3 Pharmacogenetics
The goal of drug therapy is to produce a specific pharmacologic effect in a patient without producing adverse effects. Unfortunately, this goal is not fully realized for every veterinary patient. If the same drug were administered to 10 patients with a particular disease, each may respond differently with respect to both drug efficacy and the likelihood of an adverse reaction. This variation in response to drug therapy can be caused by a number of patient factors, including the patient’s age, health or disease status, species, gender, and breed. In many instances, however, these factors cannot fully explain the degree of interpatient variation observed. Pharmacogenetics, the study of genetic determinants of response to drug therapy, is likely the ultimate way to establish the right drug and dose for each patient, thereby optimizing efficacy and minimizing toxicity. Despite the fact that this branch of pharmacology is still in its infancy as a science, a number of important discoveries have already contributed to improved pharmacotherapy in human and veterinary patients.
Genetic variation can affect both the pharmacokinetics (i.e., drug absorption, distribution, metabolism, and excretion) and pharmacodynamics (i.e., interaction with drug transporters and receptors) of pharmaceutical agents (Figure 3-1). Currently, the greatest body of knowledge with regard to pharmacogenetics involves genetic variation in drug metabolism. Indeed, the concept of pharmacogenetics originated in the 1950s as a result of the observation in human beings that two populations of individuals existed with respect to their ability to metabolize succinylcholine, followed by the realization that this trait was inherited.1 Approximately 1 in 3500 Caucasian subjects is homozygous for a mutation in the butyrylcholinesterase gene.2 Because this gene is responsible for hydrolysis and inactivation of succinylcholine, patients who have the mutation experience prolonged muscle paralysis and apnea. At the time, molecular biological techniques had not yet been discovered, so the field of pharmacogenetics was initially based purely on phenotypic observations.
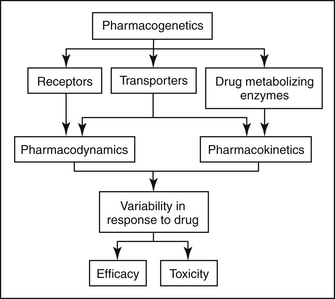
Figure 3-1 Pharmacogenetics can alter both drug efficacy and the likelihood of toxicity. Polymorphisms in drug receptors contribute to variable pharmacodynamics, and polymorphisms in drug-metabolizing enzymes contribute to variable pharmacokinetics. Interestingly, polymorphisms in drug transporters can contribute to variation in pharmacodynamics and/or pharmacokinetics, depending on the particular drug transporter involved.
Modern pharmacogenetics has progressed rapidly from those initial phenotypic observations. It currently involves identifying both the phenotype and the genetic variation, or polymorphism, responsible for it. Furthermore, the pharmacogenetic traits that were first identified were monogenic—that is, they involved a polymorphism at a single gene. It is presently known that myriad genes may be involved in determining a particular drug’s disposition, which increases the complexity of pharmacogenetics. Pharmacogenetics now involves systematic searches to identify functionally significant variations in DNA sequences in genes that affect drug disposition. Sequencing of the human, canine, and now feline genome should facilitate the progress of pharmacogenetics toward individualized drug therapy.
It is important to note that individualization of drug therapy encompasses two distinct, yet equally important, clinical implications. First is the ability to predict those patients at high risk for developing drug toxicity. These patients may have a polymorphism in a drug-metabolizing enzyme that results in low clearance rates for the drug. For such patients a lower drug dose or alternative drug should be administered. Second is the ability to predict those patients that are most likely to benefit from a particular drug because of appropriate receptor interactions. Patients with polymorphisms in drug receptors may be poor responders to certain pharmaceutical agents because of inappropriate drug–receptor interactions. Rather than using a trial-and-error approach to drug therapy, a veterinarian could select the drug most likely to produce the desired pharmacologic response in a particular patient, decreasing the amount of time in that the patient’s disease state is poorly controlled.
Described in this chapter will be several recent discoveries in pharmacogenetics and examples of pharmacogenetically based differences in drug absorption, distribution, metabolism, excretion, and drug–receptor interactions. The role of these discoveries in clinical veterinary medicine will also be presented.
Pharmacogenetics of Drug Absorption
Until recently, systemic bioavailability of orally administered drugs was considered to be a function of physicochemical characteristics of the drug and subsequent hepatic metabolism. A number of other factors have recently been shown to affect the ability of a drug to be absorbed into the systemic circulation after oral administration. Intestinal phase I drug metabolism and active drug extrusion by efflux transporters are now considered to be among the most important determinants of oral drug bioavailability. Consequently, polymorphisms in intestinal drug-metabolizing enzymes and drug transporters should dramatically affect oral drug absorption.
In humans CYP 3A enzymes are expressed at higher levels in mature villus tip enterocytes than in hepatocytes.3 Because intestinal villi make up such a large surface area, there is a high likelihood that absorbed drug will interact with intestinal CYP 3A enzyme, facilitating substantial first-pass metabolism. Interpatient variability in intestinal CYP 3A levels has been studied in a small sample of human patients. Elevenfold variations in CYP 3A protein content and sixfold variation in enzymatic activity were identified, suggesting that CYP 3A polymorphisms exist in the human population.4
Intestinal drug metabolism is thought to be important in veterinary patients also, but relatively little is known with regard to interpatient variability in enzyme activity. In dogs tissue distribution (specifically liver and duodenum) of CYP3A12 and CYP3A26 mRNA has been investigated by the author. Overall, expression of CYP3A mRNA was greater in liver than in duodenum, but the relative contributions of the two canine CYP3A isoforms differed. Hepatic expression of CYP3A26 was greater than CYP3A12 in all dogs, with CYP3A26 making up 75.2% of the hepatic CYP3A pool. Conversely, duodenal expression of CYP3A12 was greater than CYP3A26 in all dogs, with CYP3A12 composing 99.8% of the duodenal CYP3A pool.5 Additionally, there was a large amount of interindividual variability among dogs. Further research is needed in this area to determine whether genetic differences account for the variation in expression of this important drug-metabolizing enzyme in hepatic and intestinal tissue among dog breeds and/or individual dogs.
Drug transporters are also known to play an important role in drug absorption. Many drug transporters have been identified in people, but the most well-characterized drug transporter is P-glycoprotein (P-gp), the product of the MDR1 (ABCB1) gene. The potential impact of transporter pharmacogenetics on drug pharmacokinetics is dramatically illustrated by P-gp. P-gp is a transmembrane protein that was first described in highly resistant tumor cell lines.6 Tumor cells expressing P-gp were cross-resistant to various anticancer agents (anthracyclines, vinca alkaloids, taxanes, and others). P-gp has since been shown to act as an ATP-dependent pump that exports drugs from cells. P-gp is also expressed in normal mammalian tissues, where it appears to function in a protective capacity. P-gp is expressed on bile canaliculi, renal tubular epithelial cells, the placenta, brain capillary endothelial cells, and at the luminal border of intestinal epithelial cells.7 At these locations P-gp pumps transported drugs either out of the body (into the bile, urine, or intestinal lumen) or away from protected sites (brain tissue, fetus).
KEY POINT 3-1
Intestinal and hepatic drug transporters and cytochrome P450 enzymes contribute to variability in oral drug absorption.
The significant role intestinal P-gp can play in determining oral drug bioavailability has been demonstrated in rodent studies. In mdr1(-/-) knockout mice, oral bioavailability of many P-gp substrate drugs (vinblastine, taxol, digoxin, loperamide, ivermectin, cyclosporine A, others) are substantially greater than in wild-type mice.8,9 Similarly, MDR1 polymorphisms in humans have been shown to result in altered oral bioavailability of P-gp substrate drugs. Studies have shown that oral bioavailability of digoxin, a P-gp substrate, is greater in subjects with the 3435TT MDR1 genotype compared with those with the MDR1 3435CC genotype.10 Similarly, the P-gp substrate phenytoin has been shown to have lower oral bioavailability in subjects with the MDR1 3435 CC genotype.11
P-gp has been fairly well characterized in dogs. Tissue distribution of P-gp in dogs is similar to that in people,12 and it has been shown to contribute to chemotherapeutic drug resistance in vitro and in vivo.13-15 Although its role in determining oral drug bioavailability is not well characterized, there is some evidence that P-gp is important. Bioavailability of the anticancer agent (and P-gp substrate) docetaxel was increased seventeenfold when co-administered with a P-gp inhibitor.
The MDR1 polymorphism in dogs consists of a four base-pair deletion mutation. This deletion results in a shift of the reading frame that generates several premature stop codons.16 Because protein synthesis is terminated before even 10% of the protein product is synthesized, dogs with two mutant alleles exhibit a P-gp null phenotype, similar to mdr1 (-/-) knockout mice. Affected dogs include many herding breeds. For example, roughly 75% of Collies in the United States, France, and Australia have at least one mutant allele.17 Other affected herding breeds, albeit affected at a lower frequency, include Old English Sheepdogs, Australian Shepherds, Shetland Sheepdogs, English Shepherd Dogs, Border Collies, German Shepherd Dogs, Silken Windhounds, McNab Shepherds, and Longhaired Whippets.18
One group of investigators describe a modest effect of the MDR1 deletion mutation on oral drug availability.19 Three P-gp substrate drugs (fexofenadine, loperamide, and quinidine) were simultaneously administered to two groups of dogs, dogs homozygous for the MDR1 deletion mutation (MDR1 mut/mut) and MDR1 wild-type dogs (MDR1 WT/WT). Statistically, there were no differences in area under the curve for any drug between the two groups. Plasma concentrations of fexofenadine were significantly higher at two time points in MDR1(mut/mut) dogs than in MDR1(WT/WT) dogs.19 The investigators suggest that P-gp limits intestinal absorption of fexofenadine, an antihistamine (H1), in dogs.
Pharmacogenetics of Drug Distribution
Drug distribution, the delivery of drugs from the systemic circulation to tissues, can be dramatically affected by pharmacogenetics. The drug transporter P-gp serves as an important barrier to the distribution of substrate drugs to selected tissues. For example, P-gp is a component of the blood–brain barrier, the blood–testes barrier, and the placenta. Therefore distribution of P-gp substrate drugs to these tissues is greatly enhanced in MDR1(mut/mut) dogs. MDR1(mut/mut) dogs experience adverse neurologic effects after a single dose of ivermectin (120 μg/kg). Heterozygous [MDR1(WT/mut)] or MDR1(WT/WT) dogs are not sensitive to ivermectin neurotoxicity at the 120 μg/kg dose, but MDR1(WT/mut) animals may experience neurotoxicity at ivermectin doses greater than 120 μg/kg, particularly if daily doses are administered (i.e., protocols for treatment of demodectic mange). Affected dogs also appear to have increased susceptibility to neurologic adverse effects of other avermectins, including milbemycin, selamectin (Revolution package insert), and moxidectin.20 Interestingly, a retrospective study conducted by a national veterinary poison center reported that Collies were overrepresented in canine cases of loperamide-induced neurotoxicity.21 Many Collies displayed signs of neurologic toxicity after administration of routinely recommended doses of the antidiarrheal agent loperamide (Imodium). Loperamide is an opioid that is generally devoid of central nervous system activity because it is excluded from the brain by P-gp.22,23 Loperamide neurotoxicity was recently reported in a Collie that had received a routine dose (0.14 mg/kg orally).24 The dog in this report had the MDR1(mutant/mutant) genotype. MDR1 (WT/WT) dogs do not exhibit neurologic signs after receiving even higher doses of loperamide, indicating that P-gp plays a key role in modulating distribution of substrates such as loperamide to canine brain tissue. The images in Figure 3-2 illustrate the role of P-gp in the blood–brain barrier of dogs.
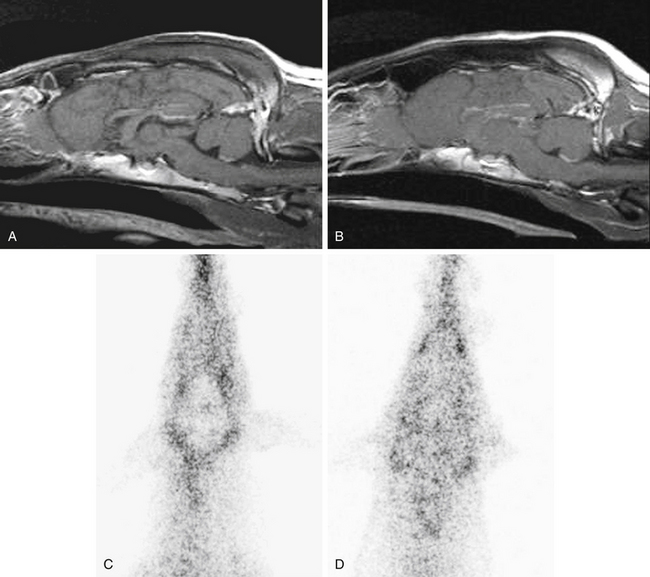
Figure 3-2 Sagittal T1-weighted post gadolinium administration magnetic resonance images displaying normal brain anatomy, tissue signal characteristics, and contrast enhancement of an MDR1 WT/WT dog (A) and an MDR1 mut/mut dog (B) showing no differences between dogs of these two genotypes. Conversely99mTc-MIBI nuclear scintigraphic neuroimaging of the brain after 99mTc-MIBI administration demonstrates the role of P-glycoprotein at the blood–brain barrier. Representative 99mTc-MIBI nuclear scintigraphic images show diminished activity in the brain compared with surrounding tissue of an MDR1 WT/WT dog (C) but similar activity in the brain compared with surrounding tissue of an MDR1 mut/mut dog (D).
(From Mealey KL, Greene S, Bagley R, et al: P-glycoprotein contributes to the blood–brain, but not blood–cerebrospinal fluid, barrier in a spontaneous canine p-glycoprotein knockout model, Drug Metab Dispos 36(6):1073-1079, 2008.)
KEY POINT 3-2
Dogs with the ABCB1-1Δ mutation experience greater brain penetration of drugs that are substrates for P-glycoprotein.
Less information is available regarding P-gp and the blood–brain barrier in cats. The author has received anecdotal reports of ivermectin toxicity in cats after standard doses, but whether the underlying cause is a result of altered P-gp expression or function is not currently known.
Distribution of some drugs to the testis and fetus may also be limited by P-gp. In human patients, this creates a problem for treating certain diseases. For example, the testes and brain are considered to be a sanctuary site for the human immunodeficiency virus (HIV).25 Because HIV-1 protease inhibitors are substrates for P-gp, the virus can remain viable in these sanctuary sites, hampering effective therapy. Similarly, therapeutic concentrations of certain chemotherapeutic agents may not be achievable for testicular cancers because of active efflux by P-gp.26 The effect of placental P-gp on distribution of drugs to the fetus is an area of active research in human medicine.27 Understanding the role of pregnancy-associated hormones in regulating P-gp expression and function is one possible key in developing strategies to deliver drugs to the mother with minimal fetal risk.
Pharmacogenetics of Drug Metabolism
Pharmacogenetic variation can affect both phase I and phase II metabolic enzyme activity. The pseudocholinesterase mutation serves as an example of how pharmacogenetic variation can result in dramatic differences in drug response among patients. Patients with a normal pseudocholinesterase genotype metabolize succinylcholine and recover from neuromuscular blockade rapidly, whereas those with the mutant genotype undergo sustained neuromuscular blockade that has resulted in prolonged apnea and the necessity for mechanical ventilation. A number of polymorphisms have been described in human CYP450 enzymes, many of these resulting in profound variations in clinical response. For example, CYP2D6 is a highly variable P450 pathway in humans with individuals ranging from undetectable activity (found in 6% to 10% of Caucasians) to “ultrarapid” activity (found in 3% to 10% of Europeans and 30% of one black population).28 The ultrarapid phenotype is due to a unique gene duplication. Drugs that are substrates for CY2D6 in people include beta-receptor antagonists (propranolol, timolol, metoprolol), antiarrhythmics (quinidine, flecainide), antidepressants (amitriptyline, clomipramine, fluoxetine, imipramine), neuroleptics, and certain opioid derivatives. Depending on the patient’s CYP2D6 genotype, the “typical” dose of a substrate drug may need to be decreased (poor metabolizers require 1⁄10 of the standard dose of nortriptyline to avoid toxicity) or increased (ultrarapid metabolizers require 5 times the standard dose to achieve therapeutic concentrations).
Relatively few polymorphisms in drug-metabolizing enzymes have been described in veterinary patients, although this is likely to change insofar as research in this area is currently in progress. However, variation in metabolism of some drugs has been documented in dogs. CYP2B11 has been shown to have at least a fourteenfold variation in activity in mixed-breed dogs.29 Greyhounds have been shown to have particularly low CYP2B11 activity, which results in sustained plasma concentrations of propofol, and delayed recovery compared with mixed-breed dogs.30 The specific genetic alteration responsible for reduced CYP2B11 in Greyhounds compared with other canine breeds has not been determined. There is some evidence to suggest that CYP 2D15 may also be polymorphic in dogs. The nonsteroidal antiinflammatory drug celecoxib is metabolized to a large degree by CYP2D15. Clearance of celecoxib in Beagles is polymorphic, with about half the population being extensive metabolizers and the remainder being poor metabolizers.31 Celecoxib has a 1.5- to 2-hour half-life in extensive metabolizers and a 5-hour half-life in poor metabolizers. One pharmacogenetic variant that has been identified in the canine CYP2D15 gene, a deletion of exon 3, results in undetectable celecoxib metabolism. The frequency and breed distribution of this polymorphism has not yet been determined. However, it is likely to have clinical significance for other drugs that are CYP2D15 substrates, including dextromethorphan, imipramine, and others.
KEY POINT 3-3
Polymorphisms in drug-metabolizing enzymes can dramatically affect drug efficacy and toxicity.
Variability in gene copy number is a phenomenon that has recently been explored in dogs.32 Among the genes that have been demonstrated to exist in copy number variant regions in canine DNA are CYP3A12 and CYP1A. Furthermore, there appear to be breed-specific copy number variants for some dog breeds. Thus variability in gene copy number may be another source of genetic variation in drug response involving phase I enzymes.
With respect to phase II metabolic enzymes, a pan-species defect in UDP-glucuronyl transferase exists in cats. Although this is not a true example of pharmacogenetics, it serves as an example of genetic variation among species, rather than within a species, that significantly affects drug disposition. Because cats have a pseudogene, rather than a functional glucuronyltransferase gene, aceptaminophen and other drugs are not conjugated with glucuronide as they are in other species. Another pan-species phase II metabolic defect occurs in dogs. N-acetyltransferase is the enzyme responsible for metabolizing sulfonamides, procainamide, hydralazine, and other drugs. Both N-acetyltransferase genes are absent in dogs, increasing the risk for hypersensitivity reactions and adverse effect from these drugs relative to other species.33
A true pharmacogenetic variation exists for the thiopurine methyltrasferase (TPMT) enzyme. TPMT is a phase II enzyme that is responsible for metabolizing azathioprine and its active metabolites to their inactive forms. A ninefold range in TPMT activity exists in dogs, and activity level appears to be related to breed. Giant Schnauzers had lower TPMT activity, and Alaskan Malamutes had high TPMT activity.34 Decreased TPMT activity has been documented to be associated with increased susceptibility to azathioprine-induced bone marrow suppression.
Pharmacogenetics of Drug Excretion
Drugs are eliminated from the body either unchanged or as metabolites. Renal and biliary excretion are the most important pathways of drug elimination, but excretion may occur by other routes as well. As noted previously, P-gp is expressed on renal tubular cells and biliary canalicular cells, suggesting that it may play a role in drug excretion. Concurrent administration of a P-gp–inhibitor decreases the biliary and renal clearance of doxorubicin in rats.35 In a separate study, biliary and renal excretion of digoxin and vincristine were increased in rats after treatment with a P-gp inhibitor.36 Further research is necessary to fully define the role of P-gp in regulating renal and biliary drug excretion in veterinary patients. However, altered biliary or renal excretion may play a role in the apparent increased sensitivity of herding breeds to chemotherapeutic drugs that are P-gp substrates. For example, MDR1(mut/mut) and MDR1(WT/mut) dogs are significantly more likely to develop hematologic toxicity, specifically neutropenia and thrombocytopenia after treatment with vincristine than ABCB1-1Δ wild-type dogs. However, these dogs tolerate cyclophosphamide at full doses.37,38 In dogs vincristine is eliminated primarily through biliary excretion of parent drug with some urinary excretion of parent drug and metabolites.39 The enzyme family responsible for metabolizing vincristine in dogs has not been identified. Biliary excretion of vincristine in other species is highly dependent on the P-gp–mediated drug transport.36 The images in Figure 3-3 illustrate the role of P-gp in the biliary excretion of substrate drugs in dogs.
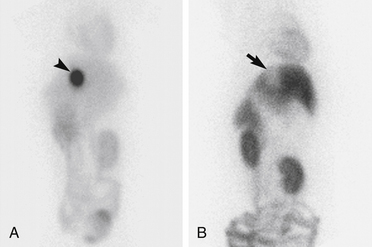
Figure 3-3 Images of the abdomen of dogs positioned in ventral recumbency on a gamma camera. Images were acquired 2 hours after intravenous injection of 99mTc-MIBI to an MDR1 WT/WT dog (A) and to an MDR1 mut/mut dog (b). Intense uptake of 99mTc-MIBI in the gallbladder (arrowhead) is present in (a). A void of activity in the location of the gallbladder (arrow) is present in (b).
(From Coelho JC, Tucker R, Mattoon J et al: Biliary excretion of technetium-99m-sestamibi in wild-type dogs and in dogs with intrinsic (ABCB1-1Δ mutation) and extrinsic (ketoconazole treated) P-glycoprotein deficiency, J Vet Pharm Ther 32:417-421, 2009.)
Pharmacogenetics of Drug Receptors
A relatively new and important area of pharmacogenetics research involves polymorphisms in genes encoding drug receptors and effector proteins. In human patients polymorphisms have been described in angiotensin-converting enzyme, beta-2 adrenergic receptors, the dopamine receptor, the estrogen receptor, and others.40 In vitro functional studies suggest that these polymorphisms have functional significance. A polymorphism in the canine dopamine receptor D4 gene has been described, but its clinical implications are not yet understood.41
Pharmacogenetics and Hypersensitivity Reactions
Pharmacogenetic differences in metabolic pathways not only can affect type A adverse drug reactions (predictable; generally correlating with plasma drug concentration) but can also affect type B adverse drug reactions (idiosyncratic). Idiosyncratic toxicity to sulfonamides is similar in dogs and humans and can be characterized by fever, arthropathy, blood dyscrasias (neutropenia, thrombocytopenia, or hemolytic anemia), hepatopathy consisting of cholestasis or necrosis, skin eruptions, uveitis, or keratoconjunctivitis sicca.42 In humans slow acetylation by NAT2 has been shown to be a risk factor for sulfonamide hypersensitivity reactions. It has been proposed that the alternative metabolic pathway in these individuals produces reactive metabolites.43 Covalent binding of reactive metabolites of these drugs to cell macromolecules results in cytotoxicity and immune response to neoantigens. Ongoing research in one veterinary pharmacology laboratory (Department of Medical Sciences, School of Veterinary Medicine, University of Wisconsin-Madison; latrepanier@svm.vetmed.wisc.edu) is under way to characterize dogs with possible idiosyncratic sulfonamide reactions, using several methodologies, including enzyme-linked immunosorbent assay (ELISA) for antidrug antibodies, immunoblotting for antibodies directed against liver proteins, flow cytometry for drug-dependent antiplatelet antibodies, and in vitro cytotoxicity assays.
Pharmacogenetics in Clinical Practice
Scientific interest in the field of human pharmacogenetics has increased each year in parallel with the knowledge of the human genome. However, interest in this field from physicians has lagged significantly behind, presumably because relatively few significant clinical consequences can be correlated to the vast number of pharmacogenetic mutations described in the literature. There are two main reasons for this discrepancy. Up to this point, polymorphisms described in human patients either have had low allelic frequencies or the clinical relevance of a particular polymorphism was not significant. For example, a highly clinically relevant polymorphism in the human TPMT gene has been described. TMPT metabolic activity in affected patients is essentially absent, so these patients experience severe neutropenia after a “normal” dose of azathioprine. Because this TPMP polymorphism affects approximately 0.3% of the Caucasian population, pharmacogenetic testing is not routinely performed in clinical practice.44 Conversely, the allelic frequency of a genetic polymorphism of the human MDR1 gene has been shown to be associated with lower levels of P-gp expression in the duodenum and other tissues. Although the allelic frequency of this particular MDR1 polymorphism is relatively high (>10%), it does not appear to have an important and predictable clinical impact on drug disposition. The vast majority of pharmacogenetics in human medicine is carried out for research purposes and is not performed in clinical medical practice.
In veterinary medicine, however, a commercial veterinary pharmacogenetics laboratory (Veterinary Clinical Pharmacology Laboratory, Washington State University, Pullman, WA; www.vetmed.wsu.edu/vcpl) is currently performing canine MDR1 genotyping for veterinarians, breeders, and owners in the United States. Such testing is available in Europe and Australia as well. The primary reasons that commercial pharmacogenetic testing is readily available for canine patients and not for human patients are because the MDR1 mutation in dogs has a very high allelic frequency (55% in Collies, 42% in Longhaired Whippets, and roughly 20% in Australian Shepherds), and because the polymorphism is highly predictive for serious adverse drug events, not just for ivermectin, but for chemotherapeutic drugs,38 other antiparasitics,45 loperamide,24 and other drugs.46 Box 3-1 contains a partial list of drugs that are substrates for P-gp.
Box 3-1 Selected P-glycoprotein Substrates
(Adapted from Fromm MF: Genetically determined differences in P–glycoprotein function: implications for disease risk, Toxicology 181–182:299–303, 2002; Marzolini C, Paus E, Buclin T, Kim RB: Polymorphisms in human MDR1 (P–glycoprotein): recent advances and clinical relevance, Clin Pharmacol Ther 75:13–33, 2004; Sakaeda T, Nakamura T, Okumura K: MDR1 genotype–related pharmacokinetics and pharmacodynamics, Biol Pharm Bull 25:1391–1400, 2002; Sakaeda T, Nakamura T, Okumura K: Pharmacogenetics of MDR1 and its impact on the pharmacokinetics and pharmacodynamics of drugs, Pharmacogenomics 4:397–410, 2003; Schwab M, Eichelbaum M, Fromm MF: Genetic polymorphisms of the human MDR1 drug transporter, Annu Rev Pharmacol Toxicol 43:285–307, 2003.)
∗ Substrate of CYP 3A
Future Directions
The field of pharmacogenetics, particularly in veterinary medicine, is still in its infancy. However, we have an ever-increasing arsenal of molecular tools that can be used to expand our knowledge of pharmacogenetics. Furthermore, with the recent completion of the canine and feline genome projects and the ongoing elucidation of their data, we may soon know the sequences of virtually all genes encoding drug-metabolizing enzymes, drug transporters, drug receptors, and other drug targets. With this information the traditional pharmacogenetics approach (phenotype to genotype) will likely give way to a pharmacogenomics approach (genotype to phenotype). In other words, genetic variation identified in a specific gene will be investigated to determine if it results in a variation in pharmacologic response. The convergence of these advances has the potential to individualize drug therapy for many veterinary patients.
1. Kalow W., Gunn D.R. Some statistical data on atypical cholinesterase of human serum. Ann Hum Genet. 1959;23:239-250.
2. Kalow W. The Pennsylvania State University College of Medicine 1990 Bernard B. Brodie Lecture. Pharmacogenetics: past and future,. Life Sci. 1990;47:1385-1397.
3. Patel J., Mitra A.K. Strategies to overcome simultaneous P-glycoprotein mediated efflux and CYP3A4 mediated metabolism of drugs. Pharmacogenomics. 2001;2:401-415.
4. Scordo M.G., Spina E. Cytochrome P450 polymorphisms and response to antipsychotic therapy. Pharmacogenomics. 2002;3:201-218.
5. Mealey K.L., Jabbes M., Spencer E., Akey J. Differential expression of CYP3A12 and CYP3A26 in canine liver and intestine. Xenobiotica. 2008;38(10):1305-1312.
6. Roninson I.B. The role of the MDR1 (P-glycoprotein) gene in multidrug resistance in vitro and in vivo,. Biochem Pharmacol. 1992;43:95-102.
7. Thiebaut F., Tsuruo T., Hamada H., et al. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci USA. 1987;84:7735-7738.
8. Schinkel A.H., Wagenaar E., van Deemter L., et al. Absence of the mdr1a P-glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin A. J Clin Invest. 1995;96:1698-1705.
9. Sills G.J., Kwan P., Butler E., et al. P-glycoprotein-mediated efflux of antiepileptic drugs: preliminary studies in mdr1a knockout mice. Epilepsy Behav. 2002;3:427-432.
10. Verstuyft C., Schwab M., Schaeffeler E., et al. Digoxin pharmacokinetics and MDR1 genetic polymorphisms. Eur J Clin Pharmacol. 2003;58:809-812.
11. Kerb R., Aynacioglu A.S., Brockmoller J., et al. The predictive value of MDR1, CYP2C9, and CYP2C19 polymorphisms for phenytoin plasma levels,. Pharmacogenomics J. 2001;1:204-210.
12. Ginn P.E. Immunohistochemical detection of P-glycoprotein in formalin-fixed and paraffin-embedded normal and neoplastic canine tissues. Vet Pathol. 1996;33:533-541.
13. Mealey K.L., Barhoumi R., Rogers K., Kochevar D.T. Doxorubicin induced expression of P-glycoprotein in a canine osteosarcoma cell line. Cancer Lett. 1998;126:187-192.
14. Page R.L., Hughes C.S., Huyan S., et al. Modulation of P-glycoprotein-mediated doxorubicin resistance in canine cell lines. Anticancer Res. 2000;20:3533-3538.
15. McEntee M., Silverman J.A., Rassnick K., et al. Enhanced bioavailability of oral docetaxel by co-administration of cyclosporin A in dogs and rats. Vet Comp Oncol. 2003;2:105-112.
16. Mealey K.L., Bentjen S.A., Gay J.M., Cantor G.H. Ivermectin sensitivity in collies is associated with a deletion mutation of the mdr1 gene. Pharmacogenetics. 2001;11:727-733.
17. Mealey K.L., Bentjen S.A., Waiting D.K. Frequency of the mutant MDR1 allele associated with ivermectin sensitivity in a sample population of collies from the northwestern United States. Am J Vet Res. 2002;63:479-481.
18. Neff M.W., Robertson K.R., Wong A.K., et al. Breed distribution and history of canine mdr1-1{Delta}, a pharmacogenetic mutation that marks the emergence of breeds from the collie lineage. Proc Natl Acad Sci USA. 2004;101:11725-11730.
19. Kitamura Y., Koto H., Matsuura S., et al. Modest effect of impaired function of P-glycoprotein on the plasma concentrations of fexofenadine, quinidine, and loperamide following oral administration in Collie dogs. Drug Metab Dispos. 2008;36:807-810.
20. Tranquilli W.J., Paul A.J., Todd K.S. Assessment of toxicosis induced by high-dose administration of milbemycin oxime in collies. Am J Vet Res. 1991;52:1170-1172.
21. Hugnet C., Cadore J.L., Buronfosse F., et al. Loperamide poisoning in the dog. Vet Hum Toxicol. 1996;38:31-33.
22. Ericsson C.D., Johnson P.C. Safety and efficacy of loperamide. Am J Med. 1990;88:10S-14S.
23. Wandel C., Kim R., Wood M., Wood A. Interaction of morphine, fentanyl, sufentanil, alfentanil, and loperamide with the efflux drug transporter P-glycoprotein. Anesthesiology. 2002;96:913-920.
24. Sartor L.L., Bentjen S.A., Trepanier L., Mealey K.L. Loperamide toxicity in a collie with the MDR1 mutation associated with ivermectin sensitivity. J Vet Intern Med. 2004;18:117-118.
25. Choo E.F., Leake B., Wandel C., et al. Pharmacological inhibition of P-glycoprotein transport enhances the distribution of HIV-1 protease inhibitors into brain and testes. Drug Metab Dispos. 2000;28:655-660.
26. Katagiri A., Tomita Y., Nishiyama T., Kimura M., Sato S. Immunohistochemical detection of P-glycoprotein and GSTP1-1 in testis cancer. Br J Cancer. 1993;68:125-129.
27. Young A.M., Allen C.E., Audus K.L. Efflux transporters of the human placenta. Adv Drug Deliv Rev. 2003;55:125-132.
28. Cascorbi I. Pharmacogenetics of cytochrome P4502D6: genetic background and clinical implication. Eur J Clin Invest. 2003;33(Suppl 2):17-22.
29. Hay Kraus B.L., Greenblatt D.J., Venkatakrishnan K. Court MH: Evidence for propofol hydroxylation by cytochrome P4502B11 in canine liver microsomes: breed and gender differences. Xenobiotica. 2000;30:575-588.
30. Court M.H., Hay-Kraus B.L., Hill D.W., et al. Propofol hydroxylation by dog liver microsomes: assay development and dog breed differences. Drug Metab Dispos. 1999;27:1293-1299.
31. Paulson S.K., Engel L., Reitz B., et al. Evidence for polymorphism in the canine metabolism of the cyclooxygenase 2 inhibitor, celecoxib. Drug Metab Dispos. 1999;27:1133-1142.
32. Nicholas T.J., Cheng Z., Ventura M., et al. The genomic architecture of segmental duplications and associated copy number variants in dogs. Genome Res. 2009;19:491-499.
33. Collins J.M. Inter-species differences in drug properties. Chem Biol Interact. 2001;134:237-242.
34. Kidd L.B., Salavaggione O.E., Szumlanski C.L., et al. Thiopurine methyltransferase activity in red blood cells of dogs. J Vet Intern Med. 2004;18:214-218.
35. Kiso S., Cai S.H., Kitaichi K., et al. Inhibitory effect of erythromycin on P-glycoprotein-mediated biliary excretion of doxorubicin in rats. Anticancer Res. 2000;20:2827-2834.
36. Song S., Suzuki H., Kawai R., Sugiyama Y. Effect of PSC 833, a P-glycoprotein modulator, on the disposition of vincristine and digoxin in rats. Drug Metab Dispos. 1999;27:689-694.
37. Mealey K.L., Northrup N.C., Bentjen S.A. Increased toxicity of P-glycoprotein-substrate chemotherapeutic agents in a dog with the MDR1 deletion mutation associated with ivermectin sensitivity. J Am Vet Med Assoc. 2003;223:1453-1455. 1434
38. Mealey K.L., Fidel J., Gay J.M., et al. ABCB1-1Delta polymorphism can predict hematologic toxicity in dogs treated with vincristine. J Vet Intern Med. 2008;22:996-1000.
39. El Dareer S.M., White V.M., Chen F.P., et al. Distribution and metabolism of vincristine in mice, rats, dogs, and monkeys. Cancer Treat Rep. 1977;61:1269-1277.
40. Tribut O., Lessard Y., Reymann J.M., et al. Pharmacogenomics,. Med Sci Monit. 2002;8:RA152-RA163.
41. Ito H., Nara H., Inoue-Murayama M., et al. Allele frequency distribution of the canine dopamine receptor D4 gene exon III and I in 23 breeds. J Vet Med Sci. 2004;66:815-820.
42. Trepanier L.A. Idiosyncratic toxicity associated with potentiated sulfonamides in the dog. J Vet Pharmacol Ther. 2004;27:129-138.
43. Spielberg S.P. N-acetyltransferases: pharmacogenetics and clinical consequences of polymorphic drug metabolism. J Pharmacokinet Biopharm. 1996;24:509-519.
44. Becquemont L. Clinical relevance of pharmacogenetics. Drug Metab Rev. 2003;35:277-285.
45. Barbet J., Snook T., Gay J.M., Mealey K.L. ABCB1 (MDR1) genotype is associated with adverse reactions in dogs treated with milbemycin oxime for generalized demodicosis. Vet Dermatol. 2009;20:111-114.
46. Henik R.A., Kellum H.B., Bentjen S.A., Mealey K.L. Digoxin and mexiletine sensitivity in a Collie with the MDR1 mutation. J Vet Intern Med. 2006;20:415-417.