metabolized by aldehyde dehydrogenase to acetate. Acetate is converted to acetyl-CoA and enters the Krebs cycle to be finally metabolized to carbon dioxide and water. Entry of acetyl-CoA in the Krebs cycle is dependent on adequate thiamine stores.2 Importantly, the ADH system is saturated at relatively low blood ethanol concentrations, which results in blood ethanol elimination moving from first-order to zero-order kinetics. The rate of ethanol metabolism in non-tolerant adults is approximately 10 g/h and blood ethanol levels fall by about 0.02 g/dL/h.3 An alternative pathway for ethanol metabolism is via the microsomal ethanol oxidizing system, the activity of which increased in response to chronic alcohol exposure. Metabolism by this route is relatively important at very high blood ethanol concentrations and in chronic alcoholics.
The mechanism of action of ethanol is poorly understood. However, ethanol acts as a central nervous system (CNS) depressant, at least partially by enhancing the effect of GABA at GABA-A receptors. Tolerance to the CNS depressant effect develops with chronic exposure.
Clinical presentation
Acute ethanol intoxication
The clinical features associated with acute ethanol intoxication predominantly relate to the CNS and progress with increasing blood alcohol level, although there is remarkable inter-individual variation, most commonly as a function of tolerance. Initial features include a sense of wellbeing, increased self-confidence and disinhibition. With increasing blood concentrations, impaired judgement, impaired coordination and emotional lability develop. At very high concentrations, ethanol can cause coma, respiratory depression, loss of airway protective reflexes and even death.
Presentation to the ED is usually as a result of the social and behavioural consequences of the alteration in higher CNS functions. Ethanol is frequently implicated in trauma, drowning, violence, self-harm, domestic and sexual abuse, and other acute social and psychiatric emergencies. Ethanol is a common co-ingestant in deliberate self poisoning. Less commonly the emergency presentation is a direct result of the CNS depressant effects of ethanol.
Many other important medical and surgical conditions that cause altered mental status may be incorrectly ascribed to ethanol intoxication or coexist with ethanol intoxication. Table 29.18.1 lists an (incomplete) differential diagnosis.
Table 29.18.1 Differential diagnosis of acute ethanol intoxication
| Encephalopathy |
| Head injury |
| Hypo-/hyperthermia |
| Intracranial infarction or haemorrhage |
| Metabolic |
| Overdose or other toxin |
| Post-ictal state |
| Psychosis |
| Sepsis |
In the absence of a clear history, the diagnosis of ethanol intoxication is only confirmed upon determination of a breath or blood ethanol concentration. Because ethanol consumption is so ubiquitous, a positive reading does not exclude coexisting pathology.
Ethanol withdrawal syndrome
A withdrawal syndrome usually develops within 6–24 h of cessation or reduction in ethanol consumption in dependent individuals.4 Symptoms can begin any time after the blood ethanol concentration begins to fall and blood ethanol is frequently still measurable in withdrawing patients. The duration of the syndrome may be from 2 to 7 days. Although the pathophysiology is not well understood, the syndrome presents as unopposed sympathetic and CNS stimulation. It is associated with a mortality of 5% and early clinical recognition of this syndrome is important.4
Patients may present to the ED already in withdrawal after deliberately abstaining from alcohol or after stopping drinking due to intercurrent illness or lack of funds to buy alcohol. Alternatively, ethanol-dependent patients may begin to withdraw whilst being treated in the ED, particularly where their stay is prolonged.
Clinical features of mild ethanol withdrawal are those of mild autonomic hyperactivity and include nausea, anorexia, coarse tremor, tachycardia, hypertension, hyper-reflexia, insomnia and anxiety.5 In more severe cases, the patient goes on to develop more pronounced anxiety, insomnia, irritability, tremor, tachycardia, hyper-reflexia, hypertension, fever, visual hallucinations, decreased seizure threshold and finally delirium. Symptoms usually peak by 50 h.6 Delirium tremens represents the extreme end of the spectrum of ethanol withdrawal. It is an uncommon but potentially lethal complication.
Wernicke’s encephalopathy
This is an acute neuropsychiatric syndrome that develops in certain alcohol-dependent individuals as a result of thiamine deficiency. It is a spectrum disorder that is classically described as a triad of:
In up to 20% of cases, the signs and symptoms of the classic triad are not evident at presentation. Less common presentations include stupor, hypothermia, cardiovascular instability, seizures, visual disturbances, hallucinations and alterations in behaviour. In extremis the condition may present with hyperthermia, hypertonia, spastic paresis, dyskinesias and coma.8
Wernicke’s encephalopathy is a clinical diagnosis and constitutes a medical emergency with significant morbidity and a mortality of 10–20 % if left untreated. For this reason, the emergency physician must maintain high index of suspicion in patients with long-term heavy ethanol intake.
Alcoholic ketoacidosis
Alcoholic ketoacidosis (AKA), also termed alcoholic acidosis, is an often unrecognized potentially life-threatening medical condition that develops in the alcoholic patient in response to starvation. The normal response to starvation is increased gluconeogenesis from pyruvate. In the alcoholic patient, pyruvate is preferentially converted to lactate. In response, fatty-acid metabolism is increased as an alternative source of energy, resulting in the production of acetyl-CoA and acetoacetate, which in turn is reduced to β-hydroxybutyrate (BOHB), producing the ketoacidotic state.
Patients with AKA usually present with a history of prolonged heavy alcohol misuse preceding a bout of particularly excessive intake, which has been terminated several days earlier by nausea, severe vomiting and abdominal pain.9-11 There may be a history of previous episodes requiring brief admissions with labels of ‘query pancreatitis’ or ’alcoholic gastritis.’12 Examination usually reveals tachypnoea, tachycardia, hypotension and diffuse epigastric tenderness on palpation. In contrast to patients with diabetic ketoacidosis, mental status is usually normal. The presence of an altered mental state should prompt consideration of other causes especially hypoglycaemia and acute ethanol intoxication.
Toxic alcohol poisoning is an important differential diagnosis. Toxic alcohol acidosis does not produce ketosis and in contrast does cause significant alteration to conscious state, visual symptoms (methanol) and renal failure/crystalluria (ethylene glycol).12
Clinical investigation
The excretion of ethanol by the lungs, although relatively unimportant in terms of ethanol elimination, obeys Henry’s law, i.e. the ratio between the concentration of ethanol in the alveolar air and blood is constant. This allows breath sampling of ethanol to reliably estimate blood ethanol concentration.
Most non-tolerant adults would be expected to develop some impairment of higher functions at blood ethanol concentrations in the range of 0.025–0.05 mg/dL (5–11 mmol/L) and to develop significant CNS depression in the range of 0.25–0.4 mg/L (55–88 mmol/L).
In a patient presenting with acute intoxication, no investigations may be necessary; however, blood or breath ethanol levels (BAL) are frequently useful to confirm the diagnosis. A BAL of zero is highly significant in a patient with an altered level of consciousness, as ethanol intoxication is excluded, and other diagnoses need to be considered. A positive blood ethanol level does not exclude alternative diagnoses.
Other investigations should be performed as clinically indicated in an effort to exclude coexisting pathologies and alternative diagnoses as detailed above.
In the patient with AKA, bedside investigations reveal a low/normal glucose, low or absent breath ethanol and urinary ketones (these may be low or absent due to the inability of bedside assays to detect all ketone moieties, especially BOHB). Laboratory investigation will reveal an anion gap (AG) acidosis (this may be severe with AG > 30) and mild hyperlactaemia insufficient to account for the AG.12
Treatment
Acute ethanol intoxication
Severe ethanol intoxication with CNS depression is life-threatening but a good outcome is assured by timely institution of supportive care. In particular, attention may need to be given to the airway and ventilation. Hypotension generally responds to intravenous crystalloid infusion. The blood sugar level must be checked and normoglycaemia maintained. Intravenous thiamine should be administered, particularly to those with chronic ethanol abuse. There is no specific antidote to ethanol intoxication.
Less severe ethanol intoxication presents a management challenge to the emergency physician when it results in a combative or violent patient threatening harm to self or staff, or threatening to discharge against medical advice. Such patients frequently require chemical sedation with titrated doses of intravenous benzodiazepines or butyrophenones in order to facilitate assessment and observation, ensure safety for patient and staff and prevent unsafe discharge.
Ethanol withdrawal
The key to management of this condition is early recognition and institution of adequate dosing of benzodiazepines. Very large doses of benzodiazepines may be required to control symptoms. The risk and likely severity of ethanol withdrawal can usually be anticipated if an accurate history of alcohol intake and previous withdrawals is obtained. Coexisting conditions should be managed on their own merits. It is important to exclude hypoglycaemia and correct if present. Thiamine 100 mg (preferably intravenously) should be immediately given to any chronic alcoholic patient who presents with or develops an altered mental status.
The management of ethanol withdrawal in the ED or observation ward is greatly facilitated by the use of ethanol withdrawal charts. These charts facilitate recognition of the first signs of ethanol withdrawal and timely administration of benzodiazepines in adequate doses. An example of such a chart is shown in Figure 29.18.1. Benzodiazepine, usually diazepam, administration is titrated to the clinical features of withdrawal. The total dose required to manage withdrawal is highly variable. Benzodiazepines are usually given orally but can be administered intravenously to the uncooperative or severely withdrawing patient. With extreme withdrawal, refractory to benzodiazepines, small aliquots of ethanol may need to be prescribed.
Wernicke’s encephalopathy
As Wernicke’s encephalopathy is a clinical diagnosis with high mortality if untreated, any known or suspected alcoholic patient who presents with altered mental status should receive thiamine 100 mg i.v. during the initial assessment. Recommended thiamine dosing in patients with suspected Wernicke’s is more aggressive with 500 mg i.v. (over 30 min) three times per day for 2–3 days reducing to 250 mg once daily (i.v. or i.m.) for 3–5 days if a response is observed.8 Parenteral administration is vital as thiamine is poorly absorbed orally. If dextrose administration is required, it must follow thiamine replacement as it may acutely worsen the neurological status of the thiamine-deficient patient. Magnesium is a co-factor for thiamine-dependent transketolase and so any magnesium deficiency should be corrected.13
Alcoholic ketoacidosis
Initial resuscitation should include administration of adequate volumes of crystalloids to treat hypovolaemia followed by thiamine and infusion of dextrose containing fluids. Potassium and magnesium supplementation should be given according to serum electrolyte results. Administration of dextrose, usually an infusion of 5% dextrose, is essential as it stimulates insulin release, inhibits glucagon release and so inhibits fatty acid oxidation. Thiamine facilitates entry of pyruvate into the Krebs cycle. Administration of insulin or bicarbonate is not necessary.14
Fluid, electrolyte and acid–base status should be closely monitored and further therapy tailored to the clinical response. Careful evaluation and treatment of the coexisting medical disorders is essential.
Disposition
The disposition of many ethanol-intoxicated patients presenting to the ED is determined by the associated medical, surgical, psychiatric or social issues. Ethanol-intoxicated patients should only be discharged from the ED when their subsequent safety can be ensured. Discharge into the care of a competent relative or friend is sometimes appropriate. Other patients, particularly if aggressive or neurologically impaired, require admission to a safe environment until such time as the intoxication resolves and they can be reassessed. An observation ward attached to the ED may be the most appropriate place if available. More severely intoxicated patients requiring airway control and support of ventilation should be admitted to the intensive care unit.
Patients in ethanol withdrawal may require admission for management of the precipitating medical or surgical illness. For those patients who wish to complete withdrawal with a view to abstinence, the remainder of the withdrawal may be managed in a general medical ward, specialized medical or non-medical detoxification centre, or at home. Medical detoxification is mandatory where a severe withdrawal syndrome is anticipated. In any case, ongoing psychosocial support will be required and it is important for EDs to have a good knowledge of the locally available drug and alcohol services to ensure appropriate referral.
Patients with Wernicke’s encephalopathy should be admitted for ongoing care and thiamine and magnesium supplementation. The ophthalmoplegia and nystagmus usually have a good response to thiamine within hours to days. Ataxia and mental changes improve more slowly if at all and have a poorer prognosis. Up to 50% of cases will show no response despite thiamine therapy.13
Patients with ethanol-induced ketoacidosis also require admission for ongoing dextrose and thiamine, monitoring of fluids and electrolytes and management of the precipitating medical condition. Mortality from ethanol-induced ketoacidosis per se is rare with early recognition and treatment, but death may occur as a result of the underlying medical condition, particularly if unrecognized.
Ideally, any patient with an ethanol-related presentation should be offered referral to drug and alcohol rehabilitation services for counselling.
Toxic Alcohols
Epidemiology
Both methanol and ethylene glycol poisoning are extremely rare in Australasia. This is primarily due to their limited availability.
Methanol is found in model aeroplane fuel and laboratory solvents. There is no methanol in ‘methylated spirits’ sold in Australia (this is in fact pure ethanol with bittering agents to minimize palatability). Methanol is more freely available in other countries where it is found in household cleaning agents and windshield de-icer. Mass poisoning incidents are reported following incorrect distillation of ethanol.
Ethylene glycol is most commonly encountered as a constituent of radiator antifreeze or coolant. It is also found in hydraulic fluids and solvent preparations. Significant poisoning in Australasia almost always occurs following deliberate ingestion.
Toxicology
Methanol and ethylene glycol are both small molecules that are rapidly absorbed from the gastrointestinal (GI) tract with a volume of distribution that approximates total body water (0.6 L/kg). Toxic alcohols are oxidized initially by hepatic cytosolic and microsomal alcohol dehydrogenases (ADH) and then further metabolized by aldehyde dehydrogenase into acidic moieties. Methanol is metabolized initially to form formaldehyde and then to formic acid. Ethylene glycol is metabolized to glycoaldehyde and then to glycolate, glyoxylate and oxylate. The plasma half-lives of the toxic alcohols are appreciably increased in the presence of ethanol because ethanol has a much higher affinity for ADH: four times that of methanol and eight times that of ethylene glycol. As a result, the presence of ethanol greatly delays the onset of clinical and biochemical features of toxicity.
Methanol toxicity is mediated through the formation of formic acid. Formic acid binds to cytochrome oxidase resulting in impairment of cellular respiration. Its half-life is prolonged (up to 20 h) and its metabolism is dependent on the presence of tetrahydrofolate. The presence of systemic acidosis enhances the movement of formic acid intracellularly. The initial acidosis is secondary to formic acid; however, as cellular respiration is disturbed and toxicity progresses a concurrent lactic acidosis is usually evident.15 Accumulation of formic acid manifests as increasing AG acidosis, gastrointestinal and neurological toxicities.
Ethylene glycol itself is a direct irritant to the GI tract and has CNS depressant effects similar to those of ethanol. The major toxicity is mediated through the acid metabolites, glycolate and oxylate.16 Oxalate complexes with calcium, leading to crystal deposition chiefly in the renal tubules and the CNS. Myocardium and lungs can also be affected. In addition, these acids appear to be inherently toxic.16 Complexing with calcium produces systemic hypocalcaemia and may manifest with prolongation of the QT interval. A profound AG acidosis develops and is principally attributed to glycolic acid accumulation although a concurrent lactic acidosis (type B) also contributes.
Toxic doses
The lethal dose of methanol is conservatively estimated as 0.5–1.0 mL/kg of a 100% solution.17 Clinical toxicity and visual sequelae may be seen with smaller doses, perhaps as little as 0.25 mL/kg.
The lethal dose of ethylene glycol is thought to be in the order of 1.0 mL/kg of a 100% solution.16
Clinical features
Methanol
Initially mild CNS depression typical of ethanol intoxication is evident. A latent period (6–24 h) is classically observed during which time the patient may appear asymptomatic. Progressive ophthalmic, GI and CNS symptoms may then develop. Hyperpnoea is usually observed secondary to the metabolic acidosis. Progressive obtundation leading to coma and seizures heralds the onset of cerebral oedema and signifies poorer prognosis.18 Those who recover from serious CNS toxicity can display extrapyramidal movement disorders.19 Retinal toxicity may be irreversible in up to a third of cases.18
Ethylene glycol
The progression of clinical features following ingestion of ethylene glycol is described in three stages: neurological, cardiopulmonary and renal. These stages are artificial and toxicity may progress in a rapid manner with concurrent toxicities being observed. Initially an intoxication syndrome analogous to ethanol occurs along with nausea and vomiting due to mucosal irritation. A progressively severe AG acidosis with renal failure and hypocalcaemia is characteristic. Crystalluria may be observed. With severe poisoning, renal failure progresses rapidly. Central nervous system depression is observed with severe manifestations including seizures, coma and cerebral oedema. Hyperpnoea occurs secondary to the metabolic acidosis.
Clinical investigation
Direct assay of methanol or ethylene glycol concentrations in serum is rarely readily available. In the absence of direct assays, the ability to exclude a potentially lethal toxic alcohol ingestion at presentation is limited. The combination of an osmolar gap (OG) and a wide AG acidosis is highly suggestive of either methanol or ethylene glycol intoxication. However a normal OG does not exclude toxic alcohol ingestion. In the presence of a profound acidotic state it is possible that a toxic alcohol has been largely metabolized and thus no longer sufficiently present to raise the OG. Additionally, baseline OGs may vary from −14 to +10 between individuals and so a ‘normal’ OG may mask a large occult increase representing a potentially lethal ingestion.19 Similarly a normal AG at presentation is not sufficient to exclude toxic alcohol ingestion. Early in the clinical course an AG may be normal, only to develop rapidly as metabolism progresses. This is particularly so in the presence of ethanol where the onset of an AG acidosis will be delayed until the ethanol itself has been preferentially metabolized.
Falls in serum bicarbonate and arterial pH correlate well with levels of toxic organic acid metabolites in the circulation and in the absence of direct assays are their chief surrogate markers.16,20 In this context, it is common practice to exclude toxic ingestion where there is a normal venous bicarbonate (>20) 8 h after the serum or breath ethanol has been documented as undetectable.21,22
When available in a clinically useful timeframe direct assays may shorten hospital assessment times especially with accidental exposures. The interpretation of serum methanol and ethylene glycol concentrations requires consideration of time since ingestion, ethanol co-ingestion and acid–base status.
Treatment
The definitive care for methanol and ethylene glycol ingestions is dialysis with concurrent ADH blockade therapy. All cases of deliberate self poisonings with a toxic alcohol need to be managed in a facility with easy access to dialysis if clinical intoxication becomes apparent. ADH blockade therapy can impede the progression of clinical toxicity and permit safe transfer to an appropriate facility.
Alcohol dehydrogenase blockade
Blockade of ADH can be achieved by the administration of either ethanol or the specific ADH antagonist fomepizole (not currently available in Australasia). These agents prevent metabolism of toxic alcohols and the accumulation of their organic acid metabolites. ADH blockade significantly increases the half-life of parent toxic alcohols and in Australasia does not represent definitive care. However, fomepizole can be used in isolation to treat toxic alcohol ingestions where sufficient supplies exist for lengthy therapy and serial levels can be obtained and tracked into safe ranges.16
Ethanol therapy can be initiated with a loading dose of 8 mL/kg of 10% ethanol intravenously or 1.8 mL/kg of 43% ethanol orally (equivalent to 3 × 40 mL shots of vodka in a 70 kg adult). Maintenance therapy requires an infusion of 1–2 mL/h of 10% ethanol or 0.2–0.4 mL/h of 43% ethanol orally (equivalent to one 40 mL shot of vodka each hour in a 70 kg adult). The ethanol concentration should be maintained in the range of 100–150 mg/dL (22–33 mmol/L) by careful titration of maintenance administration guided by frequent blood ethanol concentrations.
Haemodialysis
Haemodialysis represents definitive care for confirmed toxic alcohol ingestions. It effectively removes parent toxic alcohols and their acidic metabolites. Lactate free and bicarbonate buffered dialysates may assist the correction of acidaemia. Commonly accepted indications for haemodialysis are listed in Table 29.18.2. Endpoints for haemodialysis are listed in Table 29.18.3. Ethanol is also rapidly cleared by dialysis and ethanol infusion rates need to be increased (usually doubled) during haemodialysis.
Table 29.18.2 Indications for haemodialysis in toxic alcohol poisoning15,16
| Severe metabolic acidosis (pH<7.25) |
| Renal failure (ethylene glycol) |
| History of a large toxic alcohol ingestion and osmolar gap >10 mmol/L |
| Visual symptoms (methanol) |
| Ethylene glycol or methanol levels >50 mg/dL (if available) |
Table 29.18.3 Endpoints for haemodialysis in toxic alcohol poisoning15,16
| Correction of acidosis |
| Osmolar gap <10 mmol/L |
| Ethylene glycol or methanol level <20 mg/dL (if available) |
Supportive care and co-factor therapy
Folinic or folic acid administration is recommended in methanol poisoning (folinic acid 2 mg/kg i.v. qid) to aid in endogenous metabolism of formic acid.16 Pyridoxine and thiamine supplementation is recommended in ethylene glycol poisoning when the patient is thought to be deplete (e.g. alcoholics), again to aid endogenous metabolism of the pathogenic acids.
In methanol poisoning, systemic acidaemia enhances the movement of formic acid into the intracellular compartment. Correction with intravenous bicarbonate if pH < 7.3 is recommended.16
Calcium replacement in ethylene glycol poisoning is contentious given that it may promote calcium oxalate crystal formation. Consequentially, calcium should only be replaced if there is symptomatic hypocalcaemia (including prolongation of the QT interval) or intractable seizures.
Prognosis
Prompt ADH blockade therapy and dialysis ensures an excellent outcome in toxic ingestions who present before the development of established end-organ toxicity. Delayed diagnosis and treatment is associated with death and permanent neurological and renal sequelae, including blindness in the case of methanol poisoning.
1 Alcohol in Australia: Issues and Strategies. A background paper to the National Alcohol Strategy: A plan for Action 2001 to 2004. Commonwealth of Australia, 2001.
2 Abdulla A, Badawy B. The metabolism of alcohol. Clinical and Endocrinological Metabolism. 1978;7:247-252.
3 Brennan DF, Bertzelos S, Reed R, et al. Ethanol elimination rates in an ED population. American Journal of Emergency Medicine. 1995;13:276-280.
4 Adinoff B, Bone GH, Linnoila M. Acute ethanol poisoning and the ethanol withdrawal syndrome. Medical Toxicology. 1988;3:172-196.
5 Turner RC, Lichstein PR, Peden JG, et al. Alcohol withdrawal syndromes: a review of pathophysiology, clinical presentation and treatment. Journal of General Internal Medicine. 1989;4:432-444.
6 Berk WA, Todd K. Relationship of abstinence to the presentation and peak intensity of signs and alcohol withdrawal. Annals of Emergency Medicine. 1993;22:339-345.
7 Reuler JB, Girard DE, Cooney TG. Current concepts. Wernicke’s encephalopathy. New England Journal of Medicine. 1985;312:1035-1039.
8 Sechi G, Serra A. Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and management. Lancet Neurology. 2007;6:442-455.
9 Levy LJ, Duga J, Girgis M, et al. Ketoacidosis associated with alcoholism in non-diabetic subjects. Annals of Internal Medicine. 1973;78:213-219.
10 Cooperman MT, Davidoff F, Spark R, et al. Clinical studies of alcoholic ketoacidosis. Diabetes. 1974;23:433-439.
11 Fulop K, Hoberman HD. Alcoholic ketosis. Diabetes. 1975;24:785-790.
12 McGuire LC, Cruickshank AM, Munro PT. Alcoholic ketoacidosis. Emergency Medicine Journal. 2006;23:417-420.
13 Zuburan C, Fernandes JG, Rodnight R, et al. Wernicke–Korsakoff syndrome. Postgraduate Medical Journal. 1997;73:27.
14 Fulop M. Alcoholic ketoacidosis. Endocrinological and Metabolic Clinics of North America. 1993;22:209-219.
15 Barceloux DG, Bond GR, Krenzelok EP, et al. American Academy of Clinical Toxicology Ad Hoc Committee on the Treatment Guidelines for Methanol Poisoning. American Academy of Clinical Toxicology practice guidelines on the treatment of methanol poisoning. Journal of Toxicology Clinical Toxicology. 2002;40(4):415-446.
16 Barceloux DG, Krenzelok EK, Olson K, et al. American Academy of Clinical Toxicology practice guidelines on the treatment of ethylene glycol poisoning. Journal of Toxicology – Clinical Toxicology. 1999;37(5):537-560.
17 Jakobsen D, McMartin KE. Methanol and ethylene glycol poisonings: mechanism of toxicity, clinical course, diagnosis and treatment. Medical Toxicology. 1986;1:309-334.
18 Naraqui S, Dethlefs RF, Slobodniuk RA. An outbreak of acute methyl alcohol intoxication. Australia & New Zealand Journal of Medicine. 1979;9:65-68.
19 Hoffman RS, Smilkstein MJ, Howland MA, et al. Osmol gaps revisited: normal values and limitations. Journal Toxicology Clinical Toxicology. 1993;31:81-93.
20 Sejersted OM, Jakobsen D. Formate concentrations in plasma from patients poisoned with methanol. Acta Medica Scandinavica. 1983;1213:105-110.
21 Jolliff HA, Dart RC, Bogan GM, et al. Can the diagnosis of ethylene glycol toxicity be made without serum EG levels and osmolality values? (abstract). Journal of Toxicology –Clinical Toxicology. 2000;38(5):539-540.
22 Murray L, Daly FFS, Little M, Cadogan M, editors. Toxicology handbook. Sydney: Elsevier, 2007.
23 Huntley JS, Blain C, Hood S, et al. Improving detection of alcohol misuse in patients presenting to an accident and emergency department. Emergency Medicine Journal. 2001;18:99-104.
24 Hungerford DW, Pollock DA, Todd KT. Acceptability of emergency department-based screening and brief intervention for alcohol problems. Academic Emergency Medicine. 2000;7:1383-1392.
29.19 Carbon monoxide
Introduction
Carbon monoxide (CO) poisoning is an important cause of mortality and morbidity from poisoning. Immediate resuscitation including 100% oxygen therapy is essential, and the long-term results of most patients will be good with this simple intervention. It is unclear whether any additional intervention will reduce the low but important risk of serious long-term neurological damage.
Aetiology, pathophysiology and pathology
Carbon monoxide is a colourless, odourless, tasteless and non-irritant gas, produced by incomplete combustion of hydrocarbons.1 Small amounts are also produced endogenously by normal metabolic processes. The most common sources of significant exposure are car exhausts, cigarette smoke, fires and faulty home heaters and barbecues. Catalytic converters reduce the production of carbon monoxide and are in all cars manufactured in the last decade or two. Carboxyhaemoglobin (COHb) concentrations in cigarette smokers range as high as 10%.
The pathophysiology of CO exposure is complex and incompletely understood. Upon exposure, CO binds to haemoglobin with an affinity 210 times that of oxygen, thereby decreasing the oxygen-carrying capacity of blood. CO can also produce injury by several other mechanisms, including direct disruption of cellular oxidative processes, binding to myoglobin and cytochrome oxidases and causing peroxidation of brain lipids.1 However, the end result is tissue hypoxia, leading to varying degrees of end-organ damage and eventually death. The severity of poisoning is a function of the duration of exposure, the ambient concentration of CO and the underlying health status of the exposed individual. Although useful for diagnosis when detected, the initial COHb level correlates poorly with outcome.2
Epidemiology
Poisoning with CO is an important cause of unintentional and intentional injury worldwide. In the USA alone, an estimated 1000–2000 accidental deaths due to CO exposure occur each year, resulting from an estimated 40 000 exposures.3 In Australia and the UK it is the most common agent in completed suicide by poisoning.
Prevention
Prevention of environmental or occupational exposure is possible by use of CO air monitors. COHb concentrations are increased for any given inspired CO concentration if the person is exercising or at high altitudes (increased breathing rate and pulmonary blood flow). These considerations are relevant to acceptable levels of exposure.
The introduction of catalytic converters has reduced CO production in vehicle exhaust, and this in turn appears to be leading to a reduction in fatal suicidal poisoning in some countries.4,5
Clinical features
The signs and symptoms of acute carbon monoxide poisoning are shown in Table 29.19.16 and correlate well with the maximum COHb concentration. Initial symptoms are non-specific and probably predominantly due to compensatory mechanisms to maintain tissue oxygen delivery to vital organs (e.g. tachycardia, headache, dizziness, gastrointestinal symptoms). Signs with more severe toxicity directly reflect tissue hypoxia with central nervous and cardiovascular toxicity being the most critical manifestations. Death results rapidly when impaired oxygenation of the heart prevents the compensatory increase in cardiac output. The skin is classically cherry pink although severely ill patients are often pale or cyanosed. Pre-existing cerebral or cardiovascular disease, anaemia and volume depletion or cardiac failure increases toxicity (for a given COHb). These people all have a reduced ability to compensate by increasing cardiac output or redistributing blood supply to vital organs.
Table 29.19.1 Typical clinical symptoms and signs relative to COHb (normal = 0.5%)6
| COHb (%) | Symptoms and signs |
|---|---|
| <10 | Nil (commonly found in smokers) |
| 10–20 | Nil or vague non-descript symptoms |
| 30–40 | Headache, tachycardia, confusion, weakness, nausea, vomiting, collapse |
| 50–60 | Coma, convulsions, Cheyne–Stokes breathing, arrhythmias, ECG changes |
| 70–80 | Circulatory and ventilatory failure, cardiac arrest, death |
Due to low oxygen pressures, the high affinity of fetal haemoglobin for CO, and the much longer half-life of CO in the fetal circulation, the fetus is particularly susceptible to CO poisoning. The outcome of significant CO poisoning in the mother is often fetal death or neurological damage.
Delayed or persistent neuropsychiatric sequelae occur, largely confined to those who have loss of consciousness at some stage.7,8 Long-term follow-up is necessary as more subtle defects can develop or become apparent over a few weeks to months. The most common problems encountered are depressed mood (even in those accidentally exposed) and difficulty with higher intellectual functions (especially short-term memory and concentration).7,8 More severe problems include parkinsonism and speech problems. Neuropsychological testing may detect subtle defects not apparent on crude mini-mental state testing. The incidence of sequelae depends on the definition used – major deficits are relatively uncommon but neuropsychiatric complaints related to memory or concentration may occur in as many as 25–50% of patients with a loss of consciousness.7,8
Differential diagnosis
In suicide attempts, the diagnosis of CO poisoning is generally apparent from the circumstances when the person is found. The major diagnostic issue is whether there is some other deliberate self-poisoning as this is extremely common. In unconscious patients, the ECG, paracetamol concentration and electrolytes should be reviewed with this possibility in mind.6
A large proportion of victims of smoke inhalation also have cyanide poisoning. This rarely leads to a change in management (due to problems with administering the cyanide antidotes in this setting) but should be suspected when CNS effects are out of proportion with COHb concentrations and if there is a marked lactic acidosis.
Clinical investigation
Blood gases and oximetry
Most pulse oximeters do not measure COHb but merely the ratio of oxyHb to deoxyHb. Thus they may give very misleading results in the setting of CO poisoning. Blood gases with a co-oximeter are required to quantify COHb. COHb concentrations (plus or minus a back calculation based on estimated half-life since removal) provide a rough guide to the extent of exposure. However, it is difficult to accurately estimate the oxygen dose received pre-hospital and therefore the half-life. There is also substantial variability between individuals in the extent they are able to compensate for high COHb. Therefore, the correlation with acute and long-term clinical effects is not good. COHb may confirm (or possibly exclude) the diagnosis but should not be used to estimate long-term prognosis.
ECG
Patients should have a baseline ECG (electrocardiogram), repeated 6 h later, and ECG monitoring for at least 24 h and cardiac enzymes if the initial ECG is abnormal. The most important signs seen are those of cardiac ischaemia and these are identical to those seen in coronary artery disease.
Biochemistry
Cardiac enzymes should be measured when there is severe clinical toxicity or ECG changes. Metabolic acidosis, predominantly due to lactate will provide an indication of tissue hypoxia. Electrolytes (sodium, potassium, magnesium) should be measured as low concentrations of any of these may exacerbate cardiac toxicity. S100B concentrations, indicating acute neurological injury, have the potential to be useful in estimating neurological damage but further validation is required before they can be related to long-term severity in this setting.9,10
Criteria for diagnosis
A high COHb (>15%) with typical symptoms or signs confirms the diagnosis of acute CO poisoning.
In some parts of the world it is common to attribute many non-specific presentations to chronic carbon monoxide exposure, often despite COHb concentrations that are normal or within the range of those seen in ‘healthy’ smokers. There are no agreed on criteria for making a diagnosis of chronic carbon monoxide poisoning, but the diagnosis should not be seriously entertained without confirmation of high ambient CO concentrations in the proposed environmental source.
Treatment
Initial management is directed towards securing the airway and stabilizing respiration and circulation. If there is impaired consciousness, ensure the airway is maintained with intubation if necessary. The comatose patient should be placed on a cardiac monitor, a 12-lead ECG performed, an intravenous line inserted and blood drawn for full blood count, electrolytes, lactate, COHb, blood sugar and cardiac enzymes. If awake, the patient should be reassured and discouraged from activity, for muscle activity will increase oxygen demand.6
Metabolic acidosis should not be treated directly unless the acidosis itself contributes to toxicity (pH < 7.0). It should respond to improved oxygenation and ventilation and the net effect of acidosis on oxygen delivery is probably beneficial.
Oxygen
This decreases the biological half-life substantially from 4 h in ambient air to approximately 40 min in a 100% oxygen atmosphere (Fig. 29.19.1).6 100% oxygen should be administered with mechanically assisted ventilation if necessary. In patients able to tolerate it, continuous positive airway pressure by mask may allow 100% oxygen delivery without intubation. Four to six hours of 100% normobaric oxygen will remove over 90% of the carbon monoxide. If the only available oxygen delivery device is a Hudson mask, it should be remembered that at a flow rate of 15 L/min no more than 60% oxygen is delivered. At these concentrations the half-life of CO is still around 90 min and a longer period of oxygen may be required for severe poisonings (Fig. 29.19.1). Oxygen toxicity is unlikely with less than 24 h treatment but the risk increases with increasing exposure.
Fig. 29.19.1 Approximate decline in COHb from 50% according to the inspired oxygen concentration and pressure.
When immediately available, hyperbaric oxygen (HBO) should be considered for patients with serious CO poisoning. Oxygen at 2–3 atmospheres will further reduce the half-life of COHb to about 20 min (Fig. 29.19.1) but more importantly it causes very rapid reversal of tissue hypoxia due to oxygenation of tissue from oxygen dissolved in the plasma.
Controversy exists on the benefits, risks and indications for HBO (see controversies below). Indications for HBO commonly used by hyperbaric facilities are simply those factors that indicate a higher risk of long-term neuropsychiatric sequelae.7,8 These include:
Other treatments
Animal models suggest possible benefits from use of allopurinol and N-acetylcysteine to protect against oxidative damage during hypoxic/reperfusion injury.11 Numerous experimental (i.e. never used in humans) agents have also been suggested. Their use cannot be recommended outside of clinical trials; however, they may be a more logical treatment to prevent neurological damage from reactive oxygen species than hyperbaric oxygen.
1 Weaver LK. Carbon monoxide poisoning. Critical Care Clinic. 1999;15(2):297-317.
2 Seger D, Welch L. Carbon monoxide controversies: neuropsychologic testing, mechanism of toxicity, and hyperbaric oxygen. Annals of Emergency Medicine. 1994;24(2):242-248.
3 Hampson NB. Emergency department visits for carbon monoxide poisoning in the Pacific Northwest. Journal of Emergency Medicine. 1998;16(5):695-698.
4 Amos T, Appleby L, Kiernan K. Changes in rates of suicide by car exhaust asphyxiation in England and Wales. Psychological Medicine. 2001;31(5):935-939.
5 Mott JA, Wolfe MI, Alverson CJ, et al. National vehicle emissions policies and practices and declining US carbon monoxide-related mortality. Journal of American Medical Association. 2002;288(8):988-995.
6 Buckley NA, Dawson AH, Whyte IM. Hypertox. Assessment and Treatment of Poisoning. 2006. http://curriculum.toxicology.wikispaces.net/2.2.9.1.1+Carbon+monoxide. (accessed 4 September 2008)
7 Buckley NA, Isbister GK, Stokes B, et al. Hyperbaric oxygen for carbon monoxide poisoning : a systematic review and critical analysis of the evidence. Toxicological Review. 2005;24(2):75-92.
8 Juurlink D, Buckley N, Stanbrook M, et al. Hyperbaric oxygen for carbon monoxide poisoning. Cochrane Database Systematic Review. (1):2005. CD002041
9 Brvar M, Mozina M, Osredkar J, et al. Prognostic value of S100B protein in carbon monoxide-poisoned rats. Critical Care Medicine. 2004;32(10):2128-2130.
10 Brvar M, Mozina H, Osredkar J, et al. The potential value of the protein S-100B level as a criterion for hyperbaric oxygen treatment and prognostic marker in carbon monoxide poisoned patients. Resuscitation. 2003;56(1):105-109.
11 Omaye ST. Metabolic modulation of carbon monoxide toxicity. Toxicology. 2002;180(2):139-150.
12 Buckley NA, Isbister GK, Juurlink DN. Hyperbaric oxygen for carbon monoxide poisoning : evidence versus opinion. Toxicological Review. 2005;24(3):159-160.
13 Scheinkestel CD, Bailey M, Myles PS, et al. Hyperbaric or normobaric oxygen for acute carbon monoxide poisoning: a randomised controlled clinical trial. Medical Journal of Australia. 1999;170(5):203-210.
14 Weaver LK, Hopkins RO, Chan KJ, et al. Hyperbaric oxygen for acute carbon monoxide poisoning. New England Journal of Medicine. 2002;347(14):1057-1067.
15 Bunc M, Luzar B, Finderle Z, et al. Immediate oxygen therapy prevents brain cell injury in carbon monoxide poisoned rats without loss of consciousness. Toxicology. 2006;225(2–3):138-141.
16 Brvar M, Finderle Z, Suput D, et al. S100B protein in conscious carbon monoxide-poisoned rats treated with normobaric or hyperbaric oxygen. Critical Care Medicine. 2006;34(8):2228-2230.