Chapter 33 Chemotherapy
Throughout the centuries the sufferers of this disease have been the subject of almost every conceivable form of experimentation. The fields and forests, the apothecary shop and temple have been ransacked for some successful means of relief from this intractable malady. Hardly any animal has escaped making its contribution in hide or hair, tooth or toenail, thymus or thyroid, liver or spleen in the vain search for a means of relief. — Bainbridge, The Cancer Problem, 1914
Before appropriate treatment for a particular cancer can be instituted, the tumor must first be diagnosed by cytology or histopathology. Testing is then conducted to allow staging of the tumor—to determine if there is clinically visible evidence of metastatic spread. Lymph node cytology or biopsy, radiographs, ultrasound examinations, and computed tomography or magnetic resonance imaging scans may be used as staging procedures. The biological behavior typical for the cancer must be considered. If, as a rule, the tumor tends to remain localized, surgery or radiation therapy—or a combination of the two—may be the best way to obtain cure or control. If the tumor is likely to metastasize by lymphatics or hematogenously, however, some form of systemic therapy must be added to make a cure more likely. Systemic therapy involves the administration of biological agents, hormones, or cytotoxic chemotherapy. Theoretically, cancer chemotherapy is given to kill or suppress the growth of malignant cells without killing normal cells; in fact, however, most of the commonly used drugs are capable of killing both normal and malignant cells, depending on the dose administered. To be useful in clinical practice, an antineoplastic drug must possess selective toxicity—that is, it should be more toxic to cancer cells than to normal host cells at conventional doses. Finally, the clinician must determine whether the patient has concurrent diseases, with a complete blood count (CBC), biochemical panel, urinalysis, and possibly testing for feline leukemia virus and feline immunodeficiency virus. This section of the chapter addresses the principles of chemotherapy and the side effects seen in dogs and cats.
Drugs used in chemotherapy cause their anticancer effects by interacting with important substrates or enzymes that are related to DNA synthesis or function. Therefore most anticancer drugs are ineffective against cells that are not actively proliferating. Tumors with a high mitotic index (e.g., lymphoma) are much more likely to be sensitive to chemotherapy than those in which mitotic activity is low. Because chemotherapeutic drugs are effective principally on cells that are actively replicating, it is important to have an understanding of the phases of the cell cycle before discussion of individual drugs (Figure 33-1). The part of the cell cycle in which active mitosis occurs has been termed the M phase; it is quite short in all cells, generally lasting less than 1 hour. The period during which DNA synthesis occurs for chromosome doubling in preparation for mitosis is called the S phase and ranges from 8 to 30 hours. When scientists began to learn about cell division, they realized that there were other phases in the cell cycle; because they initially did not understand what was occurring in the cell at these times, the phases were called G (G1 and G2) for gap. G1 follows mitosis, and protein synthesis and RNA transcription occur during this phase. G1 is extremely variable in length depending on the cell type, ranging from 7 to 170 hours. G2 precedes the next mitotic event and is usually brief, ranging from 1 to 4 hours. G0 has been used to describe those cells that are not actively cycling. Certain cell types, such as myocytes and neurons, enter G0 and never cycle again. Other cell types, such as hepatocytes, proliferate in young animals and then cease cycling at maturity but are capable of beginning to cycle again if cell replacement is necessary. Fibroblasts become terminally differentiated in connective tissue, but stem cells remain in the tissue. These can become reproductively active again to repopulate the tissue if a wound occurs.
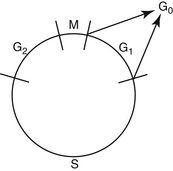
Figure 33-1 Cell generation cycle: M—mitosis; S—DNA content is being doubled in preparation for mitosis; G1 and G2—gaps. Protein synthesis and RNA transcription are occurring in these phases; G0—cells that are not actively cycling.
Chemotherapeutic drugs can be classified into three groups on the basis of their activity in the phases of the cell cycle (Box 33-1). Agents that are considered to be lethal to cells in all phases of the cell cycle, with resting cells as sensitive as proliferating cells, are called cycle nonspecific. Examples include nitrogen mustard (the first chemotherapy agent; its activity was discovered in 1919 with the effects of mustard gas on the troops in World War I) and high-dose cyclophosphamide. Agents that are capable of damaging both resting and cycling cells (although cells in cycle are much more sensitive) include conventional-dose cyclophosphamide and doxorubicin. These drugs spare resting cells and are called cycle specific. Agents that are phase specific exert their lethal effects exclusively or primarily during one phase of the cell cycle, usually S or M; resting cells and cells in the other phases of the cell cycle are not killed. Examples include methotrexate, cytosine arabinoside, vincristine, and vinblastine.
Box 33-1 Sites of Action of Chemotherapeutic Drugs
There are three phases in the development of a new chemotherapeutic drug (Box 33-2). Many artificial chemicals and naturally occurring compounds are screened (generally by the National Cancer Institute or by industry) for cytotoxicity, first in cultures of cancer cells and then in mice or rats. If a particular compound looks promising, a phase I trial is conducted, providing an initial pharmacologic evaluation. The appropriate mode of administration is established for the drug, and common side effects are discovered. Patient tolerance of increasing dosage is also determined. Phase I trials are conducted on very small numbers of patients, generally with advanced and ultimately terminal cancers for which no conventional treatment is available, and doses tolerated by these patients may be below the ultimate therapeutic range. If the compound shows no prohibitive toxicity and shows even slight efficacy (a partial response in a few patients), the phase II trial begins. In this phase screening for efficacy of the drug against a variety of tumors is conducted. After the spectrum of activity is determined, dose–response relationships are determined. The phase III trial is then used to determine drugs that work effectively together, and the new combination protocol is ultimately compared with the existing best treatment. Phase IV trials are known as postmarketing surveillance trials. They involve safety surveillance after the necessary authorities permit the drug to be sold. This surveillance may be required by regulatory authorities. The chemotherapy agent may not have been tested for interactions with other drugs, for example. The safety surveillance is designed to detect any rare or long-term adverse effects over a much larger patient population and longer time period than was possible during the phase I through phase III clinical trials. Harmful effects discovered during phase IV trials may result in a drug being removed from the market.
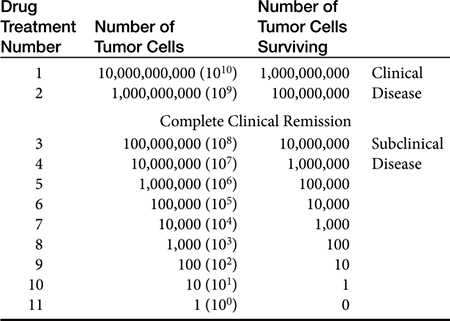
Most cancers in animals and humans are diagnosed only after they are well advanced. In the 1960s, Skipper and coworkers1 used the rodent L1210 leukemia to illustrate this point and determine cell kill kinetics in tumors. The L1210 leukemia is a rapidly growing tumor with a growth fraction of 100% and a doubling time of only 12 hours. At this rate of growth, a billion cells would accumulate in the rodent only 19 days after injection of a single cell. After treatment of the leukemia with chemotherapy, the investigators determined that cytotoxic drugs kill by log kill kinetics—that is, a given dose of an effective drug kills a constant fraction of cells and not a constant number, regardless of the number of cells present. This principle is known as the fractional kill hypothesis. For example, if a certain drug is known to kill 90% of the tumor cells present, it will kill 90% of the cells whether the beginning number is 10 cells or 10 billion cells (Table 33-1). Thus it should be theoretically possible to cure with chemotherapy, with rapid successive administration of chemotherapy drugs. In fact, antineoplastic drugs kill an extremely variable fraction of cells, ranging from a very small fraction to a maximum of 99.99%; for many tumors the fractional kill is disappointingly small. What prevents theoretical “cures” with chemotherapy is (1) the inability to give drugs in rapid succession, because of host toxicity, and (2) the development of a drug-resistant population of tumor cells during the course of treatment.
Table 33-1 Tumor Depopulation Related to Successive Drug Cycles Assuming a 90% Fractional Cell Kill in a Model System
Tumors as small as 1 g (109 tumor cells—a billion) may be detected in the body, especially if they are located in areas such as the skin or mouth. However, it is far more common for tumors to escape detection until they are 10 g (1010 tumor cells) or more. The maximum malignant tumor mass compatible with human life is about 1 kg (1012 tumor cells). If it is assumed that a given tumor originates from a single cell, then a 1-g tumor (109 cells) has gone through 30 doublings from the original cell. To get to 1 kg, only about 10 more doublings will take place.2 It should be clear from these sobering numbers that a large, unresectable tumor burden with only modest sensitivity to chemotherapy cannot be cured or, in many cases, even palliated with conventional chemotherapy administration protocols. The average volume doubling time for various human solid (e.g., nonleukemic) tumors is about 2 months. However, for certain rapidly growing tumors such as embryonal nephroma, seminoma, lymphoma, and leukemia, the volume-doubling time is less than 1 month. For other tumors the volume-doubling time is as long as 1 year. Because chemotherapy affects rapidly dividing cells, tumors with short volume-doubling times are generally chemotherapy sensitive, whereas tumors with long volume-doubling times are generally chemotherapy resistant.
With regard to the efficacy of chemotherapeutic drugs, criteria have been described for measuring response; these are called RECIST (Response Evaluation Criteria in Solid Tumors). To evaluate a tumor’s response to treatment, the clinician must have a marker lesion, a repeatably measurable tumor mass or parameter that can be periodically rechecked and remeasured (preferably with the same person doing the repeat measurements). This may be a lymph node or nodes that can be measured with calipers, a liver lesion measured by ultrasound, nodules visible on a radiograph, a biochemical value such as the calcium level, and so forth. The evaluation as to efficacy of treatment is conventionally made after two cycles of chemotherapy or an appropriate trial of another agent has been administered:
Complete response (CR)—Resolution of all measurable neoplastic disease (or return of marker to normal, if there is no measurable disease), with appearance of no new lesions. A chemotherapeutic drug that can cause a CR in a significant number of animals with a specific cancer is quite likely to increase disease-free survival, especially when used as an adjuvant agent.
Partial response (PR)— Reduction in measurable tumor dimensions of 30% or greater, with no appearance of new lesions. Although a temporary PR may provide the patient with some decrease in discomfort from the cancer, it is unlikely to have a significant effect on survival. However, this drug might be useful in other patients at an earlier stage of disease or in patients in which the tumor can be surgically removed before chemotherapy. A PR proves that the drug does have some activity against the tumor that is being treated.
Stable disease (SD)—No significant change is noted in measurable tumor dimensions, or a response is seen that is less than a PR. Actual values are less than a 30% decrease in marker lesion size and less than a 20% increase in marker lesion size.
Progressive disease (PD)—The tumor is clearly growing (20% or greater increase in lesion size), or new lesions appear.
Certain criteria may be useful for declaring a treatment protocol ineffective or unsafe in a specific patient. Progressive growth (usually greater than 20%) in a measurable tumor lesion or the appearance of new lesions after two cycles of chemotherapy would suggest that the drug or protocol is not at all useful for the tumor being treated. Severe toxicity with irreversible, cumulative, or unpredictable manifestations also generally suggests that the drug should no longer be used in this particular patient. If symptoms from the cancer cause the patient’s condition to deteriorate, with the only response to drug treatment being SD or PR, the drug should be discontinued and another treatment selected if possible.
The common cancers in dogs and cats can be broadly divided into three categories:
In principle, all cells that are actively cycling in the body should be sensitive to chemotherapy; however, the fact that chemotherapy is generally only modestly effective speaks to the fact that this is not entirely true. In many cancers the tumor cells are actually less sensitive to cytotoxic drugs than are the hematopoietic cells within the marrow cavity. This forces the clinician to give a chemotherapeutic drug and wait to evaluate the toxic effects produced before another course of treatment can be administered. During the interval of time in which the patient’s neutrophil or platelet count is too low to give another drug, endogenous hematopoietic growth factors are being produced, mediating proliferation of stem cells, the bone marrow recovers, and peripheral cell counts return to normal. Return of blood cell counts to normal after chemotherapy is the usual point at which another course of treatment may be given. It is important not to give another cycle of chemotherapy when peripheral blood counts are extremely low because stem cells are actively proliferating at this time; treatment with cytotoxic drugs administered when stem cells are actively dividing increases the chance that the stem cell population may be killed and recovery may never occur. In humans this is the point at which bone marrow or stem cell transplantation is performed.
Unfortunately, tumor cells may recover from chemotherapeutic injury and begin to proliferate again before the animal’s marrow recovers. Even when a tumor is exquisitely sensitive to drug treatment and an apparent complete response is obtained, a line of drug-resistant cells often develops. It is a common clinical experience to find that a tumor may respond quite well to the first treatment with a drug, with progressively less impressive responses as the drug is given repeatedly. Classically, multidrug resistance occurs when large numbers of the cells in a tumor overexpress a gene (the MDR1 gene) that encodes P-glycoprotein, a transmembrane protein important in cell transport. This protein pumps chemotherapeutic drugs from the inside of the cell to the extracellular environment so that they cannot act within the cell; P-glycoprotein is a transmembrane drug efflux pump. Other mechanisms leading to acquired drug resistance include (1) the development in the tumor cell of alternative metabolic pathways to avoid the chemotherapeutic drug’s mechanism of action; (2) the fraction of tumor cells actively dividing decreases after several cycles of treatment, thus protecting the remaining noncycling cells against damage; and (3) tumor cells may enter a biological “sanctuary site” in which they are protected from injury because of a lack of drug diffusion into that area (e.g., brain, eye, testicle, spinal cord).
Delaying administration of chemotherapy because of hematopoietic or gastrointestinal toxicity often results in a patient appearing to be in remission, with no visible neoplastic disease but with large amounts of microscopic tumor. Thus drug resistance sometimes develops as a result of chemotherapy being administered in a regimen that is “too little, too late.” The highest possible doses of chemotherapy given as frequently as can be tolerated by the patient, early in the course of the neoplastic disease (when smaller numbers of cells are present), should have a much higher chance of producing a cure or long-term remission than chemotherapy given after a large number of tumor cells have infiltrated various organs. In human patients the wait for marrow recovery has been overcome with the use of bone marrow or stem cell transplants, performed after chemotherapy treatment is given in doses high enough to kill tumor cells as well as ablate normal marrow cells. The extreme expense, technical difficulty, and high morbidity rates associated with this procedure do not allow for its use in dogs and cats as a routine clinical procedure, at least at this time. In veterinary oncology in the early twenty-first century, clinicians are usually limited to palliation of tumors; only rarely can they expect to cure their patients.
A regimen of chemotherapy treatment can be divided into several phases. The period of induction is the initial intensive chemotherapy intended to produce remission. Remission is defined as the point at which no measurable tumor mass can be found; for lymphoma remission is declared when the enlarged lymph nodes, liver, and spleen have returned to normal size and malignant cells have disappeared from the peripheral blood and bone marrow. This does not mean that all (or even most) tumor cells have been killed, however, and consolidation therapy with different drugs may be given after apparent clinical remission to produce a larger tumor cell kill. For some tumors such as lymphoma, the animal may be given “pulse” doses of drugs after induction and consolidation to maintain the gains obtained with the induction protocol; this is called the maintenance phase of chemotherapy. Recently, intensification protocols have been described for tumors in which drug resistance is common. These protocols are administered during the maintenance period, when the patient is in apparent remission, and are an attempt to kill developing drug-resistant clones of cells by using one or two new drugs at high doses.
Combination chemotherapy is, in general, more effective than single-agent therapy. Using multiple drugs sequentially provides additive antitumor effects without greatly increasing host toxicity, especially if drugs are selected carefully for different toxicities. Combination chemotherapy may delay tumor resistance to drugs compared with single agents. Selecting different drugs that have effects on more than one cell cycle phase may also result in a greater fractional cell kill per cycle of chemotherapy.
Traditionally, in estimating the appropriate dose of a drug to administer to a patient, clinicians have used body weight of the patient as the main criterion; the dose has been figured as the number of mg/kg to be given. In a series of studies begun in the 1880s, however, it was demonstrated that many physiologic parameters could better be estimated on the basis of body surface area (BSA). Basal metabolic rate, blood volume, cardiac output, and renal function parameters were found to correlate much more closely to the individual’s BSA than to weight. It was then found that drug doses calculated per unit of body weight were greater in smaller animals and children than in larger animals and adults, whereas doses calculated per unit of surface area were similar for all species and ages. On the basis of the findings in these studies, researchers concluded that BSA might be useful as a standard for calculating drug doses in cancer chemotherapy. The calculation for determining BSA for a given species is made by using the following formula:
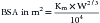
In the preceding formula, Km is a factor based on the different metabolic rate of each species; for the cat it is 10, and for the dog it is 10.1. W is the body weight in grams. Because the K values for dogs and cat are quite close, a table has been formulated that permits quick estimation of the BSA on the basis of the animal’s weight in kilograms. The appropriate dose of the chemotherapeutic agent to be administered is calculated by multiplying the dose/m2 by the patient’s BSA (m2) taken from the table. A serious and potentially fatal mistake made by some clinicians when using a nomogram or table to estimate the BSA has been to use the animal’s weight in pounds rather than in kilograms. To avoid this error, a good rule is to calculate the dose of a chemotherapeutic drug and then ask another person to calculate it again separately before the drug is administered.
More recently, the use of BSA as a means of calculating doses for all chemotherapeutic agents has been questioned. For many chemotherapy drugs, myelosuppression is the most common toxicity and is dose limiting; it has been found that BSA does not correlate well with either stem cell number in the bone marrow or with resulting hematopoietic toxicity. In fact, correlation is highly significant between bone marrow effects of the cytotoxic drugs and body weight. A phase I study in dogs was performed to evaluate toxicity of doses of intravenous melphalan calculated by BSA. A significantly greater number of small dogs experienced significant toxicity than did large dogs. Another study compared marrow toxicity induced by doxorubicin given at 30 mg/m2 to that induced by the drug given at doses calculated at 1 mg/kg. It was found that a disproportionately greater number of dogs weighing less than 10 kg developed severe myelosuppression at the 30 mg/m2 dose than at the 1 mg/kg dose.3 Limited toxicosis was seen in dogs weighing more than 10 kg with either of the dosing schemes, however. Plasma doxorubicin concentrations were less after treatment at the 1 mg/kg dose in both large and small dogs than in those given 30 mg/m2, and it is possible that 1 mg/kg may be an inappropriately low dose for treatment of animals with cancer. For drugs that may produce severe myelosuppression, measurement of hematopoietic stem cell numbers for each individual patient would clearly provide the most information to prospectively calculate doses for chemotherapeutic agents. Until such a test is available, however, clinicians must use the doses available in the literature, always carefully taking into account the individual animal’s response to the previous drug dose before administering the next treatment. If a doxorubicin dose of 1 mg/kg is well tolerated by a dog or cat weighing less than 10 kg, the next dose may be increased slightly, gradually approaching the dose calculated by BSA; this is called dose escalation. In daily clinical practice veterinarians judge the adequacy of therapy by measuring the response of visible, measurable masses; only much later are they able to evaluate the results of their treatment by survival results. In a rodent model for osteosarcoma, reduction in the dose intensity of melphalan and cyclophosphamide caused a marked decrease in the cure rate long before there was a reduction in the rate of complete clinical remission. On average, it is estimated that a dose reduction of approximately 20% leads to a loss of 50% in the cure rate. A positive relationship between dose intensity and response rate has been demonstrated in many human tumors, including lymphoma and ovarian, colon, and breast cancers. Clinicians should administer the highest dose of a chemotherapeutic drug that can be tolerated by the patient if they are attempting to cure; if palliation is the only goal, a dose that will produce clinical remission without dose escalation may be appropriate, however. Careful patient monitoring for therapeutic response and toxicity is still the best way to titrate the drug dose for each individual patient (Box 33-3).
Box 33-3 Timing of Chemotherapy (Dose–Schedule Relationships)
Because chemotherapeutic drugs are quite toxic, the following guidelines for making the decision to begin chemotherapy are critically important:
Metronomic Chemotherapy
Most chemotherapy targets rapidly dividing cells (preferably tumor cells, but inevitably some normal cells are also affected), and it is given at maximum tolerated doses (MTDs). Chemotherapy drugs also have another target: endothelial cells that form the lining of newly formed blood vessels, such as those whose creation is orchestrated by tumors to fuel their growth. There is a considerable body of evidence that even very low, almost nontoxic doses of chemotherapy drugs, when delivered frequently for a prolonged period of time, can retard tumor blood vessel growth (or angiogenesis) by destroying endothelial cells. Treatment approaches along these lines are now being tested in clinical trials, and they have been coined metronomic chemotherapy.
The treatment targets endothelial cell precursors (endothelial progenitor cells) that are recruited from the bone marrow and circulate to various sites in the body. Metronomic chemotherapy is given in very low doses repetitively (usually daily) over a long period of time compared with the MTD therapy that has been traditionally used. These repetitive low doses of chemotherapy drugs (cyclophosphamide at 10 mg/m2 per day has been used most often in veterinary medicine) are designed to minimize toxicity and target the endothelium or tumor stroma, as opposed to targeting the tumor. Thus the treatment is theoretically useful to stabilize or slow tumor growth rather than kill tumor cells. Studies conducted in cell lines and animal models have also suggested that combining metronomic chemotherapy with targeted antiangiogenesis agents (e.g., piroxicam) is more effective than metronomic chemotherapy alone.
Metronomic chemotherapy will probably be most useful in slow-growing, indolent tumors, such as soft tissue sarcomas with a low mitotic index and well-differentiated carcinomas, particularly after debulking to microscopic disease. There is a low incidence of side effects, and the treatment is relatively inexpensive. The hope is that this treatment will allow some cancers to be treated as manageable chronic conditions.
Common Side Effects of Chemotherapy
Most clients are extremely satisfied with their experience with chemotherapeutic treatment of their pet’s cancer. Fewer than one in four animals are reported to have significant side effects of chemotherapy, and only 5% will have a serious event that requires hospitalization. However, if a pet does experience a serious reaction, there are certainly adverse consequences. The animal’s quality of life is decreased, at least for a time; there may be unexpected expenses for the owner associated with hospitalization and costly treatments; and it may be necessary to delay the next scheduled chemotherapy session, which sometimes allows the cancer to visibly return in the delayed interval between treatments. All this is likely to result in clients who are less enthusiastic about the idea of chemotherapy for their pet than they were before the occurrence of the unexpected side effect.
Many adverse effects of chemotherapy can be minimized or prevented by careful management, but some animals experience unanticipated side effects that no amount of care or forethought could have prevented. Certain breeds, especially Collies and rarely Australian Shepherds and Shetland Sheepdogs, are carriers of a mutation of the P-glycoprotein multidrug resistance 1 (MDR1) gene. If dogs that are homozygous for this gene are given anthracyclines or vinca alkaloids, the cellular excretion of the drugs is diminished, and they have increased drug exposure and thus increased toxicity. It is estimated that 70% of Collies in the United States are heterozygous for this mutated gene, and 31.2% of Collies are homozygous, with much lower percentages for Australian Shepherds, Shetland Sheepdogs, and other herding dogs. A test for the mutation status of this gene is available through the Veterinary Clinical Pharmacology Laboratory at the College of Veterinary Medicine, University of Washington (http://www.vetmed.wsu.edu/depts-vcpl/). If this test is not performed, dogs of these breeds with cancer should be given conservatively low doses of anthracyclines and vinca alkaloids, or the drugs should not be given at all.
Cells of normal tissue are damaged by chemotherapy; most, given time, will recover. Tissues that are especially affected include those in which the cells have a short life span and require constant renewal (e.g., bone marrow, gastrointestinal mucosa, gonads, hair follicles). Box 33-4 provides a brief overview of the classes of body tissue and, if applicable, typical renewal properties.
Myelosuppression
Myelosuppression and subsequent infection are the most common dose-limiting toxic effects of chemotherapy. Drugs that can be particularly myelosuppressive in dogs and cats include lomustine (CCNU), cyclophosphamide, carboplatin, doxorubicin (particularly when used in combination with another chemotherapeutic agent), and vinblastine. In some cats vincristine has been noted to produce a marked and prolonged neutropenia.
Many mechanisms contribute to infection after chemotherapy. Certain chemotherapeutic agents prevent phagocyte mobilization or impair function of these cells. Some cancers infiltrate the bone marrow, producing myelophthisis and contributing to cytopenias. Suppression of leukopoiesis by chemotherapy drugs may lead to associated barrier disruptions of the skin, oral cavity, and alimentary tract mucus, and the normal pulmonary “mucociliary elevator” may not function effectively to clear bacterial organisms. Endogenous bacterial infections may develop, caused by the host’s native microbial flora; these are commonly due to aerobic and anaerobic gram-negative bacteria from the gastrointestinal tract or Staphylococcus organisms from the skin. In addition, hospitalized patients frequently develop catheter-related bacteremias, often caused by microbes transmitted to the susceptible patient from the hospital environment or from another animal; these organisms may be antibiotic resistant. Patients with absolute neutrophil numbers greater than 1500/μL are generally protected against endogenous infections. If the number is between 1000 and 1500/μL, the owner is advised to monitor the animal’s condition and report any fever or anorexia. If the number is between 500 to 1000/μL, prophylactic antibiotics are generally dispensed unless the period of neutropenia is anticipated to be very short. When the absolute neutrophil number is less than 500/μL, treatment with granulocyte colony-stimulating factor (G-CSF, filgrastim, Neupogen, Amgen) or granulocyte/macrophage colony-stimulating factor (GM-CSF, sargramostim, Leukine, Berlex) may be considered (discussed later), although many patients recover without the administration of one of these cytokines—of course, antibiotics should also be administered. If the patient is not febrile, it should probably not be hospitalized because its likelihood of acquiring a hospital-acquired resistant bacterial infection is high. If the patient is febrile, however, it should usually be hospitalized for blood cultures and intravenous antibiotic administration. In general, antibiotics should not be given prophylactically for neutropenia unless they are necessary, because they increase the risks for development of bacterial resistance and fungal infection in these immunocompromised patients. When necessary, choice of an empirical antibiotic regimen should take into account the type of infection the patient is likely to have: home acquired (probably endogenous) or hospital acquired (likely to be exogenous and possibly antibiotic resistant). Appropriate antibiotic combinations for use in the febrile neutropenic patient would be an aminoglycoside (e.g., amikacin) plus an antipseudomonal penicillin (ticarcillin, carbenicillin, piperacillin) or cephalosporin (cephalothin, cefazolin, cefoxitin). The third-generation cephalosporin ceftazidime is an antibiotic with an excellent spectrum of efficacy against gram-negative bacteria and Pseudomonas, and it is moderately effective for treatment of Staphylococcus infections. Because it has poor efficacy against anaerobic organisms, it must be combined with a drug such as clindamycin, metronidazole, or an antipseudomonal penicillin; these combinations are very useful in treating infections in neutropenic cancer patients. Imipenem is useful as a single agent in these patients, with excellent efficacy against enteric gram-negative bacteria, Pseudomonas, anaerobes, and Staphylococcus, but the high cost of this antibiotic limits its use in veterinary medicine at this time. It is also an antibiotic that should probably be reserved for use in humans with antibiotic-resistant infections.
If myelosuppression is severe and life-threatening, recombinant G-CSF or GM-CSF is often administered. These products are human glycoproteins that regulate production of neutrophils within the bone marrow; they are produced in Escherichia coli bacteria. Both stimulate neutrophil progenitor proliferation, differentiation, and functional activity with minimal toxicity. Long-term (i.e., longer than 30 days) use of human G-CSF in the dog or cat results in antibody formation, however, with significant and prolonged decreases in neutrophil counts. At a daily dose of 5 μg/kg subcutaneously, the effects of canine G-CSF on the normal canine bone marrow are rapid and predictable: Mean neutrophil counts in normal dogs increased to 26,330/μL after one injection, with a maximum count of 72,125/μL by day 19 of administration. The neutrophil counts returned to normal in these dogs within 5 days after daily therapy was discontinued.4 Canine G-CSF is not commercially available, but recombinant human G-CSF has resulted in similar elevations of neutrophil counts in the dog. In cats 10 to 14 days of human G-CSF resulted in maximum neutrophil counts ranging from 20,370 to 61,400/μL.5 Thus a short course of G-CSF may be used in dogs or cats either before aggressive chemotherapy, in an attempt to ameliorate or prevent myelosuppression, or as a rescue after chemotherapy has induced significant neutropenia.
Gastrointestinal Side Effects
Another frequent side effect of chemotherapy relates to the gastrointestinal toxicity of these drugs; anorexia, vomiting, and diarrhea may be noted in some individuals treated with cytotoxic agents. These side effects are not noted in dogs and cats as predictably as in humans, but they can occur in sensitive individuals with most of the commonly used drugs. Agents with a high potential for acute nausea after administration include cisplatin, dacarbazine, and high-dose cyclophosphamide; those with a moderate potential include carboplatin, conventional-dose cyclophosphamide, doxorubicin, mitoxantrone, and occasionally vincristine. In animals with only mild to moderate nausea, metoclopramide (0.2 to 0.5 mg/kg orally or subcutaneously thrice daily), or prochlorperazine (0.3 mg/kg orally or subcutaneously thrice daily) may be effective. Premedicating with subcutaneous administration of butorphanol at 0.4 mg/kg will sometimes block the postadministration vomiting caused by cisplatin. In animals with severe nausea and vomiting caused by chemotherapy (which is rare, fortunately), one of the serotonin 5-hydroxytryptamine (5-HT) receptor antagonists may be given either orally, subcutaneously, intramuscularly, or rectally. The most commonly available member of this class of drugs is ondansetron, but several newer antiemetics of the class are now also available (e.g., dolasetron), and the price of the drugs has dropped to make them reasonable to use now that ondansetron is available as a generic. The dose of ondansetron in the dog is 0.1 to 0.3 mg/lb intravenously or subcutaneously twice daily (oral dose is 0.5 to 1 mg/kg every 12 to 24 hours), and the dose of dolasetron is 0.5 to 0.6 mg/kg subcutaneously or intravenously every 24 hours. The serotonin receptor antagonists act more specifically than other antiemetics to prevent the vomiting induced by chemotherapy or radiation. Serotonin receptors of the 5-HT type are located on vagus nerve terminals and in the chemoreceptor trigger zone. Serotonin is released from enterochromaffin cells in the small intestine when they are severely damaged. The released serotonin stimulates vagal afferents through the 5-HT receptors, and nausea and vomiting ensue. Ondansetron and dolasetron block the 5-HT receptor site, which prevents the serotonin effect.
A new antiemetic that is proving to be very effective in the control of chemotherapy-induced nausea and vomiting is maropitant (Cerenia). It is a neurokinin-1 (NK-1) receptor antagonist and is available both in injectable form and as tablets; the dose is 1 mg/kg subcutaneously once daily or 2 mg/kg orally once daily. The NK-1 receptor antagonists drugs work at NK-1 receptors in the emetic center to block both peripheral and central stimuli that cause emesis, by inhibiting the binding of substance P. Substance P is found in significant concentrations in the nuclei that make up the emetic center and plays a central role as a neurotransmitter in the afferent pathways of the emetic reflex.
Other gastrointestinal side effects may also occur as sequelae to chemotherapy drug administration. Diarrhea occurs much less often than vomiting and nausea and is generally readily treated with loperamide (0.08 mg/kg orally thrice daily). Doxorubicin sometimes produces a severe hemorrhagic colitis in dogs, for which hospitalization and symptomatic treatment with antibiotics and intravenous fluids may be necessary; rarely, this side effect caused by doxorubicin can be life-threatening. Anorexia may be noted with several drugs, especially in cats with doxorubicin or vincristine administration. Appetite stimulation with drugs such as cyproheptadine may help in these cats, but enteral feeding is sometimes necessary.
Phlebitis and Necrosis
Several commonly used chemotherapeutic drugs will produce phlebitis or local necrosis (or both) at the site of administration if extravasated. Drugs that can be expected to produce severe reactions include the vinca alkaloids, doxorubicin, and dactinomycin; moderate reactions may be seen with bleomycin, cisplatin, dacarbazine, and mitoxantrone. These cytotoxic drugs may irritate the lining of access veins during administration, producing phlebitis, or may escape the cutaneous vasculature and spread throughout the surrounding tissues, causing a local inflammatory reaction (chemical cellulitis). Alternatively, some of the drugs will produce local tissue necrosis if extravasated. Doxorubicin produces the most dramatic and severe reactions. Extravasation will produce marked epidermal hyperplasia, with mitosis of many epidermal cells at the margins of the lesion; the reaction will contain individual necrotic keratinocytes, lobular panniculitis, and reactive fibroblasts and endothelial cells. No inflammatory reaction will be seen. In the area of direct extravasation, pan-epidermal, dermal, and subcutaneous tissue necrosis will be present. This necrosis begins 1 to 2 weeks after the drug is extravasated and may continue for up to 4 months. With all these drugs, extreme precautions should be taken to prevent extravasation, particularly with venipuncture and catheter placement. A “first-stick” catheter should always be used; infusion through a preexisting catheter is not advised. The animal should be observed closely (and possibly restrained) during the entire time of the infusion, in case movement should dislodge the catheter.
Infusion of the chemotherapy drug should be terminated immediately if the patient shows signs of pain during drug administration or if there is blebbing at the catheter or needle entrance site. If the catheter or needle is still present, the clinician should aspirate any fluid from the extravasated area. A continuing dilemma in the management of extravasation injuries is the absence of evidence-based management strategies. Almost all the recommendations in the literature are (of course) based on anecdotes; even knowing for sure that an extravasation occurred is sometimes difficult, so assessing whether there has been a positive response to a particular treatment is also questionable. However, the current recommendations for vinca alkaloid extravasation are to inject 150 units of hyaluronidase (if available; hyaluronidase has become difficult to obtain) through the patent catheter or needle and then apply local heat for 15 to 30 minutes four times daily for 48 hours. For anthracycline extravasation, the clinician should apply an ice pack for 15 to 30 minutes four times daily and 90% dimethyl sulfoxide (DMSO) topically several times daily for 7 to 14 days; surgical removal should be considered if the area of extravasation is confined. DMSO is a free radical scavenger that causes potent vasodilation and has pain reduction and antiinflammatory mechanisms. Dexrazoxane is a drug that has been used to prevent anthracycline-induced cardiotoxicity. It may also decrease free radical formation, and when given intravenously immediately after extravasation (1000 mg/m2 intravenously, repeated at the same dose the next day), it appears to prevent the tissue necrosis associated with anthracycline extravasation. Growth factors regulate and coordinate wound healing, and they may also be helpful in altering damage from chemotherapy drug extravasation. In animal models both G-CSF and GM-CSF have been shown to be significantly better than saline or no treatment in decreasing the severity and extent of necrosis with doxorubicin extravasations. In one human patient who did not respond to injected dexrazoxane, GM-CSF injected into an ulcerated area of extravasation led to tissue granulation and complete healing within 8 weeks. With any luck, clinicians will never have to use the following protocol: A 1-mL vial of GM-CSF was diluted with saline. Several small injections were then given within the borders of the ulcer. As new granulation tissue formed, GM-CSF injections were given three times weekly for 2 weeks, then twice a week for 2 weeks. Because treatment of extravasation injuries is not yet uniformly effective in preventing the local irritation and necrosis caused by extravasation of these drugs, prevention is the best answer.
Alopecia
Alopecia is common in certain breeds of dogs after chemotherapy, particularly after administration of doxorubicin or cyclophosphamide. Hair loss is predictable in Poodles and in mixed-breed dogs of Poodle lineage; it is also commonly seen in Terriers and Old English Sheepdogs. Occasionally, it may be noted in other breeds as well. It is common for cats to lose their whiskers during chemotherapy. For some owners the alopecia induced by chemotherapy is very distressing, and owners of breeds in which this is likely to occur should be prepared for this possibility. Hair regrowth begins 1 to 2 months after chemotherapy is discontinued. However, an alteration in the color or texture of the new hair may be noted; the regrown hair may be a lighter or darker shade and may be softer or curlier than the animal’s previous hair.
Reproductive Side Effects
Although generally less important in dogs and cats with cancer than in humans, the effects of chemotherapy on gonadal function should be explained to owners considering treatment, particularly if the animal is shown or has been used for breeding. Most chemotherapy drugs will cause hypofertility or infertility by impairing production of sperm and oocytes. In the male animal, loss of libido may result from Leydig cell dysfunction and decreased testosterone levels, especially with corticosteroid treatment for lymphoma. Owners should consider cryostorage of sperm from the dog before beginning chemotherapy. However, it is common to find on semen evaluation that general debility from the cancer itself has resulted in poor semen quality even before chemotherapy drugs have been given. Reversibility of gonadal dysfunction produced by chemotherapy is variable depending on the agent administered, the dose intensity of the protocol used for treatment, and the age of the patient itself. During chemotherapy and for a variable period after the treatment is completed, a male dog or cat should not be used for breeding and a female should not become pregnant because congenital malformations may result in the offspring.
Palmar–Plantar Erythrodysesthesia
Palmar–plantar erythrodysesthesia (PPES), also known as hand–foot syndrome in humans, has been seen with constant-rate infusions of doxorubicin, cyclophosphamide, ifosfamide, 5-fluorouracil, and other agents in humans. In dogs PPES has been most commonly associated with the administration of liposome-encapsulated doxorubicin (Doxil).6 PPES is a primarily a dermal toxicity characterized by reddening of the skin. It is followed by edema and eventual ulceration of the skin. PPES tends to occur in areas of friction, such as the weight-bearing portions of the feet and the axillary regions. Histologically, these lesions are described as focal areas of parakeratosis, acantholysis, and chronic active inflammation of the skin and underlying dermis. PPES often resolves quickly after discontinuation of the drug but may recur if the drug is reinstituted. Oral pyroxidine may help ameliorate some of the side effects, although it will not completely prevent PPES.7
Human Health Hazards
Exposure of hospital personnel and owners to carcinogenic, mutagenic, and teratogenic drugs and drug-containing animal waste must be considered. In the days before clinicians took appropriate precautions when mixing and administering chemotherapy, there were many reports in the human literature of fetal loss and birth of infants with congenital defects among nurses and pharmacy staff members who were frequently exposed to chemotherapy drugs.8,9 Proper storage, preparation, and administration of chemotherapeutic agents, as well as proper disposal of cytotoxic drug waste and urine and stool of the animal being treated, should be a concern of every clinician who treats an animal for cancer.8,9 Reviews on proper handling of chemotherapy in the workplace are available; the Occupational Safety and Health Administration (OSHA) publication “Controlling Occupational Exposure to Hazardous Drugs” may be downloaded from OSHA’s website (www.osha.gov/dts/osta/otm/otm_vi/otm_vi_2.html).
Drugs Used in Cancer Chemotherapy
The following sections cover drugs commonly used in veterinary oncology for the treatment of canine and feline neoplasia (Table 33-2). Included are cautionary comments regarding administration, toxicity, and effectiveness. Note that dosages for chemotherapy drugs vary greatly depending on tumor type and protocol used. It is extremely important that a clinician considering the use of one of these drugs as a single agent or in a combination protocol carefully consider all of the drug’s possible toxicities rather than merely look up a dose from a chart or formulary.
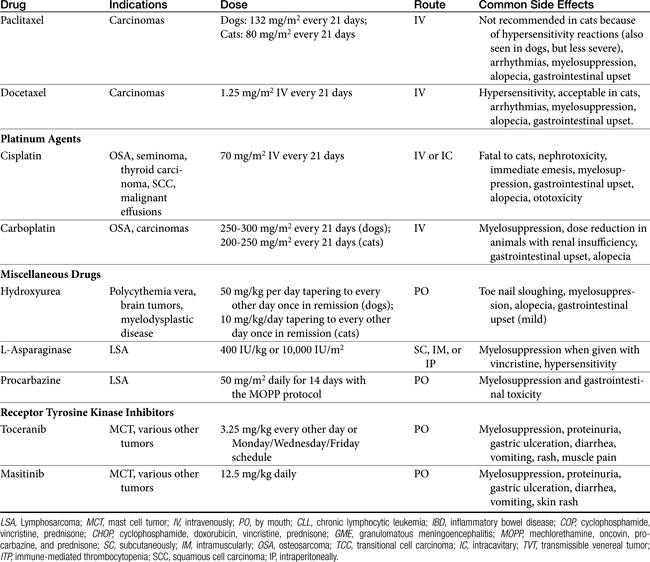
Alkylating Agents
Nitrogen Mustards
Cyclophosphamide
Mechanism of action
Cyclophosphamide is a classic alkylating agent that is extensively metabolized in the liver to the active cytotoxic metabolites phosphoramide mustard and acrolein. The metabolite phosphoramide mustard is responsible for most of the antineoplastic effects of the drug. Cyclophosphamide is cell cycle specific at normal dosing schedules but may be cell cycle nonspecific at extremely high doses. Resistance to treatment with cyclophosphamide does develop in tumor cells, probably related to increased ability of the tumor cells to produce glutathione and obtain protection from oxidative damage. Excretion takes place principally by the kidneys, and modification of dose and dose interval should be considered when a patient has significant renal disease.
Preparations
Cyclophosphamide is available as 25-mg and 50-mg tablets for oral administration as well as an intravenous preparation in vials containing 100 mg, 200 mg, or 500 mg. The drug is equally effective when given orally or parenterally. Cyclophosphamide cannot be used for intracavitary treatment because it must be metabolized to its active form in the liver.
Side effects
Cyclophosphamide has the potential for extremely dangerous marrow suppression as well as nausea and vomiting. Of all the chemotherapeutic drugs in common use in veterinary oncology, neutropenia occurs most predictably with cyclophosphamide. As a result, the drug must initially be administered with great caution; the degree of myelosuppression varies from patient to patient but may be early and profound. For this reason neutrophil counts must be carefully assessed whenever the drug is used for the first time in a patient. Some animals will develop myelosuppression after a week of cyclophosphamide; in others the drug will have to be discontinued after 3 to 5 days because of severe neutropenia or thrombocytopenia. A baseline total white blood cell count, differential, and platelet estimate should be taken before the drug is administered. Usually, depression in the absolute neutrophil count begins on the third day of administration, so the next blood count is taken on that day. From that day on during the first cycle of cyclophosphamide therapy, a total leukocyte count, differential, and platelet estimate are obtained every day until the cycle of cyclophosphamide therapy is completed. If the absolute neutrophil count drops below 3000/μL, the drug is discontinued entirely for that cycle; on the next cycle, it is reinstituted at a dose 25% less than the initial daily dose. If the neutrophil count drops to less than 1500/μL, the drug is stopped and reinstituted on the next cycle at a dose 50% less than the initial dose. If the number of neutrophils drops to less than 1000/μL and fever ensues, empirical antibiotic therapy should be begun. Recombinant human G-CSF may also be given for several days if necessary until the animal’s neutrophil count returns to normal. After the animal’s individual tolerance for cyclophosphamide is determined, fewer CBCs will need to be checked during therapy. In maintenance protocols one CBC per cycle before administration of the drug begins is generally adequate.
Hemorrhagic cystitis may result from cyclophosphamide administration, usually after long-term use; however, it has been reported after one intravenous administration.10 It is caused by the metabolite acrolein, which is excreted in urine and reaches a urine level of 100 to 200 times the serum concentration; this metabolite is extremely irritating to the bladder mucosa and produces necrosis of smooth muscle. Chronic cystitis leading to bladder fibrosis may occur with long-term use. Affected dogs and cats will present with clinical signs of gross hematuria, often with blood clots, and will be reported to be straining to urinate. Concurrent treatment with prednisone decreases the incidence of hemorrhagic cystitis, probably by causing polydipsia and polyuria. Lymphoma patients rarely develop this complication, insofar as prednisone administration is generally a part of the treatment protocol. Any animal that is to receive cyclophosphamide should have a urinalysis performed before the drug is administered to rule out preexisting hematuria caused by bacterial cystitis or prostatitis. The client should be warned to watch for hematuria during the course of treatment with cyclophosphamide, and administration should cease immediately if the problem is noted. Several precautions can help prevent this problem while an animal is receiving cyclophosphamide. The animal should be encouraged to drink more fluids; salting food and offering beef or chicken bouillon may help increase fluid intake. Because the cystitis is caused by acrolein producing local irritation on the bladder mucosa, the owner should encourage frequent urination by walking his or her dog more frequently and should make sure that the dog is allowed to urinate before retiring for the night. It is better not to administer cyclophosphamide in the evening because acrolein will then concentrate in the urine overnight. Although the free radical scavengers acetylcysteine and mesna have been reported to prevent cyclophosphamide-induced hemorrhagic cystitis in humans treated with high-dose cyclophosphamide before bone marrow transplantation, it is not clear that they are necessary in dogs and cats treated with standard chemotherapy protocols. The incidence of this side effect seems to be low with most current cyclophosphamide dosing regimens, which use intermittent “pulse” doses of cyclophosphamide rather than continuous daily dosing of the drug.
Alopecia occurs in susceptible dogs as another side effect of cyclophosphamide. When cyclophosphamide is used in combination with doxorubicin or dactinomycin, cardiotoxicity of these compounds may be potentiated.
Melphalan (phenylalanine mustard)
Mechanism of action
Melphalan is an alkylating agent that is a phenylalanine derivative of mechlorethamine, the first chemotherapeutic agent discovered. In World War I, it was noted that troops who had received poisoning with mustard gas often had aplastic anemia and severe lymphopenia, with depletion of lymphocytes in the spleen and lymph nodes. This finding caused clinicians to study the effects of nitrogen mustard on lymphoma, first in the mouse and later in humans. Many derivatives of this original compound have been discovered, but melphalan remains one of the most useful.
Spectrum of activity
In dogs and cats, melphalan is generally used for the treatment of plasma cell tumors, either plasma cell myeloma or extramedullary plasmacytoma.11
Preparations
Melphalan is available for oral use as a scored 2-mg tablet and for intravenous injection as a 50-mg vial. In dogs and cats, the drug is conventionally used orally.
Side effects
Myelosuppression is the most common side effect of melphalan, but it is not generally severe. Monitoring of a CBC should be done every 2 weeks during induction and then monthly during maintenance.
Dosing regimen
The recommended dose for melphalan is 0.1 mg/kg daily for 7 to 10 days, then 0.05 mg/kg daily until remission is achieved. The drug is then given as a maintenance agent for 7 days out of every month at 0.1 mg/kg per day. Because food can apparently decrease the oral absorption of the drug, it should be given several hours before the animal is fed.
Mechlorethamine
Mechanism of action
Mechlorethamine (Mustargen), is the prototypical nitrogen mustard. These drugs alkylate DNA by initially losing a chlorine molecule and allowing the β-carbon to react with the nucleophilic nitrogen atom to form the cyclic, positively charged, and very reactive aziridinium moiety. The reaction between this aziridinium ring and the electron-rich nucleophile creates the initial alkylated product. Cross-linking of the DNA occurs when a second aziridinium ring is formed by the remaining chloroethyl group, allowing for a second alkylation.
Spectrum of activity
In dogs it is used as a rescue agent in the MOPP protocol (mechlorethamine, oncovin, procarbazine, and prednisone) for high-grade lymphoma.
Preparations
Mechlorethamine is available in 5-g and 25-g bottles for reconstitution. It is also available in a topical preparation for mycosis fungoides; however, because of the risk of human exposure, the topical preparation is not recommended for veterinary use.
Side effects
Myelosuppression is the most common side effect of this drug; however, it is a potent vesicant and is irritating topically. The gastrointestinal side effects appear to be more severe with this drug than the other nitrogen mustards. A CBC should be performed 7 to 10 days after each dose and before each dose.
Chlorambucil
Mechanism of action
Chlorambucil is another of the derivatives of nitrogen mustard; it is the slowest acting and least toxic of the alkylating agents commonly used in veterinary medicine. The drug is easily absorbed by passive diffusion when administered orally, so any food given with it may interfere with its absorption.
Spectrum of activity
Chlorambucil is used as a mainstay for treatment of chronic lymphocytic leukemia,12 small cell lymphoma, Waldenström’s macroglobulinemia, and thymoma in dogs and cats. It has been substituted in combination chemotherapy protocols for cyclophosphamide when hemorrhagic cystitis has ensued but is not especially effective for maintenance therapy of high-grade lymphomas. Activity may also be seen against plasma cell myeloma and ovarian carcinoma.
Side effects
Marrow suppression is quite late, gradual in onset, and rapidly reversible in dogs and cats but may be profound if it is not discovered sufficiently early. In general, myelosuppression is not seen until the drug has been given daily for at least 1 month; it is recommended that a CBC be obtained once every 2 weeks during induction. As soon as remission occurs, the drug should be administered only intermittently as a maintenance protocol (i.e., alternate weeks or 1 week out of 4). Chlorambucil should not be administered with food.
Dosing regimen
The dose for chlorambucil is 0.1 to 0.2 mg/kg orally per day for 4 to 7 days, then 0.1 mg/kg daily until remission occurs. Alternatively, the drug may be given once every 2 weeks at a dose of 0.4 mg/kg. After remission is obtained, a maintenance protocol may be started, with the drug administered intermittently as indicated by the tumor treated (e.g., 0.1 mg/kg daily for 7 consecutive days, followed by 21 days off).
Nitrosoureas
Mechanism of action
CCNU (lomustine) and BCNU (carmustine) are drugs that are very lipid soluble and cross the blood–brain barrier with ease. Excretion is primarily renal, so dose modification must be considered if the patient has renal disease.
Spectrum of activity
Although CCNU and BCNU are used in humans to treat certain lymphomas, the drugs find their principal use in veterinary medicine for treatment of central nervous system (CNS) neoplasia.13,14 The two drugs are unique in their ability to attain therapeutic levels in brain tissue. Recent information suggests that CCNU may also have some efficacy in treatment of canine mast cell tumor.
Preparations
BCNU is available in a 100-mg vial for intravenous administration; CCNU is given orally and is available as 10-, 40-, and 100-mg capsules.
Side effects
Both the nitrosoureas may be quite emetogenic immediately after administration. The vomiting and nausea usually last less than 24 hours after administration, and the animal’s discomfort can generally be ameliorated with butorphanol given subcutaneously at 0.4 mg/kg thrice daily. In some cases, ondansetron will be necessary to relieve symptoms.
Prolonged bone marrow suppression is common with both of the nitrosoureas. Neutropenia may be noted as early as 1 week after administration but may persist for up to 6 weeks. In some cases neutropenia is severe enough to adversely affect the animal’s quality of life, and treatment with intravenous antibiotics and recombinant human G-CSF may be necessary if the animal becomes febrile.
Dosing regimen
BCNU must be given intravenously. The product is reconstituted with alcohol and then added to saline or 5% dextrose in water to be given as an intravenous infusion over 1 to 2 hours. Severe pain may be seen at the injection site even if no extravasation is occurring; a longer infusion time may help decrease discomfort from the administration. The conventional dose of BCNU is 50 mg/m2 given intravenously once every 6 weeks. CCNU is available as an oral preparation, and it is given as a single oral dose of 75 to 100 mg/m2 once every 6 to 8 weeks.
Dacarbazine
Mechanism of action
The major mode of action of dacarbazine against tumor cells appears to be alkylation of nucleic acids. Its complete chemical name is 5-(3,3-dimethyl-1-triazeno)-imidazole-4-carboxamide, which is why it is also called DTIC. Dacarbazine is cycle specific.
Spectrum of activity
Dacarbazine is not often used in veterinary oncology. At one time, it was suggested as a treatment for canine melanosarcoma, but results were disappointing. Dacarbazine has its major use for treatment of relapsed lymphoma in combination with doxorubicin.15
Preparations
Dacarbazine is available for intravenous administration in vials of 100, 200, and 500 mg.
Side effects
Local pain is often seen during administration; concentrated solutions of the drug are very irritating to veins, and extravasation will produce severe phlebitis. Myelosuppression is mild and does not occur until the second or third week after treatment, but a CBC should be checked before each subsequent treatment is administered. Vomiting and nausea are common during the first few days of treatment but can usually be blocked by prior administration of chemoreceptor trigger zone–blocking antiemetics. These gastrointestinal symptoms may be lessened by using a lower dose initially and gradually escalating the dose during the course of treatment, but the signs seem to subside after 1 or 2 days of treatment despite continued therapy with dacarbazine.
Mitotic Inhibitors
Vinca Alkaloids
Mechanism of action
The vinca alkaloids are extracted from the common periwinkle plant, Vinca rosea. This plant was originally investigated by pharmacologists because of its reported ability to lower blood glucose levels in several native populations. Although its efficacy as a hypoglycemic agent proved unimpressive, it was discovered that extracts of the plant had cytotoxic effects. Eventually, vincristine and vinblastine came into common clinical use as anticancer agents; the two compounds differ only slightly, with vincristine having a formyl side chain and vinblastine having a methyl side chain on the larger parent molecule. A third vinca alkaloid has recently become more popular in the veterinary market. Vinorelbine (Navelbine) acts similarly to the other vinca alkaloids, with the exception that it achieves very high concentrations within the lung parenchyma. All of the drugs appear to act as spindle poisons by binding to microtubular proteins within cells. The spindles are thus unable to act during mitosis, leading to arrest of the cell in metaphase. Generally, vincristine is thought of as a phase-specific drug effective only in the M phase of the cell cycle. Vinblastine, however, also blocks the cell’s utilization of glutamic acid, thus inhibiting purine synthesis. For this reason vinblastine acts against cells in active mitosis but also in other phases of the cell cycle.
Spectrum of activity
Vincristine and vinblastine have their major use in veterinary medicine in combination chemotherapy protocols for treatment of lymphoma and lymphoid leukemias. Some efficacy of vincristine may be seen either as a single agent16 or in combination with doxorubicin and cyclophosphamide for treatment of soft tissue sarcomas,17 and vincristine is the drug of choice for treatment of transmissible venereal tumors.18,19 Although it was previously suggested that vincristine might be effective in the treatment of mast cell tumor in the dog, a recent report has discounted the drug’s role in management of this tumor; only 2 of 27 dogs with mast cell tumors had even a partial response to vincristine given at 0.75 mg/m2.20 The principal use of vinblastine in veterinary oncology at this time is in the treatment of mast cell tumors. It has been shown to be much more effective than vincristine at treating this disease, with reported response rates of 40% when combined with prednisone.21 It may also be used as a substitute for vincristine in a combination chemotherapy protocol when a vincristine-induced neuropathy has been noted. Vinorelbine has been used primarily against primary lung tumors, especially well-differentiated ones such as bronchogenic carcinomas and bronchoalveolar carcinomas.
Preparations
Vincristine is available for intravenous use in 1-mg, 2-mg, and 5-mg vials. Vinblastine is also for intravenous administration only and is supplied in 10-mg vials. Vinorelbine comes in 10-mg or 50-mg vials of a 10 mg/mL solution.
Side effects
Because of its phase-specific effects, vincristine is not generally myelosuppressive in the dog; occasionally, it may produce significant neutropenia in the cat.22 Anorexia and nausea are sometimes seen in both dogs and cats treated with vincristine, especially at the higher levels of the dose range. Unlike vincristine, vinblastine is quite myelosuppressive, and the interval between doses is often prolonged because of the duration of neutropenia produced by the drug. Vinorelbine is similarly myelosuppressive to vinblastine.
Local phlebitis and severe pain occur if any of the vincas is extravasated. Although a catheter may be placed, conventionally a butterfly needle is used to administer vincristine, vinorelbine, or vinblastine. The vein is punctured with the butterfly needle in the usual fashion, and blood flow is observed into the tubing. Several mL of saline are infused into the vein so that leakage may be observed. The vinca alkaloid is then given as a bolus injection and is followed by several more mL of sterile saline to ensure that not a drop of the drug remains on the tip of the needle.
One of the principal limitations of long-term treatment with vincristine in clinical practice is the development of a drug-induced sensory and motor neuropathy, the pathogenesis of which is poorly understood. The cat may be more sensitive to the development of this phenomenon than the dog.23 Severe nerve fiber degeneration may be seen, as well as focal axonal swellings with secondary demyelination of peripheral nerves.24,25 Vincristine administration should be discontinued immediately as soon as any signs of neuropathy are noted because further treatment may produce severe, generalized motor weakness. The neurotoxicity will generally improve within several months after the drug is discontinued, but some of the signs may be irreversible. Although neurologic problems are rare with vinblastine administration, they may rarely occur with this drug as well.
Dosing regimen
The appropriate dose of vincristine is 0.5 to 0.75 mg/m2 intravenously once weekly according to the treatment protocol used. Treatment with vinblastine should begin at 2 mg/m2 by intravenous injection once every 2 weeks. At each cycle, the clinician should increase the vinblastine dose in increments of 0.25 mg/m2 until myelosuppression is seen (absolute neutrophil count less than 3000/μL). Then a maintenance dose of vinblastine, which is one increment smaller than the dose that produced leukopenia, should be administered. Because vinblastine is so myelosuppressive, the clinician should not administer the drug when the animal’s absolute neutrophil count is less than 3000/μL. The dose range for vinorelbine is 10 to 12.5 mg/m2 intravenously, using the same dosing schedule as vinblastine.
Taxanes
Mechanism of action
The taxanes are one of the most significant additions to the anticancer arsenal added during the twentieth century. Paclitaxel, the parent drug, was discovered by a National Cancer Institute program in which extracts from thousands of plants were evaluated for anticancer activity. Paclitaxel is derived from the bark of the Pacific yew tree (Taxus brevifolia). Paclitaxel and its semisynthetic analog, docetaxel, have demonstrated significant antitumor activity in humans. Both drugs have a unique mechanism of action compared with other microtubule inhibitors. The taxanes bind to the interior surface of the microtubule lumen at the N-terminal 1-31 amino acids and residues 217-233 of the β-tubulin subunit. Binding to this site does not interfere with the binding of other microtubule inhibitors to their respective sites. Ultimately, the taxanes disrupt microtubule dynamics by suppression of microtubule instability and treadmilling. Like the vinca alkaloids, these drugs are considered to be cell cycle specific for the M phase; however, the majority of cell death occurs in the S phase.
Spectrum of activity
In humans these drugs have been used primarily as anticarcinoma agents with Food and Drug Administration approval for carcinomas of the breast, ovarian carcinomas, prostatic carcinoma, head and neck carcinomas, gastric carcinoma, Kaposi’s sarcoma, and non–small cell lung cancer. In dogs paclitaxel has been reported to have activity against mammary tumors, squamous cell carcinoma, transitional cell carcinoma, osteosarcoma, and malignant histiocytosis.26,27 In vitro, paclitaxel has shown activity against a number of feline vaccine–associated cell lines; however, no in vivo information exists. Docetaxel has similar uses but is safer for cats. Paclitaxel has also been used with some success as an inhalation chemotherapy agent, although administration is difficult and not routinely available.28
Preparations
Paclitaxel is available in various sizes of a 20 mg/mL solution available for intravenous administration (30-mg, 100-mg and 300-mg multidose vials). This drug is not water soluble and therefore must be dissolved in a specific carrier, Cremophor. Docetaxel is available in various sizes of a 40 mg/mL solution for intravenous administration (20-mg and 80-mg single dose vials). Docetaxel must also be dissolved in a carrier solution of Polysorbate 80. These carriers are responsible for a number of the side effects seen, including hypersensitivity reactions. This drug is available in the United States only in an injectable form. It has low oral bioavailability on account of the high numbers of ABC transporters and P-glycoprotein efflux pumps within the intestinal lumen cells, as well as significant first-pass metabolism by the liver. Nonetheless, oral fomulations available in Europe have shown real promise against high-grade mast cell tumors. These formulations are given in conjunction with oral modulators of ABC transporters or cytochrome P-450 (or both).
Side effects
Side effects in dogs are fairly consistent with those seen in humans. Myelosuppression is common and typically occurs 5 to 7 days after injection. Gastrointestinal upset, including anorexia, nausea, diarrhea, and vomiting, can be seen as well. Alopecia has not been reported in the dog with this particular drug, although it stands to reason that this could be a possible sequela of drug administration. Peripheral neuropathies are more common with the vinca alkaloids, but they can be seen with administration of the taxanes as well. In humans arrhythmias are a common side effect of this drug. Indeed, the Taxus species of plants are known for causing arrythmias in cattle who ingest their bark. Though this has not been a reported side effect in the dog, it is recommended that an electrocardiogram be performed before administration. If underlying arrhythmias are noted, the clinician is advised not to use the taxanes in these patients. One of the most significant side effects of these drugs is hypersensitivity. This is related to the carriers (Cremophor and Polysorbate 80) that are necessary to keep these drugs in solution. Careful administration and monitoring during drug administration are recommended. Because of the severe hypersensitivity reaction of cats to paclitaxel, it is recommended that this drug be avoided in the species all together. Docetaxel has been safety administered to cats and is the preferred taxane for this species. This drug is not extremely myelosuppressive, and its myelosuppression is typically resolved at the next dose, 3 weeks later. The dose should be delayed if the mature neutrophil count is less than 2000 cells/μL.
Dosing regimen
The dose for paclitaxel is 165 mg/m2 intravenously every 3 weeks. The dose range for docetaxel is 25 to 30 mg/m2 given intravenously every 3 weeks. A specific premedication protocol is necessary to prevent severe hypersensitivity reactions. Animals are premedicated the night before treatment with 1 mg/kg of prednisone. Approximately 30 to 60 minutes before injection, diphenhydramine (4 mg/kg) is given intramuscularly, cimetidine (4 mg/kg) is given intravenously, and dexamethasone SP (1.5 to 2 mg/kg) is given intravenously. The diluted taxol (diluted 10:1 0.9% saline to drug) is started at a rate of 30 mL/hr for 30 minutes. If no allergic reaction is seen at that time, then the rate is increased to 60 mL/hour. The catheter is flushed with 25 to 30 mL of 0.9% saline afterwards. If signs of a hypersensitivity reaction do occur, the clinician should stop the infusion, medicate if necessary, and then restart at a slower rate. Therapy should be discontinued if severe hypersensitivity reactions occur.
Antitumor Antibiotics
Doxorubicin
Mechanism of action
Doxorubicin is an anthracycline glycoside derived from Streptomyces peucetius. It is directly cytotoxic, binding irreversibly with DNA and preventing both RNA and DNA synthesis. Cellular damage caused by doxorubicin results in enzyme-catalyzed, iron-mediated free radical formation, which produces further tissue damage. Ultimately, these effects result in induction of apoptosis in both normal and neoplastic cells. After intravenous administration doxorubicin is metabolized in the liver to active and inactive metabolites. The drug is excreted primarily in the bile but persists in plasma for prolonged periods.
Spectrum of activity
Doxorubicin has been proved effective in the treatment of a number of tumors of the dog and cat, including lymphoma, leukemias, and certain sarcomas and carcinomas.29-33 It appears that doxorubicin may be synergistic with cyclophosphamide in the treatment of some sarcomas,34 and it is combined with cytosine arabinoside as an extremely effective (although very myelosuppressive) protocol for leukemia.
Side effects
Cardiotoxicity is generally the dose-limiting factor for doxorubicin administration in dogs.35 It results from free-radical damage to the myocardium, with oxidation and death of myocardial cells in the presence of iron. Doxorubicin is an active iron chelator, and the resulting iron–doxorubicin complex catalyzes free radical reactions, leading to myocardial damage. Acute cardiac toxicity may occur at any time and after any dosage; it commonly takes the form of an arrhythmia, which resolves with time in most animals. Arrhythmias are more common if there is previous cardiac disease, previous or concurrent thoracic irradiation, or concurrent cyclophosphamide administration. The second form of cardiac toxicity induced by doxorubicin is congestive heart failure, with myocardial degeneration and cardiac muscle fibrosis leading to heart failure; this generally occurs with cumulative doses greater than 240 mg/m2. Once congestive heart failure induced by doxorubicin is present, patients may respond to aggressive therapy for heart failure, but some patients will die despite all treatment. Many attempts have been made in humans to diagnose incipient cardiac toxicity before clinical manifestations of heart failure begin, but it remains impossible to predict which patients will develop these changes. Echocardiographic measurement of ventricular fractional shortening and serial electrocardiography are probably the best methods to monitor dogs and cats that are receiving treatment with doxorubicin.
Both humans and dogs have shown a great deal of individual variation in susceptibility to doxorubicin-induced cardiotoxicity. Even though clinical cardiac disease may not be evident after treatment with doxorubicin, subclinical damage to the heart is common. In a study of 115 children with lymphoblastic leukemia treated with doxorubicin and followed for many years, 57% had abnormal cardiac function later in life.36 The EDTA-derivative drug ICRF-187 (dexrazoxane), given at 0.8 mg/kg 30 minutes before doxorubicin administration, apparently decreases cardiotoxicity without reducing cancer cell cytotoxicity,37 but no large clinical trials of this compound in dogs or cats with cancer have been reported. Dexrazoxane acts as a cardioprotectant by chelating iron, helping to prevent the free radical–induced damage caused by doxorubicin; the product is not commercially available at this writing.
Although cats do not generally show clinical cardiac disease associated with doxorubicin treatment, histologic and echocardiographic evidence of damage to the myocardium occurs in cats treated with cumulative doses of 170 to 240 mg/m2.38 Renal damage also occurs in cats after chronic treatment with doxorubicin; this is manifested by azotemia, dilute urine, and gradually decreasing creatinine clearance values during the course of administration.39 Another serious side effect in cats is the profound anorexia that may accompany administration of doxorubicin at the conventional dose given to dogs (30 mg/m2); cats given this dose do not act as though they are nauseated, but they may not eat voluntarily for weeks, sometimes requiring placement of a feeding tube. Small dogs weighing less than 10 kg may also experience unexpected nausea and anorexia at this treatment dose. For cats and small dogs, a doxorubicin dose of 1 mg/kg has proved to be much better tolerated.
Myelosuppression may occur several days after administration, usually beginning on day 4. Peak action on the bone marrow occurs from days 10 to 14. Because doxorubicin has a high affinity for mast cells, causing them to degranulate, anaphylaxis, urticaria, generalized erythema, and head shaking have been seen in the dog. To prevent this, the patient may be premedicated with antihistamines and steroids before administration. Alopecia may also occur in susceptible breeds of dogs.
Extreme phlebitis and necrosis occur if doxorubicin is extravasated. This necrosis begins 1 to 2 weeks after the drug is extravasated and continues for 1 to 4 months. Doxorubicin also produces an unusual “radiation recall” effect; if previous radiation damage has occurred, even years before, doxorubicin administration will cause its recurrence. This is not likely to be a problem in dogs and cats, given that radiation therapy to the thorax is rarely performed in these species, but it is often a serious complication of doxorubicin treatment in humans. Radiation to the thoracic cavity (when the heart is in the radiation field) may also potentiate the cardiotoxicity of doxorubicin, which could be an important consideration for dogs and cats with thymoma or mediastinal lymphoma.
Dosing regimen
Although the conventional dose of doxorubicin in medium-size to large dogs is 30 mg/m2 given every 3 weeks, in very small dogs or cats this dose may produce profound myelosuppression and anorexia. For this reason the dose in very small animals (less than 10 kg) should be reduced to 1 mg/kg every 3 weeks. Doxorubicin should be administered slowly into the tubing of a freely running intravenous infusion of saline or 5% dextrose solution. The tubing should be attached to a catheter that was placed on the first stick to prevent any leaking of drug through holes in the vein. At least 5 minutes should be taken to give the drug. Alternatively, doxorubicin may be mixed in a small volume of saline (50 to 100 mL) and dripped intravenously over 20 to 30 minutes. Because doxorubicin is physically incompatible with many other drugs, including heparin, aminophylline, cephalothin, dexamethasone, diazepam, hydrocortisone, and furosemide, care should be taken not to give other drugs through the same line during the doxorubicin infusion.
Doxorubicin Analogs
Mechanism of action
Because doxorubicin is so cardiotoxic, much effort has been devoted to development of analogs that might have less toxicity but that would maintain the level of tumor response. Epirubicin (4′-epidoxorubicin) is an analog that has been claimed to be less cardiotoxic in humans for equivalent doses. The mechanism of this drug is similar to that of doxorubicin. Idarubicin (4-demethoxydaunorubicin) is another anthracycline glycoside that is unusual in that it is the only antitumor antibiotic that can be given orally.
Spectrum of activity
The spectrum of canine and feline tumors that will respond to epirubicin therapy is probably similar to that for doxorubicin. Epirubicin’s principal use has been as a single agent in dogs with canine lymphoma; response rate and duration of response are not significantly different from what would be expected with doxorubicin therapy.40 Idarubicin has been used orally in cats for maintenance therapy of lymphoma after remission is obtained with other drugs.41
Preparations
Epirubicin is not commercially available at this time. Idarubicin is commercially available only in an injectable form, in 5-mg, 10-mg, and 20-mg vials.
Side effects
The claim that epirubicin has an advantage over doxorubicin in lessened toxicity has not proved to be particularly persuasive. Myelosuppression is similar, with the neutrophil nadir seen 10 days after administration. A significant number of dogs treated with epirubicin still show evidence of cardiotoxicity, as measured by ventricular fractional shortening on echocardiography. Idarubicin may produce gastrointestinal signs and myelosuppression in treated cats, and dose modification may be necessary. Because idarubicin is very cardiotoxic in humans, it presumably has the same effect in dogs and cats; appropriate precautions and monitoring should be considered.
Dosing regimen
Epirubicin is given intravenously at a dose of 30 mg/m2 for dogs weighing more than 10 kg and 1 mg/kg for dogs less than 10 kg. Similar precautions for administration to those taken with doxorubicin should be followed. Idarubicin has been given orally at 2 mg/cat daily for 3 days once every 3 weeks; injectable doses of idarubicin for dogs and cats have not been reported.
Dactinomycin (Actinomycin D)
Mechanism of action
Dactinomycin is one of the actinomycins, a group of antibiotics produced by various species of Streptomyces. The drug binds to DNA by intercalation and causes single-strand DNA breaks. Ultimately, dactinomycin causes apoptosis in susceptible tumor cells. As with doxorubicin, drug resistance to dactinomycin is caused by overexpression of P-glycoprotein; it is therefore unlikely that response to dactinomycin will be seen in lymphomas in which the tumor cells have become resistant to doxorubicin.
Spectrum of activity
Dactinomycin has chemotherapeutic activity against canine lymphoma; activity has been seen in some drug-resistant lymphomas but generally only when the tumor is not yet doxorubicin resistant. Partial responses have been seen with dactinomycin treatment of nephroblastoma and botryoid rhabdomyosarcoma in the dog. Certain carcinomas may also respond, including anal sac adenocarcinoma, squamous cell carcinoma, thyroid carcinoma, and transitional cell carcinoma.42 The principal use of dactinomycin, however, is as a substitute for doxorubicin when a potentially cardiotoxic cumulative dose of doxorubicin has been reached and it is desirable to continue administration of an antitumor antibiotic.
Side effects
Dactinomycin is extremely necrotizing when extravasated, similar to the effects produced by doxorubicin; it must be given through a “first-stick” catheter. Minimal myelosuppressive activity is noted when the drug is given as a solitary agent. Nausea and vomiting may occasionally occur during the first few hours after administration but may be partially prevented by administration of chemoreceptor trigger zone–blocking antiemetic agents. As with doxorubicin, dactinomycin potentiates the effects of radiation therapy and has a radiation-recall effect. Cardiotoxicity is extremely rare with this drug.
Dosing regimen
Dactinomycin is given intravenously at a dose of 0.5 to 1 mg/m2 once every 3 weeks. Because it is extremely corrosive to soft tissue, catheter placement should be meticulous. The calculated dose should be mixed with normal saline or 5% dextrose in water and dripped intravenously over 20 to 30 minutes.
Bleomycin
Mechanism of action
Bleomycin is an antitumor antibiotic derived from a strain of Streptomyces first isolated from the soil of a Japanese coal mine. Its cytotoxic effect is mediated by DNA binding and fragmentation, with single- and double-strand breaks. Interestingly, bleomycin seems to be more damaging to nonproliferating cells than to those actively proliferating. It has a unique lung toxicity in most animal species studied.
Spectrum of activity
Bleomycin is most effective for treatment of squamous cell carcinoma in cats and dogs;43 remissions are usually partial and of short duration, however. Recently, impressive results were obtained with intralesional injections of bleomycin into acanthomatous epulides in three dogs.44 These benign oral tumors are generally treated with surgical removal, sometimes involving a partial mandibulectomy or maxillectomy. The tumors were markedly smaller after three weekly bleomycin injections and had disappeared by 8 to 10 injections. No recurrence was noted in any of the cases during the follow-up periods, which ranged from 1 to 2 years. Bleomycin is also effective in humans as a sclerosing agent in the treatment of malignant pleural effusion, but there are no reports of this use in dogs or cats.
Preparations
Bleomycin is given intravenously or by intralesional or intracavitary injection; it is supplied in 15-mg or 30-mg vials.
Side effects
Unlike the other antitumor antibiotics, myelosuppression is unlikely with bleomycin. Chronic administration of bleomycin to dogs every other day for more than 8 months resulted in the development of a pneumonitis, which progressed to pulmonary fibrosis.45 The earliest symptom was dyspnea, with patchy opacities of the lung fields noted on radiographs. Microscopic changes included squamous metaplasia of the bronchiolar epithelium, fibrinous edema, and a diffuse interstitial fibrosis. Cutaneous ulceration and loss of nails also occurred in these dogs. Because bleomycin in clinical patients is principally used for short-term palliation of tumors, none of these chronic changes is likely to occur in clinical patients.
Mitoxantrone
Mechanism of action
Mitoxantrone is a derivative of anthracene and is related to doxorubicin and daunorubicin. It intercalates into DNA and causes cross-linking, with inhibition of both DNA and RNA synthesis. It is cell cycle specific but phase nonspecific.
Spectrum of activity
Partial and complete remissions have been reported when mitoxantrone is used as a solitary chemotherapeutic agent in lymphoma.46 Because the drug is very expensive, it is not generally used for induction or maintenance therapy. Its principal use is in lymphomas in which the tumor cells are resistant to other drugs; a response rate of 26% can be expected.47 Rare partial remissions and even rarer complete remissions are associated with administration of mitoxantrone to dogs and cats with various carcinomas and sarcomas, but the use of this drug in tumors other than lymphoma has been generally disappointing.
Side effects
Side effects of mitoxantrone administration are mild to moderate gastrointestinal toxicity and myelosuppression. Although the myelosuppression associated with mitoxantrone is generally not marked, some dogs and cats will develop dangerously low neutrophil and platelet counts; for this reason it may be prudent to begin treatment at the lower end of the dose range, with gradual escalation of the dose as treatment proceeds. Extravasation of the drug may result in severe local reactions, including ulceration and cellulitis. Although mitoxantrone is a relative of doxorubicin, its cardiotoxicity in dogs appears to be much less; no clinical evidence of cardiac effects was noted in a study of mitoxantrone administration in 129 dogs with different malignancies.48 However, because mitoxantrone does induce both acute and chronic congestive heart failure in humans, it probably is not a good choice for dogs or cats in which the maximum safe dose of doxorubicin has been reached or in patients with preexisting cardiac disease. A blue-green color may be noted in the sclera and urine of treated animals after therapy.
Antimetabolites
Methotrexate
Mechanism of action
Methotrexate is one of the antimetabolites, which as a class act as structural antagonists of normal metabolites or as cofactors of nucleic acids, generally having their greatest effect on cells in the S phase of the cell cycle. Methotrexate exerts its cytotoxic effect by competing for a binding site on the enzyme dihydrofolate reductase. This reversible binding prevents the synthesis of folate, which is important in production of the purine nucleotides and thymidine. An “antidote” for methotrexate cytotoxicity is leucovorin (citrovorum factor), which provides folate for biochemical activity in the cell. Methotrexate is principally eliminated in urine; in humans 80% to 90% of the administered dose is excreted unchanged in the urine within 24 hours. Assessment of renal function is important before administration of methotrexate, and dose modification in patients with compromised renal function may be necessary to prevent toxicity caused by delayed drug clearance. Because the antimetabolites have a short half-life in the body, they are most effective when given by constant-rate infusion, thus killing cells as they enter the S phase; however, a protocol for safe and effective constant-rate infusion of methotrexate has not been published for the dog.
Spectrum of activity
Although methotrexate has been widely used in human oncology, often at very high doses with “leucovorin rescue,” it has found only limited use in veterinary medicine as a part of combination protocols for lymphoma.
Preparations
Methotrexate sodium may be administered intramuscularly, subcutaneously, or intravenously; the product for injection is available in 20-mg, 50-mg, 100-mg, 200-mg, 250-mg, and 1-g vials. For oral administration it is supplied as 2.5-mg scored tablets. Leucovorin calcium is available in vials of 100 mg for parenteral administration as well as 5- and 15-mg tablets for oral administration.
Side effects
Gastrointestinal side effects are the most important toxicities produced by methotrexate, with nausea occurring commonly. Oral ulceration and diarrhea may also be seen, and methotrexate should be used with great caution or not at all in patients with ulcerative colitis. Myelosuppression is mild at low-dosage ranges. With long-term, low-dose therapy, hepatic dysfunction is a significant problem in humans, and methotrexate hepatotoxicity has been reported in the dog.49 Because nonsteroidal antiinflammatory drugs and aspirin may decrease renal excretion of methotrexate and thus increase its toxicity, these drugs should not be given along with methotrexate. Concurrent administration of methotrexate with a trimethoprin–sulfa antibiotic would be likely to lead to severe folate deficiency and therefore increase the severity of myelosuppression.
Dosing regimen
The oral dose of methotrexate is 2.5 mg/m2 given daily for 5 days, followed by a 2-day rest period. This is repeated weekly until remission is achieved; 10 mg/m2 given twice weekly followed by a 7-day rest period would be another acceptable protocol. Toxic hematopoietic effects may be reversed by 6 to 12 mg of leucovorin given subcutaneously four times a day for 4 doses.
Cytosine Arabinoside (Cytarabine, ara-C)
Mechanism of action
Cytosine arabinoside is highly specific for the S phase of the cell cycle, and its effectiveness is therefore dependent on maintaining constant drug levels; constant-rate infusion or frequent, closely-spaced doses are necessary for successful treatment of tumors using this drug. Cytosine arabinoside is transported into the cell and metabolized to 5′-triphosphate ara-C, which inhibits DNA polymerase. The metabolite is then incorporated into DNA, preventing templating from DNA and inhibiting DNA repair. Cytosine arabinoside is one of the few chemotherapeutic drugs that crosses the blood–brain barrier easily, and it can therefore be used to treat CNS lymphoma, as well as to kill leukemic cells in the cerebrospinal fluid.
Spectrum of activity
In veterinary oncology cytosine arabinoside is generally used in combination protocols for treatment of canine and feline lymphoma. It may also be used to treat acute leukemias of both lymphoid and nonlymphoid types.50
Preparations
Cytosine arabinoside is available for intravenous or subcutaneous injection in vials containing 100 mg, 500 mg, 1 g, or 2 g.
Side effects
Because of its specificity for cycling cells in the S phase, cytosine arabinoside will usually cause myelosuppression, with the degree of myelosuppression increasing with the frequency and duration of administration. When large intravenous doses are given by bolus injection intravenously rather than by infusion, nausea and vomiting are common.
Dosing regimen
For lymphoma single-agent cytosine arabinoside may be given at a dose of 600 mg/m2 intravenously once a week; lower doses of 200 to 300 mg/m2 weekly should be used if cytosine arabinoside is part of a combination drug protocol. For treatment of acute leukemias, 100 mg/m2/day given by constant-rate infusion or divided into 4 daily subcutaneous injections repeated for 5 days will produce the greatest response against leukemic cells. It is important to note that cytosine arabinoside, especially when used with doxorubicin, is extremely effective in clearing the bone marrow of tumor cells in leukemia patients. In general, patients will have a period of severe bone marrow aplasia for 7 to 21 days after the cycle is completed, often with neutrophil numbers less than 1000/μL and platelet counts less than 50,000/μL. Infection or hemorrhage may ensue during this period, and treatment with recombinant human G-CSF should ideally be used daily along with chemotherapy.
5-Fluorouracil
Mechanism of action
5-Fluorouracil is a pyrimidine analog that exerts its cytotoxic effect by inhibiting thymidylate synthetase and thus DNA synthesis and, to a lesser extent, RNA synthesis. The cytotoxic effects of 5-fluorouracil are greatest on cells in the G1 and S phases; with longer periods of exposure to the drug, cells in other phases of the cell cycle may also be killed. Because the drug is erratically absorbed from the gastrointestinal tract, it is generally given intravenously. It is also available as a topical cream.
Spectrum of activity
In humans 5-fluorouracil is the drug of choice for gastrointestinal carcinomas; it is effective in the palliative management of carcinoma of the colon, rectum, stomach, and pancreas. In the dog and cat, however, it has found limited usage because of neurotoxicity.
Preparations
For injection 5-fluorouracil is available in 500-mg vials. For topical use it is supplied in 25-g tubes.
Side effects
5-Fluorouracil treatment in dogs is often accompanied by CNS reactions (behavior changes such as barking incessantly, running in circles, aggressiveness)51-53; continuing administration of the drug in the face of such neurologic signs may lead to seizures and death.54 Mild myelosuppression and nausea are sometimes noted. Stomatitis and mucositis resembling pemphigus vulgaris may be seen in dogs receiving several weeks of treatment. Because unprovoked rage, extreme dementia, and sudden death may occur in cats treated with 5-fluorouracil, it should not be used in this species.55
Dosing regimen
5-Fluorouracil may be given intravenously at 150 to 200 mg/m2 for 3 days, then 100 mg/m2 on the fifth, seventh, and ninth days. No drug is given on the fourth, sixth, and eighth days. Blood count should be monitored at the end of the cycle and before the next cycle begins. If gastrointestinal signs, stomatitis, neurologic signs, or falling white blood cell count (less than 4000/μL) are noted, the drug should be discontinued. Generally, cycles of 5-fluorouracil are repeated monthly. In the dog 5-fluorouracil is useful for small skin carcinomas or solar keratosis as a topical cream. This is applied twice daily until there is an erosive inflammatory response with ulceration (usually 2 to 4 months), at which time use of the drug should be stopped. Healing may take several months after the topical treatment is discontinued. Owners should wear gloves while administering the cream. 5-Fluorouracil should not be used in cats.
Hydroxyurea
Mechanism of action
Hydroxyurea inhibits ribonucleotide reductase, leading to depletion of essential DNA precursors; cells accumulate in the S phase of the cell cycle.
Spectrum of activity
Hydroxyurea is used for the palliative treatment of chronic myelogenous leukemia,56 eosinophilic leukemia–hypereosinophilic syndrome in cats,57 and basophilic leukemia in dogs.58 It is also effective for management of polycythemia vera in the dog and cat.59
Side effects
Side effects of hydroxyurea are generally mild and well tolerated and include nausea and myelosuppression. In the dog loss of toenails may occur with chronic hydroxyurea administration, and a seborrhea sicca–like syndrome may be noted.
Dosage regimen
Hydroxyurea is given orally at a dose of 35 to 50 mg/kg once daily for 7-10 days, then every other day until remission. After remission is obtained (leukemic cell counts are reduced in leukemia patients, or packed cell volume is normal in patients with polycythemia vera; neutrophil and platelet counts are in the normal range), a dose of hydroxyurea is determined that will maintain remission. In some patients daily administration of hydroxyurea will continue to be necessary, but at a lower dose—20 mg/kg/day, for example. In other patients administration of a higher dose (such as 50 to 75 mg/kg) twice weekly will be adequate. The dose of hydroxyurea is titrated to the patient’s CBC results.
Platinum Drugs
Cisplatin
Mechanism of action
Cisplatin is a very useful drug in human and veterinary oncology. Its complete chemical name is cis-dichlorodiammineplatinum (DDP), reflecting the fact that it is formed by platinum surrounded by chlorine and ammonia atoms in the cis position of the horizontal plane. Its cytotoxic effects are thought to be due to alkylation of DNA.
Spectrum of activity
Cisplatin has been shown to produce objective responses in many types of carcinomas in the dog.60-63 Administration of cisplatin as an adjuvant agent with amputation in canine osteosarcoma has produced significantly longer survival times than seen with amputation alone.64,65 Complete remission has also been seen in dogs with metastatic seminoma. Intracavitary cisplatin has resulted in complete and durable palliation of pleural effusion resulting from mesothelioma and carcinomatosis of unknown origin66; intraperitoneal treatment with cisplatin for patients with carcinomatosis caused by ovarian carcinoma would also be reasonable.
Side effects
Treatment of cats with cis-platinum is contraindicated; dyspnea and death with pulmonary edema occur within 48 and 96 hours after cisplatin administration, even with only one treatment.67 Cisplatin is extremely nephrotoxic in the dog, especially if prehydration is not performed with treatment. Before each treatment, blood urea nitrogen (BUN) and creatinine evaluations and a urinalysis should be performed; elevation of BUN and creatinine in the face of a dilute urine should signal the onset of renal toxicity, and additional treatments of cisplatin should not be given. Carboplatin, which is much less nephrotoxic, may be substituted for cisplatin.
Acute gastrointestinal toxicosis with nausea, anorexia, and vomiting is common after cisplatin administration, usually beginning 2 to 4 hours after treatment (most dogs will vomit for less than 6 hours); the severity of the emesis can be decreased with administration of butorphanol or ondansetron. Hypomagnesemia, hypocalcemia, hyponatremia, hypokalemia, and hypophosphatemia may occur after repeated doses of cisplatin, probably as a result of renal tubular damage. Myelosuppression is generally mild but lengthy, with a double neutrophil nadir at 6 and 15 days after treatment. A CBC should be performed before administration of each cisplatin treatment; neutropenia may persist as long as 28 days after a single treatment, causing delay in the administration of the next course of therapy.
Dosing regimen
The dose for cisplatin in dogs is 60 to 70 mg/m2 given once every 21 days. An antiemetic such as butorphanol (0.4 mg/kg) is generally given before beginning the treatment; another dose may be given 2 hours later if vomiting becomes a problem. Intravenous saline solution should be given to prehydrate the patient at the rate of 25 mL/kg per hour for 3 hours (18.3 mL/kg per hour for 4 hours in dogs that might become volume overloaded), followed by a 20-minute intravenous infusion of cisplatin mixed in saline. This is followed by additional fluids given for an additional hour (2 hours for a heart failure patient because the fluid rate is lower) at the same rate. Needles or intravenous sets containing aluminum parts that may come into contact with cisplatin should not be used for preparation or administration because a precipitate will form, causing loss of potency. For intracavitary treatment of mesothelioma or carcinomatosis, the animal is prehydrated with intravenous saline, as previously described. For intraperitoneal therapy 50 mg/m2 of cisplatin is diluted in 0.9% NaCl to a total volume of 1 L/m2; for intrapleural delivery the dose is 250 mg/m2. The solution is warmed to body temperature and instilled through an aseptically placed catheter over 15 minutes, and posttherapy hydration is performed in the manner previously described. Treatments are repeated once every 4 weeks as needed to maintain remission. Cisplatin must not be given to cats.
Carboplatin
Mechanism of action
Because of the effectiveness of cisplatin in the treatment of many tumors, great interest developed in the search for another platinum compound that would maintain the same level of cytotoxicity without being as toxic to the patient. Carboplatin was developed at Michigan State University to fulfill these requirements; it is similar to cisplatin in pharmacology and antitumor effects. The major route of elimination of carboplatin, like cisplatin, is renal excretion; however, carboplatin causes significantly less renal toxicity, so fluid diuresis before and after administration is not required.
Spectrum of activity
In the treatment of human cancers, carboplatin and cisplatin appear to share the same spectrum of activity; this is presumed to be the case also in the dog. Because of its very different level of toxicity compared with cisplatin, carboplatin is a logical alternative to cisplatin for patients with renal disease or in patients with cardiac disease, in which the large amount of fluids administered with cisplatin treatment might be dangerous. Carboplatin is also safe to use in cats, unlike cisplatin, and intralesional injections of a carboplatin–oil emulsion into squamous cell carcinomas of the nasal planum in cats have resulted in objective responses and apparent cures in some cats.68
Side effects
Carboplatin is significantly less nephrotoxic than cisplatin and only rarely produces nausea and vomiting. However, dose-dependent neutropenia and thrombocytopenia are common, and the myelosuppression produced by the drug may be prolonged. In general, carboplatin treatment should not be repeated until neutrophil and platelet counts are in the normal range. Toxicity in the cat is principally associated with myelosuppression, as in the dog.69 Although carboplatin has limited renal toxicity, concomitant treatment with aminoglycosides may result in enhanced kidney toxicity as well as hearing loss.
When carboplatin in purified sesame oil has been used to treat squamous cell carcinomas in cats intralesionally, systemic toxicosis was not observed in any of the cats. Plasma concentrations of carboplatin did not significantly increase during the course of treatment. Water in sesame oil emulsions have been shown to be effective carriers for intratumor administration of antineoplastic agents, preserving drug activity and enhancing concentration of drug locally by allowing slow release into tissues. This allows for intensification of carboplatin chemotherapy without dose-limiting adverse effects.
Dosing regimen
Carboplatin is given as a 15- to 20-minute intravenous infusion at a dose of 250 to 300 mg/m2 once every 3 to 4 weeks for dogs; unlike cisplatin, intravenous carboplatin is safe for cats at a dose of 150 to 200 mg/m2 once every 3 to 4 weeks. As with cisplatin, aluminum reacts with carboplatin, causing a precipitate; needles with aluminum parts should not be used for the preparation or administration of carboplatin.
For intralesional injection of the nasal planum in cats with squamous cell carcinoma, treatments should be done with the animal under general anesthesia because of the pain that the injection procedure is likely to produce. Carboplatin is prepared in a water–oil emulsion that includes 10 mg of carboplatin in 1 mL of water mixed with 2 mL of sterile, purified, medical-grade sesame oil; a viscous, yellowish liquid is created by this mixture. The emulsion is injected into the tumor and surrounding borders so that approximately 1.5 mg of carboplatin is injected per cubic centimeter of tumor tissue. Four weekly doses are given.
Miscellaneous Drugs
Receptor Tyrosine Kinase Inhibitors
Mechanism of action
A new class of drugs, receptor tyrosine kinase inhibitors (RTKIs), has now entered the veterinary market. Receptor tyrosine kinases are a family of receptors expressed on the surface of all cells. These receptors play a large role in normal cell signal transduction and, when functioning normally, are tight regulators of cellular growth and differentiation. The ligands for these receptors are typically growth factors that are secreted by the cells themselves, released from the extracelluar matrix, or are secreted by other cells in the vicinity.70 Once activated, these receptors work by phophorylating proteins on tyrosine residues using adenosine triphosphate (ATP) in the process. They can activate tyrosine residues on themselves or other proteins as part of the initiation of a cell signaling process that will eventually lead to alterations in gene transcription. Small molecule inhibitors such as the RTKIs inhibit the phosphorylation of tyrosine residues, thereby stopping the signal transduction process. In many neoplastic cells these receptors are overexpressed, mutated to be constitutively turned on, or both. In most cases these small molecule inhibitors competitively bind the ATP binding site on the receptor, preventing the binding of ATP that is necessary to drive the phosphorylation of tyrosine residues.70 Two RTKIs are currently available or will soon be available to the veterinary market: toceranib (Palladia) and masitinib (Kinavet) has been recently approved by the FDA and will be available soon in the United States.
Spectrum of activity
Toceranib has been shown to have activity against members of the split kinase family of RTKs and inhibits vascular endothelial growth factor receptor, platelet-derived growth factor receptor, and c-kit (CD117).71 This drug is thought to have antiangiogenic and antitumor effects. It is labeled for use against mast cell tumors in dogs; however, anecdotal reports suggest that it may be useful against other tumors as well (anal sac adenocarcinomas, osteosarcomas, and soft tissue sarcomas). Masitinib has been shown to have activity against c-kit and platelet-derived growth factor receptor. This drug has been approved in Europe for canine mast cell tumors and is currently undergoing approval by the Food and Drug Administration for use in the United States.
Preparations
Toceranib is available in 10-mg, 15-mg, and 50-mg oral tablets. Masitnib is available in 50-mg tablets, although smaller formulations may be available soon. Currently, toceranib is available only to board-certified veterinary pathologists; however, Pfizer plans to release the drug to general practitioners sometime in 2011.
Procarbazine
Mechanism of action
The mechanism of action of procarbazine is not clearly understood, although it is thought to work through DNA alkylation and methylation, thereby decreasing DNA and RNA synthesis. The drug is metabolized by the liver and excreted almost entirely in the urine.
Spectrum of action
Procarbazine is typically used as a part of the MOPP rescue protocol for lymphoma (see the section on mechlorethamine).
Preparations
Procarbazine is available in nonscored, unbreakable 50-mg tablets. It may be necessary to compound for smaller sizes in small dogs and cats.
L-Asparaginase, Pegaspargase
Mechanism of action
L-asparaginase, an enzyme derived from E. coli, exploits a qualitative biochemical defect found in some tumor cells. In acute lymphoid leukemia and lymphoma, most malignant cells depend on an extracellular source of asparagine for survival. Normal cells, however, are able to synthesize asparagine and thus are affected less by the rapid extracellular depletion of asparagine produced by L-asparaginase. Although most susceptible tumors respond with dramatic reduction in size with the first administration of L-asparaginase, drug resistance of these cells develops quickly; a population of tumor cells is selected for in which the enzyme asparagine synthetase is present, asparagine can be made intracellularly, and the tumor cells are therefore unaffected by the enzyme’s administration. Pegaspargase is modified from L-asparaginase by covalently conjugating monomethoxypolyethylene glycol to the enzyme, forming the active ingredient PEG-L-asparaginase; pegaspargase produces fewer hypersensitivity reactions with administration than does conventional L-asparaginase.
Spectrum of activity
L-asparaginase is principally useful in the treatment of lymphoma and lymphoid leukemia. Because hypersensitivity and drug resistance develop relatively rapidly, L-asparaginase is a useful agent for induction of remission or in relapsed lymphoid malignancies, but it should not be employed as part of a maintenance protocol.
Preparations
L-asparaginase is available in vials containing 10,000 IU for parenteral administration. Pegaspargase is supplied in vials containing 3750 IU.
Side effects
Because asparaginase is a foreign protein, severe allergic reactions may be seen on repeated administration. In humans this is a significant problem, and it is recommended that an intradermal skin test be performed if the drug is to be given repeatedly; in the dog, however, anaphylactoid reactions are rare.72 Extreme facial edema and swelling or pain at the site of injection have been noted in some dogs within 24 hours after L-asparaginase administration, however, presumably as a manifestation of an allergic reaction to the drug. Pegaspargase was developed in an attempt to decrease the allergic reactions associated with administration of the drug; it is conjugated with polyethylene glycol and is indicated when L-asparaginase therapy is necessary despite a hypersensitivity reaction to previous treatment. Studies with the polyethylene glycol–modified enzyme in dogs have indicated that it is also active against lymphoma.73 The necessity for its use in veterinary medicine is limited because of the comparative rarity of allergic drug reactions with the use of conventional L-asparaginase.
Side effects associated with the administration of L-asparaginase in the dog are quite rare. Hyperamylasemia occurs in some patients and may progress to acute necrotizing pancreatitis.74 L-asparaginase administration in humans causes a temporary but fairly dramatic inhibition in protein synthesis by the liver, resulting in reduced levels of clotting factors. Levels of antithrombin III and fibrinogen in dogs with lymphoma after L-asparaginase administration have not been found to be abnormal, however, and other clotting parameters were not significantly affected either.75,76 Clinically important bleeding or thrombosis may occur in the dog but is extremely rare.77 L-asparaginase deaminates extracellular asparagine to L-aspartic acid and ammonia. In patients with preexisting hepatic disease or significant liver function abnormalities related to tumor infiltration, treatment with L-asparaginase may result in a syndrome resembling ammonia encephalopathy, with confusion and stupor. If serum ammonia levels are found to be high in these patients, treatment with lactulose should be instituted until signs abate.
Dosing regimen
Dosage is 10,000 to 20,000 IU/m2 or 400 IU/kg (maximum dose is 10,000 IU) weekly or as part of a combination protocol. The drug may be given subcutaneously, intramuscularly, or by intravenous administration. If L-asparaginase is given intravenously, the drug should be given over a period of not less than 30 minutes through the side arm of an already running infusion of sodium chloride or 5% dextrose.
Piroxicam
Mechanism of action
Piroxicam is a nonsteroidal antiinflammatory agent that has antiinflammatory, analgesic, and antipyretic properties in animals; edema, erythema, and tissue proliferation can be inhibited by the administration of the drug. Piroxicam inhibits the generation of thromboxane B2 in the blood of dogs by more than 70%, and more than 50% inhibition was maintained in most of the dogs for 48 hours.78 The drug has also been reported to have antitumor activity in animal models and in metastatic tumors in humans. The exact mechanism for the role of piroxicam in cancer treatment is not established at this time, but it is unlikely that the effects can be attributed to a direct cytotoxic effect.79
Spectrum of activity
Piroxicam has produced objective responses in several types of carcinomas, including transitional cell carcinoma,80 squamous cell carcinoma, mammary adenocarcinoma, and pulmonary metastatic carcinoma.81 Its principal use is in palliation of transitional cell carcinoma of the urinary tract; relief of stranguria and hematuria often associated with transitional cell carcinoma may be seen for 4 to 11 months after the beginning of treatment.
Side effects
Serious gastrointestinal toxicity with mucosal ulceration and bleeding, sometimes with perforation, may occur with piroxicam administration, especially if the drug is given daily. If daily administration of piroxicam is necessary, concurrent misoprostol at a dose of 5 μg/kg orally thrice daily should be considered to prevent gastrointestinal ulceration. Nephrotoxicity with renal papillary necrosis has also been reported with higher doses.
Corticosteroids
Mechanism of action
Several glucocorticoid hormones are used in the treatment of patients with cancer. In increasing order of potency, these are hydrocortisone, prednisone, and dexamethasone. The antiinflammatory effects of these hormones may help control pain in patients with terminal disease. Reduction of edema in the CNS with primary brain tumors or brain metastases occurs, especially with dexamethasone; barrier permeability within the tumor is decreased, thus reducing the rate of edema formation.
Corticosteroids are effective in lymphoid tumors by producing a direct lymphocytotoxic effect, apparently binding to intracellular receptors and inducing apoptosis. However, a population of steroid-resistant tumor cells (possibly lacking steroid receptors) develops rapidly if the steroid is used as a single agent, usually within 3 to 4 months after treatment of lymphoma begins. Because glucocorticoids are transported out of the cell by the multidrug resistance gene product P-glycoprotein, remission may be shorter and more difficult to achieve with certain other chemotherapeutic agents after steroid resistance develops.82
Spectrum of activity
Corticosteroids are most useful for their direct cytotoxicity in the management of lymphomas, lymphoid leukemias, thymomas, and plasma cell tumors. They are also important in the symptomatic management of mast cell tumors, shrinking these tumors by decreasing edema and inflammation and by reducing the eosinophilic and neutrophilic infiltrate commonly seen in these tumors. Whether neoplastic mast cells are actually killed by corticosteroid administration has not been determined. Corticosteroid administration may produce a dramatic improvement in clinical signs when used in patients with intracranial and spinal cord neoplasms, relieving signs of compression temporarily. These hormones are also useful in relieving the general debility, fever (noninfectious), and anorexia of cancer. Because corticosteroids produce a kind of euphoria, their administration to animals with terminal metastatic disease may improve quality of life transiently, even though tumor growth is not inhibited by the drug.
Preparations
Prednisone and dexamethasone are available for oral and parenteral use in a variety of tablet and solution strengths. The drugs are also available in preparations for ophthalmic administration.
Side effects
Side effects associated with the high doses of corticosteroids used in cancer treatment are numerous. For most dog owners, polydipsia and polyuria are the side effects of lymphoma or mast cell tumor treatment that are most difficult to accept; methylprednisolone, although much more expensive, may produce less polydipsia and polyuria. Owners should also be warned of the ravenous appetite often associated with steroid administration in dogs and cats. Temporal muscle atrophy, gastrointestinal ulceration and perforation, impaired wound healing, endocrine alopecia, increased incidence of bacterial infections, acute necrotizing pancreatitis, and personality changes are all occasional side effects seen with steroid administration, especially in dogs. Owners should be warned not to discontinue steroid treatment suddenly if their pet has been receiving corticosteroids for longer than 2 weeks because the hypothalamic–pituitary–adrenal axis is probably suppressed at the high doses being given.
Dosing regimen
Steroids may be administered orally, subcutaneously, intramuscularly, or intravenously, depending on the patient’s condition. A conventional dose of prednisone for treatment of lymphoma would be 30 to 40 mg/m2 orally once daily through induction, then on alternate days during maintenance. Edema induced by intracranial or spinal neoplasia may be treated with prednisone at the aforementioned dose or with dexamethasone at 0.1 mg/kg twice daily.
Monoclonal Antibodies
In humans, monoclonal antibody (Mab) therapy has been used successfully to treat non-Hodgkin’s lymphoma (NHL) and colon cancer. Monoclonal antibodies such as rituximab (an anti-CD20 antibody) and bevicizumab (an anti-VEGF antibody) have been used successfully and are approved for use in human oncology. However, in the dog, these humanized antibodies do not have broad enough specificity to cross species. Two studies in the dog have evaluated the ability of rituximab to bind to canine CD20, a B-cell marker. However, in both studies, this antibody failed to bind CD20 and no tumor cell killing was identified.83,84 Canine CD20 is not similar enough to human CD20 for cross reaction. Likewise, a study evaluating the efficacy of bevacizumab in canine mast cell tumors failed to show down regulation of VEGF and failed to decrease proliferation in this cell line.85 Therefore, specific canine monoclonal antibodies for these receptors will have to be developed before these therapies will be made available for veterinary oncology.
Immunotherapy
Currently, only one FDA-approved anti-cancer immunotherapy is available for dogs, and no therapies are approved for cats. The Merial ONCEPT melanoma vaccine is the first DNA-based vaccine for canine cancer.
Mechanism of action
The canine melanoma vaccine uses a plasmid with DNA for a non-canine tyrosinase protein inserted. Tyrosinase is a protein found ubiquitously in melanocytes and functions in the packaging of melanin granules within the cells. The foreign tyrosinase produced by the vaccine is different enough from canine tyrosinase that it can break tolerance and be recognized by the dog’s immune system as a foreign protein inducing an active immune response.86,87 Cross-reactivity between the foreign tyrosinase response and canine tyrosinase can occur leading to destruction of neoplastic melanocytes.
Spectrum of activity
This drug is only approved for canine melanoma in the microscopic disease setting. Therefore, ideally, it should be combined with more local therapies such as radiation or surgery to delay or prevent the development of metastasis. This vaccine is often used in an extra-label fashion in the gross disease setting; however, long-term survival analyses, and response rates are not available. There are anecdotal reports of the vaccines use in other species such as horses and cats with no noted adverse events, however, efficacy data in these species is lacking.
Preparations
The melanoma vaccine is available in single dose vials and is administered at a total volume of 0.4 mL per dog intradermally using the specific applicator, the Canine Transdermal Device. The vaccine is administered initially once every other week for a total of 4 doses at induction and then a booster is administered once every 6 months for the remainder of the dog’s life.
Other Immunotherapy
Many experimental treatments using immunotherapeutic principles are being explored including autologous T cell infusions, bone marrow transplantation, and dendritic cell vaccines. These therapies are only in the earliest stages of canine clinical trials and will likely not be commercially available for several years to come.
SUMMARY
Chemotherapeutic drugs can become common place in any general practice as long as the proper safety and handling precautions are instituted. These drugs can be dangerous to the veterinary hospital staff as well and the patient if handled or administered incorrectly. No one should ever administer a chemotherapy drug that they are uncomfortable handling for any reason. These drugs have varied clinical uses and side effects and a good understanding of each and every drug is necessary before administration. That being said, clinicians at veterinary hospitals who do not administer chemotherapy still need a basic knowledge of the side effects and their management for patients whom they have referred for chemotherapy elsewhere. Many clients will come to their general practitioner first for these conditions. Additionally, clients often seek advice from a trusted general practitioner regarding their options about cancer treatment in animals. The decision to treat their pet is often an emotional one, and a good understanding of the drugs and therapies that can be offered to them will put an owner at ease.
Cancer chemotherapy is a field that is constantly changing. New information is available almost daily and new drugs are approved every year. Indeed, cancer therapy is one of the most dynamic research fields. It is recommended that a practitioner who commonly uses chemotherapy frequently review the literature for new administration techniques, new drugs, and new uses for conventional chemotherapy drugs.
1. Skipper H.E., Schabel F.M.Jr., Wilcox W.S. Experimental evaluation of potential anticancer agents: XII. On the criteria and kinetics associated with “curability” of experimental leukemias. Cancer Chemother Rep. 1964;35:1-111.
2. DeVita V.T. Principles of chemotherapy. In: DeVita V.T., Hellman S., Rosenberg S.A., editors. Cancer: principles and practice of oncology. Philadelphia: JB Lippincott; 1993:276-292.
3. Arrington K.A., Legendre A.M., Tabeling G.S., et al. Comparison of body surface area-based and weight-based dosage protocols for doxorubicin administration in dogs. Am J Vet Res. 1994;55:1587-1592.
4. Obradovich J.E., Ogilvie G.K., Cooper M.F., et al. Effect of increasing dosages of canine recombinant granulocyte colony-stimulating factor on neutrophil counts in normal dogs. Proc Vet Cancer Soc 10th Ann Conf. 5, 1990.
5. Fulton R., Gasper P.W., Ogilvie G.K., et al. Effect of recombinant human granulocyte colony-stimulating factor on hematopoiesis in normal cats. Exp Hematol. 1991;19:759-767.
6. Amantea M., Newman M.S., Sullivan T.M., et al. Relationship of dose intensity to the induction of palmar-plantar erythrodysesthesia by pegylated liposomal doxorubicin in dogs. Hum Exp Toxicol. 1999;18(1):17-26.
7. Vail D.M., Chun R., Thamm D., et al. Efficacy of pyridoxine to ameliorate the cutaneous toxicity associated with doxorubicin containing pegylated (Stealth) liposomes: a randomized, double-blind clinical trial using a canine model. Clin Cancer Res. 1998;4(6):1567-1571.
8. Swanson L.V. Potential hazards associated with low-dose exposure to antineoplastic agents. Part I. Evidence for concern. Compend Cont Educ Pract Vet. 1988;10:293-300.
9. Swanson L.V. Potential hazards associated with low-dose exposure to antineoplastic agents. Part II. Recommendations for minimizing exposure. Compend Cont Educ Pract Vet. 1988;10:616-624.
10. Peterson J.L., Couto C.G., Hammer A.S., et al. Acute sterile hemorrhagic cystitis after a single intravenous administration of cyclophosphamide in three dogs. J Am Vet Med Assoc. 1992;201:1572-1574.
11. Trevor P.B., Saunders G.K., Waldron D.R., et al. Metastatic extramedullary plasmacytoma of the colon and rectum in a dog. J Am Vet Med Assoc. 1993;203:406-409.
12. MacEwen E.G., Hurvitz A., Hayes A. Hyperviscosity syndrome associated with lymphocytic leukemia in three dogs. J Am Vet Med Assoc. 1977;1977(170):1309-1312.
13. Dimski D.S., Cook J.R. Carmustine-induced partial remission of an astrocytoma in a dog. J Am Anim Hosp Assoc. 1990;26:179-182.
14. Fulton L.M., Steinberg H.S. Preliminary study of lomustine in the treatment of intracranial masses in dogs following localization by imaging techniques. Semin Vet Med Surg. 1990;5:241-245.
15. Van Vechten M., Helfand S.C., Jeglum K.A. Treatment of relapsed canine lymphoma with doxorubicin and dacarbazine. J Vet Intern Med. 1990;4:187-191.
16. Hahn K.A. Vincristine sulfate as single-agent chemotherapy in a dog and a cat with malignant neoplasms. J Am Vet Med Assoc. 1990;197:504-506.
17. Hammer A.S., Couto C.G., Filppi J., et al. Efficacy and toxicity of VAC chemotherapy (vincristine, doxorubicin, and cyclophosphamide) in dogs with hemangiosarcoma. J Vet Intern Med. 1991;5:160-166.
18. Calvert C.A., Leifer C.E., MacEwen E.G. Vincristine for treatment of transmissible venereal tumor in the dog. J Am Vet Med Assoc. 1982;181:163-164.
19. Singh J., Rana J.S., Sood N., et al. Clinico-pathological studies on the effect of different antineoplastic chemotherapy regimens on transmissible venereal tumours in dogs. Vet Res Commun. 1996;20:71-81.
20. McCaw D.L., Miller M.A., Bergman P.J., et al. Vincristine therapy for mast cell tumors in dogs. J Vet Intern Med. 1997;11:375-378.
21. Vickery K.R., Wilson H., Vail D.M., Thamm D.H. Dose-escalating vinblastine for the treatment of canine mast cell tumour. Vet Comp Oncol. 2008;6(2):111-119.
22. Hahn K.A., Fletcher C.M., Legendre A.M. Marked neutropenia in five tumor-bearing cats one week following single-agent vincristine sulfate chemotherapy. Vet Clin Pathol. 1996;25:121-123.
23. Todd G.C., Griffing W.J., Gibson W.R., et al. Animal models for the comparative assessment of neurotoxicity following repeated administration of vinca alkaloids. Cancer Treat Rep. 1979;63:35-41.
24. Cho E.S., Lowndes H.E., Goldstein B.D. Neurotoxicology of vincristine in the cat. Morphological study. Arch Toxicol. 1983;52:83-90.
25. Hamilton T.A., Cook J.R., Braund K.G., et al. Vincristine-induced peripheral neuropathy in a dog. J Am Vet Med Assoc. 1991;198:635-638.
26. Poirier V.J., Hershey A.E., Burgess K.E., et al. Efficacy and toxicity of paclitaxel (Taxol) for the treatment of canine malignant tumors. J Vet Intern Med. 2004;18(2):219-222.
27. Simon D., Schoenrock D., Baumgärtner W., Nolte I. Postoperative adjuvant treatment of invasive malignant mammary gland tumors in dogs with doxorubicin and docetaxel. J Vet Intern Med. 2006;20(5):1184-1190.
28. Hershey A.E., Kurzman I.D., Forrest L.J., et al. Inhalation chemotherapy for macroscopic primary or metastatic lung tumors: proof of principle using dogs with spontaneously occurring tumors as a model. Clin Cancer Res. 1999;5(9):2653-2659.
29. Berg J., Weinstein M.J., Springfield D.S. Results of surgery and doxorubicin chemotherapy in dogs with osteosarcoma. J Am Vet Med Assoc. 1995;206:1555-1560.
30. Moore A.S., Cotter S.M., Frimberger A.E., et al. A comparison of doxorubicin and COP for maintenance of remission in cats with lymphoma. J Vet Intern Med. 1996;10:372-375.
31. Valerius K.D., Ogilvie G.K., Mallinckrodt C.H., et al. Doxorubicin alone or in combination with asparaginase, followed by cyclophosphamide, vincristine, and prednisone for treatment of multicentric lymphoma in dogs: 121 cases (1987-1995). J Am Vet Med Assoc. 1997;210:512-516.
32. Ogilvie G.K., Reynolds H.A., Richardson R.C., et al. Phase II evaluation of doxorubicin for treatment of various canine neoplasms. J Am Vet Med Assoc. 1989;195:1580-1583.
33. Jeglum K.A., Wheareat A. Chemotherapy of canine thyroid carcinoma. Compend Contin Educ Pract Vet. 1983;5:96-98.
34. Sorenmo K.U., Jeglum K.A., Helfand S.C. Chemotherapy of canine hemangiosarcoma with doxorubicin and cyclophosphamide. J Vet Intern Med. 1993;7:370-376.
35. Mauldin G.E., Fox P.R., Patnaik A.K., et al. Doxorubicin-induced cardiotoxicosis. J Vet Intern Med. 1992;6:82-88.
36. Lipshultz S.E., Colan S.D., Gelber R.D., et al. Late cardiac effects of doxorubicin therapy for acute lymphoblastic leukemia in childhood. New Eng J Med. 1991;324:808-815.
37. Imondi A.R., Torre P.D., Mazue G., et al. Dose-response relationship of dezrazoxane for prevention of doxorubicin-induced cardiotoxicity in mice, rats, and dogs. Cancer Res. 1996;56:4200-4204.
38. O’Keefe D.A., Sisson D.D., Gelberg H.B., et al. Systemic toxicity associated with doxorubicin administration in cats. J Vet Intern Med. 1993;7:309-317.
39. Cotter S.M., Kanki P.J., Simon M. Renal disease in five tumor-bearing cats treated with Adriamycin. J Am Anim Hosp Assoc. 1985;21:405-409.
40. Vonderhaar M.A., Morrison W.B., Glickman N.W., et al. Comparison of efficacy of doxorubicin and epirubicin as single agent therapy for canine multicentric malignant lymphoma. Proc Vet Canc Soc Ann Mtg. 1993;13:46.
41. Moore A.S., Ruslander D., Cotter S.M., et al. Efficacy of, and toxicoses associated with, oral idarubicin administration in cats with neoplasia. J Am Vet Med Assoc. 1995;206:1550-1554.
42. Hammer A.S., Couto C.G., Ayl R.D., Shank K.A. Treatment of tumor-bearing dogs with actinomycin D. J Vet Intern Med. 1994;8:236-239.
43. Buhles W.C.Jr., Theilen G.H. Preliminary evaluation of bleomycin in feline and canine squamous cell carcinomas. Am J Vet Res. 1973;34:289-291.
44. Yoshida K., Watarai Y., Sakai Y., et al. The effect of intralesional bleomycin on canine acanthomatous epulis. J Am Anim Hosp Assoc. 1998;34:457-461.
45. Schaeppi U., Phelan R., Stadnicki S.W., et al. Pulmonary fibrosis following multiple treatment with bleomycin (NCS-125066) in dogs. Cancer Chemother Rep. 1974;58:301-310.
46. Ogilvie G.K., Obradovich J.E., Elmslie R.E., et al. Efficacy of mitoxantrone against various neoplasms in dogs. J Am Vet Med Assoc. 1991;23:587-596.
47. Moore A.S., Ogilvie G.K., Ruslander D., et al. Evaluation of mitoxantrone for treatment of lymphoma in dogs. J Am Vet Med Assoc. 1994;205:1903-1905.
48. Ogilvie G.K., Obradovich J.E., Elmslie R.E., et al. Toxicoses associated with administration of mitoxantrone to dogs with malignant tumors. J Am Vet Med Assoc. 1991;198:1613-1617.
49. Pond S.M. Effects on the liver of chemicals encountered in the workplace. West J Med. 1982;137(6):506-514.
50. Hamilton T.A., Morrison W.B., DeNicola D.B. Cytosine arabinoside chemotherapy for acute megakaryocytic leukemia in a cat. J Am Vet Med Assoc. 1991;199:359-361.
51. Harvey H.J., MacEwen E.G., Hayes A.A. Neurotoxicosis associated with use of 5-fluorouracil in five dogs and one cat. J Am Vet Med Assoc. 1977;171:277-278.
52. Hammer A.S., Carothers M.A., Harris C.L., et al. Unexpected neurotoxicity in dogs receiving a cyclophosphamide, dactinomycin, and 5-fluorouracil chemotherapy protocol. J Vet Intern Med. 1994;8:240-243.
53. Dorman D.C., Coddington K.A. Richardson RC: 5-Fluorouracil toxicosis in the dog. J Vet Intern Med. 1990;4:254-257.
54. Okeda R., Kimura S., Toizumi S., et al. Neuropathologic study on chronic neurotoxicity of 5-fluorouracil and its masked compounds in dogs. Acta Neuropath. 1984;63:334-343.
55. Theilen G. Adverse effect from use of 5% fluorouracil. J Am Vet Med Assoc. 1987;191:276.
56. Leifer C.E., Matus R.E., Patnaik A.K., et al. Chronic myelogenous leukemia in the dog. J Am Vet Med Assoc. 1983;183:686-698.
57. Hamilton T.A. The leukemias. In: Morrison W.B., editor. Cancer in dogs and cats. Baltimore: Williams & Wilkins; 1998:721-729.
58. MacEwen E.G., Dragner F.H., McClelland A.J., et al. Treatment of basophilic leukemia in a dog. J Am Vet Med Assoc. 1975;166:376-380.
59. Peterson M.E., Randolph J.F. Diagnosis of canine primary polycythemia and management with hydroxyurea. J Am Vet Med Assoc. 1982;180:415-418.
60. Fineman L.S., Hamilton T.A., de Gortari A., et al. Cisplatin chemotherapy for treatment of thyroid carcinoma in dogs: 13 cases. J Am Anim Hosp Assoc. 1998;34:109-112.
61. Himsel C.A., Richardson R.C., Craig J.A. Cisplatin chemotherapy for metastatic squamous cell carcinoma in two dogs. J Am Vet Med Assoc. 1986;189:1575-1578.
62. Shapiro W., Kitchell B.E., Fossum T.W., et al. Cisplatin for treatment of transitional cell and squamous cell carcinomas in dogs. J Am Vet Med Assoc. 1988;193:1530-1533.
63. Knapp D.W., Richardson R.C., Bonney P.L., et al. Cisplatin therapy in 41 dogs with malignant tumors. J Vet Intern Med. 1988;2:41-46.
64. Thompson J.P., Fugent M.J. Evaluation of survival times after limb amputation, with and without subsequent administration of cisplatin, for treatment of appendicular osteosarcoma in dogs: 30 cases (1979-1990). J Am Vet Med Assoc. 1992;200:531-533.
65. Kraegel S.A., Madewell B.R., Simonsen E., et al. Osteogenic sarcoma and cisplatin chemotherapy in dogs: 16 cases (1986-1989). J Am Vet Med Assoc. 1991;199:1057-1059.
66. Moore A.S., Kirk C., Carcona A. Intracavitary cisplatin chemotherapy experience with six dogs. J Vet Intern Med. 1991;5:227-231.
67. Knapp D.W., Richardson R.C., DeNicola D.B., et al. Cisplatin toxicity in cats. J Vet Intern Med. 1987;1:29-35.
68. Theon A.P., Van Vechten M.K., Madewell B.R. Intratumoral administration of carboplatin for treatment of squamous cell carcinomas of the nasal plane in cats. Am J Vet Res. 1996;57:205-210.
69. Hahn K.A., McEntee M.F., Daniel G.B., et al. Hematologic and systemic toxicoses associated with carboplatin administration in cats. Am J Vet Res. 1997;58:677-679.
70. London C.A. Tyrosine kinase inhibitors in veterinary medicine. Top Companion Anim Med. 2009;24(3):106-112.
71. London C.A., Malpas P.B., Wood-Follis S.L., et al. Multi-center, placebo-controlled, double-blind, randomized study of oral toceranib phosphate (SU11654), a receptor tyrosine kinase inhibitor, for the treatment of dogs with recurrent (either local or distant) mast cell tumor following surgical excision. Clin Cancer Res. 2009;15(11):3856-3865.
72. Ogilvie G.K., Atwater S.W., Ciekot P.A., et al. Prevalence of anaphylaxis associated with the intramuscular administration of L-asparaginase to 81 dogs with cancer: 1989-1991. J Am Anim Hosp Assoc. 1994;30:62-65.
73. MacEwen E.G., Rosenthal R.C., Fox L.E., et al. Evaluation of L-asparaginase: polyethylene glycol conjugate versus native L-asparaginase combined with chemotherapy. A randomized double-blind study in canine lymphoma. J Vet Intern Med. 1992;6:230-234.
74. Hansen W.E., Schulz G. The effect of dietary fiber on pancreatic amylase activity in vitro. Hepatogastroenterol. 1982;29(4):157-160.
75. Mandell C. Antithrombin III concentrations associated with L-asparaginase administration. Vet Clin Pathol. 1992;21:68-70.
76. Rogers K.S., Barton C.L., Benson P.A., et al. Effects of single-dose L-asparaginase on coagulation values in healthy dogs and dogs with lymphoma. Am J Vet Res. 1992;53:580-584.
77. Swanson J.F., Morgan S., Green R.A., et al. Cerebral thrombosis and hemorrhage in association with L-asparaginase administration. J Am Anim Hosp Assoc. 1986;22:749-755.
78. Galbraith E.A., McKellar Q.A. Pharmacokinetics and pharmacodynamics of piroxicam in dogs. Vet Rec. 1991;128:561-565.
79. Knapp D.W., Chan T.C., Kuczek T., et al. Evaluation of in vitro cytotoxicity of nonsteroidal anti-inflammatory drugs against canine tumor cells. Am J Vet Res. 1995;56:801-805.
80. Knapp D.W., Richardson R.C., Chan T.C., et al. Piroxicam therapy in 34 dogs with transitional cell carcinoma of the urinary bladder. J Vet Intern Med. 1994;8:273-278.
81. Knapp D.W., Richardson R.C., Bottoms G.D., et al. Phase I trial of piroxicam in 62 dogs bearing naturally occurring tumors. Cancer Chemother Pharmacol. 1992;29:214-218.
82 Price G., Page R., Fischer B., et al. Efficacy and toxicity of doxorubicin/cyclophosphamide maintenance therapy in dogs with multicentric lymphoma. J Vet Intern Med. 1991;5:259-262.
83. Impellizeri J.A., McKeever K.P., Crow S.E. The role of rituximab in the treatment of canine lymphoma: an ex vivo evaluation. Vet J. 2006;171(3):556-558.
84. Jubala C.M., Valli V.E., Getzy D.M., et al. CD20 expression in normal canine B cells and in canine non-Hodgkin lymphoma. Vet Pathol. 2005;42(4):468-476.
85. Rebuzzi L., Sonneck K., Gleixner K.V., et al. Detection of vascular endothelial growth factor (VEGF) and VEGF receptors Flt-1 and KDR in canine mastocytoma cells. Vet Immunol Immunopathol. 2007;115(3-4):320-333.
86. Bergman P.J., et al. Development of a xenogeneic DNA vaccine program for canine malignant melanoma at the Animal Medical Center. Vaccine. 2006;24:4582-4585.
87. Bergman P.J., et al. Long-term survival of dogs with advanced malignant melanoma after DNA vaccination with xenogeneic human tyrosinase: a phase I trial. Clin Cancer Res. 2003;9:1284-1290.