Chapter 16 Fluids, Electrolytes, and Acid–Base Therapy
Physiology
Water
Water accounts for 60% of lean body weight in adult animals, with variation between species. This percentage is affected by fat and age. Obese and older animals tend to have a smaller percentage of body weight composed of water, whereas up to 90% of body weight in neonates is water. Approximately two thirds of this water is present within cells (intracellular fluid [ICF] compartment) and one third is extracellular fluid (ECF). Of the ECF, three fourths is interstitial fluid and one fourth of the ECF volume is plasma. Thus plasma accounts for only 5% of body weight.1,2
Water freely diffuses through cell membranes. Consequently, all body compartments have approximately the same osmolality (290 to 310 mOsm/kg in dogs and 300 to 330 mOsm/kg in cats).3 Osmotic force is the prime determinant of distribution of water across cell membranes (i.e., the partitioning of water between the ECF and ICF). Plasma sodium is the primary determinant of ECF osmolality. Glucose and urea make minor contributions as well, which is reflected in the following calculation:
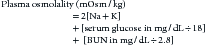
In the laboratory osmolality is measured by freezing point depression or vapor pressure osmometry. The measured value is often higher than the calculated value because this equation does not include all osmotically active particles present in plasma.
As can be seen from the preceding formula, sodium is principally responsible for ECF osmolality. Osmolality in the ICF is principally determined by potassium, magnesium, phosphates, and proteins. To promote water movement from one compartment to another, the osmolality of one or more compartments must change.4 Tonicity, or effective osmolality, refers to the osmotic pressure of a solution—Osmotic pressure is the pressure required to prevent water movement across a semipermeable membrane. When the relationship between tonicity and osmolality is under consideration, it is important to distinguish between permeant and impermeant solutes. Permeant solutes (e.g., urea) move freely across cell membranes and do not induce net water movement when introduced into a solution (i.e., they are ineffective osmoles). Impermeant solutes (e.g., sodium and glucose) induce water movement when introduced into a solution and are called effective osmoles, because they do not readily cross cell membranes. Glucose, to the extent that it causes hyperglycemia, is an impermeant solute and does contribute to ECF osmolality. However, glucose administration usually does not cause hyperglycemia because it is rapidly taken up by cells in the presence of insulin. The implication is that tonicity is less then osmolality.
The capillary walls are freely permeable to sodium, chloride, and glucose. As a result, these substances are osmotically inactive across capillary membranes. However, plasma proteins are limited in their ability to cross capillary membranes, and plasma volume is ultimately maintained by the colloid osmotic pressure (COP), COP is the force created by macromolecules present within the vasculature that prevents water from escaping to the extravascular space. Thus, COP is essentially the pressure exerted by plasma proteins. Albumin contributes to roughly 75% of COP in healthy patients, with globulins making up the remainder. At the venous end of the capillary bed, plasma proteins exert an osmotic force in excess of the hydrostatic gradient, resulting in a net fluid flux from the interstitium into the vessels. Thus albumin is important for distribution of water between the intravascular and interstitial compartments, and plasma proteins in general play a key role in the maintenance of intravascular fluid volume.1 This is in contrast to the osmotic equilibrium governing water distribution across the ECF and ICF compartments.
The amount of available water in the body is determined by a balance between intake and loss.1 Water intake occurs by drinking and eating, which are controlled by thirst and hunger. Alterations in water intake can occur with neurologic disease and congenital abnormalities. Animals normally lose water through urine, feces, and expired air. To a lesser extent, evaporation from the skin surface promotes water flux across the epidermis. Obligatory urinary and fecal losses are determined by the solute load that must be excreted. Respiratory losses of water are affected by ambient temperature and humidity. Water losses may increase as a result of vomiting, diarrhea, and polyuria. Rarely, clinically relevant water losses arise through traumatized tissues or with cavitary effusions. Fever also can be responsible for increasing loss of water.5 Fluid losses, such as those incurred by vomiting, diarrhea, and polyuria, are often classified as isotonic (i.e., small solutes are lost in proportion to their concentration in plasma), but more commonly these losses are slightly hypotonic, with sodium concentrations of 80 to 120 mg/dL. This is manifested by hypernatremia, which is commonly observed with dehydration. To maintain hydration homeostasis, urine output may exceed the obligatory urine output, the latter being the volume required for elimination of solutes. While the term free water is well recognized and accepted, this fluid resembles a hypotonic crystalloid in content, as opposed to water. Water excretion is due to modification of antidiuretic hormone (ADH) secretion. ADH is normally released from the posterior pituitary gland when osmoreceptors in the hypothalamus detect hypertonicity or when baroreceptors in the cardiovascular system sense hypovolemia. Release of ADH promotes water retention. For water to be excreted by the kidneys, there must be adequate delivery of tubular fluid to the ascending limb of the loop of Henle, and the renal collecting ducts must remain impermeable to water in the absence of ADH.6
Sodium
Sodium is the osmolar skeleton of the ECF. Under normal conditions the kidneys regulate its elimination, excreting as much sodium as is ingested. Renal sodium excretion is normally regulated by aldosterone, atrial natriuretic factors (ANF), and intrinsic renal mechanisms (e.g., renal blood flow [RBF], glomerular filtration rate [GFR], and glomerulotubular balance). Glomerulotubular balance refers to the ability of the kidney to maintain a relatively constant fractional reabsorption of sodium, despite changes in GFR. If there is expansion of the effective circulating volume (i.e., overhydration, hypervolemia), the cardiac atria distend with subsequent release of ANF. ANF and other natriuretic factors inhibit both the formation and effects of angiotensin and decrease sodium reabsorption from the renal medullary collecting ducts. Conversely, when atrial receptors detect a decrease in volume, sympathetic tone is increased and ANF release is inhibited. Further, decrements in renal perfusion and the associated decrease in delivery of sodium (and/or chloride) to the macula densa activate the renin–angiotensin–aldosterone system, promoting sodium to be reabsorbed from the renal tubules and collecting ducts.7
Potassium
The regulation of total body and plasma potassium concentration is important to the extent that potassium alterations may profoundly affect the resting membrane potential (RMP) of cardiac and neuromuscular cells.8 Hypokalemia lowers the RMP, making it more difficult to achieve an action potential with subsequent contraction of a muscle. Hyperkalemia raises the RMP, which may result in an action potential of decreased amplitude or, in extreme cases, continuous depolarization of the cell membrane. Although the majority of the body’s potassium is found within cells, it is the plasma concentration that corresponds with and contributes to relevant myocardial and neuromuscular abnormalities.
Potassium homeostasis can be conceptualized as having both an internal and an external balance. External balance refers to the total amount of potassium in the body and is determined by the balance between intake versus loss. Potassium enters the body principally through ingestion, and almost all ingested potassium is absorbed. A small amount of potassium is normally excreted in the feces, although the colon may serve as an important route for potassium excretion in disease states. Normal potassium loss occurs predominantly through the kidneys, the amount determined by ECF potassium concentration, delivery of sodium and water to the distal tubule and collecting ducts, as well as the secretion of aldosterone. Although the kidneys normally eliminate potassium efficiently, renal mechanisms cannot maintain normal plasma serum potassium concentrations when grossly excessive amounts of potassium are ingested.9
Internal potassium balance refers to the distribution of potassium between the ICF and ECF compartments. The Na-K ATPase enzyme pump found in cellular membranes actively transports potassium into the cell and maintains a high intracellular potassium concentration (i.e., approximately 150 mEq/L) relative to the ECF (approximately 4 mEq/L). In hyperkalemic states insulin and β2-adrenergic activity augment the transport of potassium into the cells, safely storing additional potassium until it can be eliminated.9 The internal balance mechanisms may be thought of as a temporary measure designed to allow the kidneys and, to a lesser extent, the colon time to restore potassium balance.8,10
Acid–Base Balance
The body must deal with very large amounts of acid (H+) that are generated daily by normal metabolic processes. This is important because H+ is very reactive, and small amounts (i.e., nanoequivalents) can have detrimental effects on protein structure and function (i.e., enzymes, cell membranes, and receptors). Normal blood pH is 7.40. Many metabolic functions are exquisitely sensitive to pH, and normal function can only occur within a very narrow pH range. If, for example, a patient’s pH falls to 7.20 (which represents an increase of approximately 20 nEq H+/L), myocardial dysfunction, vasodilation, or dysrhythmias may be observed.11,12
Changes in [H+] are opposed by buffer systems within the body. These systems consist of a buffer pair of an acid (H+ donator) and its conjugate base (H+ acceptor) as follows:
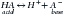
Weak acids and their conjugate bases constitute the most effective buffer pairs in the body, insofar as they are minimally dissociated and readily capable of accepting or donating H+ in the presence of changes in H+ load. This is in contrast to strong acids, which are highly dissociated in most biological fluids.
It was noted 100 years ago that the hydration of CO2 in the presence of carbonic anhydrase forms H2CO3 (carbonic acid), and carbonic acid acid will ionize into and achieve equilibrium with its conjugate base, bicarbonate (HCO3−), and H+, almost instantaneously:
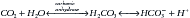
By rearranging the variables in the preceding equation, applying laws of mass action, and recognizing that H2CO3 exists in almost continuous equilibrium with dissolved CO2, the value of dissolved CO2 was substituted, giving rise to the following equation:
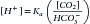
Ultimately, the partial pressure of CO2 in the blood (pCO2) was substituted for dissolved CO2, the concept of pH was defined (the negative logarithm of [H+]), and the following equation was derived:
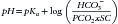
where pKa is the logarithm of the ionization constant Ka for H2CO3 and SC is the solubility coefficient of CO2 in blood (0.03).
This is the classic Henderson–Hasselbalch equation, fundamental to appreciating traditional acid–base chemistry and interpreting acid–base derangements. This equation shows that the pH of the ECF varies when either [HCO3−] or pCO2 is altered.
There are two categories of acid found in the body: nonvolatile (produced by metabolism of proteins) and volatile (derived from CO2) produced by cellular respiration throughout the body.11 Nonvolatile (also called fixed) acid (H+) is primarily excreted by the kidneys, whereas volatile acid (CO2) is eliminated by way of the lungs. On a daily basis, small pH changes within the body are counteracted by multiple complex and often opposing physiochemical processes that, for the sake of simplicity, are presented as (1) the actions of intracellular and extracellular buffering systems (chemical buffering), (2) modulation of ventilation (physiologic buffering), and (3) renal reclamation and elimination. There are many buffer systems throughout the body. The principle intracellular buffers are hemoglobin, phosphate, and proteins, which are capable of buffering both volatile and nonvolatile acids. The principle extracellular buffer systems are calcium carbonate and phosphate of bone, in addition to the bicarbonate-carbonic acid system, which reacts only with nonvolatile acid. Hemoglobin and albumin account for most of the nonbicarbonate buffering capacity. The HCO3− buffering system is capable of responding to an acute change in [H+], and the HCO3−/H2CO3/CO2 equilibrium equation (see Equation 16-1) allows changes in pH to be further modulated by changes in ventilation. This “open system” greatly enhances the buffering capacity of the HCO3− system and is capable of buffering changes in pH within minutes of an acid or alkali load. Finally, the kidneys play a major role in maintaining pH by increasing or decreasing acid elimination in the urine. Although hours to days may be required for this system’s buffering ability to reach completion, it is clinically the most reliable of all the adaptive mechanisms to normalize pH.
Acid–base physiology may be viewed as having an external and an internal balance, as described for potassium. External balance refers to the elimination of acid from the body, whereas internal balance allows for the body to safely sequester excessive acid until it can be excreted. The pH of the ECF is largely determined by how effectively these mechanisms function.
A perceived limitation to traditional acid–base physiology is that it is too simplistic, ascribing changes in pH to either respiratory (changes in pCO2) or metabolic (changes in bicarbonate) causes, thereby neglecting the contribution of other variables in solution. To address this, nontraditional or quantitative approaches to acid–base chemistry and analysis are described.13 The terms Stewart approach and Fencl–Leith approach are variants of the often confusing and misunderstood nontraditional approaches to acid–base analysis. The nontraditional approach subdivides metabolic causes into two independent variables, those associated with a strong ion difference (SID) ions that are totally dissociated (e.g., sodium, chloride, potassium, lactate) and ATOT (ions of weak acids that are partially dissociated, e.g., phosphate and albumin). The nontraditional approach defines the pCO2 (respiratory contribution) as an independent variable. According to the quantitative approach, changes in HCO3 are explained by alterations in SID, ATOT, and pCO2. It has been demonstrated that if enough ATOT and SID determinants are measured, an approximate pH can be calculated. The perceived strength of the quantitative approach is that it provides an estimate of the “contribution” of individual electrolyte derangements to an acid–base imbalance when pH is not measured. However, the quantitative approach is only a means of estimating the metabolic contribution to pH when it is not otherwise measured, but not the pH itself; the pH is also highly dependent on the respiratory component, the pCO2. The traditional system utilizes an overview parameter such as bicarbonate, total CO2, or base deficit to quantify the metabolic contribution. The Stewart approach breaks down the metabolic component into two large groups: SID and ATOT; however, it does not further define the contributions. The Fencl–Leith modification is the only truly semiquantitative approach, insofar as it it breaks down the metabolic contribution into six individual components (water [marked by serum sodium], bicarbonate [marked by chloride], albumin, lactate, ketones [if measured], phosphate, and the effect of unmeasured ions). Advocates of the nontraditional approach find that it is more accurate, avoiding some of the oversimplifications associated with the traditional, albeit more familiar, Henderson–Hasselbalch equation. Both are simplifications of a complex dynamic system, and both suffer from inherent inaccuracies.13 For practical purposes, benefits of the nontraditional approach are best appreciated when pH and a blood gas analyzer are not available to the clinician. Other advantages to this approach, when compared with traditional acid–base chemistry, are less clear and remain a topic of recurrent deliberations. Importantly the astute clinician should appreciate that all methods of acid–base analysis merely quantitate the magnitude of a disorder. Similarly, acid–base findings may provide subtle clues to pathogenetic mechanisms, but they do not characterize or define a disease or its management. Such inferences mandate evaluation of the complete history, thorough physical examination, and results of other laboratory tests. To remain consistent with concepts taught in most clinical settings, the traditional approach (Henderson–Hasselbalch) to analyzing acid–base disturbances is discussed.
The blood bicarbonate concentration is normally regulated by the kidneys. The kidneys reabsorb filtered bicarbonate and regenerate bicarbonate that has been consumed in the process of buffering acid. In this way, bicarbonate may be conceptualized as a kind of conveyer belt, combining with nonvolatile acid (i.e., H+), thereby maintaining a stable pH in the face of an acid load. When bicarbonate combines with H+, carbonic acid (H2CO3) is formed, which dissociates to form CO2 and water. CO2 is eliminated by ventilation (see Equation 16-1). When organic anions (e.g., lactate, pyruvate, gluconate, acetate, citrate, ketones) are metabolized, a hydrogen ion is carried along in the process. This alters the carbonic acid H2CO3 ↔ H+ + HCO3 equilibrium (mass action), shifting it rightward, generating new bicarbonate. Many processes in the body directly or indirectly use a chloride–bicarbonate counterporter. When chloride is excreted (e.g., into the stomach lumen), a “new” bicarbonate anion moves in the opposite direction (into the ECF). Similar counterporters exist in red blood cells, renal tubules, and small intestinal epithelial cells.
When nonvolatile acid is present in excess, it reacts with bicarbonate and other buffer systems, especially intracellular protein and phosphate.14 The plasma bicarbonate concentration decreases as the bicarbonate combines with H+ and forms H2CO3, the pH decreases, which by definition is an acidosis. More specifically, it is a metabolic acidosis because the disorder promotes the accumulation of excessive nonvolatile acid. The body will try to reestablish pH by lowering the pco2 (hyperventilation). This is the appropriate and anticipated compensatory response. Although less common, excessive loss of HCO3− (as occurs with some types of diarrhea) may cause a metabolic acidosis with a similar compensatory response.15 When the primary derangement promotes accumulation of excess volatile acid (an increase in pCO2), pH decreases and the acid–base disorder is described as a respiratory acidosis. Respiratory acidosis, by definition is an increase in pCO2,. In patients with primary respiratory acidosis, the kidneys respond by increasing excretion of acid.15
By definition, metabolic alkalosis is an elevation in pH caused by excessive plasma bicarbonate. Alkalosis decreases pulmonary ventilation, which elevates the pco2 Conversely, reduction of pco2 produces an alkalosis, the terms hyperventilation and respiratory alkalosis often used interchangeably. In response to a primary respiratory alkalosis, renal acid excretion is reduced.16
The kidneys respond to respiratory acid-base disorders over several hours to days. The pulmonary response to metabolic acid-base disorders occurs in a matter of minutes. In dogs it is possible to predict the approximate degree of a compensation; anticipated compensation is less clear or predictable in cats. Compensatory mechanisms are not efficient enough to return the pH within the normal range. These defense mechanisms do not correct the acid-base disturbance but merely minimize the change in pH imposed by the disturbance. Moreover, overcompensation for a primary acid–base disorder does not occur,11
Disease-Induced Changes
Sodium
Changes in plasma sodium concentration usually reflect changes in total body water content. Hyponatremia and hypernatremia primarily cause clinical problems by promoting neuronal edema or dehydration (ICF), respectively. The severity of clinical signs is thought to correspond with the rate of change rather than with the magnitude of hyponatremia or hypernatremia. If changes in serum sodium occur slowly, the brain can usually adjust the number of osmotically active molecules to prevent cerebral fluid gain or loss. This underscores the fact that severe hyponatremia or hypernatremia should be corrected gradually rather than quickly.17 When the serum sodium abnormality has been gradual in onset, the patient’s sodium should be corrected slowly, not lowered faster than 1 mmol/L/hr or increased at a rate exceeding 0.5 mmol/L/hr.
Hypernatremia
Naturally occurring disease processes resulting in hypernatremia are usually disorders of free water loss.7,18 Rarely, hypernatremia results from the addition of sodium (e.g., ocean water ingestion, hypertonic saline or sodium bicarbonate administration). Hypernatremia is a reflection of inadequate water relative to sodium content in the ECF. While ECF volume is determined by the total sodium content (not concentration) in the body, plasma sodium concentration is not a measurement of a patient’s volume status. The thirst mechanism is so effective that hypernatremia seldom occurs in animals that have access to adequate amounts of water and are not vomiting or regurgitating. Hypernatremia often develops in hospitalized patients, and a methodical preemptive diagnostic approach, including serial evaluation of electrolytes and quantification of fluid intake and loss, permits rapid recognition of water imbalance and prevent wide variations in serum sodium. Hypernatremia should prompt consideration of the disorders listed in Box 16-1.
Hyponatremia
Because naturally occurring diseases rarely produce sodium losses in excess of water, hyponatremia in the clinical setting is almost invariably due to excess water in the ECF.
Hyponatremia may be caused by several mechanisms, and a systematic approach to cause and correction is advised. Sodium and its attendant ions account for approximately 95% of the osmotically active substances in extracellular water. While hyponatremia is commonly associated with hypo-osmolality, plasma osmolality must be assessed to appropriately determine of the cause of hyponatremia (Box 16-2).7
Hyponatremia with concurrent normal plasma osmolality suggests laboratory error or pseudohyponatremia. Pseudohyponatremia may occur with hyperlipidemia because the lipid occupies space in the volume of serum obtained for analysis; the concentration of sodium itself is not affected. Hyponatremia with concurrent plasma hyperosmolality suggests that other osmotically active particles (e.g., glucose, mannitol) are drawing water out of the ICF and into the plasma, thus diluting the sodium that is present.7,19 This is often called translocational hyponatremia because it is caused by translocation of sodium across cell membranes.
Hypoosmolar hyponatremia in a patient producing large volumes of dilute urine reflects impaired free water excretion (e.g., inappropriate ADH secretion).
Volume and hydration status may further suggest causes of hyponatremia. Overtly hypovolemic patients often have an obvious source of fluid loss. Hyponatremia in an overhydrated patient is often observed with (but is not the cause of) cavitary effusions associated with congestive heart failure, advanced oliguric renal failure, or severe hepatic disease (usually cirrhosis). These disorders may be associated with such exuberant water retention that patients become hyponatremic in the face of total body sodium excess. Patients with congestive heart disease or cirrhosis are often described as having decreased effective circulating volume (ECV). ECV is the volume of arterial blood effectively perfusing tissue. ECV is a dynamic quantity and not a measurable, distinct compartment. The fluid retention seen with ineffective cardiac output is appropriate (because of release of ADH and angiotensin II), whereas the fluid retention observed with renal failure and cirrhosis is a component of the disease process itself. These patients have decreased ECV and consequent reduction in GFR and RBF; as a result, the body attempts, albeit ineffectively, to retain the fluid within the ECF.7
Hypoosmolar hyponatremia may be seen with chronic gastrointestinal disease. These patients may be hypernatremic early in the course of their disease, but sustained ADH release, excessive water consumption, or both may ultimately lead to hyponatremia. Diseases associated with third-space losses (e.g., pancreatitis, peritonitis) promote ADH release, and hyponatremia may be seen in affected patients. It is a common misunderstanding that hyponatremia in this setting is a result of the fluid accumulation itself. The hyponatremia, however, is due to changes in ADH, and fluid accumulation is not the proximate cause. Cutaneous fluid losses (e.g., burns) may be isonatremic with little change in plasma sodium or may be associated with hyponatremia.7,19
Similarly, sodium may be lost from the body as a result of hypoadrenocorticism or following administration of thiazide diuretics. Diuretics decrease the kidney’s diluting capacity and increase sodium excretion. These patients may be distinguished from those with gastrointestinal loss of hypotonic fluid by evaluating the urinary sodium concentration (fractional excretion of sodium [FE Na+]). FE Na+ is not a test but rather a calculation based on the concentrations of sodium and creatinine in blood and urine.
Under normal circumstances, the body’s response to hypovolemia and hyponatremia is to impede renal sodium losses as a means of restoring ECF. Consequently, little sodium would be found in the urine (FE Na+ <1%). Animals in which renal losses are the cause of the hyponatremia will, however, have substantial urinary sodium concentrations (FE Na+ >3%).19
Hyponatremia and hypoosmolality occur uncommonly in well-hydrated euvolemic dogs and cats. Although rare, primary polydipsia (also called psychogenic polydipsia) syndromes associated with inappropriate ADH release or overzealous administration of hypotonic fluids are described, and are possible causes of hyponatremia in this setting.7
Chloride
Most often, changes in plasma chloride concentration are clinically relevant because they affect acid-base status. In particular, hypochloremia is associated with metabolic processes that are coupled with alkalosis.20 Similarly, hyperchloremia tends to be associated with acidosis.
Hypochloremia
The major causes of hypochloremia are increased loss caused by vomiting of gastric contents or excessive administration of loop diuretics (e.g., furosemide).21 Chloride losses caused by gastric vomiting are usually associated with hypokalemia and metabolic alkalosis. Occasionally, a paradoxical aciduria is also seen. Physical examination and laboratory findings often corroborate a history of vomiting. Rarely, administration of large doses of sodium without corresponding administration of chloride (e.g., high doses of sodium penicillin or sodium bicarbonate) may cause hypochloremia. When a hypochloremic patient is also hyponatremic, hyperkalemic, or both, the clinician should consider hypoadrenocorticism. If, however, the clinician corrects the plasma chloride concentration to account for changes in plasma free water, for example:
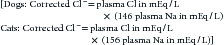
patients with hypoadrenocorticism may have a normal or increased corrected chloride concentration.20 Changes in chloride are often interpreted with changes in free water, which alters sodium and chloride concentrations proportionally. This can be done by correcting the chloride concentration for changes in sodium using the preceding formulas. Primary disorders of chloride (i.e., acid–base disturbances) have abnormal corrected chloride values, whereas disorders of free water do not; determination of corrected chloride values seldom affects diagnostic or therapeutic decisions in a given patient. Its determination is only to permit the interested clinician to quantitatively assess the contribution of bicarbonate (marked by changes in chloride) to the metabolic component of an acid–base disturbance.
Hyperchloremia
Hyperchloremia is principally found in animals that are simultaneously hypernatremic as a result of the loss of free water and in patients that have received fluids containing proportionally more chloride than sodium relative to plasma values. Examples include administration of 0.9% saline, hypertonic saline, parenteral nutrition, and especially 0.9% saline supplemented with potassium chloride. Hyperchloremia may also be observed in patients that have a metabolic acidosis associated with a normal anion gap (i.e., hyperchloremic metabolic acidosis). This biochemical finding is commonly seen in animals with small bowel diarrhea caused by loss of bicarbonate-rich, chloride-poor fluid.20 It can also be seen with regurgitation or vomiting of duodenal contents and in patients with renal tubular acidosis.
Potassium
Changes in the plasma potassium concentration are clinically important because of the effect of potassium on cellular metabolism, RMPs, and the subsequent strength of contraction when an action potential is generated.9 Hypokalemia and hyperkalemia both cause muscular weakness and predispose to cardiac dysrhythmias. Although there is variation between patients, plasma potassium concentrations ≥ 8.0 mEq/L or ≤ 2.0 mEq/L are generally considered life threatening, although less dramatic changes can be dangerous if there are concomitant electrolyte derangements (e.g., hyponatremia, hypocalcemia, or hypercalcemia) or if the changes occur rapidly.10 Potassium is highly labile, and changes in pH of the ECF may cause significant alterations in serum potassium, particularly in critically ill patients.
Hyperkalemia
Hyperkalemia may be artifactual (i.e., pseudohyperkalemia) or real. If hyperkalemia is real, it is either iatrogenic (increased intake or administration) or spontaneous. The major cause of spontaneous hyperkalemia is decreased urinary excretion caused by renal, urinary tract or adrenal disease (Box 16-3).10
Box 16-3 Causes of Hyperkalemia
Artifactual
Pseudohyperkalemia, an in vitro increase in potassium concentration, may be observed with marked thrombocytosis (generally ≥ 800,000/ul). Because the elevation in potassium in this setting is due to leakage from disrupted or dying cells, it is important to measure plasma or serum potassium concentrations shortly after a sample is obtained to avoid drawing erroneous conclusions about a patient’s electrolyte status. Similarly, animals with white blood cell counts equal to or greater than 100,000/ul (e.g., leukemia) may rarely have pseudohyperkalemia because of transcellular leakage of potassium.22,23 Neonates, Akitas, and English springer spaniels are known to have a high potassium content within their red blood cells and hemolysis may cause hyperkalemia 24 If there is doubt as to whether the hyperkalemia is artifactual or real, one should obtain a lithium heparin–anticoagulated blood sample and promptly harvest the plasma.21
The list of drugs that may cause hyperkalemia is extensive.10 With the exception of overexuberant infusion of potassium chloride in intravenous fluids and administration of drugs that inhibit production or activity of aldosterone (e.g., enalapril, spironolactone), significant hyperkalemia caused by drug administration is rare and generally only occurs if there is underlying renal or adrenal dysfunction. Only the more common causes are provided in Box 16-3.
Decreased urinary potassium excretion may be due to severe primary renal dysfunction (e.g., typically anuric or oliguric primary renal failure), postrenal causes (e.g., uroperitoneum, uretheral obstruction) or to secondary renal dysfunction resulting from aldosterone deficiency (i.e., hypoadrenocorticism). Disorders promoting development of edema or cavitary effusions or enteritis (especially whipworm infection)25-27 are sometimes associated with the development of mild hyperkalemia, the exact pathogenetic mechanism of which remains unclear. The clinician should rule in or rule out hypoadrenocorticism early in the diagnostic evaluation because it has an excellent prognosis if recognized and treated promptly. An adrenocorticotropic hormone (ACTH)–stimulation test is required for definitive diagnosis, although resting serum cortisol concentrations may be used to screen for its presence. Dogs with resting cortisol levels of more than 2 ug/dL rarely have hypoadrenocorticism,28 and in such cases the submission of a post-ACTH (stimulated) cortisol sample may not be indicated.
Hypokalemia
Hypokalemia results from reductions in dietary intake or increased losses through the urinary or gastrointestinal tract. Decreased intake is unlikely to cause hypokalemia unless the diet is severely deficient or when potassium-free intravenous fluids are administered to an inappetent patient. Potassium translocation from ECF to ICF may be seen in patients receiving parenteral nutrition, insulin, sodium bicarbonate, or glucose-containing fluids.8,9 Similarly, alkalemia may reduce serum potassium levels as extracellular potassium moves intracellularly in exchange for hydrogen ions.
Increased loss of potassium is the most important cause of hypokalemia in dogs and cats. Vomiting and diarrhea lead to gastrointestinal losses, whereas drug therapy (e.g., furosemide) and renal failure or polyuria result in renal losses. The latter is commonly observed in older cats and may be detected in the absence of azotemia. Hyperaldosteronism is an uncommon disorder of cats that may cause profound hypokalemia. If the cause of hypokalemia is unclear after routine evaluations, the clinician may calculate the fractional excretion of potassium (FEK). Animals that are hypokalemic as a result of nonrenal causes should have normal (approximately 4% to 6%) to decreased FEK, whereas cats with potassium-losing nephropathies usually have values above 6% to 10%.8,29,30
Miscellaneous Minerals
Miscellaneous minerals that occasionally are of concern to the clinician are phosphorus and magnesium. Severe hypophosphatemia capable of causing clinical signs is principally seen in ketoacidotic diabetic patients that are overtreated with insulin and occasionally in emaciated cats receiving enteral or parenteral nutrition,31,32 although hypophosphatemia can be seen from time to time in patients with other diseases.33 The most clinically relevant consequence of hypophosphatemia is hemolysis. Hyperphosphatemia is commonly seen in patients with renal disease but seldom requires special considerations when formulating fluid therapy. Magnesium is primarily absorbed in the jejunum and ileum, while the kidneys regulate magnesium balance. Hypomagnesemia is primarily due to renal or gastrointestinal losses.34 Hypomagnesemia may cause cardiac dysrhythmias and neuromuscular irritability. Hypomagnesemia is increasingly recognized feature of ketoacidotic diabetes mellitus. It is also seen in dogs with severe protein-losing enteropathy.35 Depletion of magnesium has a permissive effect on potassium exit from the ICF leading to ECF accumulation of potassium, which is subsequently lost from the body. This potassium deficiency is refractory to supplementation until the magnesium deficit is also corrected. Similarly, hypocalcemia may occur as a secondary electrolyte abnormality when there is a magnesium deficit. Hypermagnesemia is primarily associated with renal disease. Excessive provision of magnesium (e.g., in diet or intravenous fluids) is also reported.36,37
Acid–Base Status
Traditional evaluation of acid–base status is best accomplished by blood gas analysis, integrating information provided by the pH, pCO2, and bicarbonate. When evaluating blood gas values, consideration is first given to pH.11 The normal pH of blood analyzed at 37° C is 7.35-7.45. If the pH is abnormal, is the patient acidemic (pH < 7.35) or alkalemic (pH > 7.45) If it is abnormal, by definition an acid–base disorder exists. If the pH is normal, an acid–base disorder is unlikely but should not be ruled out, because there may be a mixed acid-base disturbance (i.e., more than one primary acid–base disturbance occurring concurrently) or (uncommonly) a fully compensated acid–base disturbance. These circumstances are not always readily distinguishable. The reader is referred to other references for information on mixed disorders.38
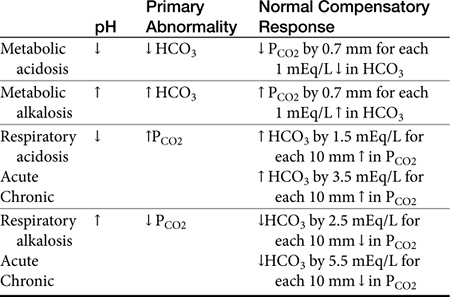
If the pH is abnormal, the clinician should try to determine which component (respiratory or metabolic) is the primary contributor (Table 16-1). Subsequently, evaluation for appropriate compensation of the primary disorder is undertaken. Generally, the pH will vary in the direction of the primary disorder. The other component is the secondary or compensatory component attempting to restore pH to normal. When both components vary in the same direction at the pH, both disorders are primary, i.e., a mixed disorder is present. In the dog there are guidelines for expected compensation in acid-base disorders (see Table 16-1); however, it is not possible to extrapolate these guidelines from the dog to the cat. Results of blood gas analysis are interpreted in light of anamnesis and clinical examination findings to most appropriately determine the most probable cause of the acid–base disorder.11
Metabolic Acidosis
Metabolic acidosis is probably the most widely recognized canine and feline acid–base disorder. The most common causes of metabolic acidosis in these species are listed in Box 16-4. Acidosis from accumulation of lactic acid is thought to be the most common cause of metabolic acidosis. In the setting of poor peripheral perfusion, anaerobic metabolism often predominates, with subsequent production of lactic acid.39 The acidemia is not actually due to dissociation of the lactate; the lactate simply reflects the anaerobic metabolism that has released protons. Therefore lactic acidosis may be seen in conjunction with other disorders. Lactic acidosis is usually diagnosed by elimination of other causes of acidosis, presence of elevated anion gap, and physical examination findings suggesting ischemia or volume depletion. Blood lactate levels may be measured (i.e., hand-held monitors or standard blood gas analyzers).20,21,40 The anion gap, although reported with other serum chemistry measurements, is actually a calculated value [(Na + K) – (HCO3 + Cl)]. The electrolytes used in the formula are called measured cations (Na, K) and measured anions (HCO3, Cl). Ions not included in the formula are called unmeasured cations and unmeasured anions.
Most dogs and cats with diarrhea do not develop a significant acidosis. Occasionally, patients with profuse diarrhea lose excessive amounts of bicarbonate in the feces and consequently develop an inorganic (hyperchloremic) metabolic acidosis. This acidosis may occur simultaneously with lactic acidosis in some patients with concomitant dehydration or hypovolemia.20
Renal failure causes acidosis when the kidneys cannot adequately excrete H+, regenerate HCO3−, or both. Renal ammonium excretion is the principle means of eliminating protons, and this is usually adequate until renal failure becomes severe. Acidosis from renal dysfunction may have a normal anion gap initially, but an increased anion gap is expected in more severely uremic patients as unmeasured anions accumulate. Hypoadrenocorticism causes an acidosis in part because aldosterone is needed for the secretion of H+ into collecting duct fluid; thus it may be thought of as a functional renal disease (i.e., without structural nephron damage). Specific renal tubular defects (renal tubular acidosis) are uncommon in dogs and cats but should be on the list of differential diagnoses for patients with a hyperchloremic acidosis and normal anion gap.20
Diabetic ketoacidosis occurs when there is excessive production of ketone bodies, especially b-hydroxybutyrate, generally causing an increased anion gap acidosis.41
When ethylene glycol is ingested, it is metabolized by the liver to glycolic acid. This often results in severe acidemia that is refractory to standard treatments, largely because acid metabolites continue to be produced until all of the ethylene glycol is metabolized.42 The acidosis typically occurs before there is any evidence of renal injury. History may be informative, and several quantitative colorimetric in-house test kits are available. Calcium oxalate crystalluria often precedes overt renal failure. Both monohydrate (i.e., “picket fence”) and dihydrate (i.e., “Maltese cross”) calcium oxalate crystals may be observed in urine sediment. Markedly increased anion and osmolol gaps are seen shortly after exposure.43 A high anion gap acidosis may be documented within 3 hours and persists for at least 24 hours.
Metabolic Alkalosis
Clinically significant metabolic alkalosis in dogs and cats is usually due to vomiting of gastric contents, diuretic therapy, or excessive administration of bicarbonate. Other causes of alkalemia (e.g., severe hypokalemia, severe hypomagnesemia) are clinically less important.16,20
Vomiting of gastric contents causes loss of H+ as well as loss of Cl− and water. Hypovolemia and concurrent hypochloremia prevent the relative excess of bicarbonate from being eliminated in the urine, reflecting the body’s priority of restoring ECF volume. To restore ECF volume, the body reclaims sodium and water from the renal tubules. To efficiently reabsorb sodium (a cation), a negatively charged ion(s) must be reabsorbed (generally Cl− or HCO3−) simultaneously to maintain electroneutrality. If insufficient amounts of Cl− are present to accompany the reclaimed sodium, bicarbonate will be reabsorbed instead, even though total body bicarbonate is in excess.20 Similar pathogenic mechanisms may be observed when excessive furosemide administration promotes a brisk diuresis with chloride-rich urine. Both of these circumstances are considered chloride-responsive alkaloses because complete resolution can only occur if chloride-replete fluids are a component of therapy. Clinically important chloride-resistant alkalosis is uncommon in dogs and cats.20
Respiratory Alkalosis
Respiratory alkalosis, or primary hypocapnia, is relatively common, occurring when alveolar ventilation exceeds the amount required to eliminate the CO2 produced by metabolism; hyperventilation from any cause (e.g., excitement, pain, fear, hypoxemia, sepsis, liver disease) causes PCO2 to decrease and can result in an alkalosis.44,45 The alkalosis itself, however, rarely causes detrimental effects in the patient, but this should not dissuade the clinician from seeking out the underlying cause.
Respiratory Acidosis
Respiratory acidosis, or primary hypercapnia, is occasionally diagnosed in dogs or cats with naturally occurring diseases; more commonly it is associated with sedation and anesthesia. Carbon dioxide diffuses more rapidly across the alveolar membrane than oxygen, and hypoventilation is for all practical purposes the only cause of, and therefore defines, respiratory acidosis. Primary hypoventilation may be seen in patients with upper airway obstruction, neuromuscular weakness that impairs breathing (e.g., myasthenia gravis, hypokalemia, botulism), muscular rigidity that prevents breathing (e.g., tetanus, seizures), chest wall or pleural cavity disorders (e.g., pneumothorax, pleural effusion, flail chest), and chronic obstructive pulmonary disease.46
Blood Gas Analysis
Blood gas analysis mandates precise technique and thus properly trained support staff.11,21 Hand-held “point-of-care” units have made blood gas analysis feasible for many practices.47 However, the total serum CO2 (TCO2) available on most chemistry panels from commercial veterinary laboratories may be used as a rough surrogate marker for HCO3− and as such may be used as an approximation of the metabolic contribution to acid–base derangements when HCO3 is not measured.21
Although Tco2 is less expensive and is more widely available than blood gas analyzers, relying on this measurement requires the clinician to make assumptions that may be incorrect for a given patient.21 One must speculate whether an abnormal bicarbonate concentration is due to a primary metabolic or a respiratory derangement. Suppositions should be supported by careful evaluation of history, physical examination, and ancillary laboratory data. Major alterations in bicarbonate concentrations are often but not invariably primary events (i.e., low or high TCO2 representing primary metabolic acid–base disorders). It is reasonable to rely on the Tco2 provided that the apparent acid–base abnormality is consistent with what the clinician infers from the history and physical examination findings. However, compensation of primary disturbance cannot be assessed, particularly if a pH value is not available.
Fluids
Calculation
Fluid therapy is a complex topic and this section is not all encompassing. However, basic principles of fluid therapy are discussed. Planning fluid therapy requires consideration of maintenance needs, estimated hydration deficit, and assessment of ongoing losses.
Maintenance Fluid Needs
Maintenance fluid requirements (i.e., the amount of fluid necessary to replace insensible losses and obligatory urinary losses) vary with the animal’s size, age, and occasionally diet (i.e., eating a diet containing excessive solute such as sodium increases the patient’s water requirements for maintenance). Ambient temperature and humidity also affect maintenance requirements. Studies have been performed evaluating basal metabolic rate in companion animals. The optimal formula for determining the maintenance needs for dogs and cats is controversial. In general, smaller animals need more milliliters per kilogram daily than do larger animals. For most domestic species, estimates of daily water requirements range from 40 mL/kg/day (large dogs) to 60 mL/kg/day (cats and small dogs).48,49 The daily maintenance water requirement for a healthy cat or dog weighing between 6 and 60 kg is as follows: (30 x BW[kg]) + 70. Studies using indirect calorimetry suggest that for animals weighing less than 10 kg or more than 50 kg, a more precise formula is as follows: 97 × body weight (0.655).50 Water is present in foods and is generated as foods are metabolized. Maintenance requirements should be provided to patients not consuming sufficient quantities of water on their own.
The combination of variables (e.g., age, hydration status, perfusion, cardiac and renal function) makes it difficult to calculate exactly how much fluid an individual patient needs. In general, as long as the estimated amount of required fluids is provided and the patient is assessed frequently, clinically significant problems referable to fluid therapy are likely to be avoided. With the exception of the conditions listed in subsequent sections, providing slightly more fluid than is believed necessary is in most cases recommended insofar as it is more common to underestimate fluid needs than to overestimate them. However, patients with myocardial dysfunction, oliguric renal disease, severe anemia, severe hypoalbuminemia, or pulmonary edema can be seriously harmed by excessive fluid administration.
Deficit Fluid Needs
Fluid deficit should be estimated when patients are initially examined. Although there are guidelines for estimating degree of dehydration (Table 16-2), there are many pitfalls in this approach. Any excited or dyspneic animal that is breathing through its mouth may have dry, tacky oral mucous membranes, whereas nauseated animals may have moist membranes in the face of dehydration. Weight loss causes some degree of decreased skin turgor, while but obese animals may not have changes in skin turgor even when they are 8% to 10% dehydrated.51
Table 16-2 Determination of Degree of Dehydration
| Manifestation | Degree (%) |
|---|---|
| Loss of skin elasticity | 5 |
| Oral mucous membranes becoming tacky | 6-7 |
| Prolonged capillary refill time | 6-8 |
| Skin tenting that persists | 8-10 |
| Eyes sunken back into orbits | 10 |
| Cool extremities, early shock | 10-12 |
Findings of increased hematocrit and plasma proteins support the clinical suspicion of dehydration (i.e. animals that are hemoconcentrated and hyperproteinemic are usually dehydrated). However the clinician seldom knows what these parameters were shortly before the animal became ill, and the hematocrit and total protein values by themselves are not a reliable means to assess hydration status. Many animals with chronic illness have anemia of chronic disease; dehydration may cause them to have a spuriously normal hematocrit value. Likewise, patients can be hypoproteinemic, and dehydration may cause them to have a seemingly normal serum protein concentration. There are many possible causes for a well-hydrated dog or cat to be hyperproteinemic, generally due to elevated globulin fraction (e.g., heartworm disease, ehrlichiosis, chronic dermatitis, feline infectious peritonitis). Alterations in body weight may be useful in assessing changes in hydration status because changes in body weight generally reflect changes in fluid content. When treating dehydrated patients, the clinician may interpret an increase in body weight as an encouraging sign that hydration is being restored. However, an increase in body weight in a well-hydrated patient may be an early indicator of fluid overload. Patient history should not be overlooked. Any animal that is not drinking or eating but has ongoing losses (e.g., vomiting, diarrhea, polyuria, tachypnea) is or will become dehydrated.
In general, because of the problems associated with determining the degree of dehydration, slightly overestimating the deficit is recommended unless the patient has syndromes mentioned in the discussion of maintenance fluid requirements. The dehydration deficit is determined by the following formula:
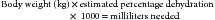
Deficit fluids can be administered more rapidly than maintenance fluids when patients are closely and frequently monitored. One half of the dehydration deficit can be administered as a bolus, with the remainder replaced as a constant rate infusion over 12 to 24 hours. Cats have smaller blood volumes per body weight than dogs (i.e., 50 to 60 mL/kg as opposed to 80 to 90 mL/kg in the dog) and can become fluid overloaded with smaller fluid doses than dogs. Once the estimated deficit is replaced, the animal should be re-examined, assessing for evidence of further deficits.51
Ongoing Losses
Ongoing losses can be divided into normal, insensible losses (e.g., from respiration, normal urine and fecal losses, skin evaporation) and those that are not normal (e.g., vomiting, diarrhea, polyuria, fever, tachypnea). In general, insensible respiratory losses involve water but not electrolytes (i.e., “free water”), whereas abnormal losses often involve electrolytes as well as water. Therefore the clinician should characterize the nature of fluid losses when choosing a fluid to administer. Careful assessment of body weight (i.e., with an accurate scale that measures ounces or tenths of a pound) remains a reliable means of monitoring for ongoing fluid balance in an individual patient. It is also important to weigh the animal after it has urinated and before it is fed. Because approximately 60% of body weight, rapid changes in body weight usually reflect changes in body water content as opposed to muscle mass or fat. One kilogram represents approximately 1000 mL of water. Frequent measurement or close estimation and matching of “ins and outs” (e.g., oral intake, parenteral fluids, urine production, vomiting, and diarrhea) can help prevent gross errors in approximation of patient requirements.
Choice of Fluid
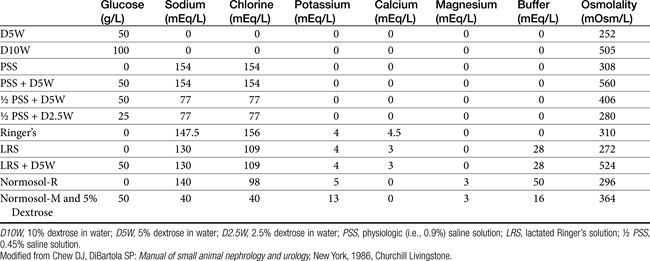
There are several categories of fluids and additives available (Box 16-5). Crystalloid solutions (e.g., physiologic saline solution [PSS; 0.9% saline solution], 5% dextrose in water [D5W], Ringer’s solution) are composed of electrolytes and nonelectrolytes that can pass freely out of the vascular space. Isotonic replacement crystalloids have an electrolyte composition similar to that of extracellular fluid, with a relatively high sodium and low potassium concentration. The terms high and low refer to the concentrations of sodium and potassium relative to each other in the fluid bag itself; it is important to note that the sodium and potassium are both essentially normal when compared with ECF and plasma. The most widely available isotonic replacement solutions are 0.9% saline, lactated Ringer’s solution (LRS), Plasmalyte A (Baxter Healthcare Corp), and Normosol-R (Hospira, Inc). In true hypovolemia a very small concentration gradient exists between ECF and ICF spaces. Consequently, water shifts do not occur across the cell membrane. Intravascular crystalloid equilibrates with the interstitial space, with 20% to 25% of the infused volume remaining within the intravascular space 1 hour after infusion. Metabolism of the lactate in LRS or acetate and gluconate in Normosol-R or Plasmalyte A provides base to the body. When used for long-term therapy, there is a tendency for patients to develop mild to moderate hypokalemia. Normal ongoing losses typically have lower sodium and higher potassium than normal ECF, and therefore administering replacement solutions does not provide adequate potassium. Hypernatremia is less often encountered with use of replacement solutions for ongoing losses, unless renal sodium excretion is impaired. Thus potassium supplementation is almost always indicated, but switching to a commercially available isotonic maintenance fluid with a lower sodium concentration (e.g. Normosol-M [Hospira, Inc]) for long-term therapy after rehydration is not always necessary. Commercially available maintenance solutions (Table 16-3) are designed to fulfill the electrolyte requirements of patients with normal daily losses that are unable to maintain adequate fluid and electrolyte intake, and may be used to replenish obligate and insensible net ongoing losses. As these fluids have a relatively high potassium concentration (13 mEq/L), they should not be administered rapidly (0.5 mEq/kg/hr [suggested maximum rate of potassium administration] × 13 mEq/L = 38.5 mL/kg/hr = maximum rate of administration). Hypotonic solutions include Normosol M (Hospira), D5W, and 0.45% saline. Although the osmolality of D5W is 252 mOsm/L (which is close to being isotonic), the glucose is rapidly taken up by cells and metabolized after it is administered to the patient. Therefore administering glucose solutions is essentially equivalent to giving free water (unless hyperglycemic diuresis with subsequent dehydration results). Administration of D5W does not significantly contribute to meeting caloric needs (D5W has 170 kcal/L) and it is not intended to treat severe hypoglycemia. Symptomatic hypoglycemia is initially treated with bolus injection of 25% or 50% dextrose. Subsequently, adding a sufficient volume of 50% dextrose to a balanced isotonic crystalloid to produce a final dextrose concentration of 2.5% to 5% is usually sufficient for maintaining glycemic status on a non-emergent basis. Rapid infusion of hypotonic solutions can cause severe dilution of serum electrolytes, especially sodium. Although there are rare indications for hypotonic fluids (e.g., recalcitrant hypernatremia or as a vehicle for delivery of specific drugs), they are seldom required in most clinical situations. Infusion of hypertonic (7.5%) saline creates a large osmotic gradient, and water is drawn from the intracellular and, to a lesser extent, interstitial compartment causing a rapid, albeit transient (<30 min), expansion of intravascular volume. Combining hypertonic saline with 6% hetastarch or dextran 70 prolongs the beneficial effects. Hypertonic solutions are reported to be safe and effective for the treatment of hypovolemic hypotension and may have a role in minimizing intracranial pressure. Their use is contraindicated with hypernatremia and dehydration, and as is true with all crystalloids, excessive volume is associated with cardiac failure. Crystalloids containing preservatives (e.g., benzyl alcohol) are not recommended because they may have adverse effects, especially in cats.52
Colloidal solutions (e.g., dextran 70, hetastarch 6%, Oxyglobin) are retained in the intravascular space to a greater degree than crystalloids and draw fluids from the cellular and interstitial compartments into this compartment.51 Although both colloids and isotonic crystalloids can be used to rapidly expand intravascular volume in animals in shock, colloids are generally more effective (and effective at lower doses) than crystalloids.53 It generally requires two to four times as much isotonic crystalloid solution to expand the ECF compartment as it does with dextran or hetastarch. Hetastarch and dextrans are primarily indicated for rapid volume expansion. Dextrans and hetastarch may also be used to maintain plasma COP in patients with severe hypoalbuminemia. Anaphylactic reactions have been reported with both hetastarch and dextrans, but they are uncommonly observed. Low-molecular-weight dextrans (i.e., dextran 40) have been reported to cause renal failure, and they are not routinely employed in clinical practice. Both hetastarch and dextrans may also cause coagulation abnormalities when administered at high doses. Clinically significant hemorrhage is rare.
Purified hemoglobin both acts as a strong colloid and increases the oxygen-carrying capacity of the blood. Stroma-free, hemoglbin-based, oxygen-carrying solutions (e.g., Oxyglobin) are used to treat anemia in a variety of species. Indications also include volume resuscitation in hypovolemic states. The greatest advantage of Oxyglobin is its ability to carry oxygen to tissues and offload oxygen more effectively than blood because it is not limited by red cell flow; it has also been shown to improve microvascular perfusion, thus improving oxygen tension in injured tissues. Human serum albumin (HSA) obtained from purified human plasma is available commercially and has been used in critically ill companion animals with pancreatitis, peritonitis, acute hepatic failure, and protein-losing enteropathy. In addition to its role in maintaining colloid osmotic pressure, albumin has many other properties that may benefit critically ill patients, including maintenance of the selective permeability of the microvascular barrier, and as a carrier of a number of substances, including bilirubin, fatty acids, hormones, and drugs. Most commercially available albumin products are prepared as 5% or 25% solutions. The chemical structure of human albumin is not identical to canine or feline albumin, and type III hypersensitivity reactions, which are sometimes severe and fatal, are reported after the administration of HSA in healthy dogs.54 Until further studies suggest otherwise, routine use of HSA is discouraged unless all other means of restoring albumin or colloid osmotic pressure have failed. Lyophilized canine albumin has recently become available commercially. If this product is shown to have a favorable side effect profile (compared with human albumin products), it would be expected to prove useful in the management of severely ill hypoalbuminemic dogs with correspondingly low colloid osmotic pressure.
Clinicians often need to tailor a fluid for a specific patient. This is usually done by adding potassium chloride, 50% glucose, potassium phosphate, calcium gluconate, calcium chloride, magnesium sulfate, sodium bicarbonate, or a combination thereof. The specific amounts are discussed under specific disorders.
Route of Administration
Fluids may be administered orally, intravenously, subcutaneously, intraosseously, or peritoneally.55 Whenever oral intake is inadequate or not feasible (e.g., subject refuses to drink adequate volumes, vomits, does not absorb fluids sufficiently quickly for desired effect), parenteral administration is preferred. Intravenous administration is the quickest way to replenish an underexpanded ECF and is the standard of care for acute replacement of fluid losses (e.g., due to shock) or maintaining ECF volume during anesthetic procedures. This route allows uninterrupted infusion, which is expected to be advantageous to severely ill, hemodynamically unstable animals. Venous catheter access can be difficult because of the patient’s size or temperament, vascular collapse, or prior use of veins for catheters or venipuncture. Routine catheter care is mandatory to prevent infection, phlebitis, extravasation, and clotting within the catheter lumen.
Subcutaneous fluid administration is technically easier than intravenous administration and is usually adequate when the need for fluids is not severe or acute. Owners can usually be taught to administer subcutaneous fluids at home. From 50 to 200 mL may be injected per site, and several sites may be injected at one time. Poor absorption may be observed if excessive fluids are administered at one site. Although more commonly administered once daily, subcutaneous fluids may be given as frequently as every 6 hours if clinically indicated and tolerated by the patient. If fluid from the prior injections has not been absorbed, however, it is necessary to determine the reason before more fluids are administered. Severely dehydrated animals may absorb subcutaneous fluids very slowly because of poor peripheral vascular perfusion. Furthermore, administration of hypertonic or irritating solutions under the skin is discouraged, because fluids may be drawn from the central compartment or cause injury to local and adjacent tissues.55 Subcutaneous fluids are often considered to be relatively safe in patients with heart disease, but this is an incorrect assumption and fluid overload may rapidly ensue.
Intraperitoneal administration permits the infusion of large volumes of fluids, and absorption generally occurs more quickly than with subcutaneous administration. This route is occasionally used for neonates whose veins are not readily accessible with a catheter. The clinician can administer a warmed (not hot) isotonic fluid aseptically with a 23- to 20-gauge needle or similar bore size over-the-needle catheter. Fluids given by this route may be administered over a short period (i.e., less than 5 minutes) until the abdomen becomes obviously distended (usually about 20 mL/kg). Patient discomfort and respiratory distress indicate that an excessive volume was administered into the peritoneal cavity. Aseptic technique must be used so that bacteria are not introduced. On balance, intraperitoneal administration does not offer clear advantages over intravenous, intraosseous, or subcutaneous routes, which are preferred.
Intraosseous administration56 is accomplished by using either a specifically designed needle, a bone marrow aspiration needle, or an 18- to 20-gauge spinal or hypodermic needle. The needle is inserted into the marrow cavity of the humerus or femur; less common sites are the tibia and ilium. Fluids are then administered as for intravenous administration. Gravity drip rates of approximately 10 mL/min may be reached. Absorption occurs more quickly than with subcutaneously administered fluids, and it may be easier to obtain access to the marrow cavity in very small animals than to the jugular or cephalic vein.55-58 In rare cases pain may be seen as a consequence of infection, administration of cold solutions, or excessive administration rate.
The administration of crystalloid solutions rectally as an enema is described, because the normal colon will avidly absorb intraluminal water. This technique is seldom used in the clinical setting for provision of fluid deficits. Moreover, intravenous administration is mandatory for volume-depleted patients, and subcutaneous fluids are so easy to administer that there is essentially no reason for rectal administration. However, it may be used in select emergencies to modify core body temperature in a severely hypothermic or hyperthermic patient.
Determining Adequacy of Fluid Therapy
Fluid requirements should be evaluated and adjusted regularly. Generally fluid therapy is not abruptly discontinued but rather is gradually tapered to maintenance, or lower than maintenance rates over 12 to 24 hours. A common mistake with long-term intravenous fluid therapy is failing to adjust fluid rates as the condition of the pet changes. Close monitoring of body weight is useful to corroborate the observed efficacy of fluid therapy. Physical examination parameters compatible with dehydration (e.g., skin turgor, dry oral mucous membranes) should improve with appropriate fluid therapy, assuming there are no intercurrent disease processes associated with weight loss or development of tachypnea. Frequent observation of respiratory rate and effort and thoracic auscultation are expected to permit early detection of pulmonary edema. Similarly, new cardiac murmurs or gallop rhythms not audible before fluid therapy was instituted often presage overt clinical signs of fluid overload.
Because urine output is affected by myriad factors, it is not possible to define what constitutes normal urine output for many patients. As a general rule, urine output should be 1 to 2 mL/kg/hr. Although urine output is seldom quantified, it should be apparent if a patient is producing reasonable volumes of urine that is not extremely concentrated. This is not reliable for patients with renal failure, and some attempt must be made to quantify urine output when oligoanuria is a concern (e.g., weighing urine-soaked diapers, catching all urine in ambulatory patients) if a urinary catheter and closed collection system is not in place. Central venous pressure (CVP) is a measure of the hydrostatic pressure within the central venous compartment that provides an assessment of intravascular blood volume and cardiac function. It is typically measured by a percutaneously placed jugular catheter, which has its tip in the cranial vena cava. The catheter can be attached to either an electric pressure transducer or a water manometer. When interpreted in concert with other diagnostic findings, CVP is most useful for guiding fluid therapy in seriously ill animals with oliguric renal disease or myocardial dysfunction.55
Electrolytes
Considerations for Therapy
Therapy for electrolyte disorders primarily consists of supplementing electrolytes or decreasing their plasma concentrations by promoting excretion, dilution, or sequestration. The major electrolyte disturbances capable of causing clinical signs in dogs and cats, and thus requiring therapy, are hypokalemia and hyperkalemia. It is rarely necessary to address hyponatremia specifically, except with regard to some patients with hypoadrenocorticism. Hypoadrenocorticism is relatively uncommon, but volume replacement and management of hyperkalemia are usually the major goals for such patients in a crisis. Similar to hyponatremia, hypernatremia usually does not require specific treatment and often will correct itself as the underlying disease is being treated.
Hyponatremia
Hyponatremia is almost universally due to retention of free water except in patients with hypoadrenocorticism. Primary sodium loss (e.g. diarrhea) is not common. Unless the hyponatremia is severe enough to cause clinical signs, the clinician should first identify and correct the underlying disorder. Neurologic clinical manifestations (due to an osmotic shift of water into brain cells) are rarely observed with acute hyponatremia. To prevent rapid changes in the patient’s sodium, the initial fluid chosen should have a sodium concentration similar to the serum sodium. It is recommended that sodium be raided no faster than 1 mEq/L/hour, although there is debate regarding the pace and degree of sodium correction. There is a risk of neurologic complications (myelinolysis), which typically occurs 3 to 4 days after aggressive correction of hyponatremia and may include weakness, ataxia, quadriparesis, and hypermetria.59,60 Buffered isotonic crystalloids such as LRS are generally preferable to PSS for this purpose because plasma normally has approximately 145 mEq Na/L and 110 mEq Cl/L. PSS contains 154 mEq of each per liter, whereas LRS has 130 mEq Na/L and 109 mEq Cl/L. The sodium concentration in Normosol-R (140 mEq/L) is slightly greater than that of LRS. Therefore administration of PSS adds too much chloride relative to the amount of sodium, although this is seldom clinically significant. It is rare that hypertonic saline solutions are needed to replace sodium in small animals.7,17
Hypernatremia
Spontaneous hypernatremia is almost always due to loss of free water, and is not a reflection of total body sodium content. Sodium gain (e.g., ocean water ingestion or overzealous fluid administration) is less common. If the patient is hypovolemic, rapid administration of isotonic crystalloid (including saline) is likely to cause rapid changes in sodium and therefore is discouraged. The initial fluid for correcting volume deficits should have a sodium equal to (or slightly below) that of the patient so as not to change sodium too rapidly. Subsequently, slow administration of a hypotonic crystalloid (e.g., D5W, 0.45% NaCl) may be appropriate. Hypernatremia by itself is seldom associated with signs, and lowering the plasma sodium concentration too quickly is likely, be more detrimental to the patient than the actual hypernatremia. If the hypernatremia existed for more than a few hours, it is often appropriate to administer PSS, a mixture of physiological saline solution plus 5% dextrose in water, or 0.45% NaCl/2.5% dextrose. Conservative administration of either solution decreases serum tonicity slowly enough to prevent neurologic complications. To minimize the risk of neuronal overhydration, serum sodium should not be lowered at a rate exceeding 0.5 mEq/L/hr.7,61
Hypochloremia
Hypochloremia is principally found in patients with excessive losses caused by gastric vomiting or diuretic administration and is of clinical relevance principally because of its effects on systemic acid–base balance. Administration of PSS with or without potassium chloride is usually adequate to replace the chloride and resolve the problem.
Hyperchloremia
Hyperchloremia may be managed by administration of an alkalinizing fluid such as LRS (Cl = 98 mEq/L), Plasmalyte, or Normosol-R (Cl = 110 mEq/L). Isotonic saline (Cl = 154 mEq/L) is seldom indicated for treating hyperchloremia, even for the initial management of patients with hyponatremia caused by hypoadrenocorticism. Even in patients with true hyperchloremia (i.e., not corrected for water), hypotonic fluids should not be administered without careful assessment of sodium status.
Hypokalemia
Supplemental administration of potassium is routine in small animal medicine because hypokalemia is a common abnormality, especially in inappetent hospitalized patients receiving more than 2 to 3 days of intravenous fluids. With rare exceptions, all anorexic animals on maintenance fluids should receive potassium in excess of that contained in most replacement fluids because of obligatory losses of potassium into the urine. Animals with polyuria or other avenues of potassium loss may have even greater needs. Only pets that have or are prone to hyperkalemia should not receive potassium supplementation. It is not possible to predict with confidence exactly how much potassium a particular patient will need; therefore the patient’s plasma potassium concentration should be periodically monitored when receiving supplemental potassium.
If the patient can accept oral fluids and is not vomiting, oral administration of potassium gluconate is usually an efficient means of replenishing plasma potassium.62 There is great interpatient variability in the amount of enteral potassium needed to correct hypokalemia.29,30,63
Intravenous supplementation of potassium is common but mandates frequent monitoring of both the patient and the serum potassium concentration. As a general rule, potassium should not be infused at a rate exceeding 0.5 mEq/kg per hour, although if necessary, carefully monitored patients may receive greater rates without consequence. If it is necessary to administer 0.5 mEq/kg per hour or more, continuous electrocardiographic monitoring for cardiotoxic effects (i.e., bradycardia, tall T waves, small P waves, ventricular arrhythmias, ventricular fibrillation, and asystole) is recommended. Table 16-4, in addition to the other tables in this chapter, provides guidelines for determining how much potassium to add to fluids for intravenous administration. Many clinicians routinely start by adding 15 to 20 mEq K/L to maintenance fluids while periodically monitoring the animal’s plasma potassium concentration.
Table 16-4 Approximate Amount of Potassium to Add to Fluids for Intravenous Administration
| Serum Potassium (mEq/L) | mEq Potassium to Add to Fluids | Maximum Rate of Infusion (mL/kg/H) |
|---|---|---|
| 3.5-4.0 | 15/L | 30 |
| 3.0-3.5 | 28/L | 16 |
| 2.5-3.0 | 40/L | 12 |
| 2.0-2.5 | 60/L | 8 |
| <2.0 | 80/L | 6 |
Potassium may also be added to fluids intended for subcutaneous administration, the safe amount depending on the total osmolality of the solution and not the potassium concentration in the fluid bag. General guidelines suggest that solutions containing potassium in quantities greater than 35 mEq/L should not be given subcutaneously.8 If the clinician experiences difficulty correcting hypokalemia despite seemingly appropriate potassium supplementation, the serum magnesium concentration should be checked.64,65
Hyperkalemia
Therapy for hyperkalemia depends on the severity of clinical signs and the magnitude of the hyperkalemia. The clinician should always look for the cause of hyperkalemia, because it often indicates significant renal or adrenal disease. If hyperkalemia is mild (e.g., 5.5 to 6.5 mEq/L) and is not expected to rise rapidly, it is appropriate to screen for common associated syndromes (e.g., hypoadrenocorticism, renal failure, iatrogenic potassium administration). Administration of potassium-free fluids (e.g. 0.9% NaCl) is often advised without qualification. However, isotonic replacement solutions (K ≤4 mEq/L) will similarly “dilute” a hyperkalemic patient and augment renal potassium excretion. Therapy with any balanced, buffered isotonic crystalloid may be appropriately selected for treating mild hyperkalemia, particularly insofar as many patients will be somewhat dehydrated. If the patient is also hyponatremic (i.e., hypoadrenocorticism), 0.9% saline (higher concentration of sodium compared with buffered isotonic solutions) may be administered if the clinician monitors for excessive rate of lowering of plasma sodium. If the patient has hyperkalemia of sufficient magnitude to cause cardiotoxicity (i.e., usually ≥8 mEq/L), additional therapy may be warranted (Table 16-5).10 The routine use of sodium bicarbonate for managing hyperkalemia is generally discouraged. However, this drug may be given in an acute setting to a patient that has concurrent metabolic acidosis. Administration of dextrose and insulin decreases blood potassium concentration but can also decrease serum phosphorus concentration, which only rarely causes problems (i.e., hemolytic anemia).66 Slow administration of 10% calcium gluconate may be administered for rapid correction of hyperkalemic cardiotoxicity. It does not lower the potassium level but transiently protects the heart until other measures (i.e., fluid administration) decrease the plasma potassium concentration.8
Table 16-5 Symptomatic Therapy for Hyperkalemia
| Treatment | Dose |
|---|---|
| Potassium-free fluids | Physiologic saline solution or 5% dextrose |
| Calcium gluconate | 0.5-1.0 mL 10% calcium gluconate/kg intravenously (10-15 minutes) |
| Dextrose and insulin | 0.5 U regular insulin/kg + 2 g dextrose per unit insulin |
| Sodium bicarbonate | Based on blood gas analysis or 1-2 mEq/kg |
Hypophosphatemia
Optimal management of hypophosphatemia depends somewhat on whether the clinician is trying to prevent hypophosphatemia or to treat associated problems (e.g., hemolytic anemia) caused by hypophosphatemia. Severe hypophosphatemia is often seen as a complication of diabetic ketoacidosos. Other causes include hyperparathyroidism and hyperalimenation. If the patient has a dangerously low serum phosphorus concentration (e.g., 1.0 to 1.5 mg/dL) but does not yet have clinical signs, a simple rule of thumb is to provide one quarter of the maintenance potassium being given in the intravenous fluids (assuming maintenance rates are being used) as potassium phosphates and the remainder as potassium chloride.66 The patient’s serum phosphorus concentration is then monitored two to three times daily until it is out of the danger zone (i.e., serum phosphorus >2 mg/dL), at which time the phosphorus supplementation is stopped. If the animal is experiencing hemolysis, then a constant-rate infusion of 0.01 to 0.03 mmol phosphate/kg per hour may be given.31,32,66 Greater rates of phosphorus adminstration (e.g., 0.05 to 0.1 mmol/kg per hour) are described but seldom required to treat hypophosphatemia.31 The patient must be monitored three to four times daily, however, to ensure that the serum phosphorus concentration is increasing and that the serum calcium concentration is not decreasing. Severe, symptomatic hypocalcemia might result from excessive phosphorus administration.
Hypomagnesemia
If hypomagnesemia needs to be treated or prevented, magnesium sulfate or magnesium chloride should be administered in D5W or 0.9% NaCL. Initial dosing guidelines are 0.75 to 1 mEq/kg/day given intravenously by constant-rate infusion, although 0.15 to 0.30 mEq/kg may be given over 10 to 15 minutes for life-threatening cardiac arrhythmias.67 Because magnesium is a divalent ion, 1 mEq is equivalent to 0.5 mmol. Magnesium should not be added to LRS, or given in the same fluid line as calcium-containing fluids or insulin.
Acid–Base Status
Considerations for Therapy
In general, the clinician should always attempt to determine the underlying cause of the acid–base abnormality and correct it. If the acidemia or alkalemia is so severe that it puts the patient at significant risk, however, symptomatic therapy is needed.
Metabolic Acidosis
A blood pH below 7.20 puts a patient at risk for arrhythmia, hypotension, and myocardial dysfunction. For such patients administration of sodium bicarbonate may be considered. A patient with blood pH below 7.1 is considered to be at high risk for cardiovascular complications, and prompt bicarbonate therapy should be considered. The goal of such therapy is to raise the pH to approximately 7.20 or slightly greater, not to correct the pH so that it returns to the normal range. Sodium bicarbonate should not be given to patients with respiratory acidosis or decreased respiratory drive. If too much base is administered, a patient may become alkalemic after the cause of the acidemia (e.g., diabetic ketoacidosis, lactic acidosis) is corrected.20
Metabolic Alkalosis
Alkalosis is important because of the chloride abnormalities that may be associated with it. The alkalosis does not cause the hypochloremia but may potentiate it and therefore enhance the effects of high pH on the myocardium. Most dogs and cats with clinically important metabolic alkalosis have a chloride-responsive condition (e.g., vomiting gastric contents, excessive furosemide administration in anorexic animals). Correction of volume depletion and supplementation with chloride (e.g., PSS occasionally with potassium chloride supplemented) is usually adequate to correct the problem.20
Respiratory Acidosis and Alkalosis
Respiratory acidosis and alkalosis require therapy directed at the cause of the problem and seldom require symptomatic therapy.
Drugs Used to Correct Acid-Base Abnormalities
The main drugs used to alter blood and body pH are sodium bicarbonate and fluids containing bicarbonate precursors. In particular, LRS has often been used to treat acidosis. Under normal circumstances, hepatic metabolism of lactate causes consumption and elimination of protons (i.e., H++), thus raising the pH.20 If the lactate is not metabolized (e.g., the patient already has lactic acidosis), LRS may cause a mild dilutional acidosis similar to saline or will not affect acid–base status. Moreover, the lactate in LRS will never cause a lactic acidosis because it is a salt. Even when the lactate is not metabolized, however, expanding the ECF may improve peripheral perfusion. Similar fluids such as Normosol-R and Plasmalyte are considered to be just as effective. Sodium bicarbonate (NaHCO3) can be used to correct acidemia. It works rapidly by providing base, which titrates H+. Sodium bicarbonate is not always beneficial and is seldom required for acidotic patients. Its use is particularly controversial for patients with lactic acidosis.68,69 When NaHCO3 is added to the blood, a small percentage of it is converted to CO2 almost immediately. Traditionally, if this CO2 cannot be exhaled, it may exacerbate preexisting acidemia. More commonly, however, the respiratory acidosis offsets the bicarbonate-induced metabolic alkalosis, with negligible, if any, increase in pH. Clinicians should appreciate that when NaHCO3 is given to patients with diabetic ketoacidosis, they may become alkalotic after the ketone bodies are metabolized (rebound phenomenon). Physiologically, most patients can deal with alkalosis better than with acidosis.
Various formulas can be used to help the clinician decide how much bicarbonate (HCO3) to administer.20 The following is suggested for most situations when NaHCO3 is indicated:
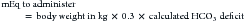
The body weight is multiplied by 0.3 to approximate the ECF volume, which is where the HCO3 will first be distributed. There will eventually be diffusion of HCO3 into the cells, and additional HCO3 may have to be administered to retain the desired effect in the ECF. For that reason some clinicians utilize a factor of 0.5 instead of 0.3.
The calculated HCO3 deficit to treat is not the same as “base deficit” (a calculation provided by a blood gas analysis). To calculate the HCO3 deficit to treat, the clinician must first decide what plasma concentration of HCO3 is desired. It is appropriate to aim for a plasma concentration of approximately 14 to 15 mEq/L if the HCO3 concentration is less than that initially. The patient’s HCO3 concentration is then subtracted from the desired HCO3 concentration (e.g., 15 mEq/L). The resulting number is the calculated HCO3 deficit to treat for this patient at this time. Once the total amount of HCO3 to be administered is calculated, it is administered intravenously, usually over 1 to 4 hours. After several hours, this HCO3 will distribute to the ECF and later to the ICF, and the patient should be reevaluated to determine whether more HCO3 is needed.
NaHCO3 is not a benign drug, although severe adverse effects are rare and complications can be minimized if it is used judiciously. Hypernatremia, hypervolemia, hypokalemia, ionized hypocalcemia, hypotension, nausea, and paradoxical intracellular acidosis, are possible.70 The recognition of potential adverse effects of NaHCO3 led investigators to develop other forms of base that could be administered to restore acid–base status while circumventing the problems associated with NaHCO3. THAM (tris hydroxymethyl aminomethane) is sodium free and has a free amino group that buffers protons. THAM administration doesn’t result in generation of CO2. This drug never gained widespread acceptance, in part because of reported side effects including respiratory depression, hyperkalemia, and hepatotoxicity. Carbicarb (1:1 mixture of sodium bicarbonate and sodium carbonate) has been shown in animal studies of metabolic acidosis to be superior to NaHCO3 in preserving or improving cardiac output and intracellular pH. However, further development of this drug was abandoned, in large part because it was not convincingly demonstrated to be superior to NaHCO3. Ethylene glycol intoxication often necessitates aggressive NaHCO3 therapy. Large amounts of acid are produced as ethylene glycol is metabolized to glycolic acid20; therefore it may be difficult to give NaHCO3 in sufficient amounts to maintain a safe pH, depending on how much ethylene glycol was ingested.
Special Considerations
Shock
Shock is a syndrome characterized by the presence of severe clinical signs, including altered mental status, mucous membrane color, capillary refill time, heart rate, and pulse quality. Shock may be classified as hypovolemic, cardiogenic, or distributive. Obstructive shock, as described, is less commonly discussed as a distinct entity as its pathophysiology is believed to overlap with other classifications. Classification schemes, including this one, tend to be oversimplifications because relatively few global clinical parameters represent many complex dynamic processes occurring at the cellular level. Hypovolemic shock occurs when loss of blood volume causes a severe decrease in tissue perfusion and oxygenation. Causes include hemorrhage and severe dehydration. Cardiogenic shock is caused by myocardial failure with or without arrhythmias that decrease cardiac output. In obstructive shock inadequate tissue perfusion results from obstruction of blood flow within the vasculature. Obstructive shock may be seen in animals with massive pulmonary thromboembolism, pericardial effusion, or gastric dilation/volvulus. Distributive shock is characterized by nonuniform loss of peripheral vascular resistance. Resistance in specific tissue beds may be increased, decreased, or normal. Although cardiac output may be increased, the clinical picture is generally one of vasodilation. The vascular and cellular events result in the global release of inflammatory mediators, leading to the systemic inflammatory response syndrome (SIRS). Sepsis may lead to distributive shock. If there is refractory hypotension, it is further defined as septic shock. Further, patients may have distributive shock subsequent to trauma or other severe inflammatory processes in the absence of an infectious cause (e.g., pancreatitis, anaphylaxis).71 Appropriate treatment of a patient in shock requires characterizing the shock syndrome present in a given patient.53,72
Hypovolemic Shock
The immediate need is to reestablish effective circulating blood volume. Appropriate resuscitation fluids are isotonic crystalloids (e.g., LRS, PSS, Normosol R) with or without the addition of colloids or hypertonic saline. If isotonic crystalloids are used alone in patients with ongoing blood loss, a decrease in vascular volume may be expected after initial resuscitation as redistribution occurs across the extracellular space. With colloids a more steady state of vascular volume expansion is expected. Hypertonic saline should be used cautiously or not at all for patients that are already hypernatremic. Colloids (e.g., dextran 70, 6% hetastarch) are sometimes used with hypertonic saline; or they may be used alone. Hetastarch is given at a dose of 5 to 20 mL/kg in dogs and 5 mL/kg in cats over 5 to 10 minutes, although doses of up to 40 mL/kg per day are described. To prevent hypervolemia, the rate of infusion of isotonic crystalloids should be decreased by 40% to 60% after colloids are started. In the face of ongoing blood loss, optimal therapy ideally should include blood products. If the patient is symptomatic for anemia and blood products are not available, Oxyglobin may be given.53,58,72-74
Unless there is suspicion for hypoadrenocorticism, steroids are generally not recommended. Vasopressors are not generally indicated for hypovolemic patients. In particular, clinicians should avoid α-agonists that increase blood pressure by causing vasoconstriction, unless patients are severely hypotensive and the clinician must time while other interventions are implemented.53
Obstructive Shock
Gastric dilation/volvulus (GDV) is the prototype disorder characterized by obstructive shock. Distention of the stomach with food and air compresses the portal vein and vena cava. This compression can occur with dilation and does not require volvulus. Fluids for resuscitation should be administered through large-bore cephalic catheters because fluid administration through saphenous catheters will be ineffective. Balanced electrolyte solutions are administered at 60 to 90 mL/kg in the initial hour of treatment. Hypertonic saline/dextran solutions can also be used initially (5 mL/kg over 5 to 10 minutes) but must be followed by isotonic crystalloids to maintain perfusion.75 Once resuscitation is under way, the stomach is decompressed by carefully passing an orogastric tube. If passage of the tube is not possible, the stomach is trocarized. Because ischemia and reperfusion appear to be important mechanisms of injury, administration of free radical scavengers may eventually become important in the early treatment of this disorder. There is no evidence supporting the use of glucocorticoids or nonsteroidal inflammatory agents in gastric dilation/volvulus patients.
Cardiogenic Shock
Treatment of cardiogenic shock is directed toward improving myocardial contractility, reducing afterload and preload and controlling dysrhythmias. The clinician should examine the patient frequently to ensure that fluid therapy does not lead to decompensation because these patients are often volume intolerant. It is typically inadvisable to administer crystalloids in edematous cardiac patients. For the rare patient that must receive fluids (e.g., as a vehicle for drug administration), limited amounts of sodium should be given (e.g., 0.45% NaCl).76 Maintenance rates or less are then carefully administered. CVP measurement can be particularly useful in monitoring these patients.
Distributive Shock
Distributive shock is principally seen in animals with SIRS. In SIRS release of various inflammatory mediators causes abnormal blood flow regulation with subsequent tissue hypoxia, even when there is normal to increased cardiac output (hence the term hyperdynamic shock). Increased vascular permeability may cause plasma volume to leak into interstitial tissues, further reducing tissue perfusion.77-80
It can be difficult to effectively treat some patients with SIRS. Blood volume should be maintained as for hypovolemic shock. Either isotonic or hypertonic crystalloids may be used. The clinician must also identify and eliminate the cause of the inflammation, which usually includes broad-spectrum antibiotic therapy. Pharmacologic therapies directed at specific inflammatory pathways have been investigated, but none have been found to be effective to date.81 There is limited evidence supporting the administration of nonsteroidal antiinflammatory agents and corticosteroids, and their use is generally discouraged. A syndrome of relative adrenal insufficiency (RAI) is described in critically ill humans and animals, and patients with RAI may benefit from low (i.e., physiologic) doses of corticosteroids. Criteria for the diagnosis of RAI in animals and humans have been published, but at present the syndrome is not widely recognized.82
Hypoglycemia is relatively common in critical illness, and intravenous glucose supplementation may be needed. Most patients require fluid support, and adding hypertonic glucose to balanced isotonic fluids to create a fluid with a final glucose concentration of 5% is recommended. Although intermittent boluses of hypertonic glucose may be administered (0.5 gm/kg of 10% to 25% dextrose solution), continuous infusion of hypertonic solutions through a peripheral vein may cause thrombophlebitis. The addition of hypotonic glucose–containing fluids (e.g., D5W) at maintenance rates can usually maintain blood glucose concentrations of more than 100 mg/dL in volume-sensitive patients.
Renal Disease
Fluid therapy for patients with renal disease should first correct significant fluid, electrolyte, and acid–base abnormalities and subsequently produce a diuresis to eliminate normally excreted substances that have been retained. It is important to distinguish patients with oliguric renal disease from those with polyuric renal disease and also to determine if pets have chronic kidney disease or acute on chronic kidney injury. Severely hypoalbuminemic patients with proteinuric nephropathy present special challenges. Oliguria describes a patient that is producing less than 1 to 2 mL urine/kg per hour, although some clinicians define oliguria as less than 0.25 mL/kg per hour.83 Anuria is a more severe situation indicating negligible urine production. Polyuria means that greater than normal amounts of urine being produced. Most patients with chronic kidney disease are polyuric until they are preterminal, when they may become oliguric. Patients with acute renal injury similarly may be polyuric or oliguric–anuric.
Polyuric Renal Failure
The first goal of fluid therapy for patients with polyuric renal failure is to correct dehydration (which is commonly present with decompensated renal failure) and severe electrolyte or acid–base abnormalities. If the patient is seriously ill, intravenous fluids are usually preferred to prevent delayed uptake from subcutaneous depots or vomiting of orally administered fluids. The next step is to provide sufficient fluids to match urinary losses. Occasionally, pharmacologic attempts are made to further augment renal function and urine production with diuretics (e.g., furosemide). This should not be done unless the patient is euhydrated and on intravenous fluids or is overhydrated. The use of appropriate and aggressive crystalloid therapy is generally adequate, and although this makes sense intuitively, there is limited evidence to support the use of diuretics in this setting.
As much fluid as possible should be administered without overhydrating the patient. The amount of fluid administered is slowly increased as damaged kidneys recover and adapt to the increased demand placed on them. As a general rule of thumb, therapy is begun by replacing deficits and weighing the patient. Fluids are then administered at 1.5 to 2 times the calculated maintenance rate or more, if necessary to keep up with renal losses. If the patient does not gain weight inappropriately and does not have signs of fluid overload (i.e., gallop rhythm, murmur, pulmonary crackles), the clinician can slowly increase the amount of fluids administered each day, usually by 15% to 30%. Although loop diuretics (e.g., furosemide) are sometimes also required, vigilant therapy with balanced crystalloids often produces an adequate diuresis. When fluid-induced diuresis is brisk, the rate of intravenous fluid administered is slowly decreased (e.g., 10% to 25%) each day, and the body weight is closely monitored to prevent the patient from becoming dehydrated.84 Recording the volume of urine produced over a given time (e.g., 24 hours) is advisable. Measurement of CVP is not useful in this setting.
Oliguric and Anuric Renal Failure
Oligoanuric renal failure is more difficult to manage than polyuric renal failure. Oligoanuria is observed with severe acute renal injury and occasionally in preterminal chronic renal disease.83 In the latter situation there is usually little that can be offered to help the patient besides renal replacement therapy or transplantation. In the former situation, it is important to try to quickly convert the oliguria to polyuria by replacement of the fluid deficit and then administration of furosemide, osmotic diuretics, or dopamine, although none of these drugs has been proved to benefit or alter the prognosis of anuric renal failure patients. Furosemide, administered either as a bolus or as a constant-rate infusion, is typically the first way that the clinician attempts to convert an oliguric patient to a polyuric state. Although once highly regarded, dopamine is generally of dubious value in patients with established oliguric, acute renal failure.
If furosemide therapy fails to convert an oliguric patient to a polyuric state, osmotic diuresis may be attempted by administering a bolus of mannitol (1 gm/kg intravenous bolus) followed by additional boluses or a constant-rate infusion (1 to 2 mg/kg/minute). Alternatively, 10% to 20% dextrose may be administered. The use of dextrose is favored by some clinicians because it may be metabolized if it cannot be excreted. The clinical significance of this is uncertain. However, if this is the case, the clinician should be able to detect glucose in newly formed urine, with the recognition that some patients with acute renal failure already have glucosuria as a result of proximal renal tubular damage. The body weight should be monitored closely to prevent excessive overhydration. Serum electrolytes should also be monitored. It is important that maintenance fluids be initiated immediately after infusing dextrose or mannitol to prevent renal hypoperfusion. If urine production is not increased by these measures, infusions should be discontinued before overhydration, pulmonary edema, and hypertonicity occur (i.e., before half of the total calculated volume is administered).83
Great care must be taken not to volume overload oliguric patients. If life-threatening overhydration occurs, all intravenous therapies are discontinued. Diuretics may be administered, but these patients may not improve without hemodialysis or hemofiltration. CVP should be monitored. CVP values consistently greater than 10 to 12 cm H2O are compatible with but not diagnostic for fluid overload. CVP values between 5 and 10 cm H2O suggest that the patient may tolerate additional fluid challenge. 85 Another technique is to measure urine output and administer fluids accordingly, using a technique sometimes called “ins and outs.”84 The patient is initially rehydrated over 6 to 8 hours. The clinician next divides the day into six 4-hour intervals. The amount of urine produced over each interval is given back to the patient over the subsequent interval.
Oligoanuric renal failure patients are predisposed to hyperkalemia and severe acidosis, Serum magnesium abnormalities are also common. If the patient is well hydrated and remains oliguric or anuric, all intravenous fluids are discontinued. Alternatively, the clinician may elect to replace only insensible losses with intravenous fluids at this time, but this has inherent risks. Body weight is assessed several times daily. Consideration of referral for hemodialysis is appropriate in persistently (i.e., duration longer than 24 hours) oliogoanuric patients.
The healing phase of acute renal failure may be accompanied by an intense diuresis that can require administration of several times the calculated daily maintenance volume. If an oliguric or anuric patient becomes polyuric, it is necessary to guard against dehydration. The rate of fluid administration must be slowly decreased when the patient is being weaned off intravenous fluids.83 Excessive losses of potassium may occur during the healing, polyuric phase.
Urethral Obstruction
The clinical condition of dogs and cats with urethral obstruction depends on the duration and severity of the obstruction. Less commonly, bilateral ureteral obstruction will result in oligoanuria; in this situation the bladder is small or empty. When urethral obstruction is diagnosed, intravenous fluids should be instituted immediately, with the rate based on the degree of hypoperfusion and dehydration present. Hyperkalemia, if severe and associated with bradydysrhythmias, should be treated aggressively before relieving the obstruction. If acidosis is present and hyperkalemia is not associated with life-threatening signs, fluid resuscitation alone will often be sufficient to correct these values. A balanced isotonic electrolyte solution is recommended for initial therapy in most cases.86 These patients often develop severe polyuria after the obstruction is relieved (e.g., postobstructive diuresis), the severity of which generally depends on the degree of renal tubular damage or loss of urine-concentrating ability. It is common for severely affected patients to have daily fluid needs that exceed their calculated maintenance values during this healing period.86
Proteinuric Nephropathies
Glomerular diseases that have progressed in duration or severity to cause hypoalbuminemia are recognized in companion animals. Some patients with severe proteinuria will have clinical and clinicopathologic features of nephrotic syndrome. By definition, nephrotic syndrome implies glomerular protein loss sufficient to cause hypoalbuminemia, in addition to edema (or ascites) and hypercholesterolemia. Although it is postulated that patients with nephrotic syndrome have more severe glomerular lesions and a less favorable prognosis than patients that do not fulfill these criteria, this is not the case for dogs.
When choosing fluid therapy, the clinician must consider both the need for diuresis and the effects of further dilution of serum albumin concentrations by aggressive administration of crystalloids in addition to effects on blood pressure.87 The patient may already have edema, ascites, or pleural effusion resulting in part from the hypoalbuminemia. Further dilution of plasma albumin concentrations with intravenous fluids may unmask or exacerbate fluid retention. Uncommonly, cavitary effusions will be of sufficient severity to require removal by paracentesis. Judicious administration of a loop diuretic can be used at this time; the diuretic causes fluid loss from the vascular compartment that is ideally replaced by fluid from the third space. If too much fluid is removed from the vascular compartment, renal hypoxia and damage may result.87 Removal of cavitary effusions is generally discouraged. Such withdrawal, especially if repeated, will further lower the body albumin concentration and make fluid reaccumulation more likely.
If the patient’s cardiovascular status is rapidly deteriorating because of severe hypoalbuminemia or if it is necessary to perform a procedure (especially one requiring anesthesia) in a patient with marginal cardiovascular status, a synthetic colloid (e.g., hetastarch, dextrans) can ge given. Alternatively, plasma may be administered. However, synthetic colloids are more effective in maintaining osmotic pressure in the vascular space. Moreover, the amount of plasma required to replace the deficit in albumin is relatively large, and the ability of administered albumin to maintain vascular expansion is usually gone within 24 to 72 hours (as it is with artificial colloids). Plasma is indicated only for patients with clinically significant coagulopathies that require invasive procedures. Many patients with glomerular disease are hypertensive, and blood pressure must be monitored carefully lest fluid therapy cause worsening of the hypertension with subsequent retinal or central nervous system injury.
Cardiac Disease
Most animals with cardiac failure do not need parenteral fluid therapy. Animals that are eating and drinking can usually be managed by offering distilled water and using diuretics as needed. However, if the patient is severely dehydrated, is anorexic, or has concurrent renal failure or other organ failure, fluid therapy may be needed. Treatment of such animals in severe cardiac failure is similar to the treatment of those with oliguric renal failure; the clinician must be careful not to overhydrate the patient. Measurement of CVP is useful; however, pulmonary artery catheters (i.e., Swan–Ganz) provide information (e.g., pulmonary wedge pressure) that is not available from CVP catheters. It is important to note that pulmonary artery catheters are most useful for monitoring cardiovascular status but not fluid balance in critically ill patients. Fluid administration is controversial with many cardiac diseases. Because of their lower sodium content, hypotonic solutions may be more appropriate when fluids are deemed necessary because patients with heart disease are often unable to appropriately excrete sodium. However, it is not the sodium concentration of the fluid, per se, but the volume of fluid that contributes to this situation. Similarly, sodium bicarbonate administration is often discouraged because of the relatively high sodium load.88
Electrolyte abnormalities in patients with cardiac disease must be treated carefully. Oral potassium supplementation is best for hypokalemic animals that are not vomiting. Hypokalemic patients are more prone to digitalis toxicity and supraventricular arrhythmias.8 Marked repeatable hyponatremia is usually an indication that cardiac disease is advanced. Cautious sodium administration is appropriate only for patients with severe, symptomatic hyponatremia (rare). Even then, only enough sodium to alleviate or prevent central nervous system signs should be administered.
Patients with simultaneous cardiac and renal failure are among the most difficult to manage. The clinician must identify the cause of the renal and cardiac diseases and treat each. If the underlying causes cannot be determined or treated, it is best to monitor CVP and “ins and outs” while giving small amounts of fluids with minimal amounts of sodium.88 In general, fluids are administered as rapidly as possible without excessively increasing the CVP (e.g., >10 to 12 cm H2O).
Gastrointestinal Disorders
Acute Vomiting
Acute vomiting commonly causes fluid deficits.89 The severity of the deficit will depend on the severity and cause of the vomiting and on whether the animal will eat or drink. Although vomiting is considered an alimentary tract disorder, vomiting is often caused by extragastrointestinal disorders (e.g., renal failure, adrenal failure, hepatic failure, hypercalcemia, pancreatitis, diabetic ketoacidosis). The therapy for most animals with moderate to severe acute vomiting includes not being fed or watered until the vomiting diminishes, which means that the patient has ongoing losses (both insensible and from vomiting) and no intake. Therefore, even if such an animal is not dehydrated at the time of examination, it will usually become so shortly.
If mild dehydration is present or anticipated, subcutaneous fluids are often adequate. If the dehydration is or will probably become severe or if the animal appears to be going into shock, intravenous fluids are indicated. Until the electrolyte and acid–base status are known, 0.9% saline, Normosol-R, or LRS are generally acceptable choices. Although hypokalemia is common in animals with acute vomiting, supplemental potassium should not be administered until it is known that the patient is not hyperkalemic as a result of adrenal or renal dysfunction. One of the most common causes of severe vomiting in young animals is parvoviral enteritis, which will be discussed later with acute diarrhea.
Chronic Vomiting
Most patients with chronic vomiting (i.e., that lasting 2 weeks or longer) are not overtly dehydrated when presented to the veterinarian. The fact that the animals have had the disease for so long usually means they were able to compensate for their fluid losses. If unable to compensate, they typically would have been been brought in earlier as emergencies (e.g., in hypovolemic shock). Some of these patients will be dehydrated because of a recent worsening of their disease. Even when the animal’s fluid status is acceptable, however, there may be significant electrolyte and acid–base abnormalities.
Fluid therapy must be tailored for each individual. Hypokalemia is common if the patient does not have adrenal insufficiency or severe renal failure, both of which can cause chronic vomiting. It is not possible to predict with accuracy what a given patient’s acid–base status is, even when the cause of the vomiting is known. Animals with high intestinal obstruction occasionally have a hypokalemic, hypochloremic, metabolic alkalosis that is thought to reflect gastric outflow obstruction, whereas some animals with seemingly pure gastric fluid losses can have a normal pH or are acidemic. If therapy must begin before the electrolyte and acid–base status are known (i.e., the patient is severely dehydrated), then balanced isotonic solutions (e.g., Normosol-R, PSS, LRS) are usually acceptable. Potassium supplementation is not recommended until a potassium deficit is confirmed. If renal failure and hypoadrenocorticism have been eliminated and hypokalemia is present, it is appropriate and generally safe to add 14 to 20 mEq/L of potassium chloride to crystalloid solutions and administer it at a maintenance rate.8
Acute Diarrhea
Many animals with acute diarrhea are not dehydrated unless the diarrhea is profuse, vomiting is present, or the patient is anorexic. Acute parvoviral enteritis is an important cause of acute diarrhea, vomiting, and anorexia. In severe cases intravenous fluid therapy is indicated regardless of the cause.89 Treatment initially should correct hypovolemia and hydration deficits. Subsequently, when the serum potassium is determined, 20 mEq/L of potassium chloride may be added to isotonic crystalloids. If hyperkalemia is marked, up to 40 mEq/L may be added to intravenous fluids. With parvoviral enteritis, the clinician must also be concerned with hypoglycemia (discussed later), which may result from sepsis or diminished capacity for glycogenolysis and gluconeogenesis in a sick puppy.90
Chronic Diarrhea
Most patients that are brought to the veterinarian because of chronic diarrhea are not overtly dehydrated. The fact that the animals have had diarrhea for so long and are just now being examined suggests that they are able to compensate for their fluid losses. If they had been unable to maintain hydration during the previous 2 or more weeks, they probably would have been brought in earlier in hypovolemic crisis. A recent worsening of the disease may cause dehydration, in which case it is reasonable to start fluids (usually a balanced isotonic crystalloid supplemented with 20 mEq/L potassium chloride). Hypokalemia is the most common electrolyte problem in these patients, but acute renal failure and hypoadrenocorticism, two of the most common causes of noniatrogenic hyperkalemia, occasionally cause disease characterized predominantly by diarrhea. The acid–base status cannot be predicted, and the clinician should not administer alkalinizing or acidifying solutions until at least the Tco2 (and ideally the blood pH) is known.
Another major concern for animals (especially dogs) with chronic diarrhea is hypoalbuminemia. Protein-losing enteropathies may produce serum albumin concentrations of less than 1.5 g/dL. Not all such dogs have ascites or edema.89 The clinician should avoid excessively diluting the remaining albumin in severely hypoalbuminemic patients because it can cause further pooling of fluid in third spaces and depletion of effective circulating volume. If the animal needs increased oncotic pressure, synthetic or natural colloids may be administered. However, albumin that has been administered intravenously can be lost rapidly into the intestines of animals with protein-losing enteropathies. Synthetic colloids (e.g., hetastarch) are typically more effective for management of these patients, unless they are simultaneously coagulopathic.
Pancreatic Disease
Acute Pancreatitis
Acute pancreatitis is a potentially fatal disease that usually has no specific therapy, although cats can have pancreatitis caused by bacterial, viral, or protozoal infections. Affected dogs usually are anorexic and yet vomit gastrointestinal secretions, leading to severe dehydration. The primary therapies available are fluid therapy (to enhance pancreatic perfusion) and initially withholding oral intake in patients that are vomiting.89 If an inflamed pancreas is poorly perfused, the inflammatory state may progress from a relatively mild, edematous one to a severe hemorrhage or necrotic condition with a poorer prognosis.
The most common mistakes in treating dogs with pancreatitis are thought to be premature oral feeding and inadequate administration of fluids.91 The former concern is now being debated. Not only must the fluid deficit be replaced, but a normal effective circulating volume also must be attained so that visceral perfusion is improved. Severe abdominal inflammation may cause fluid to pool in the abdomen or peripancreatic tissues and thus be unavailable for organ perfusion. Furthermore, there may be pooling of fluid in the intestines because of poor motility and serosal peritoneal inflammation.
It is important to monitor the serum albumin concentration. If the albumin concentration drops and the patient is judged to be inadequately perfused, plasma or hetastarch may be useful.92 Although contentious, there is some thought that administration of plasma may replenish circulating proteases, thus offering some protection against enzymes released from the diseased pancreas,89 as well as supply antithrombin, which may be helpful in treating disseminated intravascular coagulation.
Diabetic Ketoacidosis
Animals with diabetic ketoacidosis are often, but not invariably, severely dehydrated and acidotic when first examined. Fluid administration and judicious use of insulin generally produce favorable outcomes if the patient does not have severe pancreatitis or other severe concurrent illness.41 Most dogs and cats with severe diabetic ketoacidosis are treated initially with fluids until dehydration and electrolyte/acid–base abnormalities are at least partially corrected before beginning therapy with insulin, but this is not a universally accepted tenet of therapy. A balanced isotonic replacement solution is used. It is generally not necessary to administer NaHCO3 to these patients, and clinicians are discouraged from its use unless it is determined that a profound metabolic acidosis exists after fluid deficits have been replaced. Many ketoacidotic patients will become hypokalemic once therapy is begun; therefore it is reasonable to start supplementing potassium with the initial fluids once it is determined that the patient is not hyperkalemic.41 Hypophosphatemia is a potential problem of aggressive insulin therapy, and serum phosphate concentrations must be monitored, especially if anemia is developing. If the patient becomes severely hypophosphatemic, the intravenous fluids should be supplemented with potassium phosphate (see previous discussion on phosphorus). The patient must be monitored to prevent hypocalcemia and hyperphosphatemia.66 Hypomagnesemia should be anticipated in ketoacidotic diabetic patients.
Hepatic Disease
The main concerns regarding administration of fluids to a patient with hepatic disease are hypoalbuminemia, hypoglycemia, fluid and salt retention, and electrolyte abnormalities. Fluids restricted in sodium (e.g., half-strength PSS plus 2.5% or 5% dextrose) should be used to help avoid fluid retention in dogs with chronic hepatic disease prone to ascites (e.g., cirrhosis). Addition of glucose to the fluids (i.e., 2.5% to 5%) guards against hypoglycemia. If hypoalbuminemia is severe, administration of colloids in addition to crystalloids should be considered. Large volumes of plasma often are required to significantly increase plasma albumin concentrations, and hypoalbuminemia is not necessarily an indication for plasma administration. Plasma is primarily administered to patients that are coagulopathic. The administration of human albumin may effectively increase serum albumin, but its use is controversial because some patients have died after using it. Hypokalemia predisposes the patient to hepatic encephalopathy; supplemental potassium chloride in the intravenous fluids prevents this occurrence. Acid–base abnormalities are difficult to predict and must be assessed in each patient. In general, the clinician should prevent alkalemia because it predisposes the patient to hepatic encephalopathy.93
Hypoglycemia
Hypoglycemia may be caused by various diseases (e.g., septicemia, hypoadrenocorticism, hepatic insufficiency, insulinoma, starvation of a neonate, iatrogenic overdose of insulin). Regardless of its cause, symptomatic hypoglycemia that is causing weakness, coma, or convulsion should be treated.94 For emergencies 1 mL/kg of 50% dextrose/kg is drawn up, diluted 1:1 with PSS, and administered intravenously, slowly to effect. For a more prolonged effect, dextrose may be added to maintenance fluids. In most cases 2.5% dextrose given at maintenance rates will maintain the blood glucose concentration in a safe range (i.e., >80 mg/dL). In rare cases it may be necessary to administer 5% instead of 2.5% dextrose to achieve this goal.
Hypocalcemia
Symptomatic hypocalcemia is usually caused by puerperal tetany, hypoparathyroidism, resection of parathyroid tumors, ethylene glycol intoxication, or inappropriate administration of a hypertonic phosphate enema. Regardless of its cause, hypocalcemia causing clinical signs (i.e., tetany, convulsions) should be treated.95 While calcium gluconate 10% and calcium chloride 10% may be obtained, calcium chloride is thought to be more potent (10% calcium gluconate has 0.46 mEq Ca/mL, whereas 10% calcium chloride has 1.36 mEq Ca/mL); therefore there is potential for overdosage and cardiotoxicity may, in addition to the phlebitis and perivascular tissue injury observed if extravastion occurs. Similar tissue injury is observed if calcium chloride is given by the subcutaneous route. Calcium gluconate is therefore selected over calcium chloride for treating hypocalcemia, except in extremely rare circumstances (i.e., hepatic disease impairing metabolism of gluconate). Approximately 0.5 to 1.5 mL of 10% calcium gluconate is administered per kilogram, or 5 to 15 mg/kg may be administered slowly to effect over 10 to 30 minutes. Such a treatment usually lasts a few hours, although some animals will need additional therapy within 1 to 3 hours. Once the crisis is over, calcium may be added to the intravenous fluids to provide constant infusion. In general, 10 mL of 10% calcium gluconate is added to 500 mL of 0.9% NaCl and administered at maintenance rates. Then the serum calcium level and clinical signs are monitored, and the amount of calcium in the intravenous fluids is increased if necessary. Alternatively, the clinician may administer 1 to 2 mL calcium gluconate/kg (diluted 1:1 in sterile PSS) subcutaneously.96
Hypercalcemia
Symptomatic hypercalcemia in dogs is usually due to hypercalcemia of malignancy, vitamin D intoxication, primary hyperparathyroidism, hypoadrenocorticism, granulomatous disease, or chronic renal disease causing tertiary hyperparathyroidism.97 In the first two conditions, it is important to rapidly decrease the plasma concentration of calcium to prevent renal injury. This is ultimately best accomplished by elimination of the cause (e.g., removing or treating the malignancy causing hypercalcemia of malignancy, removing the parathyroid adenoma). While searching for the cause, however, the clinician may administer 0.9% NaCl to promote diuresis and enhance urinary calcium excretion. However, potassium concentrations must be monitored to prevent hypokalemia.98 Furosemide promotes natriuresis and calciuresis if the patient is adequately hydrated. Administration of 1 mg prednisolone/kg daily may enhance calcium excretion and reduce bone resorption as well as intestinal calcium absorption. However, steroid administration may make it more difficult to diagnose the cause of the hypercalcemia if it is due to a lymphoid malignancy. Severe cases may be treated with salmon calcitonin, pamidronate, or clodronate. Saline diuresis and furosemide are successful in controlling hypercalcemia in the short term in most patients. Prognosis depends on the underlying disease.
Surgery
Dehydration should be corrected before surgery and anesthesia, if possible. Even after deficits have been corrected, animals undergoing prolonged general anesthesia usually benefit from intravenous fluid support. Unless the patient has cardiac disease or is hypoproteinemic or anemic, isotonic crystalloids (e.g., PSS, LRS) are usually administered at 4 to 5 mL/kg per hour. Severe hypoproteinemia or anemia should be corrected with blood products or synthetic colloids before the patient is anesthetized.
If the clinician anticipates significant loss of blood during the procedure, he or she may need to increase the basal rate by 5 to 10 mL/kg per hour. If there is substantial blood loss or the hematocrit falls to less than 20% to 25% during the procedure, strong consideration should be given to administering red blood cells (either as packed red cells or whole blood) or administering purified hemoglobin. If one is attempting to correct for blood loss during the procedure with crystalloids, approximately three times the estimated volume of blood lost should be given.
1. Rose B.D., Post T.W. Regulation of water and electrolyte balance. Clinical physiology of acid-base and electrolyte disorders. ed 5. 2001. McGraw-Hill. New York. p 239
2. Edelman I.B., Leibman J. Review: anatomy of body water and electrolytes. Am J Med. 1959;27:256.
3. Hardy R.M., Osborne C.A. Water deprivation test in the dog: maximal normal values. J Am Vet Med Assoc. 1979;174:479.
4. Guyton A.C. Partition of the body fluids: osmotic equilibria between extracellular and intracellular fluids. In Textbook of medical physiology. ed 11. 2006. Saunders. Philadelphia. p 286
5. Rose B.D., Post T.W. Regulation of plasma osmolality. In Clinical physiology of acid-base and electrolyte disorders. ed 5. 2001. McGraw-Hill. New York. p 285
6. Sterns R.H., Spital A. Disorders of water balance. In: Kokko J.P., Tannen R.L., editors. Fluids and electrolytes. ed 2. Philadelphia: Saunders; 1990:139.
7. DiBartola S.P. Disorders of sodium and water: hypernatremia and hyponatremia. In: DiBartola S.P., editor. Fluid, electrolyte, and acid-base disorders in small animal practice. ed 3. St Louis: Saunders; 2006:47-79.
8. DiBartola S.P. Autran de Morais HS: Disorders of potassium: hypokalemia and hyperkalemia. In: DiBartola S.P., editor. Fluid, electrolyte, and acid-base disorders in small animal practice. ed 3. St Louis: Saunders; 2006:91-121.
9. Rose B.D., Post T.W. Potassium homeostasis. In Clinical physiology of acid-base and electrolyte disorders. ed 5. 2001. McGraw-Hill. New York. p 372
10. Willard M.D. Disorders of potassium homeostasis. Vet Clin North Am. 1989;19:241.
11. DiBartola S.P. Introduction to acid-base disorders. In: DiBartola S.P., editor. Fluid, electrolyte, and acid-base disorders. ed 3. St Louis: Saunders; 2006:229.
12. Rose B.D., Post T.W. Acid-base physiology. In Clinical physiology of acid-base and electrolyte disorders. ed 5. 2001. McGraw-Hill. New York. p 299
13. Autran de Morais HS. Strong ion approach to acid base disorders. In: DiBartola S.P., editor. Fluid electrolyte and acid base disorders in small animal practice. Philadelphia: Saunders; 2006:311.
14. Rose B.D., Post T.W. Regulation of acid-base balance. Clinical physiology of acid-base and electrolyte disorders. ed 5. 2001. McGraw-Hill. New York. p 325
15. Rose B.D., Post T.W. Introduction to simple and mixed acid-base disorders. In Clinical physiology of acid-base and electrolyte disorders. ed 5. 2001. McGraw-Hill. New York. p 535
16. Rose B.D., Post T.W. Metabolic alkalosis. In Clinical physiology of acid-base and electrolyte disorders. ed 5. 2001. McGraw-Hill. New York. p 551
17. Rossi N.F., Schrier R.W. Hyponatremic states. In Maxwell M.H., Kleeman C.R., et al, editors: Clinical disorders of fluid and electrolyte metabolism, ed 4, New York: McGraw‑Hill, 1987. p 461
18. Hardy R.M. Hypernatremia. Vet Clin North Am. 1989;19:231.
19. DiBartola S.P. Disorders of sodium and water. In: DiBartola S.P., editor. Fluid electrolyte and acid base disorders in small animal practice. Philadelphia: Saunders, 2006. p 53
20. DiBartola S.P. Metabolic acid base disorders. In: DiBartola S.P., editor. Fluid electrolyte and acid base disorders in small animal practice. Philadelphia: Saunders, 2006. p 269
21. DiBartola S.P., Green R.A., Autran de Morais H.S., et al. Electrolytes and acid-base. In: Willard M.D., Tvedten H., et al, editors. Small animal clinical diagnosis by laboratory methods. ed 4. Philadelphia: Saunders; 2004:117.
22. Bellevue R., Dosik H., Spergel G., et al. Pseudohyperkalemia and extreme leukocytosis. J Lab Clin Med. 1975;85:660.
23. Reimann K.A., Knowlen G.G., Tvedten H.W. Factitious hyperkalemia in dogs with thrombocytosis. J Vet Intern Med. 1989;3:47.
24. Degen M. Pseudohyperkalemia in Akitas. J Am Vet Med Assoc. 1987;190:541.
25. Willard M.D., Fossum T.W., Torrance A., et al. Hyponatremia and hyperkalemia associated with idiopathic or experimentally induced chylothorax in four dogs. J Am Vet Med Assoc. 1991;199:353.
26. Bissett S.A., Lamb M., Ward C.R. Hyponatremia and hyperkalemia associated with peritoneal effusion in four cats. J Am Vet Med Assoc. 2001;218:1590.
27. Graves T.K., Schall W.D., Refsal K., et al. Basal and ACTH-stimulated plasma aldosterone concentrations are normal or increased in dogs with Trichuris-associated pseudohypoadrenocorticism. J Vet Intern Med. 1994;8:287.
28. Lennon E.M., Boyle T.E., Hutchins R.G. Use of basal serum or plasma cortisol concentrations to rule out a diagnosis of hypoadrenocorticoism in dogs: 123 cases (2000–2005). J Am Vet Med Assoc. 2007;231:413-416.
29. Dow S.W., Fettman M.J., LeCouteur R.A., et al. Potassium depletion in cats: renal and dietary influences. J Am Vet Med Assoc. 1987;191:1569.
30. Dow S.W., LeCouteur R.A., Fettman M.J., et al. Potassium depletion in cats: hypokalemic polymyopathy. J Am Vet Med Assoc. 1987;191:1563.
31. Justin R.B., Hohenhaus A.E. Hypophosphatemia associated with enteral alimentation in cats. J Vet Intern Med. 1995;9:228.
32. Adams L.G., Hardy R.M., Weiss D.J., et al. Hypophosphatemia and hemolytic anemia associated with diabetes mellitus and hepatic lipidosis in cats. J Vet Intern Med. 1993;7:266.
33. Harkin K.R., Braselton W.E., Tvedten H. Pseudohypophosphatemia in two dogs with immune-mediated hemolytic anemia. J Vet Int Med. 1998;12:178.
34. Khanna C., Lund E.M., Raffe M., et al. Hypomagnesemia in 188 dogs: a hospital population-based prevalence study. J Vet Int Med. 1998;12:304.
35. Kimmel S.E., Waddell L.S., Michel K.E. Hypomagnesemia and hypocalcemia associated with protein-losing enteropathy in Yorkshire terriers: five cases (1992-1998). J Am Vet Med Assoc. 2000;217:703.
36. Brautbar N., Massry S.G. Hypomagnesemia and hypermagnesemia. In: Maxwell M.H., Kleeman C.R., et al, editors. Clinical disorders of fluid and electrolyte metabolism. ed 4. New York: McGraw-Hill; 1987:831.
37. Toll J., Erb H., Birnbaum N., et al. Prevalence and incidence of serum magnesium abnormalities in hospitalized cats. J Vet Int Med. 2002;16:217.
38. Autran de Morais H.S. Mixed acid-base disorders. In: DiBartola S.P., editor. Fluid electrolyte and acid base disorders in small animal practice. ed 3. Philadelphia: Saunders; 2000:298.
39. Hindman B.J. Sodium bicarbonate in the treatment of subtypes of acute lactic acidosis: physiologic considerations. Anesthesiology. 1990;72:1064.
40. Rose B.D., Post T.W. Metabolic acidosis. In Clinical physiology of acid-base and electrolyte disorders. ed 5. 2001. McGraw-Hill. New York. p 578
41. Nelson R.W. Diabetes mellitus. In: Ettinger S.J., Feldman E.C., editors. Textbook of veterinary internal medicine. ed 7. St Louis: Saunders; 2010:1782-1796.
42. Clay K.L., Murphy R.C. On the metabolic acidosis of ethylene glycol intoxication. Toxicol Appl Pharmacol. 1977;39:39.
43. Grauer G.F., Thrall M.A., Henre B.A., et al. Early clinicopathologic findings in dogs ingesting ethylene glycol. Am J Vet Res. 1984;45:2299.
44. Rose B.D., Post T.W. Respiratory alkalosis. In Clinical physiology of acid-base and electrolyte disorders. ed 5. 2001. McGraw-Hill. New York. p 673
45. Johnson R.A. Autran de Morais H: Respiratory acid base disorders. In: DiBartola S.P., editor. Fluid, electrolyte, and acid-base disorders. ed 3. St Louis: Saunders; 2006:293.
46. Rose B.D., Post T.W. Respiratory acidosis. Clinical physiology of acid-base and electrolyte disorders. ed 5. 2001. McGraw-Hill. New York. p 647
47. Shiroshita Y., Tanaka R., Shibazaki A., et al. Accuracy of a portable blood gas analyzer incorporating optodes for canine blood. J Vet Int Med. 1999;13:597.
48. Muir W.W., DiBartola S.P. Fluid therapy. In: Kirk R.W., editor. Current veterinary therapy VIII. Philadelphia: Saunders; 1983:28.
49. Bell F.W., Osborne C.A. Maintenance fluid therapy. In: Kirk R.W., editor. Current veterinary therapy X. Philadelphia: Saunders; 1992:37.
50. Rivers J.P.W., Burger L.H. Allometry in dog nutrition. In In Nutrition of the dog and cat. Waltham Symposium No. 7, Cambridge: Cambridge University Press; 1989. p 67
51. DiBartola S.P., Bateman S. Introduction to fluid therapy. In DiBartola S.P., editor: Fluid, electrolyte, and acid-base disorders, ed 3, St Louis: Saunders, 2006.
52. Ryan C.P. Toxicity associated with lactated Ringer’s solution containing preservatives. Fel Pract. 1982;12:7.
53. Day T.K., Bateman S. Shock syndromes. In: DiBartola S.P., editor. Fluid, electrolyte, and acid-base disorders in small animal practice. ed 3. St Louis: Saunders; 2006:540-564.
54. Francis A., et al. Adverse reactions suggestive of type III hypersensitivity in six healthy dogs given human albumin. J Am Vet Med Assoc. 2007;239:873-879.
55. Hansen B.D. Technical aspects of fluid therapy. In: DiBartola S.P., editor. Fluid, electrolyte, and acid-base disorders in small animal practice. ed 3. St. Louis: Saunders; 2006:344-376.
56. Otto C.M., Crowe D.T. Intraosseous resuscitation techniques and applications. In: Kirk R.W., Bonagura J.D., editors. Current veterinary therapy XI. Philadelphia: Saunders; 1992:107.
57. Fiser D.H. Intraosseous infusion. N Engl J Med. 1990;322:1579.
58. Okrasinski E.B., Krahwinkel D.J., Sanders W.L. Treatment of dogs in hemorrhagic shock by intraosseous infusion of hypertonic saline and dextran. Vet Surg. 1992;21:20.
59. O’Brien D.P., Kroll R.A., Johnson G.C., et al. Myelinolysis after correction of hyponatremia in two dogs. J Vet Int Med. 1994;8:40.
60. Brady C.A., Vite C.H., Drobatz K.J. Severe neurologic sequelae in a dog after treatment of hypoadrenal crisis. J Am Vet Med Assoc. 1999;215:222.
61. Rose B.D., Post T.W. Hyperosmolal states: hypernatremia. In Clinical physiology of acid-base and electrolyte disorders. ed 5. 2001. McGraw-Hill. New York. p 746
62. Fournier G., Pfaff-Poulard C., Methani K. Rapid correction of hypokalaemia via the oral route. Lancet. 1987;2:163.
63. Dow S.W., Fettman M.J., Curtis C.R., et al. Hypokalemia in cats: 186 cases (1984-1987). J Am Vet Med Assoc. 1989;194:1604.
64. Whang R. Refractory potassium repletion: a consequence of magnesium deficiency. Arch Intern Med. 1992:152.
65. Bateman S. Disorders of magnesium. In: DiBartola S.P., editor. Fluid, electrolyte, and acid-base disorders. ed 3. St Louis: Saunders; 2006:217.
66. Willard M.D., Zerbe C.A., Schall W.D., et al. Severe hypophosphatemia associated with diabetes mellitus. J Am Vet Med Assoc. 1987;190:1007.
67. Dhupa N. Magnesium therapy. In: Kirk R.W., Bonagura J.D., editors. Current veterinary therapy XII. Philadelphia: Saunders; 1995:132.
68. Narins R.G., Cohen J.J. Bicarbonate therapy for organic acidosis: the case for its continued use. Ann Intern Med. 1987;106:615.
69. Stacpoole P.W. Lactic acidosis. Ann Intern Med. 1986;105:276.
70. Hartsfield S.M. Sodium bicarbonate and bicarbonate precursors for treatment of metabolic acidosis. J Am Vet Med Assoc. 1981;179:914.
71. Brady C.A., Otto C.M. Systemic inflammatory response syndrome, sepsis, and multiple organ dysfunction. Vet Clin N Am. 2001;31:1147.
73. Schertel E.R., Tobias T.A. Hypertonic fluid therapy. In: DiBartola S.P., editor. Fluid therapy in small animal practice. ed 2. Philadelphia: Saunders; 2000:496.
74. Rudolff E., Kirby R. Colloid fluid therapy. In: Bonagura J.D., Twedt D.C., editors. Kirk’s Current veterinary therapy XIV. St Louis: Sanders; 2009:61-67.
75. Allen D.A., Schertel E.R., Muir W.W., et al. Hypertonic saline/dextran resuscitation of dogs with experimentally induced gastric dilatation-volvulus shock. Am J Vet Res. 1991;52:92.
76. Ware W.A. Myocardial diseases of the dog. In Nelson R.W., Couto C.G., editors: Small animal internal medicine, ed 3, St Louis: Mosby, 2003. p 106
77. Haskins S.C. Management of septic shock. J Am Vet Med Assoc. 1992:1915. 200
78. Jafari H.S., McCracken G.H. Sepsis and septic shock: a review for clinicians. Pediatr Infect Dis J. 1992;11(9):739.
79. Weeren F.R., Muir W.W. Clinical aspects of septic shock and comprehensive approaches to treatment in dogs and cats. J Am Vet Med Assoc. 1992;200:1859.
80. Bone R.C. Toward an epidemiology and natural history of SIRS (systemic inflammatory response syndrome). J Am Med Assoc. 1992;268:3452.
81. Lefering R., Neugebauer E.A. Steroid controversy in sepsis and septic shock: a meta-analysis. Crit Care Med. 1995;23:294.
82. Burkitt J.M., Haskins S.C. Relative adrenal insufficiency in dogs with sepsis. J Vet Internal Med. 2007;21(2):226-231.
83. Cowgill L.D., Elliot D.A. Acute renal failure. In: Ettinger S.J., Feldman E.C., editors. Textbook of veterinary internal medicine. ed 5. Philadelphia: Saunders; 2000:1615.
84. Chew D.J., Gieg J.A. Fluid therapy during intrinsic renal failure. In: DiBartola S.P., editor. Fluid, electrolyte, and acid-base disorders in small animal practice. ed 3. St Louis: Saunders; 2006:518-540.
85. Allen D.G. Special techniques. In: Allen D.G., editor. Small animal medicine. Philadelphia: JB Lippincott, 1991. 1991, p 1035
86. Stone E.A., Barsanti J.A. Urologic surgery of the dog and cat. Philadelphia: Lea & Febiger; 1992. p 119
87. Polzin D., Osborne C.A., O’Brien T. Diseases of the kidneys and ureters. In: Ettinger S.J., editor. Textbook of veterinary internal medicine. ed 3. Philadelphia: Saunders; 1989:1962.
88. Bonagura J.D., Lehmkuhl L.B., Autran de Morais H. Fluid and diuretic therapy in heart failure. In: DiBartola S.P., editor. Fluid, electrolyte, and acid-base disorders in small animal practice. ed 3. St Louis: Saunders; 2006:490-518.
89. Simpson K.W., Birnbaum N. Fluid and electrolyte disturbances in gastrointestinal and pancreatic disease. In: DiBartola, editor. Fluid electrolyte and acid-base disorders. ed 3. St Louis: Saunders; 2006:429.
90. Heald R.D., Jones B.D., Schmidt D.A. Blood gas and electrolyte concentrations in canine parvoviral enteritis. J Am Anim Hosp Assoc. 1986;22:745.
91. Williams D.A. Exocrine pancreatic disease. In Ettinger S.J., Feldman E.C., editors: Textbook of veterinary internal medicine, ed 5, Philadelphia: Saunders, 2000. p 1345
92. Logan J.C., Callan M.B., Drew K., et al. Clinical indications for use of fresh frozen plasma in dogs: 74 dogs (October through December 1999). J Am Vet Med Assoc. 2001;218:1449.
93. Schenker S., Breen K.J., Hoyumpa A.M. Hepatic encephalopathy: current status. Gastroenterology. 1974;66:121.
94. Feldman E.C., Nelson R.W. Beta-cell neoplasia: Insulinoma. In Canine and feline endocrinology and reproduction. ed 3. 2004. Saunders. Philadelphia. p 613
95. Feldman E.C., Nelson R.W. Hypocalcemia and primary hypoparathyroidism. In Canine and feline endocrinology and reproduction. ed 3. 2004. Saunders. Philadelphia. p 716
96. Feldman E.C. Disorders of the parathyroid glands. In: Ettinger S.J., Feldman E.C., editors. Textbook of veterinary internal medicine. ed 7. St Louis: Saunders; 2010:1722-1751.
97. Elliott J., Dobson J.M., Dunn J.K., et al. Hypercalcemia in the dog: a study of 40 cases. J Small Anim Pract. 1991;32:564.
98. Green T., Chew D.J. Calcium disorders. In: Silverstein D.C., Hopper K., editors. Small animal critical care medicine. St Louis: Saunders; 2009:56.