Cardiac Action Potentials
 Describe the genesis of the resting membrane potential in cardiac muscle.
Describe the genesis of the resting membrane potential in cardiac muscle.
Describe the cardiac action potential in terms of:
differences in conducting and contractile tissues.
phases of the action potential.
ion fluxes contributing to each phase of the action potential.
Describe the actions of natural neurotransmitters (acetylcholine/norepinephrine) on the cardiac action potential.
Describe the effect of ions on the cardiac action potential.
Refer to potentials seen in contractile and conducting cells which are characterized by rapid depolarization due to the opening of fast sodium channels.
These potentials are characterized by a slow upstroke because of influx of calcium, and are of lower amplitude and slower conduction than the fast action potentials; typically seen in the SA and AV nodes.
Also called diastolic depolarization/prepotential. It is the slow increase in membrane potential to threshold level that occurs between the end of one action potential and the start of another in the SA node.
There are essentially two different types of potentials that are seen in the heart. Slow potentials are seen in the pacemaker areas, i.e., the sinoatrial (SA) node and atrioventricular node. Fast action potentials are seen in contractile and conducting cells. These two potentials differ in many ways, but one is the speed with which depolarization occurs, hence the names.
Resting Membrane Potential for the SA Node and Muscle Fibers
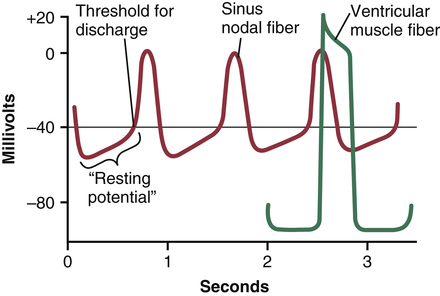
The “resting membrane potential” of the sinus nodal fiber between discharges has a negativity of about −55 to −60 millivolts in comparison with −85 to −90 millivolts for the ventricular muscle fiber. Figure 31-1 shows action potentials recorded from inside a sinus nodal fiber for three heartbeats and, by comparison, a single ventricular muscle fiber action potential. The cause of the lesser negativity in the sinus nodal fiber is that the cell membranes of the sinus fibers are naturally leaky to sodium and calcium ions, and positive charges of the entering sodium and calcium ions neutralize some of the intracellular negativity.
The Slow Action Potential of the SA Node
Cardiac muscle has three types of membrane ion channels that play important roles in causing the voltage changes of the action potential. They are (1) fast sodium channels, (2) slow sodium–calcium channels, and (3) potassium channels.
Opening of the fast sodium channels for a few 10,000ths of a second is responsible for the rapid upstroke spike of the action potential observed in ventricular muscle because of rapid influx of positive sodium ions to the interior of the fiber. Then the “plateau” of the ventricular action potential is caused primarily by slower opening of the slow sodium–calcium channels, which lasts for about 0.3 second. Finally, opening of potassium channels allows diffusion of large amounts of positive potassium ions in the outward direction through the fiber membrane and returns the membrane potential to its resting level.
But there is a difference in the function of these channels in the sinus nodal fiber because the “resting” potential is much less negative—only −55 millivolts in the nodal fiber instead of the −90 millivolts in the ventricular muscle fiber. At this level of −55 millivolts, the fast sodium channels mainly have already become “inactivated,” which means that they have become blocked. The cause of this is that any time the membrane potential remains less negative than about −55 millivolts for more than a few milliseconds, the inactivation gates on the inside of the cell membrane that close the fast sodium channels become closed and remain so. Therefore, only the slow sodium–calcium channels can open (i.e., can become activated) and thereby cause the action potential. As a result, the atrial nodal action potential is slower to develop than the action potential of the ventricular muscle. Also, after the action potential does occur, return of the potential to its negative state occurs slowly as well, rather than the abrupt return that occurs for the ventricular fiber.
Self-Excitation of Sinus Nodal Fibers
Because of the high sodium ion concentration in the extracellular fluid outside the nodal fiber, as well as a moderate number of already open sodium channels, positive sodium ions from outside the fibers normally tend to leak to the inside. Therefore, between heartbeats, influx of positively charged sodium ions causes a slow rise in the resting membrane potential in the positive direction. Thus, as shown in Figure 31-1, the “resting” potential gradually rises and becomes less negative between each two heartbeats. When the potential reaches a threshold voltage of about −40 millivolts, the sodium–calcium channels become "activated," thus causing the action potential. Therefore, basically, the inherent leakiness of the sinus nodal fibers to sodium and calcium ions causes their self-excitation.
Why does this leakiness to sodium and calcium ions not cause the sinus nodal fibers to remain depolarized all the time? The answer is that two events occur during the course of the action potential to prevent this. First, the sodium–calcium channels become inactivated (i.e., they close) within about 100 to 150 milliseconds after opening, and second, at about the same time, greatly increased numbers of potassium channels open. Therefore, influx of positive calcium and sodium ions through the sodium–calcium channels ceases, while at the same time large quantities of positive potassium ions diffuse out of the fiber. Both of these effects reduce the intracellular potential back to its negative resting level and therefore terminate the action potential. Furthermore, the potassium channels remain open for another few tenths of a second, temporarily continuing movement of positive charges out of the cell, with resultant excess negativity inside the fiber; this is called hyperpolarization. The hyperpolarization state initially carries the “resting” membrane potential down to about −55 to −60 millivolts at the termination of the action potential.
Why is this new state of hyperpolarization not maintained forever? The reason is that during the next few tenths of a second after the action potential is over, progressively more and more potassium channels close. The inward-leaking sodium and calcium ions once again overbalance the outward flux of potassium ions, and this causes the “resting” potential to drift upward once more, finally reaching the threshold level for discharge at a potential of about −40 millivolts. Then the entire process begins again: self-excitation to cause the action potential, recovery from the action potential, hyperpolarization after the action potential is over, drift of the “resting” potential to threshold, and finally re-excitation to elicit another cycle. This process continues indefinitely throughout a person’s life.
The Fast Action Potential in Cardiac Muscle
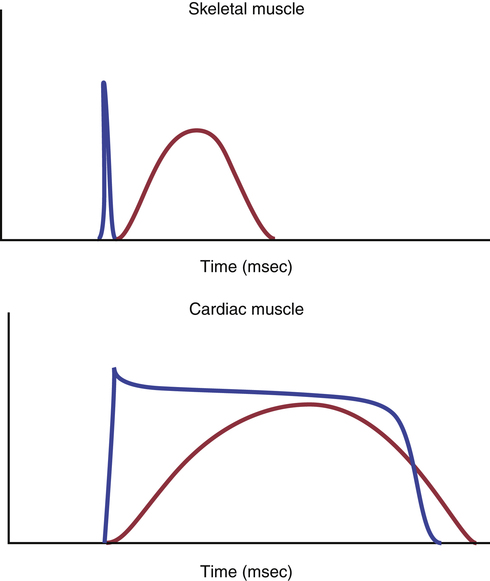
The action potential recorded in a ventricular muscle fiber, rises from a very negative value, about −85 millivolts rapidly to a slightly positive value, about +20 millivolts, during each beat. After the initial spike, the membrane remains depolarized exhibiting a plateau and is followed at the end of the plateau by abrupt repolarization. The presence of this plateau in the action potential causes ventricular contraction to last as much as 15 times as long in cardiac muscle as in skeletal muscle.
What Causes the Long Action Potential and the Plateau?
In cardiac muscle, the action potential is caused by opening of two types of channels: (1) the same fast sodium channels as those in skeletal muscle and (2) another entirely different population of slow calcium channels, which are also called calcium–sodium channels. This second population of channels differs from the fast sodium channels in that they are slower to open and, even more important, remain open for several tenths of a second. During this time, a large quantity of both calcium and sodium ions flow through these channels to the interior of the cardiac muscle fiber, and this maintains a prolonged period of depolarization, causing the plateau in the action potential. Further, the calcium ions that enter during this plateau phase activate the muscle contractile process, while the calcium ions that cause skeletal muscle contraction are derived from the intracellular sarcoplasmic reticulum. A comparison of the temporal association of the action potentials and contractile responses in skeletal and cardiac muscles is provided in Figure 31-2.
Immediately after the onset of the action potential, the permeability of the cardiac muscle membrane for potassium ions decreases about fivefold, an effect that does not occur in skeletal muscle. This decreased potassium permeability may result from the excess calcium influx through the calcium channels just noted. Regardless of the cause, the decreased potassium permeability greatly decreases the outflux of positively charged potassium ions during the action potential plateau and thereby prevents early return of the action potential voltage to its resting level. When the slow calcium–sodium channels do close at the end of 0.2 to 0.3 second and the influx of calcium and sodium ions ceases, the membrane permeability for potassium ions also increases rapidly; this rapid loss of potassium from the fiber immediately returns the membrane potential to its resting level, thus ending the action potential.
Control of Cardiac Action Potentials by the Sympathetic and Parasympathetic Nerves
Sympathetic Effect
Stimulation of the sympathetic nerves releases the hormone norepinephrine at the sympathetic nerve endings. Norepinephrine in turn stimulates beta-1 adrenergic receptors, which mediate the effects on heart rate. The precise mechanism by which beta-1 adrenergic stimulation acts on cardiac muscle fibers is somewhat unclear, but the belief is that it increases the permeability of the fiber membrane to sodium and calcium ions. In the sinus node, an increase of sodium–calcium permeability causes a more positive resting potential and also causes increased rate of upward drift of the diastolic membrane potential toward the threshold level for self-excitation, thus accelerating self-excitation and, therefore, increasing the heart rate.
In the A-V node and A-V bundles, increased sodium–calcium permeability makes it easier for the action potential to excite each succeeding portion of the conducting fiber bundles, thereby decreasing the conduction time from the atria to the ventricles.
The increase in permeability to calcium ions is at least partially responsible for the increase in contractile strength of the cardiac muscle under the influence of sympathetic stimulation, because calcium ions play a powerful role in exciting the contractile process of the myofibrils.
Vagal (Parasympathetic) Effects
The acetylcholine released at the vagal nerve endings greatly increases the permeability of the fiber membranes to potassium ions, which allows rapid leakage of potassium out of the conductive fibers. This causes increased negativity inside the fibers, an effect called hyperpolarization, which makes this excitable tissue much less excitable.
In the sinus node, the state of hyperpolarization decreases the “resting” membrane potential of the sinus nodal fibers from −65 to −75 millivolts rather than the normal level of −55 to −60 millivolts. Therefore, the initial rise of the sinus nodal membrane potential caused by inward sodium and calcium leakage requires much longer to reach the threshold potential for excitation. This greatly slows the rate of rhythmicity of these nodal fibers. If the vagal stimulation is strong enough, it is possible to stop entirely the rhythmical self-excitation of this node.
In the A-V node, a state of hyperpolarization caused by vagal stimulation makes it difficult for the small atrial fibers entering the node to generate enough electricity to excite the nodal fibers. Therefore, the safety factor for transmission of the cardiac impulse through the transitional fibers into the A-V nodal fibers decreases. A moderate decrease simply delays conduction of the impulse, but a large decrease blocks conduction entirely.
Effect of Potassium and Calcium Ions on Cardiac Potentials and Heart Function
Earlier in this chapter we indicated that apart from fast sodium channels, cardiac muscle also has potassium channels and slow sodium–calcium channels. Therefore, it is to be expected that the concentration of potassium and calcium ions in the extracellular fluids should also have important effects on cardiac pumping.
Effect of Potassium Ions
Excess potassium in the extracellular fluids causes the heart to become dilated and flaccid and also slows the heart rate. Large quantities also can block conduction of the cardiac impulse from the atria to the ventricles through the A–V bundle. Elevation of potassium concentration to only 8 to 12 mEq/L—two to three times the normal value—can cause such weakness of the heart and abnormal rhythm that death occurs.
These effects result partially from the fact that a high potassium concentration in the extracellular fluids decreases the resting membrane potential in the cardiac muscle fibers. That is, high extracellular fluid potassium concentration partially depolarizes the cell membrane, causing the membrane potential to be less negative. As the membrane potential decreases, the intensity of the action potential also decreases, which makes contraction of the heart progressively weaker.
Effect of Calcium Ions
An excess of calcium ions causes effects almost exactly opposite to those of potassium ions, causing the heart to go toward spastic contraction. This is caused by a direct effect of calcium ions to initiate the cardiac contractile process, as explained in Chapter 30.
Conversely, deficiency of calcium ions causes cardiac flaccidity, similar to the effect of high potassium. Fortunately, calcium ion levels in the blood normally are regulated within a very narrow range. Therefore, cardiac effects of abnormal calcium concentrations are seldom of clinical concern.
Effect of Drugs on the Cardiac Action Potential
It is beyond the scope of this chapter to discuss the comprehensive role of drugs acting on the cardiac action potential. Table 31-1, however, provides a framework for the actions of some drugs which the student will be able to relate to based on the ionic currents that have been discussed in this chapter.
Table 31-1
Effect of Some Drugs on the Cardiac Action Potential
| Channel / Receptor | Nature of Action | Effect |
| Sodium channel | Blocker | Reduce the maximum rise of the action potential and decrease in conduction through the conducting system and atrial and ventricular muscle |
| Beta receptor | Blocker | Reduces sodium–calcium entry in the SA and AV nodes; reduces heart rate and slows conduction |
| Potassium channel | Blocker | Prolongation of the action potential duration |
| Calcium channel | Blocker | Depression of the plateau phase of the action potential; negative inotropism |