Diseases of the Adrenal Glands
Hypoadrenocorticism
Pathophysiology
Which hormones are deficient in hypoadrenocorticism (Addison’s disease) varies according to the underlying pathophysiology. The most common form of hypoadrenocorticism is primary adrenal failure. Glucocorticoids (e.g., cortisol) alone or both glucocorticoids and mineralocorticoids (e.g., aldosterone) may be deficient, depending on which adrenocortical zones have been destroyed. Primary hypoadrenocorticism can be spontaneous or iatrogenic as a result of mitotane, ketoconazole, or trilostane administration. Secondary hypoadrenocorticism is due to pituitary failure to secrete adrenocorticotropic hormone (ACTH). Because ACTH has minimal effects on aldosterone secretion, ACTH deficiency causes isolated glucocorticoid insufficiency. ACTH deficiency can be idiopathic, caused by head trauma or neoplasia, or iatrogenic, secondary to chronic suppression of ACTH caused by administration of glucocorticoids197 or progestins.198,199 Exogenous glucocorticoids of any form, even topical, can feed back and turn off ACTH secretion. With chronic ACTH deficiency, adrenocortical atrophy occurs and cortisol secretion falls. Even though cats are considered to be relatively resistant to most side effects of glucocorticoids, they are just as susceptible as dogs to the adrenal suppressive effects. Administration of megestrol acetate (Ovaban) or other progestins to cats200 can also suppress ACTH secretion. Certain breeds of dogs (e.g., Nova Scotia Duck Tolling retrievers, Standard Poodles, and Bearded Collies) can have a genetic component to development of hypoadrenocorticism.201-203
Hypoadrenocorticism can be a life-threatening condition requiring immediate life-saving therapeutic intervention. The acute life-threatening effects generally reflect mineralocorticoid deficiency and, less commonly, glucocorticoid deficiency. Glucocorticoids affect almost every tissue; many effects are critical to normal homeostasis and become more critical in stressed patients. Glucocorticoids stimulate gluconeogenesis and glycogenolysis by direct hepatic effects and by stimulating protein and fat catabolism peripherally. They also have a permissive effect on adrenergic receptors, enhancing tissue response to alpha- and beta-receptor stimulation. Mineralocorticoids are crucial to maintaining sodium, potassium, and water balance.
Lack of cortisol secretion may cause depression, lethargy, anorexia, vomiting, abdominal pain, shaking or shivering, and weight loss. In severe cases cardiovascular collapse may result. Clinical signs indicative of mineralocorticoid deficiency include collapse and bradyarrhythmias. On routine blood work in patients with mineralocorticoid deficiency, hyponatremia, hyperkalemia, and hypochloremia are usually present. Hypoadrenocorticoid patients often are azotemic. Hypoglycemia will be present due to glucocorticoid deficiency in a small percentage of cases. Hypercalcemia occurs in about 25% of patients. Mild to moderate metabolic acidosis may be present, particularly if mineralocorticoid secretion is impaired.
Diagnosis
Diagnosis of hypoadrenocorticism can be suspected or ruled out on the basis of a baseline cortisol but must be confirmed by performance of an ACTH stimulation test. In one study dogs with basal cortisol concentrations above 55 nmol/L (2 μg/dL) that were not receiving corticosteroids, mitotane, or ketoconazole were highly unlikely to have hypoadrenocorticism; however, if the basal cortisol concentration was less than 55 nmol/L, an ACTH stimulation test was needed for further evaluation.204 Likely, the same applies to dogs receiving progestins or trilostane. The protocol for performance of an ACTH stimulation test is the same as for diagnosis of hypoadrenocorticism (discussed below). In patients with primary or secondary hypoadrenocorticism, baseline cortisol concentration will be low, with minimal to no response to ACTH stimulation. The test does not distinguish spontaneous from iatrogenic disease and only confirms cortisol insufficiency. In a cortisol-deficient patient, if moderate to marked hyponatremia or hyperkalemia are present, aldosterone is assumed to be lacking as well. Serum aldosterone concentrations can be measured, but this is often not necessary and interpretation can be problematic.205
To differentiate primary from secondary hypoadrenocorticism, plasma endogenous ACTH concentration can be measured, as described below (see section on hyperadrenocorticism). In primary hypoadrenocorticism, negative feedback on the pituitary is lost and endogenous ACTH concentrations will be greatly increased; secondary hypoadrenocorticism, by comparison, is, by definition, a lack of ACTH. Evaluation of endogenous ACTH concentration can be considered if a patient has glucocorticoid deficiency only. If aldosterone secretory ability is impaired, the disease must be primary to the adrenal glands. Distinguishing primary from secondary disease in patients with spontaneous isolated glucocorticoid deficiency can be prognostic. If the disease is primary, aldosterone secretion is likely to be lost in the future and serum electrolyte concentrations should be monitored regularly; if the hypoadrenocorticism is secondary, mineralocorticoid secretion will remain normal.
Therapy
Therapy for hypoadrenocorticism focuses first on acute management of a hypoadrenal crisis, if present, and then on long-term maintenance therapy. The goals of therapy for treatment of a crisis are to replace fluid volume and the needed hormones, correct cardiovascular collapse, and rectify electrolyte and acid–base imbalances. Once the acute crisis is resolved, patients with hypoadrenocorticism can lead normal lives as long as medication is used appropriately.
Before therapy is initiated, if hypoadrenocorticism is suspected and patient status permits, an ACTH stimulation test should be performed for diagnosis. If emergency care that includes glucocorticoid administration is required, dexamethasone is the first choice to use. Exogenous glucocorticoid administration can affect ACTH stimulation testing in two ways. First, certain glucocorticoids—prednisone, prednisolone, methylprednisolone, and hydrocortisone—cross-react on cortisol assays and, if present, will artificially elevate apparent cortisol concentrations. If any of these four glucocorticoids are administered, an ACTH stimulation test should not be performed for 12 hours, whereas if dexamethasone is given, an ACTH stimulation test can still be performed immediately. (If methylprednisolone acetate [e.g., Depo-Medrol] is administered, testing may need to be delayed much longer.) Second, any glucocorticoid can feed back and suppress ACTH and cortisol secretion. The degree of suppression and the length of duration depends on which glucocorticoid was administered, as well as the route, dose, and duration. For example, depending on the dose, a single dexamethasone injection given to treat an Addisonian crisis may, within a few days, completely suppress basal cortisol concentration and suppress post-ACTH cortisol concentrations up to 33%. However, such suppression is easily distinguishable from spontaneous hypoadrenocorticism in which serum cortisol concentration is nondetectable before and after administration of ACTH. Thus, if after a single dexamethasone injection, post-ACTH serum cortisol concentration is nondetectable, a diagnosis of hypoadrenocorticism is made; if the post-ACTH serum cortisol concentration is just below the reference range, the patient’s suppressed adrenal function is likely due to administration of an exogenous glucocorticoid, not to hypoadrenocorticism.
For treatment of an Addisonian crisis, fluid therapy is paramount and the primary priority. Although use of 0.9% saline was once advocated, rapid correction of hyponatremia has now been recognized to lead to central nervous system dysfunction.206,207 During chronic hyponatremia, the brain adapts to prevent cerebral edema. With rapid correction of serum sodium concentration, osmotic shifts and cerebral dehydration occur, with a possible resultant pontine myelinosis and neurologic signs such as disorientation, dysphagia, weakness, and quadriparesis. Thus, although balanced solutions such as Normosol-R or lactated Ringer’s solution contain potassium, they are currently the fluid of choice, with the latter possibly being the best insofar as the sodium is the lowest. Hypertonic saline administration is contraindicated. For treatment of an Addisonian crisis, shock doses of fluids should be given initially and then rehydration corrected over 6 to 24 hours, depending on patient stability. Fluid therapy should be adjusted to increase serum sodium concentration at a rate of 0.5 mEq/L/hr. Frequent measurement of serum sodium concentration is important to ensure that the rate of correction of hyponatremia is appropriate. Fluid type and rate can be adjusted accordingly. If hypoglycemia is present, dextrose should be added to the fluids to make a 5% solution.
A rapid-acting glucocorticoid such as prednisolone sodium succinate (1 to 2 mg/kg over 2 to 4 minutes; can be repeated in 2 to 6 hours), dexamethasone or dexamethasone sodium phosphate (0.5 to 2 mg/kg intravenously, every 2 to 6 hours), or hydrocortisone hemisuccinate or hydrocortisone phosphate (2-4 mg/kg intravenously over 2 to 4 minutes, every 8 hours) should be administered. Alternatively, hydrocortisone sodium succinate can be infused at a rate of 0.5 to 0.625 mg/kg/hr intravenously. Although dexamethasone can be used to replace glucocorticoid deficiency, mineralocorticoid deficiency will not be affected; thus prednisolone, which has some mineralocorticoid activity, might be preferred, at least initially. However, the effect of glucocorticoid administration on diagnostic testing should also be considered (discussed previously). Mineralocorticoid therapy is needed only if aldosterone is deficient and is not recommended until serum sodium concentration is in the reference range or slightly below. Mineralocorticoid administration can correct serum sodium concentration fairly rapidly.
Dilutional effects and increased GFR secondary to fluid administration will begin to correct life-threatening hyperkalemia. Very rarely does hyperkalemia fail to respond rapidly to volume replacement. In such instances, or if the hyperkalemia is immediately life-threatening, 10% (100 mg/mL) calcium gluconate (0.5-1 mg/kg) can be given intravenously slowly. The calcium protects the myocardium from the effects of the potassium. An electrocardiogram must be monitored during calcium infusion and treatment stopped if new arrhythmias occur or bradycardia worsens. Alternatively, regular insulin (0.06-0.125 U/kg, plus 20 mL of a 10% glucose solution for every unit of insulin given) can be administered. Insulin causes glucose to move intracellularly, and potassium will follow. Glucose is infused in an attempt to prevent hypoglycemia. However, given the abnormal glucose metabolism in hypoadrenocorticism, hypoglycemia still often results. If a patient is already hypoglycemic, just giving dextrose alone is much safer and will also cause intracellular movement of potassium. Lastly, bicarbonate can be given to address acidosis but is rarely required because the other treatments usually resolve the acidosis.
Response to initial therapy of hypoadrenocorticism should occur in 1 or 2 hours in patients suffering from hypoadrenocorticism. In general, cats take longer to respond than dogs. Because sodium deficiency may result in a washout of the medullary interstitium, renal function may not return to normal quickly, and the patient may be diuresing for several days. Care must be taken to balance fluid input with excessive output.
Mineralocorticoid replacement therapy
Maintenance therapy for hypoadrenocorticism begins when vomiting, diarrhea, weakness, and depression have resolved. Mineralocorticoid replacement is needed only for patients that are aldosterone deficient and is available in oral or depot preparations. The initial recommended dose of fludrocortisone in dogs is 0.01 to 0.02 mg/kg orally daily208 and in cats 0.1 mg/cat daily,200,209 much higher doses than required in humans. Dosage adjustments, if necessary, are made on the basis of serum electrolyte concentrations. Ideally, sodium and potassium should be within reference ranges. Sodium and potassium should be monitored every 1 to 2 weeks after initiating therapy until a patient is stable. In dogs the daily dosage is adjusted by 0.05 to 0.1 mg increments. Once electrolyte concentrations have stabilized, a patient should be reevaluated monthly for the first 3 to 6 months and every 3 to 6 months thereafter, as long as no clinical signs are apparent. In cats timing of monitoring is the same, and adjustments are made in 0.05-mg increments. In the authors’ experience, however, fludrocortisone fails to normalize sodium in a number of patients no matter how high the dose.
In dogs the final required fludrocortisone dose varies greatly between patients; in one study the median final required dose was 0.023 mg/kg/day (range approximately 0.008 to 0.75 mg/kg daily).210 Required doses often increase over the initial 6 to 18 months of therapy,205,210 possibly as a result of ongoing destruction of the adrenal cortex or changes in drug absorption or metabolism.
Overall, fludrocortisone therapy is effective. In 33 dogs, the response to treatment was considered good to excellent in 78.8%, fair in 9.1%, and poor in 12.1%.211 The most common side effects are polyuria and polydipsia, but polyphagia, hair loss, and weight gain may be seen. Most of the adverse effects occur when prednisone and fludrocortisone are administered concurrently and resolve when glucocorticoid therapy is discontinued, but polyuria and polydipsia can be seen with fludrocortisone alone.205,210,211 Although fasting hypercholesterolemia and hypertriglyceridemia have been noted with fludrocortisone administration,211 the significance of these changes remain unknown.
The recommended starting dose of desoxycorticosterone pivalate (DOCP; Percorten, Novartis) for dogs is 2.2 mg/kg intramuscularly every 25 days. For cats the dose of DOCP is 10 to 12.5 mg/cat intramuscularly monthly.209 The subcutaneous route, however, can be used, at least in dogs.212 For the majority of dogs, the dosing regimen will be effective. Although one study initiated before a manufacturer’s recommended dose was chosen found that some dogs did not need that high a dose, starting at 2.2 mg/kg is safe.213,214
To decrease cost, it may not be necessary to administer DOCP at the full label dose. With DOCP, clinicians typically assume a 28-day interval and start at a dose of 2.2 mg/kg. Electrolytes should be measured on days 14 and 28, and if they are within the reference range on day 28, the DOCP dose can be decreased 10%. When a dose is found that no longer maintains serum sodium and potassium concentrations in the reference range for the full 28 days, the lowest DOCP dose that lasted 28 days can be used. An alternative is to administer 2.2 mg/kg DOCP, lengthen the interval by 3 days with each injection until the interval is too long, and then use the longest interval during which serum electrolyte concentrations were in the reference range. However, it is probably harder for owners to remember the injections on a long interval, and the authors prefer lowering the dose and maintaining a 28-day interval instead.
A small percentage of dogs, however, do require either injections more frequently than every 25 days or more than 2.2 mg/kg to keep a 25-day or longer interval.210,215 If the patient is hyponatremic or hyperkalemic at day 14, the next dose should be increased by 10%. If the electrolytes are normal on day 14 but abnormal on day 28, the interval between injections should be decreased by 2 days205 or the dose increased 10%. In dogs that require DOCP more frequently than every 28 days, clinical signs of Addison’s disease may recur before the recheck on day 28. If return of the hypoadrenal state is suspected, the dog should be seen immediately and serum electrolytes measured. If hyponatremia and hyperkalemia are documented, the DOCP injection can be given at that time. If the dosing interval is shortened, the timing of monitoring should be changed accordingly for the next treatment period. Two rechecks should be performed during each dosing interval until good control of the Addison’s disease on the last day of the dosing interval is demonstrated.
DOCP is a highly efficacious treatment for hypoadrenocorticism with minimal side effects. Adverse effects reported include depression, polyuria, polydipsia, anorexia, skin and coat changes, diarrhea, vomiting, weakness, weight loss, incontinence, and pain on injection, but all are uncommon. Some of the adverse effects, such as polyuria and polydipsia, are more likely caused by concurrent glucocorticoid administration than by DOCP itself. Treatment failures also occur rarely.
Any recheck, whether monitoring fludrocortisone or DOCP therapy, should include a full physical exam, complete history, and determination of BUN concentration, as well as measurement of electrolytes. If at any recheck the serum electrolyte concentrations are within the reference range but problems, sometimes quite vague, such as anorexia, vomiting, diarrhea, or unwillingness to play exist, glucocorticoid deficiency is the likely cause, and the prednisone dose should be adjusted accordingly. An elevated BUN concentration can be a sign of dehydration caused by insufficient therapy.
Advantages and disadvantages exist with the use of either fludrocortisone or DOCP. For fludrocortisone the major advantage is the ease of diagnosing and adjusting an incorrect dosage because daily administration is easily altered. Daily therapy also constantly reminds owners that their pet is afflicted with a life-threatening disease and needs constant therapy and monitoring. Lastly, the medication is readily available at most pharmacies. However, fludrocortisone can be quite expensive despite the availability of a generic product, especially if higher doses are required; some patients may not be adequately controlled and side effects may occur, even when used without concomitant glucocorticoid therapy. If expense, existence of side effects, or lack of efficacy necessitates discontinuation of fludrocortisone, DOCP becomes the only choice.
For DOCP advantages include a low incidence of adverse effects if used alone, less common treatment failures than with fludrocortisone therapy, and need for infrequent administration. A subcutaneous injection can be given by owners if trained properly, but great care should be taken in selection of owners for this task. Missing an injection or giving one inappropriately and not realizing the mistake could be fatal for the patient. Apparent failures may be due to the owner’s difficulty in providing injections; improper technique should always be ruled out. If a patient truly does not respond to DOCP, fludrocortisone therapy should be instituted.
Glucocorticoid replacement therapy
For all dogs and cats that have either iatrogenic or spontaneous secondary hypoadrenocorticism or primary Addison’s disease without mineralocorticoid deficiency, only glucocorticoid replacement therapy is required. It should be remembered, however, that the disease of animals with primary glucocorticoid deficiency may progress to include mineralocorticoid insufficiency as well, and therapy must be adjusted accordingly. For an animal lacking both types of adrenocortical hormones, the need for daily maintenance glucocorticoid replacement therapy depends in part on which mineralocorticoid supplement is being administered. (For animals with hypoadrenocorticism under stress, excess glucocorticoids are always recommended; this is discussed later at more length.). Fludrocortisone has both glucocorticoid and mineralocorticoid activity, whereas DOCP has only mineralocorticoid properties. Thus approximately 50% of dogs receiving fludorocortisone may not require concomitant exogenous glucocorticoid administration.205,208 Although some dogs on DOCP have not received glucocorticoid therapy,211,215 this practice is not recommended205 insofar as the patient will be glucocorticoid deficient on a daily basis.
All animals beginning maintenance therapy for spontaneous hypoadrenocorticism should receive prednisone or prednisolone at a “physiologic” dose of 0.1 to 0.22 mg/kg once daily; prednisolone may be the preferred form in cats. If the animal is on fludrocortisone, once a dose that maintains serum electrolyte concentrations within the reference range has been determined, the glucocorticoid can be tapered to alternate days and then discontinued to see whether continued glucocorticoid therapy will be required.208 If the dog or cat is lethargic, dull, or unwilling to exercise or play or if clinical signs of hypocortisolism such as weakness, anorexia, vomiting, and diarrhea are apparent, glucocorticoids should be reinstituted at the lowest dosage that does not produce glucocorticoid-associated adverse effects and keeps the patient free of clinical signs. Patients receiving DOCP should always receive daily glucocorticoid replacement therapy, similarly at the lowest dosage possible. In cats methylprednisolone acetate (Depo-Medrol, 10 mg/month intramuscularly) can be administered if giving them pills is difficult,209 but complications of glucocorticoid therapy such as DM may be more likely.200 Depo-Medrol is not recommended for use in dogs.
In non-Addisonian patients receiving exogenous glucocorticoids chronically, every-other-day administration is recommended to minimize resultant adrenal atrophy. As patients with spontaneous hypoadrenocorticism already have significant adrenocortical destruction or atrophy, atrophy secondary to glucocorticoids is not a concern. Therefore, if a patient with spontaneous hypoadrenocorticism is deemed to need physiologic glucocorticoid replacement therapy, the medication should be given daily to make sure the patient is never glucocorticoid deficient.
During adverse periods such as illness, surgery or trauma, glucocorticoid requirements increase and additional glucocorticoid supplementation at 2 to 10 times the physiologic levels should be administered;208 if the patient does not receive daily glucocorticoid supplementation, it should be given during such times. Working dogs such as hunters and field trial participants should be allowed to complete their usual activities, but owners must be instructed to monitor their pets more closely than normal and discontinue activity if a dog appears unduly fatigued. On days of planned increased exercise or stress, the daily glucocorticoid dose should be doubled,205 or, if the animal is not receiving any glucocorticoids, a dose of 0.1 to 0.2 mg/kg can be given.
If the possibility of complete iatrogenic suppression of adrenal glucocorticoid secretion exists in patients receiving long-term exogenous glucocorticoids, the ideal way to assess adrenal reserve is by performing an ACTH stimulation test. If the response to an injection of ACTH is low, the patient should be tapered off the glucocorticoid supplementation until their adrenal gland function recovers. Once the decision to end steroid therapy is made, the dose of glucocorticoid should be decreased to physiologic doses of prednisone over 1 to 2 weeks.205 If adrenal suppression is secondary to topical glucocorticoid administration, topical administration should be stopped and oral prednisone initiated at physiologic doses. If this dose of prednisone is tolerated for a week without clinical signs of cortisol deficiency, the dosage schedule should be reduced by administering the drug every other day. After 2 weeks at this dose, the dosage should be further reduced by giving the medication every third day. After 2 to 3 weeks, prednisone most likely can be discontinued.205 Ideally, however, before discontinuation, an ACTH stimulation test should be performed 12 hours after the last dose of prednisone to ensure that the patient has a normal adrenal reserve. Cats should be placed on physiologic doses of prednisolone and tapered off as are dogs.
In older reference sources, salt supplementation was advocated for treatment of patients with mineralocorticoid deficiency,216 but this may not be necessary for dogs being fed a standard diet.205 Salt supplementation may still be helpful, however, in an occasional dog that requires large doses of an exogenous mineralocorticoid or that remains hyponatremic despite being normokalemic on an appropriate dose of mineralocorticoid.205,210 If salt is administered, the initial dose should be 0.1 mg/kg/day, divided over two or three meals.216 After initiating salt supplementation, serum sodium and potassium concentrations should be measured and the sodium chloride dose adjusted accordingly.
Prognosis
Prognosis for patients with Addison’s disease is excellent. Median survival is approximately 5 years, and patients typically die as a result of other diseases. The cause of hypoadrenocorticism, the mineralocorticoid used for treatment (i.e., fludrocortisone versus DOPCP), and signalment do not affect survival.210 Owners should be aware, however, that their pet has a potentially life-threatening disease when not treated appropriately and continuous, lifelong therapy for spontaneous hypoadrenocorticism and appropriate monitoring are essential. If the disease is iatrogenic, lifelong therapy may not be required, depending on the cause, but therapy is nonetheless important until adrenal function has recovered.
Hyperadrenocorticism
Pathophysiology
Canine and feline hyperadrenocorticism can be either pituitary or adrenal dependent. The pituitary form is more common in dogs and cats than the adrenal form, accounting for approximately 80% to 85% of cases of hyperadrenocorticism. In pituitary-dependent hyperadrenocorticism (PDH), a corticotroph tumor secretes ACTH. The excess ACTH secretion leads to increased release of cortisol from the adrenal glands. In adrenal-dependent hyperadrenocorticism, an adrenal tumor (AT) autonomously secretes cortisol. In dogs and cats, ACTH-secreting pituitary tumors are almost 100% benign, whereas cortisol-secreting ATs are approximately 50% benign and 50% malignant. In either form of the disease, the majority of the clinical signs are caused by hypercortisolemia. The pathophysiology leading to the clinical sequelae is complex because of the large number of body tissues influenced by endogenous glucocorticoids (see chapter on glucocorticoid therapy). Large tumors, either adrenal or pituitary, can also lead to clinical signs because of their mass-occupying effects.
Diagnosis
Diagnosis of hyperadrenocorticism is made on the basis of positive screening test results in patients with the appropriate history, clinical signs, and biochemical test results. Three screening tests are designed to help determine whether a patient has hyperadrenocorticism: the urinary cortisol:creatinine ratio (UCCR), the ACTH stimulation test, and the low-dose dexamethasone suppression test (LDDST). Measurement of a UCCR is a highly sensitive but very nonspecific test. Almost all (>95%) cats and dogs with hyperadrenocorticism have an elevated UCCR, but the ratio is very often elevated in dogs and cats that do not have hyperadrenocorticism (approximately 80% to 85%).217 Thus the best use of the UCCR is as a means to rule out the diagnosis of hyperadrenocorticism. If a patient has a normal UCCR, it is highly unlikely to have hyperadrenocorticism; however, because of the very high rate of false-positive results, if a UCCR is elevated, another screening test must be done to confirm the presence of hyperadrenocorticism. One advantage of the UCCR is ease of testing: A single urine sample is required. As even the stress of being in a hospital can elevate a UCCR,218,219 it should be measured on a sample collected at home.
The ACTH stimulation test is recommended for patients with minimal clinical signs of hyperadrenocorticism, patients that are receiving phenobarbital, or patients that have a nonadrenal illness present (e.g., a diabetic dog that is also suspected of having hyperadrenocorticism).217 In addition, the ACTH stimulation test is the only screening test that can differentiate between spontaneous and iatrogenic hyperadrenocorticism. The recommended form of ACTH is cortrosyn (Cosyntropin, Amphastar Pharmaceuticals, Rancho Cucamonga, Calif.). To perform the test, the clinician injects cortrosyn at a dose in dogs of 5 μg/kg intravenously220 or intramuscularly221 or 125 μg in cats.222 Blood samples are taken before and 1 hour after injection. In animals with spontaneous hyperadrenocorticism, the response to ACTH should be greater than in healthy patients.
Overall, the ACTH stimulation test will be positive in approximately 80% of dogs and cats with hyperadrenocorticism.217,223 If the forms of hyperadrenocorticism are considered separately in dogs, for PDH the sensitivity is 87%, whereas for AT the sensitivity is 61%.217 In PDH false-negative results may be attributable to early disease where adrenocortical hyperplasia is minimal. In AT the tumor tissue may not have ACTH receptors and therefore might not respond to an ACTH injection. Nonadrenal illness can affect the ACTH stimulation test in dogs and cats.224,225 In one study 14% of dogs with nonadrenal illness had an ACTH stimulation test consistent with hyperadrenocorticismeven although they did not have the disease.224 Infrequently, a subnormal ACTH response is seen in dogs with AT. A low baseline cortisol concentration with little to no response to exogenous ACTH suggests iatrogenic hyperadrenocorticism. The ACTH test also should be used to monitor patients receiving mitotane, ketoconazole, or trilostane therapy for treatment of hyperadrenocorticism.
Owing to issues related to the cost and availability of cortrosyn, interest has been raised in use of compounded ACTH. Two studies have assessed a total of five compounded forms in dogs. The gels studied appear to be effective in normal dogs.226,227 However, the protocols recommended by the manufacturers may not be appropriate, and, if a compounded ACTH gel is being used, samples should be taken before injection and at both 60 and 120 minutes after injection, so the peak response is not missed.227 Whether the gels are as effective at diagnosing hyperadrenocorticism as is cortrosyn has not been rigorously assessed; they may not be.226
In normal animals a low dose of dexamethasone suppresses ACTH secretion, and, as a result, blood cortisol concentration decreases. Patients with pituitary- or adrenal-dependent hyperadrenocorticism should continue to secrete cortisol despite being given a low dose of dexamethasone, and suppression will not occur. Overall, the LDDST shows inadequate suppression (i.e., is positive) in approximately 95% of dogs with hyperadrenocorticism.217 In general, the LDDST is recommended in patients with moderate to severe clinical signs consistent with hyperadrenocorticism.217 A disadvantage of the LDDST in dogs is that nonadrenal illness can cause the test to give false-positive results in a high percentage of dogs. As many as 56% of ill dogs that do not have hyperadrenocorticism may have a positive LDDST test result.224 In cats 6 weeks of uncontrolled DM does not affect LDDST test results,228 but whether longer or more severe illness may do so is unknown.
What is considered a “low-dose” test for screening varies between dogs and cats. In both species dexamethasone is administered with blood samples being taken before and 4 and 8 hours after injection. However, in dogs the dose used is 0.01 to 0.015 mg/kg, whereas in cats it is 0.1 mg/kg.
Which test is best, the ACTH stimulation test or LDDST, for diagnosing hyperadrenocorticism in cats is unknown. In one literature review, in cats with hyperadrenocorticism 81% of ACTH stimulation tests were positive (n=37), whereas 79% of cats (n=28) showed inadequate suppression at 8 hours after dexamethasone.223 Interestingly, in three cats, two ACTH stimulation tests were performed, with one being negative and one positive. Other reports have not shown as high a sensitivity for the ACTH stimulation, and some authors prefer the LDDST.229
Once a diagnosis of hyperadrenocorticism is made, the underlying cause—pituitary or adrenal —must be delineated because this provides information on prognosis and treatment options. The UCCR or ACTH stimulation test can never be used to differentiate between PDH and AT. In up to 60% of dogs, the LDDST can provide the differentiation as well as the diagnosis of hyperadrenocorticism.230 If the 8-hour postdexamethasone concentration is not fully suppressed (check with the laboratory for their definition of suppression; in most labs it is a serum cortisol concentration of less than approximately 30 nmol/L or 1 to 1.5 μg/dL), the results are consistent with a diagnosis of hyperadrenocorticism. If, in addition, the 4-hour postdexamethasone concentration is fully suppressed or if one or both postdexamethasone concentrations is less than 50% of baseline, PDH is present.230 However, if the baseline cortisol is already below 30 nmol/L, these guidelines do not apply.231 If both postdexamethasone concentrations are above 30 nmol/L and neither of these values is less than 50% of baseline, either PDH or AT is possible. In rare cases dogs with an AT may meet one of these criteria for diagnosing PDH.231 Whether differentiation can be done with the LDDST in cats is not known.
If the LDDST was not done as the screening test or did not delineate the form of hyperadrenocorticism present, further tests available to differentiate between PDH and AT are measurement of plasma endogenous ACTH (eACTH) concentration; the high-dose dexamethasone suppression test (HDDST); and imaging such as abdominal ultrasound, CT, and MRI. An advantage of eACTH measurement is that only a single blood sample is required, but special handling is needed. Samples should be collected with EDTA and centrifuged within 15 minutes, and the plasma separated and placed in plastic tubes. Addition of aprotinin facilitates accuracy of measurement232 as it prevents eACTH degradation. If aprotinin is used, the plasma sample must remain cool only until it arrives at a laboratory; if aprotinin is not used, the sample must remain frozen until analysis.
In dogs with PDH, eACTH concentration should be normal to elevated owing to secretion from the pituitary tumor. In dogs with an AT, the autonomous secretion of cortisol by the tumor will turn off pituitary ACTH secretion so eACTH should be below normal. Values to be used for test interpretation vary with the laboratory and assay used. An advantage of this test is that it can confirm the presence of an AT, whereas the HDDST can never do so. Unfortunately, nondiagnostic values exist. For example, at the Auburn University Endocrine Diagnostic Service, a concentration of less than 10 pg/mL is consistent with an AT, whereas one greater than 15 pg/mL is consistent with PDH. The area between 10 and 15 pg/mL is a “gray zone,” in which differentiation is impossible. Although other laboratories may have different cutoffs, a similar gray zone will exist. Gray zone results occur in approximately 18% of canine submissions. However, with repeat testing when the initial result is in the gray zone, a definitive differentiation can be achieved in approximately 96% of dogs.217 Unfortunately, there is no way to predict when a blood concentration will be in the diagnostic range.
The basis of the HDDST is that high doses of dexamethasone are generally sufficient to cause pituitary gland tumors to decrease ACTH secretion, and, as a result, cortisol concentration falls. In comparison, ATs secrete cortisol autonomously, and because eACTH concentrations are already low, dexamethasone administration does not suppress serum cortisol concentrations. After collection of a baseline cortisol concentration, 0.1 mg/kg dexamethasone is given intravenously in dogs and 1 mg/kg in cats, and then samples are collected 4 and 8 hours later. Suppression is defined as a 4- or 8-hour cortisol concentration less than approximately 30 nmol/L or 1.0 μg/dL (check with the laboratory for the specific ranges) or less than 50% of baseline. Because approximately 25% of dogs with PDH do not suppress on a HDDST, lack of suppression does not mean a patient has an AT. If the criteria for suppression are met, a patient has PDH. If the criteria are not met, a 50/50 chance still exists that the patient has PDH or AT. Thus the HDDST can never confirm the presence of an AT.
Imaging can be helpful in diagnosing hyperadrenocorticism but can never be used as a screening test. Changes associated with hyperadrenocorticism that may be seen on radiographs include hepatomegaly; a pendulous abdomen; calcinosis cutis; osteopenia; and dystrophic mineralization of bronchi, the renal pelvis, liver, gastric mucosa, and abdominal aorta. Abdominal radiography can be helpful in differentiation if an adrenal mass is found. Of 94 ATs in 88 dogs (six dogs had bilateral adenomas or carcinomas), 50 ATs (53%) were detected because of tumoral calcification (n=40) or visualization of a mass (n=17).217 ATs can often be visualized by radiography in cats.223 Both adenomas and carcinomas can contain mineral densities or appear as a mass cranial to the kidney. Mineralization itself, however, is not definitive for a tumor in either species, and as many as one third of normal cats can have adrenal calcification.
Ultrasonography may have more application as a differentiating tool than radiography because both adrenal glands can be visualized. Small or noncalcified ATs can be detected, and bilateral adrenal enlargement can be visualized in dogs with PDH. Ultrasonography defines location, size, and organ involvement of adrenal masses more precisely than radiography alone, but ATs are not always seen. A small degree of asymmetry exists normally. In 71 dogs, 68 of 79 (86%) tumors were found (eight had bilateral tumors).217 Differentiation between an adrenal adenoma and carcinoma is unlikely with ultrasound insofar as they can have a similar appearance. Neither echogenicity nor the presence of mineralization can be used. Lesions suggestive of metastasis may be found, especially in the liver.233 Evidence of invasion into the vena cava is suggestive of a carcinoma but can be difficult to judge by ultrasound. With ATs atrophy of the contralateral gland will not always be detectable by ultrasound.
Use of ultrasonography as a screening test for hyperadrenocorticism is not recommended. First, measurements of adrenal gland length and minimum and maximum diameter overlap between dogs with PDH and either dogs with non-endocrine disease or even normal dogs, so ultrasound cannot always distinguish between them. Second, the finding of an AT is not synonymous with hyperadrenocorticism. Ultrasonography cannot distinguish a functional adrenocortical tumor from a nonfunctional tumor, a pheochromocytoma, a metastatic lesion, or a granuloma.
Abdominal CT is an even more sensitive assessment of adrenal gland structure. Standard and dynamic CT can also be used for pituitary evaluation. Dynamic CT is more sensitive than conventional contrast-enhanced CT. If hypophysectomy is being considered for therapy, the extra sensitivity of dynamic CT may be helpful to ensure that the correct treatment is being provided. In other cases dynamic CT may not be warranted. MRI has been used not to differentiate but to assess the size of a pituitary mass in known cases of PDH. Biochemical testing cannot readily differentiate tumor size.
Although radiation therapy is not a great means of controlling hyperadrenocorticism,234,235 radiation done for local control of a pituitary mass is more effective and provides a better outcome and prognosis the smaller the mass and with no or minimal neurologic signs present.234,236 Compared with untreated dogs, radiation therapy for a pituitary mass, with or without the presence of hyperadrenocorticism or with or without the presence of neurologic signs, significantly increased survival. In one study median survival with treatment was not reached, but mean survival was 1405 days versus 359 for those not treated. Radiation also significantly increased control of neurologic signs.237
The true incidence of pituitary macroadenomas is unknown but has been estimated to be as high as 25% in dogs. The clinical progression of pituitary tumors is also widely unknown. Out of 21 dogs recently diagnosed with but untreated for PDH that had no neurologic signs, 11 had a pituitary mass visible on MRI.238 At 1 year 13 of the 21 had follow-up imaging. Five had no visible tumor originally, whereas eight did of 4- to 11-mm greatest vertical height. One of the dogs with a visible mass had not been treated, whereas the rest had been treated with mitotane.239 None of the five dogs with no visible mass originally had neurologic signs at follow-up; two had a visible pituitary tumor.239 Of the eight dogs that had a visible mass at first imaging, four had no apparent change in their tumor size, whereas in four the tumor had enlarged. The untreated dog was in the latter group. Overall, four developed signs of central nervous system dysfunction (19% of the original 21 and 36% of the 11 dogs with tumors visible at the first scan). Tumor size appeared to correlate with the development of signs, insofar as no dog with a mass smaller than 10 mm had neurologic dysfunction. There was no apparent correlation between pituitary mass size before treatment and increase in size over the year. 239
On the basis of this information, clinical experience, and theoretical considerations, recommendations for routine imaging of the pituitary in patients with hyperadrenocorticism have recently been formulated. With the recognition that the number of dogs studied is small, the suggestions are as follows: All dogs with PDH should have CT or MRI scans at the time of diagnosis. If no mass is visible, medical treatment should be implemented and no follow-up imaging is needed. If a mass 3 to 7 mm in greatest vertical height is seen, medical therapy should be implemented, with a repeat scan in 12 to 18 months. If a mass 8 mm or larger in greatest vertical height is seen, radiation therapy should be done and medical therapy used only if clinical hyperadrenocorticism fails to resolve within 3 to 6 months of finishing radiation. No studies have been performed to date to assess the validity of the guidelines.
Therapy for Canine Hyperadrenocorticism
Surgical and medical options exist for treatment of canine and feline hyperadrenocorticism. By whatever means, the ultimate goal of therapy is to eliminate hypersecretion of cortisol. In the United States, medical therapy is most often used for dogs with PDH. In Europe hypophysectomy is available and has been successful.240,241 For cats with PDH, owing to the limited success of medical therapy, bilateral adrenalectomy may be the treatment of choice, followed by lifelong therapy for hypoadrenocorticism.242 For dogs and cats with ATs, surgery is recommended.
Mitotane
Currently, there is no drug therapy that will cure PDH. Lifelong therapy should be anticipated. Mitotane, or o,p`-DDD (Lysodren) has long been the mainstay of medical therapy for canine PDH. A chlorinated hydrocarbon, mitotane is adrenocorticolytic, causing selective necrosis of the zona fasciculata and zona reticularis, the adrenocortical zones that secrete cortisol and sex hormones. The toxin is specific for the adrenal glands, particularly hyperplastic glands, with one exception. In normal animals mitotane has caused fatty degeneration and centrolobular atrophy of the liver, and hepatotoxicity secondary to mitotane therapy for hyperadrenocorticism has occurred.243 Although the disposition of mitotane has not been well characterized in dogs, safety has been studied in a small number of animals. Normal animals tolerated the drug at 50 mg/kg administered 5 days out of 7 for months with no apparent adverse effects. Adrenocortical function was, however, impaired.
Therapy for hyperadrenocorticism with mitotane occurs in two phases: an induction (loading) phase and a maintenance phase. For treatment of PDH, a starting dose of 40 to 50 mg/kg divided twice daily (i.e., 20 to 25 mg/kg twice daily) and administered orally should be used.244 In smaller dogs division may be impossible because of the 500-mg pill size, and the drug can be given in one dose. Doses higher than 50 mg/kg (administered daily) increase the risk of complete cortisol deficiency.244 Mitotane should always be given with food because this increases the bioavailability of intact tablets.245 Loading should end when appetite decreases, vomiting or diarrhea occurs, the patient becomes listless, water intake drops to less than 60 mL/kg daily (1 cup = 240 mL and 1 oz = 30 mL) or for a maximum of 8 days (Figure 21-6). Feeding twice-daily during loading allows better assessment of appetite, which may be the most common early sign that control has been achieved. To closely monitor the patient, best judge the endpoint, and impress on an owner the seriousness of overdosing, the clinician may find it helpful to make daily calls to the owner.242 When signs suggest that loading is complete or at the end of 8 days if no changes have occurred, adrenal reserve is assessed by ACTH stimulation testing. If the signs of hyperadrenocorticism have not changed, daily therapy can continue until the results of the ACTH stimulation test are known; otherwise, mitotane should be discontinued while awaiting the laboratory report.242
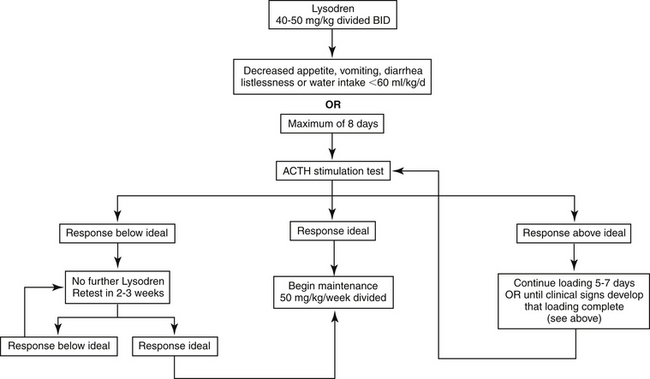
Figure 21-6 Protocol for mitotane induction therapy for canine pituitary-dependent hyperadrenocorticism.
The goal of the induction phase is to have serum cortisol concentrations before and after administration of ACTH stimulation in the normal resting range (e.g., cortisol concentration of 30 to 150 nmol/L or 1-5 μg/dL before and after ACTH). Dogs with PDH that continue to have responses to ACTH in the range for normal dogs (e.g., post-ACTH cortisol concentration of 220 to 560 nmol/L or 8 to 20 μg/dL) tend to have ongoing clinical signs. If pre- and post-ACTH cortisol concentrations are within the ideal range, maintenance therapy should begin. If cortisol concentrations are above the desired range, loading should continue for another 5 days or until clinical signs occur that suggest loading has been completed. The mean time required to achieve adequate control is 11 days, but up to 2 months is possible.244 In general, smaller dogs (<12.5 kg) and those receiving phenobarbital may require greater than average induction times. Approximately 33% of dogs will have a serum cortisol concentration less than ideal (e.g., post-ACTH cortisol concentration <30 nmol/L) after induction; mitotane therapy should be discontinued and an ACTH stimulation test performed after 2 weeks to assess adrenal function. Prednisone should be administered at physiologic doses during that time, but none should be given in the 12 hours before performing an ACTH stimulation test. In most dogs serum cortisol concentrations will rise into the ideal range within 2 to 6 weeks, but up to 18 months may be required.244
Special consideration should be given to patients with concomitant hyperadrenocorticism and DM. If a diabetic has insulin resistance secondary to hyperadrenocorticism and requires large doses of insulin for adequate glycemic control, treatment with mitotane removes the cause of insulin resistance and can lead to a rapid decrease in daily insulin requirement. Consequently, insulin overdosage and hypoglycemia may occur if the insulin administration is not adjusted accordingly. To try to slow the return to insulin sensitivity and avoid hypoglycemia, the recommended induction dose for dogs with concurrent hyperadrenocorticism and DM is 25 mg/kg once daily. Furthermore, although administration of prednisone during induction therapy for PDH is discouraged in general by some authors, prednisone (0.4 mg/kg once daily) should be given to diabetics receiving induction phase mitotane, again to help avoid hypoglycemia. Even with these precautions, diabetic patients should be monitored more closely than usual during induction.
Adverse effects of mitotane are generally gastrointestinal or neurologic. One or more adverse effects occur in approximately 25% of dogs with PDH during loading and include weakness, vomiting, anorexia, diarrhea, and ataxia.244 These develop as serum cortisol concentration falls rapidly and typically resolve quickly with appropriate therapy. If adverse effects occur, mitotane administration should be discontinued. Prednisone should be administered (0.2 to 0.5 mg/kg) until the dog can be examined, an ACTH stimulation test performed, and serum electrolytes measured. Most dogs show a clinical response to glucocorticoid administration within 2 to 3 hours. Persistence of apparent adverse effects may signify the presence of another medical problem.
If a dog does not respond to the induction protocol after 14 days, the following factors that could contribute to mitotane resistance should be considered:242 (1) The patient may have an AT, which is more resistant to mitotane. (2) The patient may be inherently resistant to mitotane; some dogs with PDH have required as many as 30 to 60 days of daily therapy or doses of 100 to 150 mg/kg daily. (3) The induction dose is too low. Dogs receiving less than 40 mg/kg daily are less likely to be adequately controlled after 10 days. (4) The drug is not being absorbed well. Ensure that the medication is being given with food, preferably a fatty meal. (5) The diagnosis may be incorrect or the patient is suffering from iatrogenic hyperadrenocorticism. Neither an animal with iatrogenic hyperadrenocorticism nor one that does not have hyperadrenocorticism will respond to mitotane. (6) The owner may not be giving the medication as directed.
Maintenance therapy will be necessary for the remainder of the animal’s life, although the dose and frequency vary among patients and can vary in an individual patient over time (Figure 21-7). In the absence of maintenance therapy, the adrenal glands will once again become hyperplastic in response to continued ACTH secretion from the pituitary gland. The maintenance phase uses a much lower overall mitotane dose of 50 mg/kg/week orally244 given as 2 to 3 smaller fractions over the course of the week, if division is possible. Because approximately 60% of dogs with PDH on maintenance mitotane therapy, especially those receiving less than 50 mg/kg weekly, relapse within 12 months of starting therapy,244 an ACTH stimulation test should be performed 1, 3, and 6 months after initiating maintenance therapy and approximately every 3 to 6 months thereafter to ensure continued control. If the pre- and post-ACTH serum cortisol concentrations are in the ideal range, therapy can remain as is. If the post-ACTH cortisol concentrations is mildly elevated (e.g., 150 to 250 nmol/L or 4-9 μg/dL), the maintenance dose can be increased by 25%, and the dog retested after 1 month to determine if adequate control has been achieved. If so, maintenance therapy should continue at the new dose. If serum cortisol concentrations are still above ideal, reinstitution of daily loading therapy for 5 to 7 days should be considered. If the post-ACTH cortisol concentration is moderately to greatly increased (e.g. greater than 250 nmol/L or 9 μg/dL), loading therapy should be reinstituted for 5 to 7 days. If induction therapy is reinitiated, the decision to end loading should be based on the same clinical signs as during the initial induction phase or should be done for a maximum of 7 days. Once the serum cortisol levels are again within the ideal range, maintenance therapy should be reinstituted at a 50% higher mitotane dosage.246
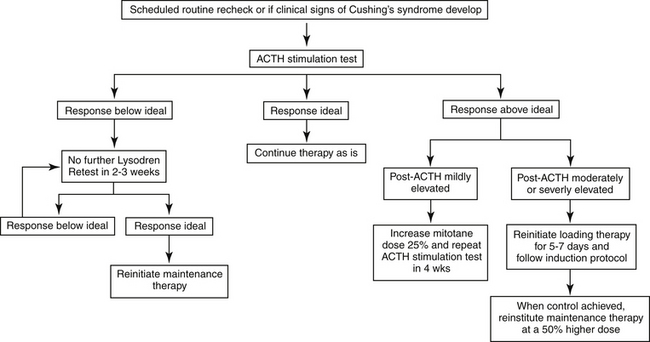
Figure 21-7 Protocol for mitotane maintenance therapy for canine pituitary-dependent hyperadrenocorticism.
In 184 dogs with PDH treated with mitotane for a mean of 2 years, the final maintenance dosage required ranged from 27 to 330 mg/kg weekly, with the two highest doses required by dogs also receiving phenobarbital. Median survival time was 1.7 years (range 10 days to 8.2 years), with the response judged as excellent in 83%, fair in 16%, and poor in 0.6%.244
Approximately 33% of dogs on maintenance mitotane therapy will develop adverse effects including anorexia, vomiting, weakness, diarrhea, and ataxia, typically shortly after initiation of the maintenance dosage or during periods of relapse when daily therapy is reinstituted. If these develop, mitotane therapy should be discontinued, physiologic doses of prednisone administered, an ACTH stimulation test performed, and serum electrolyte concentrations measured. Presence of glucocorticoid deficiency with or without mineralocorticoid deficiency can be documented using these tests and differentiated from direct drug toxicity. If the clinical signs are due either to a hypoadrenal state or to a direct effect of mitotane, they should resolve quickly with prednisone administration. If the signs do not abate, presence of a nonadrenal illness should be suspected. If glucocorticoid deficiency is documented (e.g., before and after ACTH serum cortisol concentration <30 nmol/L or 1 μg/dL), mitotane therapy should be discontinued and physiologic prednisone replacement therapy continued until serum cortisol concentrations before and after ACTH increase into the ideal range, which usually requires 2 to 6 weeks. Complete mineralocorticoid and glucocorticoid deficiency is seen in approximately 6% dogs from 1 month to years after initiation of maintenance therapy and is usually permanent.244 If mineralocorticoid and glucocorticoid deficiency is present, the patient needs to be treated for hypoadrenocorticism. Appearance of neurologic signs such as disorientation, dullness, or inappetence may be due to direct drug toxicity or may suggest the presence of a pituitary macroadenoma; CT or MRI is required to confirm the presence of a large tumor. Mitotane dose reduction may be necessary for animals that develop adverse reactions or an alternate dosing scheme can be used (e.g., the dose may be divided into smaller amounts to be given more frequently during the course of the week).
If therapy for hyperadrenocorticism is successful, most clinical signs of the disease or its complications resolve over time. Polyuria, polydipsia, and polyphagia should resolve as soon as cortisol secretion is adequately controlled. Resolution of some clinical signs (e.g., skin manifestations, nonhealing wounds, anestrus) may take 3 to 6 months or longer; calcinosis cutis may never fully resolve. Development of a puppy coat or a change in hair color may occur with mitotane treatment. Resolution of clinical laboratory changes (i.e., elevated liver enzyme activities and serum cholesterol concentration) may take up to 18 months.
An alternative protocol for treating PDH is aimed at nonselective adrenocorticolysis and complete destruction of adrenocortical tissue, with substitution therapy for ensuing adrenocortical insufficiency. Mitotane is given for 25 days at a dosage of 50 to 75 mg/kg daily and up to 100 mg/kg daily for toy breeds, divided into 3 or 4 approximately equal and equally spaced portions and given with food. Lifelong glucocorticoid and mineralocorticoid substitution is begun on the third day of mitotane administration. Prednisone should be initiated at a temporarily high dose of 1 mg/kg twice daily. Fludrocortisone (0.0125 mg/kg daily) and sodium chloride (0.1 mg/kg/day, divided over 2 or 3 meals) should also be administered.216
During the first month, owners should report by telephone at least weekly and as problems arise and should stop mitotane administration if any inappetence develops.216 In this regimen appetite change, if seen, is a direct toxic effect of the medication; mild cortisol deficiency is offset by the glucocorticoid replacement therapy and should not cause any adverse effects. If mitotane therapy continues despite a diminished appetite, a hypoadrenocortical crisis can ensue. Glucocorticoid dosage may be increased temporarily if appetite diminishes. Usually, mitotane can be resumed after 4 or 5 days when the appetite returns without further problem.216
The first follow-up visit should be 1 week after completion of mitotane administration. Serum electrolytes should be measured to ascertain whether the fludrocortisone and salt doses are correct.216 Performance of an ACTH stimulation test may be wise to ensure adequate control of the Cushing’s syndrome. The original protocol recommends that patients be treated for hypoadrenocorticism with prednisone (1 mg/kg daily, divided either in 2 equal portions or two thirds in the morning and one third in the evening), fludrocortisone at a dosage that maintains normal serum electrolyte concentrations, and salt.216 However, salt may not be necessary; lower prednisone and fludrocortisone doses may be sufficient (discussed later). Furthermore, DOCP may be used as an alternative to fludrocortisone.
The protocol was assessed in 129 dogs.247 In 30% of the dogs, mitotane administration had to be stopped temporarily because of the development of anorexia, vomiting, weakness, neurologic abnormalities, depression, and diarrhea, but it could be resumed within days (median 7 days, range 1 to 63). Convincing signs of partial or complete remission of the hyperadrenocorticism such as hair regrowth, decreased water intake and appetite, and diminished abdominal size were noted in 86%. Relapse occurred in 33%; the median disease-free interval (time until recurrence, death, or last follow-up) was 450 days from the day therapy began (range 25 to 1885).247 Adrenal testing (i.e., ACTH stimulation) is recommended only if clinical signs of hyperadrenocorticism recur, but routine measurement of serum urea nitrogen and electrolyte concentrations is required to ensure adequate control of the hypoadrenocorticism. Median survival time was 1.6 years (range 1 day to 6.1 years).247 In another study that used a daily mitotane dose of 75 to 100 mg/kg daily in 46 dogs, median survival was approximately 2 years. Although recurrence rate of hyperadrenocorticism was only 29%, perhaps owing to a higher mitotane dose, 15 dogs suffered an Addisonian crisis at some point during therapy. Overall incidence of side effects was 24%.248
Although treatment of hypoadrenocorticism may appear easier than that of hyperadrenocorticism, two main disadvantages exist for the alternative protocol, and its use has not been strongly recommended. First, treatment of an Addisonian dog can be expensive, and mineralocorticoid with or without glucocorticoid replacement therapy will be required for life. Second, and more important, failure to give medication to an Addisonian patient can be fatal, whereas missing a dose of mitotane will not put a patient in life-threatening danger. 242
If an AT is being treated, the mitotane protocol used is different from that for PDH; the goal is complete destruction of tumor tissue with serum cortisol concentrations before and after ACTH below the normal resting range (e.g., <10 nmol/L or 0.3 μg/dL on both samples).249 Although approximately 20% of dogs with ATs respond to PDH induction protocols, higher induction dosages and longer induction times are generally required for control of an AT.250,251 The cumulative induction dose of mitotane for PDH is usually 400 to 500 mg/kg, whereas that for dogs with an AT is often up to 10 times higher.250Thus initial mitotane induction dosage for treatment of an AT is 50 to 75 mg/kg daily. Because the goal is complete destruction of glucocorticoid-secreting tissue, physiologic doses of prednisone should be administered concurrently.250 The same clinical signs can be used to judge the endpoint of induction as when treating PDH, with a maximum treatment span of 14 days. At the conclusion of a loading period, an ACTH stimulation test should be performed (Figure 21-8).
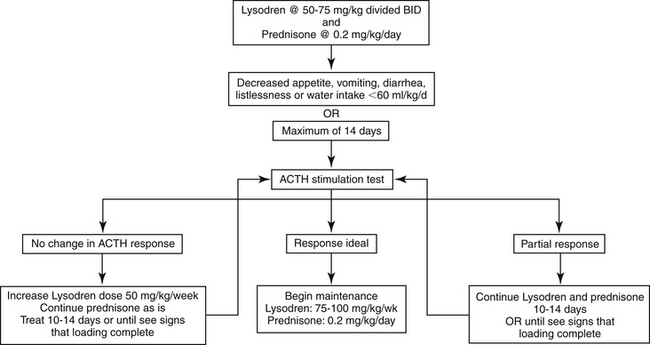
Figure 21-8 Protocol for mitotane induction therapy for canine adrenal-dependent hyperadrenocorticism.
If a partial response is seen but adequate control has not been achieved (i.e., pre- and post-ACTH cortisol concentration are lower than before treatment but not in the ideal range), mitotane should be continued at the same dosage and an ACTH stimulation test repeated every 10 to 14 days until serum cortisol concentrations fall within the ideal range. If after the initial loading dose the ACTH response is unaltered, the daily mitotane dosage should be increased in 50 mg/kg increments daily every 10 to 14 days as necessary, until an ACTH stimulation test demonstrates a response to the medication or drug intolerance occurs. Therapy is then continued at the dosage at which a response was seen or at the highest tolerated dosage, and ACTH stimulation testing is again performed every 10 to 14 days or if clinical signs suggest an endpoint has been reached. In 31 dogs with an AT, total induction time ranged from 10 days to 11 weeks with a mean of 24 days.250
Once cortisol concentrations before and after ACTH administration are within the ideal range, maintenance therapy should begin at an initial mitotane dosage of 75-100 mg/kg/week.250 Daily physiological doses of prednisone should also be administered, as these dogs have subnormal basal serum cortisol concentrations. An ACTH stimulation test should be performed after 1 month of maintenance therapy to determine whether serum cortisol concentrations have remained adequately suppressed. If pre- or post-ACTH cortisol levels are within the normal resting range (i.e., 10 to 160 nmol/L or 1 to 4 μg/dL), the mitotane maintenance dose should be increased 50% and the dog retested in 1 month. If the cortisol levels are still above the resting range at that time, induction therapy should be reinstituted; once ideal cortisol levels are again achieved, maintenance should be restarted at a 50% higher dosage than previously used. An ACTH stimulation test should again be performed 1 month after a dose adjustment to assess control (Figure 21-9). Once ongoing successful therapy is documented, an ACTH stimulation test should be done every 3 to 6 months or if clinical signs recur. Relapse occurs during maintenance in approximately 66% of cases, usually because of either too low an initial maintenance dose or tumor growth.250
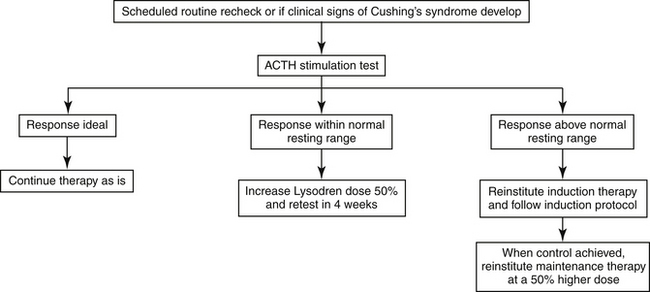
Figure 21-9 Protocol for mitotane maintenance therapy for canine adrenal-dependent hyperadrenocorticism.
As are induction doses, maintenance doses required for adequate control of an AT are higher than for PDH. In 32 dogs with an AT, the final mean maintenance dose required was 159 mg/kg weekly, slightly more than double the average maintenance dose required to control PDH. Approximately 25% of dogs with ATs require maintenance doses greater than 150 mg/kg weekly. Adverse effects, as previously described, occur in approximately 60% of dogs with ATs treated with mitotane. They can develop as long as 16 months after initiation of therapy, are more common during the maintenance rather than the induction phase, and are due either to direct toxicity of medication or to adrenocortical insufficiency, with the former being approximately twice as likely.250
If severe side effects occur, mitotane should be stopped, the prednisone dose increased to 0.4 mg/kg daily, and the dog reevaluated as soon as possible with an ACTH stimulation test and measurement of serum electrolyte concentrations to determine whether complete mineralocorticoid and glucocorticoid deficiency exists. If serum electrolytes are normal but pre- and post-ACTH serum cortisol concentrations are below 10 nmol/L, it is likely that only glucocorticoids are deficient. Mitotane therapy should be restarted, and prednisone administration continued at a dosage of 0.4 mg/kg daily to exclude cortisol deficiency as the cause of the side effects. If adverse effects recur when mitotane is reinstituted despite an increased glucocorticoid dosage, direct drug toxicity is likely. If the adverse effects are due to the mitotane, its administration can be temporarily discontinued and then reinstituted at a 25% to 50% lower dosage once signs of toxicosis have resolved. If hyponatremia, hyperkalemia, and subnormal cortisol concentrations are present, both mineralocorticoids and glucocorticoids are deficient. Loss of both types of adrenocortical hormones is likely to be permanent. Replacement therapy for both hormones should be instituted, and mitotane should not be administered until adrenal recovery can be documented by an ACTH stimulation test.
Treatment of AT with mitotane can provide good results. Of 32 dogs with an AT treated with mitotane, 66%, 28%, and 6% were judged by their owners to have a good to excellent, fair, and poor response, respectively. Mitotane does not appear to arrest metastatic tumor growth, and the response in dogs without evidence of metastatic disease is better than that in dogs with metastases. Mean survival time of dogs with ATs treated with mitotane is approximately 16 months, with a reported range of 20 days to 5.1 years.250
Ketoconazole
Ketoconazole is a triazole antifungal drug widely used for the treatment of disseminated fungal diseases. The drug inhibits cytochrome P450 enzymes responsible for synthesis of gonadal and adrenal steroids and has been used to treat hyperadrenocorticism in people. In addition, ketoconazole may antagonize glucocorticoid receptors. In normal dogs ketoconazole administration decreases serum cortisol and testosterone, but not mineralocorticoid, concentrations.252,253
Dosing of ketoconazole should be initiated at 5 mg/kg orally twice daily for 7 days, a low dosage to allow an evaluation period for development of side effects such as gastroenteritis or hepatitis. Light feeding may ameliorate gastritis resulting from ketoconazole administration. If no ill effects are observed during the first week, the dosage should be increased to 10 mg/kg orally twice daily for 14 days, after which an ACTH stimulation test should be performed. The ideal ranges for serum cortisol concentrations before and after ACTH administration are the same as when mitotane is used. If serum cortisol concentrations are above ideal, the ketoconazole dosage should be increased to 15 mg/kg orally twice daily and the dog monitored every 14 to 60 days.254 Dosages equal to or greater than 20 mg/kg twice daily may be required.255,256 If no response is seen or the disease progresses despite therapy, ketoconazole should be discontinued and alternative therapy begun.
The efficacy of ketoconazole may be less than that of mitotane. After ketoconazole therapy, basal and post-ACTH cortisol concentrations may actually be higher than those pretreatment in some dogs.242 Of 132 veterinary internists and dermatologists surveyed, specialists likely to treat Cushing’s syndrome, 52% considered ketoconazole to be effective in less than 25% of cases, 19% reported effectiveness in 25% to 49% of cases, and 14% each believed ketoconazole to be efficacious in 50% to 74% and 75% to 100% of cases.256 A recent report suggested a higher efficacy of 70% in 48 dogs,257 but the follow-up on treated dogs was inconsistent and the ideal post-ACTH cortisol concentration was not as low as recommended by most authors. Thus, although ketoconazole may lower serum cortisol concentration in dogs with PDH and clinical improvement can be seen,254,257 whether therapy is truly adequate in such a high percentage is unclear.
Ketoconazole appears to be relatively safe, with a low incidence of side effects. When seen, adverse effects may include anorexia, vomiting, elevated liver enzymes, diarrhea, and icterus.256,257 Side effects believed to occur secondary to ketoconazole administration in a small number of cases include depression; weakness; lethargy and trembling; liver failure; polyuria and polydipsia; thrombocytopenia; and dermatologic changes such as altered coat color, poor coat condition, and scaling.256 Lightening of the hair coat may also occur. The effect of ketoconazole on reproductive status has not been addressed, but it does decrease testosterone synthesis in healthy dogs252 and should be used cautiously in male dogs intended for breeding.
Despite possibly limited efficacy and high cost compared with mitotane, ketoconazole therapy may occasionally be warranted. First, ketoconazole can be used in dogs that cannot tolerate mitotane. Second, it may be used as a diagnostic aid when the diagnosis of hyperadrenocorticism is unclear. If an ACTH stimulation test shows that ketoconazole therapy has adequately controlled cortisol secretion, then any clinical signs present that were caused by hyperadrenocorticism should resolve. If the disease is placed in remission and the diagnosis thus confirmed, mitotane treatment can be initiated instead. If no resolution of clinical signs is seen, hyperadrenocorticism can be ruled out as a diagnosis and ketoconazole discontinued. Ketoconazole provides a better alternative to trial therapy than mitotane or trilostane because the adrenolytic effects of mitotane or trilostane may be irreversible, whereas cortisol levels normalize within 24 hours of discontinuing ketoconazole.254 Third, because ATs may be mitotane-resistant or the high doses of mitotane required to treat an AT may cause unacceptable side effects, ketoconazole may be used for medical treatment of ATs or before an adrenalectomy to prepare the patient for surgery. However, no study has evaluated ketoconazole efficacy in a large number of dogs with ATs.
Bromocriptine and selegiline
Dopamine clearly affects ACTH secretion from the pituitary; both elevated dopamine levels and dopamine agonism suppress ACTH secretion, at least from the intermediate lobe of the pituitary. Thus increasing dopamine concentrations or activity may inhibit ACTH oversecretion (Figure 21-10) and be useful for treatment of PDH. Raising dopamine levels can be effective only for PDH, however. Because endogenous ACTH secretion is suppressed in patients with an AT, dopamine agonism or alteration of dopamine metabolism would have little, if any, further effect on ACTH release. Moreover, because ATs function autonomously of ACTH, lowering ACTH levels would not alter cortisol secretion.
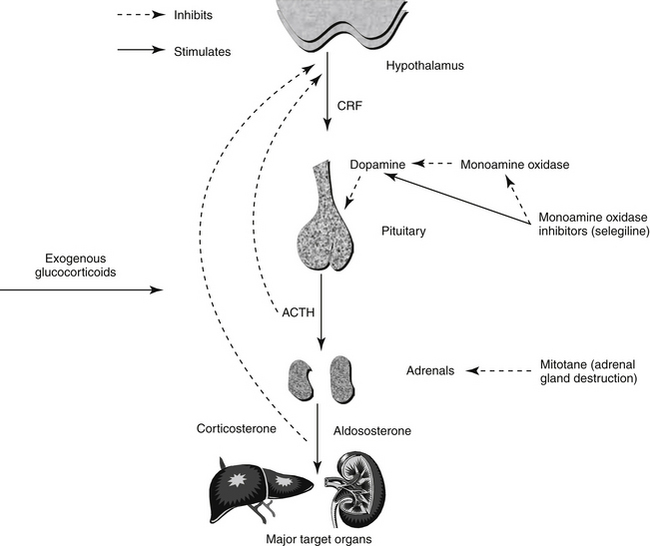
Figure 21-10 Diagram of the hypothalamic–pituitary–adrenal axis and its relationship to medications that affect the axis.
Bromocriptine, a dopamine agonist, has met with limited success for treating PDH. In one study bromocriptine was administered to seven dogs with PDH,258 and 40 dogs were included in another.259 Vomiting was a limiting side effect and cause for treatment discontinuation in a large proportion, and only 1 of the 47 responded clinically. Thus bromocriptine is not recommended for treatment of canine PDH.
Monoamine oxidase inhibitors, including selegiline (l-deprenyl), inhibit degradation of biogenic amines, most notably dopamine, and are used to treat dopamine-deficient conditions such as human Parkinson’s disease. Unlike other monoamine oxidase inhibitors, selegiline is specific for cerebral forms (i.e., monoamine oxidase B). An important question in treating canine PDH, however, is whether the ACTH-lowering effect of dopamine is on the anterior or intermediate lobe of the pituitary (or both). If dopamine inhibits only intermediate lobe ACTH secretion, as is generally believed, then selegiline use would be efficacious only in cases of PDH caused by intermediate lobe tumors (i.e., approximately 20% of canine PDH cases). Indeed, one study of 10 dogs suggested a 20% response rate.260 Unfortunately, only histopathology can differentiate anterior and pituitary lobe tumors. Furthermore, a more recent study found selegiline to be ineffective for treatment of PDH.261
If selegiline is used to treat PDH, as with other drugs, therapy must be for the lifetime of the animal.262 Treatment should begin at 1 mg/kg orally once daily for 30 days. If no response is seen, the dose should be doubled for an additional 30 days. Failure to respond at that time indicates the need for an alternative therapy. Selegiline therapy is relatively safe. Side effects are uncommon and usually mild, including vomiting, diarrhea, and ptyalism.260,261 Severe neurologic disturbances and pancreatitis possibly have been caused by selegiline therapy,260 but the neurologic problems also may have been due to the presence of a large pituitary mass. Chronic selegiline therapy does not result in glucocorticoid insufficiency263 and, based on its mechanism of action, would not be expected to affect aldosterone secretion. One disadvantage of selegiline is cost. Several generic versions are available, but although the bioequivalency of the generic preparations is the same among themselves, they are less bioavailable than the original product, l-deprenyl (Eldepryl); comparisons with the animal product Anipryl are not available. Thus, it may be wise to avoid the generic products until studies have established the appropriate canine dose. Another disadvantage of using selegiline for treating PDH is that monitoring of efficacy is based solely on relatively subjective findings. The results of the ACTH stimulation test do not change while dogs are receiving selegiline. Thus other objective measures of effect, such as quantification of water intake or measurement of urine specific gravity, should be used.
In general, use of selegiline to treat PDH is not recommended because of its low efficacy, but it could be tried in dogs with PDH that cannot tolerate mitotane and trilostane or as a diagnostic aid, as with ketoconazole. However, trial therapy with selegiline would be hard to judge. The sole measure by which to judge efficacy of therapy is resolution of clinical signs. If an animal does not respond clinically, it would be impossible to tell if the dog did not have hyperadrenocorticism or had an AT or l-deprenyl–resistant PDH. With ketoconazole therapy the ACTH stimulation test provides an objective measure of when adequate biochemical control has been achieved. If cortisol secretion is suppressed by ketoconazole and clinical signs continue, the diagnosis of hyperadrenocorticism can be ruled out.
Trilostane
Trilostane (Vetoryl) has been used to treat hyperadrenocorticism for a number of years in Europe and is now approved by the Food and Drug Administration for treatment of canine hyperadrenocorticism in the United States. A synthetic steroid analog that inhibits the adrenal enzyme 3β-hydroxysteroid dehydrogenase, trilostane suppresses production of progesterone and its end products, including cortisol and aldosterone. Additional enzymes such as 11β-hydroxylase and 11β-hydroxysteroid dehydrogenase may also be affected.264 Trilostane appears to be highly effective in suppressing cortisol secretion and controlling clinical signs in the majority of patients.248,265-270 As with mitotane, clinical signs of hyperadrenocorticism typically quickly resolve with control of cortisol concentrations, but certain ones such as dermatologic abnormalities can take up to 3 months. Other abnormalities such as calcinosis cutis or pseudomyotonia may not fully resolve. However, a small proportion of dogs with PDH (<10%) are not well controlled with trilostane.267,270,271
Trilostane is available as 10-, 30- and 60-mg capsules in the United States and may need to be compounded for smaller dogs. In approximately 50% of dogs, dosage adjustments, either up or down, will be required during the course of treatment. Authors of one study noted that in most dogs an initial sensitivity to the drug existed followed by a need for a dose increase. After time, the dose required often hit a plateau.267 Interestingly, the final dose required for control has varied greatly between studies. Part of the discrepancy may relate to the differences in what was considered the ideal post-ACTH serum cortisol concentration. However, in one study the median final dose was 6.1 mg/kg,270 and in another study it was 16 to 19 mg/kg.267 In any case, each dog should be started on the recommended dose, and then the dose should be adjusted according to ACTH stimulation test results. The duration of survival is at least as good as that achieved with mitotane therapy.268
Reported adverse effects for the most part are relatively mild, including lethargy and vomiting, but fatality has occurred.267,270,271 Although some studies found relatively low incidence of side effects, one non–peer-reviewed report described mild, self-limiting side effects such as diarrhea, vomiting, and lethargy in 63% of treated dogs.272 Safety has not been evaluated in lactating dogs and males intended for breeding. Trilostane should not be given to pregnant females.
As with mitotane therapy, excessive adrenal gland suppression can occur and warrants discontinuing medication temporarily (discussed later) and lowering the dose. Trilostane can affect aldosterone secretion as well as cortisol, so a hypoadrenocortical crisis can occur. Caution should be used in administering trilostane with an angiotensin-converting enzyme inhibitor or an aldosterone antagonist (e.g., spironolactone) because the suppressive effect on serum aldosterone concentration may be cumulative.
Although, in theory, the effects of trilostane as an enzyme inhibitor should be rapidly reversible (e.g., within a couple days), suppression can last weeks to years.266,267,273 One dog developed hypocortisolism after only 3 doses of trilostane; glucocorticoid replacement therapy was needed for at least 1 year.273 Surprisingly, adrenal necrosis can occur secondary to trilostane administration as well.274 The hypoadrenocorticism reported after complete adrenocortical necrosis in one dog lasted for at least 3 months but likely would be permanent.274 How often acute iatrogenic hypoadrenocorticism will occur in dogs treated with trilostane is unknown but is likely more common than originally believed. In one study four of six dogs with PDH and one of one with AT treated with trilostane had some degree of adrenal necrosis at necropsy. In two dogs the damage was sufficiently severe to cause hypoadrenocorticism. Both dogs had received therapy with mitotane before trilostane but had been on trilostane for 15 and 22 months.275 Thus the contribution of each drug is unclear. Adrenal rupture, possibly secondary to adrenal necrosis, may have occurred (see Vetoryl package insert).
Current guidelines are for a lower starting dose than previously recommended. The package insert states that therapy should be initiated at 2.2 to 6.7 mg/kg once daily with food. These authors recommend starting as close to the low end of the range as possible. If minor side effects are seen, drug administration should be stopped for 3 to 5 days until side effects resolve and then restarted, giving trilostane every other day for 1 week before continuing with the initial dosing scheme. It is important to differentiate minor adverse effects from hypocortisolism, and ACTH stimulation testing may be needed.
Postpill timing is crucial for dogs receiving trilostane, and the test should be initiated at 4 to 6 hours after the pill was administered. Keeping the timing consistent for each patient may also be important. The first scheduled recheck should be after 10 to 14 days of therapy. Lately, recommendations are being made to not increase the trilostane dose for at least 4 weeks after initiating therapy. An initial ACTH stimulation test should be performed after 10-14 days of therapy to ensure hypocortisolemia is not present. If at any time the post-ACTH cortisol concentration is below 40 nmol/L (1.45 μg/dL), the trilostane should be suspended and restarted at a decreased dose after 3 to 7 days (see package insert) or, ideally in the authors’ opinion, after recovery of adrenocortical function has been demonstrated. At the 2-week recheck, the dose should only be raised in cases where the owner reports that no improvement has been seen, the clinical signs are still striking, and the post-ACTH cortisol concentration is markedly above ideal. Then, in such cases at 2 weeks or if waiting until the 4-week recheck, if the post-ACTH cortisol concentration is 40 to 150 nmol/L (1.45-5.4 μg/dL), the dose should continue as is. If the post-ACTH cortisol is 150 to 250 nmol/L (5.4-9.1 μg/dL), the dose can be continued if the dog is doing well clinically; if not, twice-daily therapy should be used. The same dose that was given once daily should be given twice (e.g., if the regimen is 60 mg once daily, it can be doubled to 60 mg twice daily), or a lower dose can be given in the evening (e.g., from 60 mg once daily to 60 mg in the morning and 30 mg in the evening). If the post-ACTH serum cortisol concentration is above 250 nmol/L (9.1 μg/dL), the trilostane dose should be increased. An ACTH stimulation test should be performed 10 to 14 days after every dose adjustment. Once the clinical condition of the dog and the dose have stabilized, an ACTH stimulation test should be performed 30 and 90 days later and then every 3 months thereafter. Size of dosage adjustment, either up or down, will likely be dictated by available capsule size but ideally should be approximately 25%.
Despite recommendations in the package insert for once-daily dosing, trilostane may begin to lose effectiveness 8 to 10 hours after the pill is given.265,276,277 Indeed, trilostane has been administered twice daily.248,265,266 In one study of 44 dogs with PDH, the initial dose of trilostane was 15 mg orally twice daily for dogs less than 5 kg, 30 mg twice daily for dogs 5 to 20 kg, 60 mg in the morning and 30 mg in the evening for dogs 20 to 40 kg, and 60 mg twice daily for dogs larger than 40 kg. At the first recheck (day 7), an ACTH stimulation test was performed starting at 4 to 6 hours after the pill was given. Good control was judged on the basis of clinical signs and a serum cortisol concentration before and after ACTH administration of 30 to 110 nmol/L. Dose was adjusted in 25% to 50% increments. On further rechecks, the ACTH stimulation test was initiated 8 to 12 hours after the pill was given. Good control was believed to be a post-ACTH cortisol concentration of 30 to 250 nmol/L.266
Mean initial dose of trilostane was 6.2 mg/kg (range 2.4 to 15) divided into twice-daily doses. The dose was not changed over the course of the study in 10 dogs, increased in 19, reduced in 5, and both increased and reduced at different times in 10. At all rechecks the mean dose was between 6 and 8 mg/kg divided into twice-daily doses, but the range was approximately 2 to 20. Adverse reactions were seen in 25% of cases related to low cortisol concentration. In 11% trilostane therapy was discontinued because of prolonged suppression of serum cortisol concentration. In four dogs adrenal function returned to normal and no further treatment was needed; one dog was treated as an Addisonian.266Another study using the same protocol found high efficacy with relatively few side effects, and survival times were longer compared with those of dogs treated with a nonselective adrenocorticolytic protocol.248Special consideration should be given to twice-daily dosing in dogs in which breaks in control of the hyperadrenocorticism could be detrimental (e.g., in dogs with concurrent DM or dogs with pulmonary thromboembolism secondary to hyperadrenocorticism). The recommended starting dose is 1 mg/kg twice-daily. If using twice-daily therapy, although one study recommended performing an ACTH stimulation test 8-12 hrs post-pill,266 such timing has not been critically evaluated.
An early paper suggested that monitoring of the urine cortisol:creatinine ratio (UCCR) before a trilostane dose could be an indication of duration of action.261 However, two studies yielded conflicting results. In 22 dogs with HAC, a morning UCCR pre-dose was lower in dogs with good control compared to those with poor control (14.8 vs. 47.5); however, overlap existed between the 2 groups.265 On the other hand, in 18 PDH dogs, the UCCR was measured in samples collected before or 6 hours after trilostane administration; a pair of urine samples was collected once weekly for 4 weeks. Although basal UCCRs were significantly higher than the 6-hour values, neither the basal nor the 6-hour samples were significantly different between dogs with adequate vs. inadequate control. Post-ACTH cortisol concentrations did not correlate with UCCR. Basal UCCRs in 13 dogs and 6 hours post-trilostane UCCRs in 12 were still above the reference range 8 weeks after the dose was judged to be optimal.265a
Interestingly, in one study, of 6 dogs that had UCCRs within the reference range most of the times, three developed hypocortisolism and in three hypocortisolism was suspected based on clinical signs but was not confirmed. The hypocortisolism was documented or suspected between 16 weeks and 21 months after the trilostane dose was judged to be optimal.265a
How best to switch a dog from mitotane to trilostane is unknown. According to the Vetoryl package insert, a post-ACTH cortisol concentration should be above 250 nmol/L (9.1 μg/dL) before giving trilostane. It may be ideal to wait at least 30 days after discontinuing mitotane, but if the clinical signs of hyperadrenocorticism are severe, trilostane can be started sooner as long as the post-ACTH cortisol concentration is high enough. Dogs previously treated with mitotane may have greater response to trilostane, so careful monitoring of adrenal function is required.
Trilostane has been used to treat five dogs with ATs.278-280 Insufficient information is available to ascertain whether the treatment protocol or efficacy varies if treating dogs with PDH versus those with an AT. One dog did receive a maximum dose of 17.2 mg/kg.280 Clinical signs were controlled, at least transiently, and survival prolonged. Survival in one dog was only 117 days.278 In the other four dogs survival was at least 10 to 18 months; interestingly, in three trilostane therapy was initiated when an AT accompanied by pulmonary metastases was identified.279,280 Response of ATs to trilostane, at least in these five dogs, compares favorably to that achieved with mitotane. However, in dogs with ATs mitotane theoretically may be the preferred treatment. Mitotane is truly a chemotherapeutic drug in this instance, killing primary neoplastic cells and, perhaps, metastatic cells as well. Trilostane simply would control tumoral secretion, not growth. In fact, in dogs with PDH and in one dog with an AT treated with trilostane, the size of the adrenal glands increased.275,281
Retinoic acid
9-cis Retinoic acid has been used to treat a total of 27 dogs with PDH in two studies. Unfortunately, endogenous ACTH and α-melanocyte-stimulating hormone concentrations and UCCR were used to evaluate treatment, which makes the results difficult to interpret insofar as these tests are not typically used to monitor therapy. Interestingly, pituitary tumor size significantly decreased in 14 dogs that were treated for 180 days.282,283 Although results were promising, much more work is necessary before retinoic acid therapy can be recommended, especially when proven therapies such as mitotane or trilostane are available. In addition, retinoic acid at the doses used in the study would likely be cost prohibitive to most owners.
Therapy for Feline Hyperadrenocorticism
Hypercortisolism
Medical therapy for feline classic hyperadrenocorticism (i.e., excessive cortisol as compared to excess of other adrenocortical hormones), appears not to be as successful as in dogs; however, very little information has been published. Unilateral adrenalectomy is the treatment of choice for an AT. Given the limited success of medical therapy, some authors believe bilateral adrenalectomy with subsequent lifelong therapy for hypoadrenocorticism is the treatment of choice for feline PDH. However, cats with severe hyperadrenocorticism may not be good surgical candidates because of the debilitating effects of the disease (e.g., thin skin, immunosuppression, poor wound healing), and medical control for stabilization before surgery would be ideal. Although not available in the United States, transsphenoidal hypophysectomy has been reported for treatment of PDH in seven cats.284 Radiation therapy may be effective for aiding in the control of feline PDH.285 (See also the section on acromegaly later in this chapter.)
Early reports suggested mitotane was not highly effective in cats at doses of 25 mg/kg or 50 mg/kg for up to 90 days,286-288 but one case report suggested that mitotane therapy may be successful with higher doses and longer induction periods. A cat was treated with mitotane at 25 mg/kg once daily for 18 days with little effect. At 69 days, after a dose increase to 37.5 mg/kg once daily, there was still minimal effect, but by day 111 the hyperadrenocorticism was controlled.289
In one cat with an adrenocortical carcinoma, ketoconazole caused transient (3.5 months) resolution of polyuria and polydipsia and a reduction in the insulin dose required to control concurrent DM. Interestingly, although basal serum cortisol concentrations decreased during treatment, adrenocortical response to ACTH stimulation was not altered.290 Other mechanisms of action of ketoconazole, such as antagonism of glucocorticoid receptors, may account for the clinical improvement, or it may have been coincidental. In four other cats with hyperadrenocorticism, two responded to ketoconazole doses of 5 mg/kg twice daily for 7 days, followed by 10 mg/kg twice daily indefinitely; one had no response after 2 months; and one developed severe thrombocytopenia after 7 days of treatment, necessitating withdrawal of therapy.291
Metyrapone affects cortisol synthesis by inhibiting the adrenal enzyme 11-β-hydroxylase. Clinical response without side effects other than hypoglycemia has been achieved in six cats using 30 to 70 mg/kg twice daily.288,292,293 In two cats treated with an unknown dose, one had a slight clinical improvement (partial hair regrowth and slight resolution of polyuria and polydipsia) after 6 months, whereas the other showed no improvement after 1 month.294 Although doses as high as 65 mg/kg three times daily have been used, a maximum of 70 mg/kg twice daily has been recommended to avoid gastrointestinal complications.288 The ACTH stimulation test should be used to ensure adequate control of cortisol secretion with the same goal as for dogs treated with mitotane for PDH. Interestingly, however, one cat had clinical improvement without marked reduction in cortisol concentrations.292 However, the availability of metyrapone varies.
In a total of six cats with PDH, trilostane administration reduced clinical signs and improved endocrine test results, but all continued to have some signs of hypercortisolemia, especially dermal changes.295,296 In cats with PDH and DM treated by other means, the DM resolved in about 50% of cases;223 however, insulin requirements did not change in four cats with PDH and DM treated with trilostane.295,296 Thus trilostane ameliorates clinical signs of feline hyperadrenocorticism, but more research is needed before it can be recommended for treatment. Whether a higher dosage or longer duration of therapy will improve efficacy remains unclear.295Additionally, the ideal ranges for basal and post-ACTH serum cortisol concentrations and testing time (i.e., performance of ACTH stimulation tests beginning 4 to 6 hours after administration of the pill) for dogs were used. The pharmacokinetics and pharmacodynamics of trilostane are unknown in cats, and it may be necessary to optimize the testing protocols for cats.
Sex hormone excess
Progesterone-secreting adrenal masses have been reported in three cats.288,297,298 Clinical signs are as those for hypercortisolism. With such a mass, the cortisol response to an ACTH stimulation test is blunted, suggestive of iatrogenic hypoadrenocorticism despite a lack of a history of exogenous glucocorticoid administration. Suppression is due to the ability of progestins to feed back on the pituitary and inhibit ACTH secretion as do glucocorticoids.199 Measurement of sex steroids before and after ACTH stimulation should reveal marked hyperprogesteronemia. Abdominal ultrasonography may reveal an adrenal mass. Treatment of choice is unilateral adrenalectomy. Two cats with progesterone-secreting adrenal tumors were treated with aminoglutethimide, an inhibitor of adrenocortical steroid synthesis, preoperatively. One cat showed improvement at 2 weeks, but the response had diminished by week 6.298 The second cat improved dramatically at first, but significant mammary gland enlargement ensued and aminoglutethimide administration was discontinued; the rapid decrease in progesterone potentially stimulated prolactin secretion.288
One spayed female cat with an AT secreting estradiol and testosterone has been identified, with the most notable clinical signs being aggression and male-type behavior, malodorous urine, and vulvar hyperplasia. Therapy with trilostane (30 mg once daily) was effective in reducing clinical signs; partial improvement was seen by day 28 and almost complete resolution by day 84. Interestingly, basal concentrations of both hormones as well as that of 17-hydroxyprogesterone and progesterone increased with therapy. Clinical signs returned by day 174 despite continued therapy. The reason for clinical improvement in the face of increased measured hormone concentrations is unknown.299 Possibilities include a placebo effect (believed unlikely by the authors of the case report), false elevations of the apparent concentrations of the measured hormones by hormones that cross-react on the assays used,299 or another nonmeasured hormone that did decrease in response to therapy and was responsible for causing the clinical signs. Although unlikely, the sex hormone elevations may have been false-positive test results for a state of excessive sex hormone secretion.
Hyperaldosteronism
Primary hyperaldosteronism can occur in cats from an aldosterone-secreting tumor or from idiopathic non-tumorous secretion of excess aldosterone. Although aldosterone-secreting AT were previously believed to be rare, the incidence may be increasing.300 Affected cats are middle-aged to older, with no sex or breed predisposition. Clinical symptoms of hyperaldosteronism are typically nonspecific and result from potassium depletion, mainly causing hypokalemic myopathy. Historical complaints and physical examination findings include polyuria and polydipsia, nocturia, generalized weakness, collapse, anorexia, weight loss, a pendulous abdomen, and blindness.300-303 One cat presented with respiratory distress; the respiratory failure was believed to be a manifestation of hypokalemic polymyopathy.304 Some cats have bilateral retinal detachments and hypertension.300,301 Adrenocortical tumors secreting more than one hormone (e.g., aldosterone and progesterone) have been reported,305 and clinical signs associated with the other hormone (i.e., hyperprogesteronism) may also be present.
The hallmark of primary hyperaldosteronism is hypokalemia with an elevated serum aldosterone concentration but a normal or subnormal plasma renin activity. Measurement of an elevated serum aldosterone concentration alone is not truly adequate, but a feline plasma renin activity assay is not currently commercially available. In humans serum aldosterone concentration is measured after dietary intake of a specific sodium amount. Whether such standardization is necessary in veterinary medicine is unknown, making diagnosis of primary hyperaldosteronism more difficult. The presence of renal failure presents a particular dilemma, insofar as renal failure in and of itself can lead to hypokalemia and hyperaldosteronemia. The magnitude of aldosterone elevation may be the key. With primary hyperaldosteronism, serum concentrations were approximately 3000 pg/mL in two cats,301 whereas it was a maximum of 518 pg/mL in azotemic cats.306 Identification of an AT by radiography or ultrasonography in cats with appropriate clinical signs and laboratory results would also be highly suggestive of primary hyperaldosteronism. Until a feline plasma renin assay is available, diagnosis of primary hyperaldosteronism requires that all secondary causes of hyperaldosteronism (e.g., states associated with peripheral edema or liver failure) be ruled out.
Initial treatment of primary hyperaldosteronism is directed at controlling hypokalemia or hypertension (or both). Potassium supplementation using oral potassium gluconate at doses of 2 to 6 mmols twice daily has been used, and intravenous potassium chloride may be required in more severely hypokalemic cases. Amlodipine besylate (0.625 to 1.25 mg per cat once daily) is the initial treatment of choice for hypertension while test results are pending.307 Spironolactone, a competitive aldosterone receptor antagonist, is also recommended (2 to 4 mg/kg once daily), assisting in the control of both hypokalemia and hypertension. Severe facial dermatitis has been reported in Maine Coon cats receiving spironolactone for management of hypertrophic cardiomyopathy.308 Medical management may not be successful long-term or for normalization of serum potassium concentration. The role of antineoplastic chemotherapy has yet to be determined.
Treatment of choice for an AT is adrenalectomy. Given that these tumors are malignant in approximately 50% of cases, surgery may not be curative. In addition, the procedure has been associated with high perioperative mortality; approximately 33% of reported cases died intraoperatively or postoperatively, most commonly as a result of severe acute hemorrhage from the caudal vena cava.307 Medical therapy preoperatively to stabilize a patient is ideal, especially if the tumor is secreting another hormone such as cortisol or progesterone that can create complications such as poor wound healing. A detailed description of perioperative and intraoperative management of an aldosterone-secreting adrenocortical carcinoma with attached caval thrombus has been recently reported.309 Approximately 5% of human patients develop postoperative hyperkalemia, requiring transient administration of fludrocortisone; although not reported in the literature, the same may occur in cats.307
With medical management with combinations of potassium supplementation, amlodipine, and spironolactone, reported survival times in four of five cats with aldosterone-secreting AT ranged from 7 months to 984 days, with cats most commonly succumbing to chronic renal failure or a thromboembolic episode.307 Poor response in the fifth cat may have been due to owner compliance issues. In some cases hypertension becomes refractory to medical management. For cases that undergo adrenalectomy and survive the immediate perioperative and postoperative periods, the prognosis is good; 8 of 17 adrenalectomized cats survived for at least 1 year, and two cats were alive 3.5 and 5 years postoperatively.300 Cats with malignant tumors can do well; one cat with a carcinoma survived 1045 days after adrenalectomy.
An alternate form of hyperaldosteronism now recognized in cats is similar to the idiopathic or bilateral hyperplasia form in humans.310 Clinical presentation is similar to the tumorous form with respect to signalment and presenting complaints, but retinal detachment and blindness, perhaps due to the presence of higher blood pressures in general, is more common with idiopathic hyperaldosteronism. Ultrasonography or CT examination of the adrenal glands reveals no changes or only subtle abnormalities such as an increase in adrenal echogenicity or areas of calcification and thickening or rounding of one pole of one or both adrenal glands. Therapy consists of medical management (as discussed previously), and affected cats tend to respond well.307