Chapter 28 Antiprotozoal natural products
Diseases caused by protozoa are responsible for considerable mortality and morbidity, especially in the developing world. Many plant species are used in the preparation of traditional medicines for the treatment of protozoal diseases and plants are the source of the clinically used antimalarial drugs quinine (Fig. 28.1 (1)) (from Cinchona spp.) and artemisinin (Fig. 28.3 (8)) (from Artemisia annua). Other examples of natural-product-derived antiprotozoal agents are the nitroimidazoles, which are based on the antibiotic azomycin (Fig. 28.1 (2)) produced by a species of Streptomyces that was collected on the Island of Réunion. Azomycin was found to be active against the protozoan Trichomonas vaginalis, the causative agent of trichomoniasis, and the synthetic analogue metronidazole was the first effective treatment for this disease. Later, it was found that the latter was also highly effective in the treatment of infections caused by anaerobic bacteria. A more recent example of a natural-product-derived antiprotozoal drug is the antimalarial atovaquone, which was derived from lapachol (Fig. 28.4 (14)), a naphthoquinone found in S. American species of the Bignoniaceae. Natural products have made a significant contribution to the chemotherapy of protozoal diseases and it is possible that the continued investigation of natural-product-derived compounds will provide new antiprotozoal drugs in the future. As shown in the following sections, many of the available antiprotozoal agents have serious limitations due to their toxicity and/or the development of drug-resistant parasites, so that new drugs are urgently needed.
DISEASES CAUSED BY PROTOZOA
Malaria
In the mid-1950s it was confidently anticipated that malaria would be eradicated, but by the 1990s the disease had reached epidemic proportions throughout the tropical world. The failure to eradicate malaria was due to a number of factors, including the emergence of malaria parasites resistant to the antimalarial chloroquine, resistance of the vector female Anopheline mosquitoes to insecticides such as DDT and the avoidance of insecticide use because of toxiciological and ecological considerations. It is estimated that between 1 and 2 million people, mostly children under the age of 5 years, die from the disease each year, and that some 300–800 million people contract malaria annually. Malaria caused by Plasmodium falciparum is the most serious type because it often proves fatal due to the complication of cerebral malaria unless prompt treatment is given. There is now widespread resistance of P. falciparum to chloroquine and to some other antimalarial drugs. P. vivax is also a common cause of malaria but it is usually sensitive to treatment with chloroquine (followed by a course of primaquine to eradicate parasites, which lie dormant in the liver and may later cause relapses—these do not occur with P. falciparum malaria). Recently, chloroquine-resistant strains of P. vivax have been reported. The other species of malaria parasite which (less commonly) infect man are P. malariae and P. ovale.
Trypanosomiasis
African sleeping sickness (African trypanosomiasis) is caused either by Trypanosoma brucei gambiense (W. African form) or by T. brucei rhodesiense (E. African form). The disease is transmitted by the tsetse flies (Glossina species) and initially causes a feverish illness. Later stages of the disease are characterized by effects on the central nervous system, including movement disorders and convulsions, excessive sleepiness and finally coma. About 50 million people live in areas where the disease is endemic and, in addition to the threat to humans, it is also a majorproblem for livestock. Current drug treatment is limited as suramin and pentamidine are only effective in the early stages of the disease while the arsenical drug melarsoprol, which is used in the late stages, is toxic. Treatment has improved with the development of eflornithine (α-difluoromethylornithine), an inhibitor of polyamine synthesis but this drug is not effective against the E. African form of the disease.
In S. America, Chagas’ disease results from infection with T. cruzi, which is transmitted by house bugs living in the cracks of mud-walled houses. Chagas’ disease causes heart failure and other complications; it is estimated that some 20 million people are infected with T. cruzi. Only the acute stages of the disease are amenable to treatment but the available drugs, nifurtimox and benznidazole, are poorly tolerated.
Leishmaniasis
Leishmaniasis affects more than 20 million people worldwide and is caused by various species of Leishmania, which are transmitted by female sand-flies of the genus Phlebotomus. In S. America, cutaneous leishmaniasis (infections of the skin and mucous membranes) is caused by L. mexicana and L. braziliensis; in the Old World it is caused by L. tropica and L. major. Another form of the disease, known as visceral leishmaniasis (kala azar), occurs in northern India and is caused by L. donovani, which infects the reticuloendothelial system; unless treated, this condition is rapidly fatal. Immunocompromised patients, such as those with AIDS, are susceptible to infection with L. infantum in Mediterranean countries. The antimonial drugs diamidines and amphotericin B are used in the treatment of severe forms of leishmaniasis but toxic effects are common and it is hoped that treatment will improve with the introduction of a new drug, miltefosine, which has given encouraging results in clinical trials.
Gastrointestinal diseases
Diarrhoeal disease is often the result of protozoal infections in the gastrointestinal tract. Giardiasis, caused by Giardia intestinalis (also known as G. duodenalis and G. lamblia) is thought to infect 200 million people each year and is responsible for an estimated 10,000 deaths. Amoebic dysentery is caused by Entameoba histolytica, which, if untreated, may give rise to serious complications including liver abscesses. There are some 42 million cases annually and an estimated 75,000 deaths. Both diseases are treatable with metronidazole, although this drug is poorly tolerated by some patients. As a result of the AIDS pandemic, the importance of Cryptosporidium parvum as a common cause of diarrhoea in both normal and immunocompromised patients has been recognized; currently there is no effective treatment for cryptosporidiosis.
METHODS OF INVESTIGATION
The study of antiprotozoal compounds from plants has required the development of bioassay techniques, especially in vitro methods that allow large numbers of plant extracts to be screened for activity against pathogenic species of protozoa. In vitro assays are particularly useful for bioassay-guided fractionation of plant extracts. It is not always possible to test against the species or stages of the lifecycle that actually infect man because they cannot be cultured or will not infect animal models; for example, in vitro tests against Trypanosoma spp. are often carried out using epimastigotes, which are found in the insect vector. An in vitro assay for activity against Plasmodium falciparum was developed in 1979 following the development of in vitro methods for the cultivation of this parasite, and it is not possible to infect animal models so that the most common in vivo antimalarial assays utilize the rodent malaria parasite P. berghei in mice. Brief descriptions of the antimalarial and antiamoebic tests are given here to illustrate some of the techniques that are employed.
Antimalarial assays
In vitro (antiplasmodial) assays
Plasmodium falciparum is cultured in human red blood cells in 96-well microtitre plates. The inhibition of parasite growth in the presence of drugs may be assessed by measuring the incorporation of [3H]-hypoxanthine into the parasite. More recently, a new method has been developed that does not require the use of radiolabelled compounds. Instead, parasite growth is assessed by measuring parasite lactate dehydrogenase (LDH) activity by adding a reagent containing an analogue of NAD, acetylpyridine adenine dinucleotide (APAD) and a tetrazolium compound. APAD is reduced by parasite LDH (but not by red cell LDH) and then the reduced APAD in turn reduces tetrazolium to give a blue colour, which is measured spectrophotometrically; the intensity of the colour is proportional to parasite growth.
In vivo assays
Two different tests are commonly employed utilizing P. berghei in mice. In the 4-day suppressive test (Peters’ test), mice are inoculated with red blood cells infected with P. berghei. The plant extract or compound under test is administered daily for 4 days, starting on the day of infection, and may be given orally or by subcutaneous or intraperitoneal injection. Different dose levels are administered to groups of five mice and, on the fifth day, a blood sample is taken from each mouse. Blood films are prepared and stained (e.g. using Giemsa’s stain) so that malaria parasites may be observed and counted microsopically. The percentage of parasitized red blood cells in the test groups and control group of mice are determined and the ED50 value (i.e. the dose of extract/compound that causes a 50% reduction in parasitaemia) is calculated. If any test animals die before the end of the assay, death is considered to be due to the toxicity of the substance under test.
The Rane test utilizes an alternative procedure in which mice are given a standard incoculation of P. berghei, which would normally be expected to kill the animals within 6 days. On day 4, a single dose of the extract/compound under test is given at a series of different dose levels. The test material is considered to be active if the mice survive for 12 days or more. The minimum effective dose is compared with the maximum tolerated dose (i.e. the dose that produces no more than one in five toxic deaths). In this way, a measure of the difference between the effective dose and the toxic dose is obtained.
Antiamoebic assays
In vitro assays
The development of axenic cultures of E. histolytica has enabled the development of in vitro assays. Before the development of axenic media it was only possible to grow amoebae in the presence of bacteria (polyxenic culture), which made interpretation of test results extremely difficult. E. histolytica is grown in 96-well microtitre plates in the presence of serial dilutions of extracts/compounds. Following a suitable incubation time, the growth of amoebae may be assessed by visual observation with a microscope. Alternatively, a colorimetric method may be used, in which the culture medium is removed leaving healthy amoebae attached to the bottom of the wells while dead amoebae are washed away. A measure of the number of amoebae remaining in the wells is obtained by fixing and staining the parasites. The quantity of stain taken up is proportional to the number of amoebae and is determined spectrophotometrically.
In vivo assays
Rats are used for the determination of activity against intestinal infections and hamsters are used for assessing activity against hepatic infections. E. histolytica is introduced into the caecum via the rectum and the intestine is examined for the presence of amoebae and ulceration. Liver infections are inititiated by injection of amoebae into the lobes. In practice, such in vivo tests are difficult to perform, are time consuming and are unpleasant for the animals.
MODES OF ACTION OF NATURAL ANTIPROTOZOAL AGENTS
The effectiveness of any chemotherapeutic agent is dependent upon a favourable therapeutic ratio, i.e. the drug must kill or inhibit the parasite but have little or no toxicity to the host. Although a large number of natural products have been shown to be able to inhibit the growth of one or more species of protozoa, very few have been shown to be selectively toxic to the parasite. Selectivity depends on differences in biochemistry between the parasite and the host, such that a drug can act on a biochemical target in the parasite that is either absent in, or significantly different from, that in the host. Marked differences in metabolism from that in mammalian cells have been found in some species of pathogenic protozoa. For example, Trypanosoma spp. are unique in that glycolysis takes place in an organelle known as the glycosome, rather than in the cytosol as in all other organisms. The unusual pathways found in the glycosome may provide viable biochemical targets. The following sections describe some of the biochemical differences between parasites and host cells that are exploited by antiprotozoal drugs. However, the modes of action of many natural products with antiprotozoal activities are, at present, unknown and it is possible that some of these may act on biochemical targets unique to protozoa.
EXAMPLES OF ANTIPROTOZOAL NATURAL PRODUCTS
ALKALOIDS
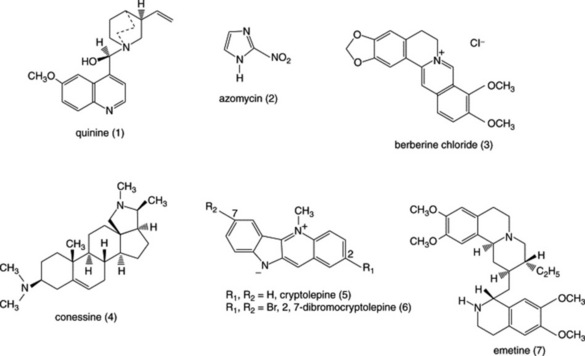
Berberine
This benzylisoquinoline alkaloid, common in members of the Menispermaceae, has been used clinically in the treatment of cutaneous leishmaniasis and has been shown to eliminate L. major amastigotes from macrophages, although it was less effective in animal models of cutaneous leishmaniasis. Berberine (Fig. 28.1 (3)) and a number of related alkaloids, including palmatine and jattrorhizine, have potent in vitro activities against P. falciparum but they have little activity against P. berghei in mice; by contrast, berberine has been reported to be effective against intestinal amoebiasis in mice but it is only weakly active against E. histolytica in vitro. These findings illustrate the limitations of in vitro tests for the prediction of activity in vivo, as compounds may need to be metabolically activated in vivo and thus may be inactive in in vitro tests. Conversely, drugs that are active in vitro might also be metabolized to inactive metabolites in vivo and lack of activity in vivo may also be due to problems related to the absorption and distribution of the drug.
Conessine
In India, the bark of Holarrhena pubescens (Apocynaceae) known as kurchi bark has been used traditionally for the treatment of amoebic dysentery. A number of steroidal alkaloids have been isolated from the bark and a few of these, including the major alkaloid conessine (Fig. 28.1 (4)) have been shown to have in vitro activity against E. histolytica.
Cryptolepine
A decoction of the roots of Cryptolepis sanguinolenta (Asclepiadaceae), a climbing plant, is used in West Africa for the treatment of malaria and various other infectious diseases. The indoloquinoline alkaloidcryptolepine (Fig. 28.1 (5)) is the main alkaloid present and this has potent in vitro activity against P. falciparum but it is also cytotoxic as a result of intercalation into DNA as well as inhibition of DNA synthesis and of topisomerase II. Cryptolepine is toxic to mice when given by intraperitoneal injection, but not toxic when given orally, and has little in vivo antimalarial activity when given by this latter route. The reasons for this appear to be related to poor absorption and/or metabolism to inactive metabolites. It has been shown that cryptolepine is oxidized by the liver enzyme aldehyde oxidase to form cryptolepine-11-one, a metabolite that is inactive against P. falciparum in vitro. A number of synthetic derivatives of cryptolepine have been made that have more potent activities against malaria parasites in vitro but that do not have the DNA-interacting properties of cryptolepine. One of these, 2,7-dibromocryptolepine (Fig. 28.1 (6)), has been shown to have promising in vivo activity against P. berghei in mice when given by intraperitoneal injection without causing toxic effects to the mice. The antiplasmodial mode of action of cryptolepine and its derivatives appears to involve the inhibition of β-haematin formation (see under quinine, below), but it appears that 2,7-dibromocryptolepine has another mode of action that explains its higher potency compared to cryptolepine. Recently, 2,7-dibromocryptolepine has been shown to have potent activity against T. brucei in vitro as well as activity in vivo.
Emetine
This alkaloid was discovered as a result of investigating Cephaelis ipecacuanha (Rubiaceae), a plant used by S. American Indians as a treatment for dysentery. Emetine (Fig. 28.1 (7)) is highly active against E. histolytica in vitro and is effective in the treatment of both hepatic and intestinal amoebiasis but has toxic effects, especially on the heart. It inhibits protein synthesis, which is probably responsible for its antiamoebic action and the toxic effects seen in man. The related (synthetic) compound dehydroemetine is less toxic, probably because it is eliminated from the body more rapidly than emetine.
Quinine
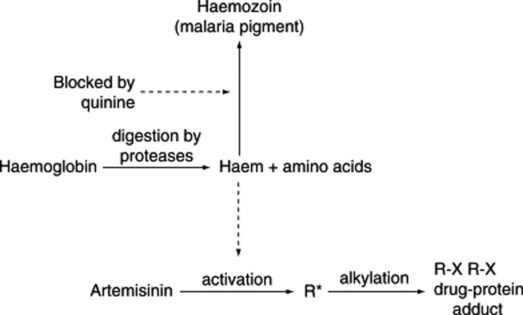
The barks of various species of Cinchona originating from S. America were used for the treatment of fevers including malaria from about 1630 until they were superseded by quinine (Fig. 28.1 (1)) (isolated from Cinchona bark in 1820). For more than a century, quinine was the only effective antimalarial available until the development of chloroquine and other synthetic antimalarials, following which the use of quinine declined. (Note: chloroquine was not, as is sometimes stated, derived from quinine but derived from products of the synthetic dyestuffs industry.) With the development of P. falciparum strains resistant to chloroquine and other antimalarials, there has been a resurgence in the use of quinine for the treatment of chloroquine-resistant malaria, although this may decline with the increasing use of the safer artemisinin derivatives. Although quinine is a relatively toxic drug, it has a selective action against Plasmodia due to differences between the host and the parasite; two distinct mechanisms are involved. First, the drug is able to accumulate in the parasite to concentrations higher than those in the host cells. Malaria parasites growing in red blood cells digest haemoglobin in an acid food vacuole; quinine, being a basic drug is concentrated in the acidic vacuole by an ‘ion trapping’ mechanism. Second, in the food vacuole haemoglobin is broken down into amino acids (which may be utilized by the parasite) leaving the haem ring as an unwanted residue. Haem is toxic and in mammalian cells it is broken down by the enzyme haem oxidase, but this enzyme is not present in malaria parasites. To detoxify haem, the parasite converts it into a polymeric substance, haemozoin, also known as malaria pigment. Quinine and some other antimalarials such as chloroquine and mefloquine, as well as cryptolepine (see above), bind to haem, thus preventing the formation of haemozoin; the haem–drug complex is toxic and causes parasite death (Fig. 28.2).
Other alkaloids
In addition to the above, many other alkaloids have been investigated for their antiprotozoal properties. Bisbenzylisoquinoline alkaloids have been isolated from a number of Menispermaceous plant species used traditionally for malaria treatment and some of them, e.g. tetrandrine and phaeanthine, have in vitro antiplasmodial activities; interestingly, these compounds were found to be more active against chloroquine-resistant than against chloroquine-sensitive strains. The above alkaloids have also been shown to reverse chloroquine-resistance in P. falciparum, an action that might be related to their calcium-channel-blocking activity, whereas others, such as gyrocarpine, daphnandrine and obaberine, have activities against promastigotes of Leishmania sp. The African medicinal plants Ancistrocladus abbreviatus (Ancistrocladaceae) and Triphyophylum peltatum (Dionchophyllaceae) contain naphthylisoquinoline alkaloids that have in vitro and in vivo antimalarial activity; dionchophylline C appears to be a promising lead for further investigations.
A number of alkaloids related to emetine have been shown to have antiprotozoal activities; two alkaloids isolated from Pogonopus tubulosus (Rubiaceae), cephaeline and tubulosine, showed potent in vitro antiplasmodial activities and tubulosine was active against malaria in mice. Several alkaloids isolated from Strychnos usambarensis were found to be active against E. histolytica, P. falciparum and Giardia intestinalis in vitro but in contrast to emetine they were less toxic to cells. An interesting series of 2-substituted quindoline alkaloids isolated from Galipea longifolia (Rutaceae), a species used in Bolivia to treat cutaneous leishmaniasis and fever, have been assessed for in vitro and in vivo antileishmanial activities. Chimanine D, (2-[1′,2′-transepoxypropyl]quinoline) was more potent than meglumine antimonate (Glucantime) against cutaneous disease caused by L. amazonensis and was effective in suppressing parasitaemia in mice with visceral leishmaniasis due to L. donovani. Another constituent, 2-n-pentylquinoline was not active against Leishmania spp., but has in vitro and in vivo antimalarial activity. The β-carboline alkaloid harmaline, a constituent of Peganum harmala and some related tryptamine derivatives, have been tested against L. amazonensis; harmaline has potent in vitro activity but α-ethyltryptamine was orally active in mice infected with the same organism. The related compound harmine was found to have activity against T. cruzi epimastigotes. Ellipticine, a constituent of Ochrosia elliptica, and a number of derivatives havebeen investigated for their effects against T. cruzi. These compounds interact with DNA but it appears that in trypanosomes the kinetoplast DNA is more susceptible than the nuclear DNA to these compounds and may therefore be a potential biochemical target. Several alkaloids of the acridone type have been shown to have in vitro antiplasmodial activity; atalaphillinine isolated from Atalantia monophylla (Rutaceae) was also active in mice infected with P. berghei. The aporphine alkaloid (−)-roemrefidine from Sparattanthelium amazonum (Hernandiaceae) also has in vitro antiplasmodial activity (against chloroquine-sensitive and chloroquine-resistant strains) and is active in vivo against P. berghei in mice.
Species of Alstonia (Apocynaceae) are widely used in traditional medicine for the treatment of malaria but the monoterpenoid indole alkaloids they contain have, with few exceptions been found to have little or no activity against malaria parasites. Those that have been shown to have in vitro antiplasmodial activity, such as villastonine and macralstonine (present in A. angustifolia), are dimeric alkaloids, whereas monomeric Alstonia alkaloids such as alstonerine, echitamine and pleiocarpamine are only weakly active. It is possible that the major alkaloids present in Alstonia species have other effects, e.g. antipyretic properties which may explain the widespread use of these species in malaria treatment. In this context it is interesting to note that the 4-quinazole derivative, febrifugine, found in the Chinese antimalarial plant Dichroa febrifuga (Saxifragaceae) is effective against malaria in mice and also enhances nitric oxide production in activated macrophages. It is suggested that the antimalarial activity of febrifugine is due to enhanced host defence mechanisms.
TERPENES
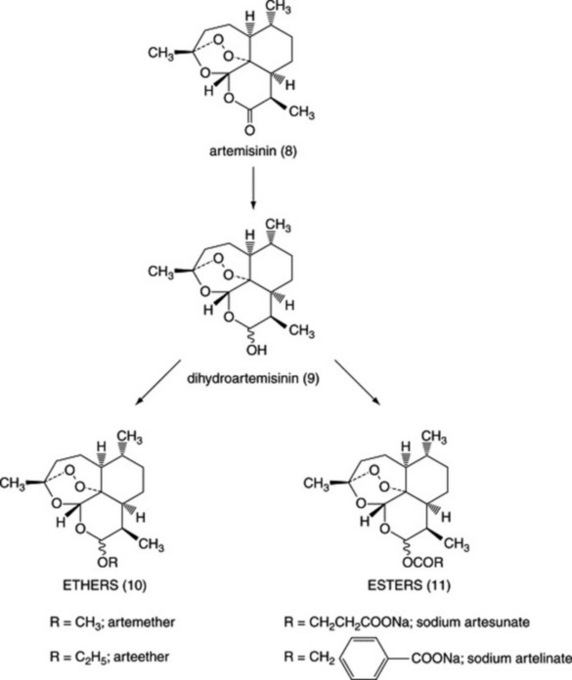
Artemisinin
For hundreds of years the Chinese have used the herb known as Qing Hao (Artemisia annua, Asteraceae) for the treatment of fevers including malaria but it was not until 1971 that Chinese scientists isolated the sesquiterpene lactone artemisinin (Fig. 28.3 (8)) and showed that it was highly active against P. falciparum. Clinical trials showed that the drug was effective in treating malaria including chloroquine-resistant malaria as well as the often fatal complication of cerebral malaria. However, after 1 month many patients had a recurrence of malaria (known as recrudescence), because not all of the parasites in the red cells had been killed by the drug. In an attempt to overcome this problem and to develop drugs with improved formulation characteristics, a number of derivatives have been prepared by reduction of the lactone carbonyl to give dihydroartemisinin (Fig. 28.3 (9)) followed by the preparation of ether (Fig. 28.3 (10)) or ester (Fig. 28.3 (11)) derivatives. The methyl ether, artemether, is soluble in oil and is given by intramuscular injection, whereas esters such as sodium artesunate and sodium artelinate are water soluble and can be given orally or by intravenous injection. Recrudescence is still a problem with these derivatives and, more importantly, malaria parasites with reduced sensitivity to artemisinin derivatives have recently been isolated from malaria patients. To prevent the possibility of artemisinin-resistant malaria parasites developing the WHO has directed that artemisinin and its derivatives should never be used alone for malaria treatment and that a second drug (e.g. mefloquine) should be given in addition. This also has the advantage of reducing the risk of recrudescence. In the body, all of the artemisinin derivatives are metabolized to dihydroartemisinin, which is more active against malaria parasites than artemisinin. Some derivatives such as sodium artesunate are so rapidly hydrolysed that the latter may be considered to be a ‘pro-drug’, whereas artemether is metabolized more slowly so that both the parent and the metabolite contribute to the antimalarial action.
Artemisinin and its derivatives are the most rapidly acting antimalarials known and are highly selective against malaria parasites. As with quinine, haem is involved in their mode of action but the mechanism is quite different (Fig. 28.2). Artemisinin is unusual in that the molecule contains an endoperoxide group and this reacts with the iron in haem, giving rise to highly reactive free radicals. Parasite death is believed to result from the reaction of these free radicals with parasite molecules, such as proteins and nucleic acids. Artemisinin does not react with the iron in haemoglobin so that uninfected red cells are unaffected. In the clinic, artemisinin derivatives are remarkably non-toxic and although animal experiments raised fears that neurotoxicity might be a problem, this has not yet been shown in malaria patients. Artemisinin may, however, be embyrotoxic so that the use of these drugs is not recommended in early pregnancy. One interesting feature of the artemisinin derivatives is that they are active against the gametocyte form of the malaria parasites, which is responsible for the transmission of the disease from man to mosquito during feeding; thus these drugs not only cure the patient but may help to reduce transmission of the disease.
Although artemisinin has been synthesized, the process is complex and not economically viable, so that artemisinin is extracted from A. annua herb and then derivatized as required. The amount of artemisinin present in the plant is about 0.5% of the dry weight of the herb but some strains of A. annua have higher amounts and plant breeding programmes are being carried out to increase the yield further. In addition, plants contain larger amounts of the compound artemisinic acid, which may be converted chemically into artemisinin, thus increasing the yield. Currently there is interest in Africa in growing A. annua for local use as a herbal antimalarial but at the present time there is little clinical evidence to support this practise. In China, a clinical trial in which malaria patients received capsules containing crude alcoholic extracts of A. annua found that while patients did respond to treatment initially, recrudescence rates were very high. As it is imperative to prevent the emergence of parasites resistant to artemisinin, it would seem unwise to use herbal preparations that could result in parasites being exposed to sub-lethal concentrations of drug, thus encouraging the development of resistance.
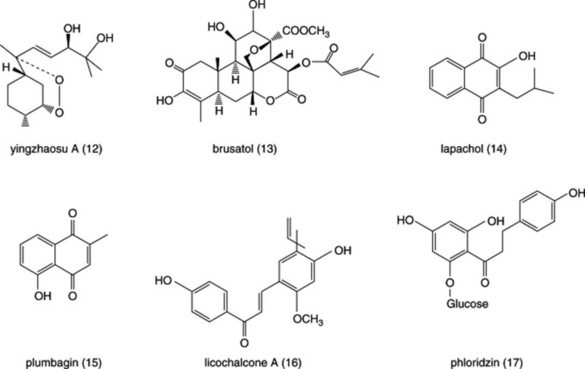
As artemisinin is a complex molecule, much effort has been put into synthesizing compounds based on the 1,2,4-trioxane ring of artemisinin. Many compounds have been made, some of which have promising in vivo activities in animal models. Endoperoxide-containing sesquiterpenes have also been found in another Chinese species, Artrobotrys unciatus (Annonaceae), also known as Ying Zhao. Yingzhaosu A (Fig. 28.4 (12)) and C were active against P. berghei in mice, although they were less potent than artemisinin. A synthetic derivative known as arteflene was evaluated in clinical trials but was abandoned due to high rates of recrudescence.
Quassinoids
The quassinoids are derived biosynthetically from triterpenoid precursors and are bitter principles present in species of the Simaroubaceae, which were found to be active against avian malaria in a screening programme in the 1940s. Quassin itself is not active but a number of quassinoids possessing an unsaturated A-ring, a lactone ring and a methylene oxygen bridge in the C-ring, such as in brusatol (Fig. 28.4 (13)) (a constituent of Brucea javanica), have very potent antiprotozoal properties against several species, including P. falciparum, E. histolytica and G. intestinalis. Unfortunately, these compounds are also very toxic to mammalian cells and attempts to improve their selectivity by making structural changes have not so far been very successful. Both the antiprotozoal and cytotoxic activities are due to the inhibition of protein synthesis and, as these processes appear to be similar in protozoa and mammalian cells, it is likely to be difficult to improve selectivity.However, it is of interest to note that one quassinoid, glaucarubinone, found in Simarouba glauca, was formerly used in France for the treatment of amoebic dysentery. The Meliaceae is a plant family related to the Simaroubaceae that produces bitter terpenoids chemically related to the quassinoids, known as limonoids. The neem tree, Azadirachta indica, is widely used in Asia for malaria treatment and contains a number of limonoids, such as gedunin, which have in vitro antiplasmodial activities but they are less potent than the quassinoids.
Other terpenoids that have reported antiprotozoal activities include the triterpene tingenone, a red–orange pigment present in some species of the Celastraceae and Hypocrataceae. It is highly active against T. cruzi epimastigotes in vitro and it appears to interact with DNA but is also an inhibitor of mitochondrial electron transport and has antineoplastic properties. Jatrophone is a diterpene found in Jatropha isabelli (Euphorbicaceae), which has activity against Leishmania promastigotes and T. cruzi epimastigotes and was effective in treating mice infected with L. amazonensis. The polyoxygenated monoterpene iridoid, arbortristoside A, from Nycanthes arbortristis (Oleaceae) was as effective against L. donovani infection in mice as a standard drug, although it was only weakly active against L. donovani amastigotes in vitro. Interest in natural products isolated from marine sources is growing and a number of diterpenes (isocyanoisocylcoamphilectanes) have been reported from the sponge Cymbastella hooperi, which possesses potent in vitro activities against chloroquine-sensitive and chloroquine-resistant P. falciparum with relatively low cytotoxicity.
QUINONES
Lapachol derivatives
Lapachol (Fig. 28.4 (14)) is a naphthoquinone found in the heartwood of S. American species of Bignoniaceae, for example Tabebuia rosea. Although lapachol itself has only weak antiprotozoal properties, a number of synthetic derivatives are more potent. β-Lapachone and allyl-β-lapachone were found to be active against T. cruzi epimastigotes and the latter was shown to reduce the infectivity of trypomastigotes inoculated into mice. β-Lapachone was not effective in vivo as it reacts with haemoglobin with the formation of methaemoglobin. The action of these compounds appears to depend on their intracellular reduction to free radical species that stimulate the production of hydrogen peroxide by acting as electron carriers between NADH or NADPH and oxygen. Hydrogen peroxide is especially toxic to trypanosomes as they have unsusual antioxidant pathways; T. cruzi is particularly vulnerable as it lacks the enzyme superoxide dismutase. Research on lapachol derivatives has led to the development of atovaquone, a synthetic naphthoquinone now licensed for use (in combination with proguanil) for the treatment of falciparum malaria.
Plumbagin
The S. American species Plumbago benensis (Euphorbiaceae) is used traditionally for the treatment of leishmainiasis and contains the naphthoquinones plumbagin (Fig. 28.4 (15)) as well as the dimers 3,3′-plumbagin and 8,8′-plumbagin. These compounds are active against Leishmania species in vitro, and some activity in vivo has also been shown. Plumbagin has also been reported to have in vitro activities against P. falciparum and epimastigotes of T. cruzi. Diospyrin, a constituent of Diospyros montana (Ebenaceae), is another naphthoquinone dimer and has been shown to inhibit the growth of L. donovani promastigotes in vitro. However, the tetraacetoxy and tetrahydroxy derivatives of diospyrin were much more active than the parent compound against P. falciparum (multi-drug-resistant strain K1) and were generally more active against L. donovani amastigotes, T. b. brucei trypomastigotes and T. cruzi amastigotes. It is likely that the above compounds have a similar mode of action to that of the lapachol derivatives.
Other quinones with antiprotozoal properties include the prenylated preanthraquinones, vismiones H and D, constituents of Visnia guineenis (Guttiferaceae) which have potent in vitro antiplasmodial activities. The quinones, 2-(1-hydroxyethyl)naphtha[2,3b]furan-4,9-quinone and isopinnatal isolated from Kigelia pinnata (Bignoniaceae) were found to possess potent in vitro activities against T. b. brucei and T. b. rhodesiense trypomastigotes.
PHENOLIC COMPOUNDS
Lichochalcone A
The Chinese liquorice species (Glycyrrhiza uralensis glabra and G. inflata) are the source of an oxygenated chalcone, licochalcone A (Fig. 28.4 (16)), which has in vitro activities against P. falciparum as well as against amastigotes and promastigotes of L. major and L. donovani. Activity has also been shown in animal models of both the cutaneous and visceral forms of leishmaniasis, as well as in mice infected with P. yoellii. Studies have shown that this compound alters the ultrastructure and function of mitochondria in Leishmania parasites. Licochalcone A is considered to be a promising lead compound for the development of new antileishmanial agents but to date no clinical studies in man have been carried out.
Phloridzin
This naturally occurring flavonoid glycoside is of interest as it was once used as a treatment for malaria, as, like quinine, it has a bitter taste. It inhibits the growth of malaria parasites in vitro by inhibiting the increase in permeability of the red cell membrane which occurs in infected cells, thus depriving the parasite of glucose and other nutrients. Unfortunately, phloridzin (Fig. 28.4 (17)) also blocks the re-absorption of glucose by the kidney tubules so that it is not suitable for clinical use.
Examples of some other phenolic compounds active against protozoa include the polyphenol gossypol, a constituent of cottonseed oil which is well known for its use as a male contraceptive in China, but is also active against P. falciparum, T. cruzi and E. histolytica in vitro. In T. cruzi gossypol inhibits the oxidoreductase enzymes α-hydroxyacid dehydrogenase and malate dehydrogenase. In the body, gossypol is concentrated in the liver and colon as a result of being excreted in the bile and therefore may be worth investigating for the treatment of amoebiasis where amoebae may be present both in the liver and in the gut lumen. A few flavonoids have in vitro activity against P. falciparum including the exiguaflavones isolated from Artemisia indica (Asteraceae). The methoxylated flavones artemetin and casticin have been shown to act synergistically with artemisinin in vitro and it has been suggested that flavonoids present in A. annua might contribute to the antimalarial action of extracts or herbal teas prepared from this species but there is no evidence to support this.
CONCLUSIONS
In recent years, the artemisinin derivatives have become important antimalarial drugs and this illustrates very well the potential of natural products to provide highly effective antiprotozoal agents. This chapter illustrates the wide range of compounds found in plant species that possess antiprotozoal activities and it is likely that natural products will continue to provide novel compounds, which may lead to new antiprotozoal drugs. However, many of the people who are afflicted with protozoal diseases do not have the resources to afford pharmaceuticals and it is important to assess locally used traditional remedies to determine their efficacy and safety. One initiative that has been set up to encourage this is the Research Initiative on Traditional Antimalarial Methods (RITAM) which is a network of researchers and others interested in the study and use of traditional, plant-based antimalarials.