Diseases of Bone of Questionable Etiology
Infantile Cortical Hyperostosis (Caffey’s disease, Caffey-Silverman syndrome, familial infantile cortical hyperostosis, sporadic infantile cortical hyperostosis)
Infantile cortical hyperostosis was originally described independently by Caffey and Silverman and by Smyth and his coworkers as a syndrome of unknown etiology in which unusual cortical thickening occurred in certain bones of infants. It was soon discovered that none of a variety of diseases which may produce cortical thickening, such as scurvy, rickets, syphilis, bacterial osteitis, neoplastic disease and traumatic injury are present in this condition. Infantile cortical hyperostosis is a self-limited disorder that affects infants and causes bone changes, soft tissue swelling, and irritability. Although the etiology is not completely understood, familial and sporadic forms appear to exist.
Etiology
This is an inflammatory process of unclear etiology. In early stages, inflammation of the periosteum and adjacent soft tissues is observed. As this resolves, the periosteum remains thickened and subperiosteal immature lamellar bone is observed. Mature specimens show hyperplasia of lamellar cortical bone without inflammation or subperiosteal changes. While the etiology is not clear, evidence of genetic transmission exists. Some believe that transmission may occur via an infectious agent with a long latency period. Other theories include a primary arterial abnormality and allergic reaction.
Clinical Features
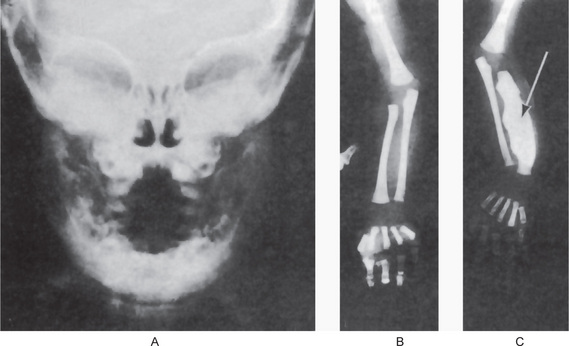
Infantile cortical hyperostosis is a selflimited condition. No sex predilection has been established. Infantile cortical hyperostosis is now believed to exist in two forms, familial and sporadic. These forms differ in their onset and presentation. The familial form seems to have an earlier onset; 24% of these cases are present at birth. Incidence of mandibular involvement is lower, and incidence of lower extremity involvement is higher in the familial form than that observed in the sporadic form. The tibia is the most frequently involved bone. The average age at onset is six to eight weeks. The disease appears to be inherited in an autosomal dominant fashion with variable penetrance. The sporadic form is becoming less common. It has a higher incidence of mandibular involvement than does the familial form. The average age at onset is 9–11 weeks. The classic presentation includes a triad of irritability, swelling, and bone lesions. The swelling appears suddenly. It is deep and firm and may be tender. Fever may occur. Babies may refuse to eat, especially if they have mandibular involvement, creating an appearance of failure to survive. Almost all cases are evident in infants by age of five months.
The mandible and the clavicles are the bones most frequently affected, the jaw involvement usually being manifested as a facial swelling. In fact, mandibular involvement is such a constant and striking feature of the disease that the question has been raised as to whether the diagnosis of the disease should ever be made in its absence. However, Saul and his coworkers reported that in the familial form of the disease mandibular involvement is less frequent and lower extremity involvement more frequent. Other bones which commonly demonstrate hyperostosis are the calvarium, scapula, ribs and tubular bones of the extremities, including the metatarsals. The soft-tissue swellings are associated with deep muscles and occur in general in the locations in which the hyperostoses subsequently arise. These swellings have been described in the scalp, face, neck, thorax and extremities.
Other signs and symptoms of the condition which have been described in some patients, but which are not inevitably present, include fever, pseudoparalysis, dysphagia, pleurisy, anemia, leukocytosis, monocytosis, elevated sedimentation rate and increased serum alkaline phosphatase.
Oral Manifestations
The oral aspects of the disease have been studied by Burbank and his associates in a series of patients who had suffered from the condition during infancy. After careful follow-up examinations they found that some patients manifested a residual asymmetric deformity of the mandible, usually in the angle and ramus area, even several years after the disease had subsided. A few patients with this deformity also had severe malocclusion. Despite the febrile component of the disease, no cases of enamel hypoplasia were observed. Care must be taken not to confuse this disease with cherubism, in which bilateral enlargement of the mandible also occurs.
Radiographic Features
Radiographic examination reveals periosteal new bone formation that can be quite florid and subsequently becomes compact causing pronounced cortical thickening (Fig. 17-28). The periosteal new bone is seen in bones underlying areas of soft tissue swelling. The distribution is patchy and asymmetric but is multifocal, although cases of monostotic involvement have been reported. The mandible is almost invariably involved and other commonly affected areas include the clavicles, ribs and long bones of the limbs. Typically, the periosteal new bone or periosteal ‘cloaking’ is confined to the diaphyses of the long bones, sparing the metaphyses and epiphyses. There are a few reports of lytic areas affecting the skull vault and facial bones but this is uncommon. The spine, phalanges and pelvis are hardly ever involved. Increased uptake of radioisotope from a radioisotope bone scan shows areas of involvement before radiographic changes are present.
Histologic Findings
In early stages, inflammation of the periosteum and adjacent soft tissues is observed. As this resolves, the periosteum remains thickened and subperiosteal immature lamellar bone is observed. The bone marrow spaces contain vascular fibrous tissue. Mature specimens show hyperplasia of lamellar cortical bone without inflammation or subperiosteal changes.
Treatment
No specific treatment exists for infantile cortical hyperostosis. The disease ultimately resolves without sequelae in six to nine months. Some periods of exacerbations and remissions may occur during the course. Corticosteroids may be helpful in alleviating symptoms in severe cases, but they do not have any effect on bone lesions. NSAIDs may also be used for symptoms. On occasion, residual skeletal changes may persist into adult life. In addition, occasional cases of recurrence of pain and cortical thickening of bone in later childhood have been reported.
Paget’s Disease (Paget’s disease of bone, osteitis deformans)
Paget’s disease, which is characterized by excessive and abnormal remodeling of bone, is a common disorder in middle-aged and elderly patients. The excessive remodeling gives rise to bones that are extensively vascularized, weak, enlarged, and deformed with subsequent complications. Paget’s disease is named after Sir James Paget, an English surgeon who described the clinical course of this disorder and originally named the condition osteitis deformans, as he believed the disease was caused by chronic inflammation.
Etiology
The etiology of Paget’s disease is still unknown. Evidence exists of a genetic link, as a 7-fold to 10-fold increase in incidence of Paget’s disease was observed in relatives of patients diagnosed with the condition. The overall pattern of apparent transmission suggests an autosomal dominant inheritance. Another possible etiology is related to viral infection. Some studies have shown the presence of viral inclusion particles in pagetic osteoclasts. Furthermore, dense fibrillar material associated with some inclusions is similar to that found in the nuclei of virus-infected cells. Certain immunocytologic data and viral antibody titers against the measles virus reinforce the viral hypothesis. The presence of minimal inflammation and few inflammatory cells in bone and peripheral blood is consistent with a chronic infectious process. Other suggested etiologies include an inflammatory cause, which is supported by evidence of clinical improvement after treatment with anti-inflammatory medications. Elevated parathyroid hormone in Paget’s disease also has been observed; however, no firm evidence links the two disorders. Furthermore, one case of Paget’s disease was diagnosed in a patient with idiopathic hypoparathyroidism. Autoimmune, connective tissue, and vascular disorders are proposed as other possible etiologies.
Paget’s disease of bone is characterized by enhanced resorption of bone by giant multinucleated osteoclasts with formation of disorganized woven bone by osteoblasts. This process evolves through various phases of activity, followed by a quiescent stage. Hence, Paget’s disease typically consists of the following three phases:
Clinical Features
The prevalence of Paget’s disease increases with age. Paget’s disease is recognized most commonly after age 50 years and is rarely diagnosed in people younger than 20 years. By the ninth decade of life, prevalence reaches nearly 10% of the peer group. The male-to-female ratio is approximately 1 : 1. There is also a marked geographic predilection for occurrence, the disease being common in England, France and Germany but rare in certain other European countries, Africa, and the Middle and Far East.
Many individuals with Paget’s disease are asymptomatic. Clinical features are extremely variable and depend on which bones are affected. The diagnosis most commonly is made incidentally during an unrelated radiographic or biochemical investigation. On occasion, the disease manifests with severe musculoskeletal impairments with neurologic and cardiovascular complications. Paget’s disease has a predilection for the axial skeleton and may be widespread at the time of diagnosis. The condition commonly affects the pelvis and spine, particularly the lumbar spine with a frequency of 30–75%. The sacrum is involved in 30–60% of cases and the skull in 25–65% of cases. The proximal long bones, especially the femur, also are affected frequently (in 25–35% of cases). Involvement of the shoulder girdle and proximal humerus is not uncommon. Though any bone may be affected, the fibula, ribs, and bones in the hands and feet are involved only infrequently. Paget’s disease may affect one bone and then remain limited in its course or progress from a few localized areas to the rest of the skeleton. The most common presenting complaint is pain. The bone pain is perceived as a dull constant aching pain deep below the soft tissues. It may persist or exacerbate during the night. The involved bones become warm to the touch because of the increased vascularity. Other typical findings and complaints of patients with Paget’s disease may include the following: pathologic fractures commonly result from weakened pagetic bone, nonspecific headaches, impaired hearing, and tinnitus are common symptoms of Paget’s disease with skull involvement. The patient’s hat size may increase or change due to skeletal deformity and enlargement, especially of the skull bones. Cranial nerve palsies can affect nerves other than the auditory nerve; however, this development is uncommon. Changes in vision may occur secondary to optic nerve involvement. Back and neck pain are common complaints, as Paget’s disease frequently affects the spine, especially the lumbar and sacral regions. Softened bone at the base of the skull may lead to platybasia, the descent of the cranium onto the cervical spine. Progressive pain, paresthesias, limb paresis, gait difficulties (waddling gait), or bowel and bladder incontinence may be caused by compression of the spinal cord or spinal nerve secondary to platybasia or vertebral fractures. Nausea, dizziness, syncope, ataxia, incontinence, and dementia can be observed with hydrocephalus, basilar invagination, and cerebellar or brainstem compressive syndromes.
Involvement of the facial bones is occasionally seen. It has sometimes been called leontiasis ossea (lion-like facies) but because this term is nonspecific, Drury advocated discontinuing its use in referring to this disease.
Oral Manifestations
Involvement of the jaws in osteitis deformans is a rather common occurrence. Stafne and Austin reported 20 cases involving the maxilla and three cases involving the mandible in a series of 138 cases of generalized osteitis deformans, an incidence of 17% jaw involvement. This predilection for the maxilla has also been noted in most other studies. A number of cases have been reported in which both jaws of a patient were involved. Cooke also provided an excellent study of 15 cases of Paget’s disease of the jaws, while Tillman has reported 24 cases. Smith and Eveson also have reviewed this disease with particular reference to dentistry, analyzing 152 cases involving the jaws previously reported in the literature. Of these, 98 involved the maxilla, 28 the mandible and 26 both jaws. Thus, the ratio of involvement of maxilla to mandible was approximately 2.3 : 1.
The maxilla exhibits progressive enlargement, the alveolar ridge becomes widened and the palate is flattened (Figs. 17-29, 17-30). If teeth are present, they may become loose and migrate, producing some spacing. When the mandible is involved, the findings are similar, but not usually as severe as in the maxilla. As the disease progresses, the mouth may remain open, exposing the teeth, because the lips are too small to cover the enlarged jaw.
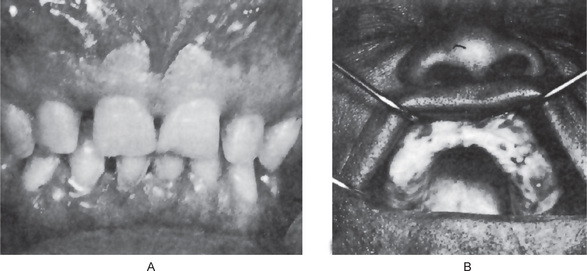
Figure 17-29 Osteitis deformans.
There is diffuse enlargement of the maxilla and thickening of the dentulous (A) and edentulous (B) alveolar ridge. In addition, tipping of the teeth due to enlargement of the maxilla is obvious. (B, Courtesy of Dr Robert J Gorlin)
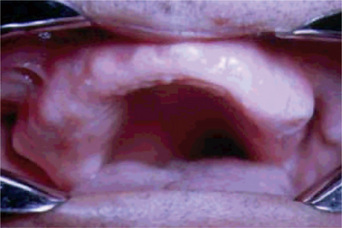
Figure 17-30 Paget’s disease.
Note the enlargement on the right maxilla. The patient was unable to use his denture.
Edentulous patients with dentures, commonly complain of an inability to wear their appliance because of increasing tightness due to expansion of the jaw. The dentures may be remade periodically to accommodate this increase in size of the jaws.
When the jaws are involved by Paget’s disease, there is usually involvement of the skull as well. But there have been some cases reported in which the skull showed no evidence of the disease.
Radiographic Features
The radiographic features of osteitis deformans are varied and depend upon the stage of the disease encountered. Paget’s disease has sometimes been described as a disorder characterized by an initial phase of deossification and softening, followed by a bizarre, dysplastic type of reossification not related to functional requirements, the two processes taking place simultaneously or alternately. With this in mind, the protean radiographic manifestations can be easily reconciled. Thus osteolytic areas of the skeleton are commonly associated with areas of osteoblastic activity. These destructive lesions may be multiple and diffuse or isolated. The isolated lesion in the skull, when large, is sometimes referred to as osteoporosis circumscripta.
The osteoblastic phase of osteitis deformans is the more commonly recognized one and inevitably occurs regardless of pre-existing osteolytic lesions. The osteoblastic areas, which appear as opacities in the radiograph, tend to be patchy in distribution, eventually becoming confluent, but often still showing minute areas of variation in radiodensity. This patchiness has been termed a ‘cotton-wool’ appearance and is especially well demonstrated in the skull and jaws (Fig. 17-31A).
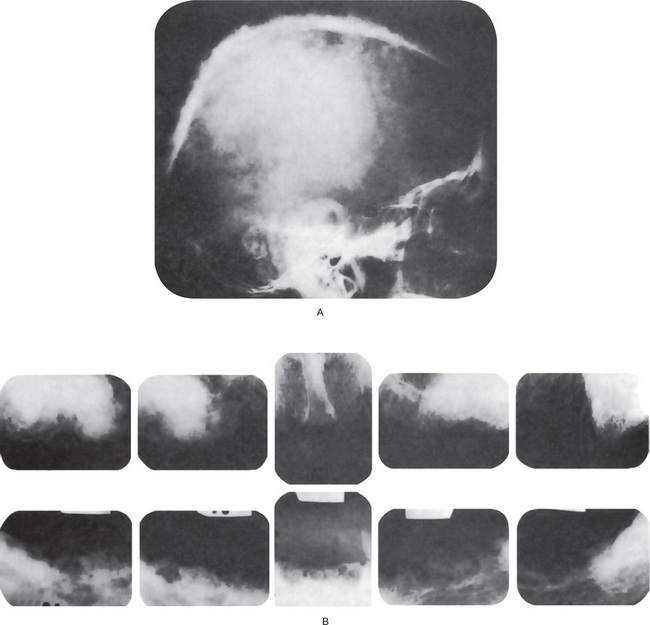
Figure 17-31 Osteitis deformans.
The radiographs of the skull (A) and the jaws (B) demonstrate the typical ‘cotton-wool’ appearance. (A, Courtesy of Dr John A Campbell)
Radiographs of the jaws may demonstrate even very early phases of the disease, although such phases may not be so specific as to be pathognomonic. An excellent description of the oral manifestations of early osteitis deformans has been provided by Spilka and Callahan. In such cases, poorly defined areas of osteoporosis may be noted, although of more diagnostic significance is the finding of loss of normal trabeculation and the appearance of irregular osteoblastic activity, again giving rise to the typical cotton-wool appearances of ‘Paget’s bone’ (Fig. 17-31B). Although the disease is usually bilateral, it may show radiographic evidence of only unilateral involvement of the jaw, especially early in the course of the disease. This may closely simulate chronic, diffuse, sclerosing osteomyelitis.
The teeth themselves and adjacent bone present significant radiographic changes suggestive of osteitis deformans also. These consist characteristically of a rather pronounced hypercementosis, and often, loss of a well-defined lamina dura around the teeth. Root resorption has been reported in some cases, but this is unusual.
Laboratory Findings
The serum calcium and serum phosphorus levels are usually within normal limits, even in cases of advanced osteitis deformans. The serum alkaline phosphatase level may be elevated; however, to extreme limits. Values as high as over 250 Bodansky units have been reported, particularly in patients in the osteoblastic phase of the disease, when there is rapid formation of new bone and when there is polyostotic involvement. In fact, there is no other disease of bone in which the serum alkaline phosphatase level may be as high as in Paget’s disease. In the monostotic form of the disease, the alkaline phosphatase level seldom exceeds 50 Bodansky units. In the very early stage of the disease this phosphatase level may not be significantly elevated, although it is simply a matter of time before this does occur. The serum acid phosphatase level is not increased. In Paget’s disease, urinary hydroxyproline levels are elevated as they reflect increased osteoclastic activity and bone resorption. Hydroxyproline is a product of collagen breakdown. More recently, measurement of the urinary excretion of bonespecific pyridinium collagen cross-links has been found to be a sensitive and specific index of bone resorption. Urinary N-telopeptide (NTX) and alpha-C telopeptide (CTX) have emerged recently as sensitive biochemical markers for bone resorption. An abnormally high alpha-CTX: beta-CTX ratio is present with active Paget’s disease. This ratio returns to the reference range following treatment with bisphosphonates.
Histologic Features
The microscopic appearance of the bone in cases of osteitis deformans varies remarkably, depending upon the stage of the disease encountered. The initial osteolytic phase is marked by disordered areas of resorption by an increased number of overtly large osteoclasts. These abnormal osteoclasts may contain as many as 100 nuclei. The subsequent osteoblastic phase follows with haphazard laying of new bone matrix and formation of woven bone without regard to the patterns of stress. Repeated episodes of bone removal and formation results in the appearance of many small irregularly shaped bone fragments that appear to be joined in a jigsaw or mosaic pattern with deeply staining hematoxyphilic reversal lines (Fig. 17-32). This pattern is the histologic hallmark of Paget’s disease. As the disease progresses, the osteoblastic phase predominates, and excessive abnormal bone formation occurs, causing more compact and dense bone. The pagetic bone is coarse and fibrous, with an avidity for calcium and phosphorus. Marrow spaces are filled with loose highly vascularized connective tissue. The hypervascular bone combined with cutaneous vasodilation causes an increase in the regional blood flow and accounts for the rise in skin temperature seen clinically. The hypervascularity consists of an increased number of patent capillaries and dilated arterioles, as well as of larger venous sinuses (Fig. 17-33).
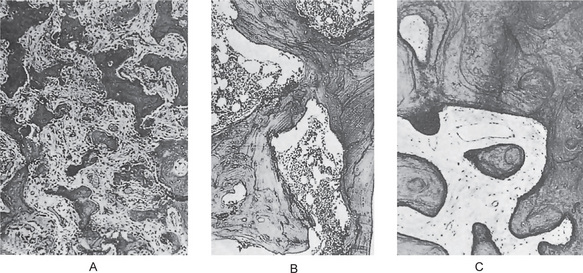
Figure 17-32 Osteitis deformans.
Photomicrographs of bone in different stages showing (A) reactive phase, (B) mosaic pattern, and (C) resting phase. Note the prominent resting and reversal lines in B.
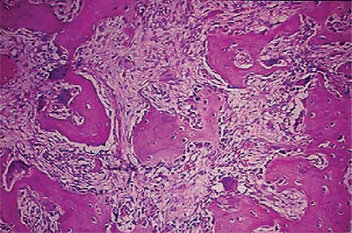
Figure 17-33 This photomicrograph taken from a bone biopsy from a patient with Paget’s disease of bone shows several bone spicules in a highly vascularized connective tissue stroma.
These areas of bone formation alternate with areas of bone resorption characterized by the presence of osteoclasts. Typically, the osteoclasts are seen inside of Howship lacunae.
The normal trabecular appearance is distorted with a mosaic pattern of irregular cement lines joining areas of lamellar bone. Pagetic bone shows no tendency to form Haversian systems or to center on blood vessels; the bones are very hard and dense. Eventually, the osteoblastic activity diminishes, and an osteoporotic or burned-out phase predominates (Figs. 17-34, 17-35). The new bone is disordered, poorly mineralized, and lacks structural integrity. The proliferation of bone and concomitant hypercementosis sometimes result in obliteration of the periodontal ligament.
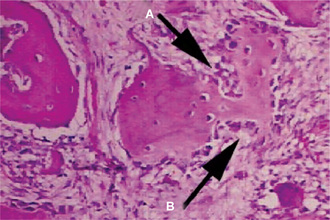
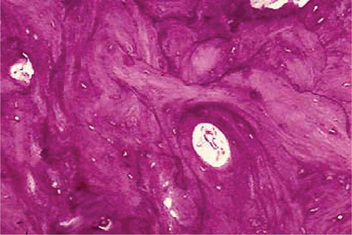
Treatment and Prognosis
There is no specific treatment for osteitis deformans. Vitamin, hormone and radiation therapy have all been utilized with sporadic reports of cures, but these have not been confirmed. Very promising results have recently been obtained in the treatment of this disease by the use of calcitonin, the parathormone antagonist produced by the thyroid gland which suppresses bone resorption. Biphosphonates have also been used with some success, since they also inhibit bone resorption as well as bone mineralization. Finally, one of the cytotoxic antibiotics, mithramycin, has been used therapeutically but has serious side effects. The use of these agents has been reviewed in detail by Smith and Eveson. The general outlook for patients with Paget’s disease is good, especially if treatment is administered before major changes in the bones have occurred. Treatment does not cure Paget’s disease but can control it. Patients with severe polyostotic Paget’s disease have a less favorable prognosis than those with monostotic disease. Patients with polyostotic disease are at higher risk for complications.
Complications
Incomplete stress fractures frequently occur in Paget’s disease. Mild injuries may cause acute true pathologic fractures in weakened pagetic bone. Pathologic fractures are more common in women than men. The most frequent site of these fractures is the femur, but fractures commonly occur in the tibia, humerus, spine, and pelvis. Sarcomatous degeneration of pagetic bone is a deadly complication. Pagetic sarcoma follows a rapid and fatal course. Sarcomatous degeneration may occur in 5–10% of patients with extensive pagetic skeletal involvement. In less widespread involvement, osteosarcoma occurs in fewer than 1% of patients with Paget’s disease. Men are affected with sarcomatous degeneration slightly more frequently than women. Peak incidence is in the seventh and eighth decades of life. The femur (most common) and proximal humerus are the sites most commonly affected; however, no bone is exempted, including sites of previously healed fractures.
Degenerative joint disease is associated with Paget’s disease. Cardiovascular abnormalities such as increased cardiac output has been observed in patients with widespread Paget’s disease. Left ventricular hypertrophy is an associated finding. Increased soft tissue and pagetic bone vascularity has been implicated as a contributing factor. High-output congestive heart failure may occur, but it is rare.
Massive Osteolysis (Vanishing bone, disappearing bone, phantom bone, progressive osteolysis, Gorham syndrome)
Massive osteolysis is an unusual and uncommon disease characterized by spontaneous, progressive resorption of bone with ultimate total disappearance of the bone. Disappearing, or ‘Phantom’ bone disease, also called Gorham’s disease, may be a form of hemangioma of bone. This relatively rare condition, usually occurring in children or young adults, is characterized by the dissolution, in whole or in part, of one or several adjacent bones. A cavernous, angioma-like permeation may be a prominent pathologic feature of the affected bones. The process is self-limited, but the extent of progression is unpredictable. It is not genetically transmitted.
Clinical Features
Massive osteolysis is most common in older children and young and middle-aged adults, affecting both genders equally. About 50% of all patients report an episode of trauma before the diagnosis, but this is often trivial in nature. Usually only one bone is affected in a given patient, although polyostotic cases have been reported. The most commonly affected bones are the clavicle, scapula, humerus, ribs, ilium, ischium, and sacrum.
The disease, which may or may not be painful, begins suddenly and advances rapidly until the involved bone is replaced by a thin layer of fibrous tissue surrounding a cavity. All laboratory values are usually normal.
Oral Manifestations
A number of cases have been reported involving the mandible and other facial bones, and these have been reviewed by Ellis and Adams and by Murphy and his coworkers. In only two of these cases was there destruction of the entire mandible. In at least three cases, there was concomitant involvement of the maxilla. The patient may present with pain or facial asymmetry, or both. One of the consistent findings in the disease has been pathologic fracture following minor trauma.
Histologic Features
The typical histologic findings is replacement of bone by connective tissue containing many thinwalled blood vessels or anastomosing vascular spaces lined by endothelial cells. It does not represent a hemangioma of bone, which remains a localized lesion, although the term ‘hemangiomatosis’ has been applied. Most authorities do not believe that the disease is due to increased osteoclastic activity, although osteoclasts may often be found in the tissues. On the other hand, their absence in areas of active resorption is often quite striking.
Cementoblastoma (True cementoma)
The benign cementoblastoma is probably a true neoplasm of functional cementoblasts which form a large mass of cementum or cementum-like tissue on the tooth root. It is quite distinctive but relatively uncommon.
Clinical Features
The benign cementoblastoma occurs most frequently under the age of 25 years, with no significant gender predilection. More than half of the tumors have occurred in persons under 20 years of age, although the range has been 10–72 years. When the site has been specified, the mandible is affected three times more frequently than the maxilla. The mandibular first permanent molar is the most frequently affected tooth. In fact, only one case is reported involving the deciduous dentition. Involvement of this mandibular first molar has accounted for approximately 50% of all reported cases. However, other teeth involved have included mandibular second and third molars, mandibular bicuspids, maxillary bicuspids and first, second and third molars. The associated tooth is vital unless coincidentally involved. The lesion is slow-growing and may cause expansion of cortical plates of bone, but is usually otherwise asymptomatic. Pain has been reported, but this may have been related to associated caries rather than to the lesion.
Radiographic Features
The tumor mass is attached to the tooth root and appears as a well-circumscribed dense radiopaque mass often surrounded by a thin, uniform radiolucent line. The outline of the affected root is generally obliterated because of resorption of the root and fusion of the mass to the tooth.
Histologic Features
The main bulk of the tumor mass is composed of sheets of cementum-like tissue, sometimes resembling secondary cellular cementum but other times being deposited in a globular pattern resembling giant cementicles. Reversal lines scattered throughout this calcified tissue are often quite prevalent. There is a variable soft-tissue component consisting of fibrillar, vascular and cellular elements. Many of the cemental trabeculae in area of activity are bordered by layers of cementoblasts. Away from these trabecular surfaces, cementoclasts may be evident. In such active areas, the lesion is frequently microscopically indistinguishable from the benign osteoblastoma or giant osteoid osteoma, and this relationship has been discussed by Larsson and his associates. In fact, some areas are so cellularly active that they bear strong resemblance to osteosarcoma.
This calcified mass will be found united to the tooth root through obliteration of the periodontal ligament, resorption of portion of the root and replacement by the tumor tissue. The periphery of the tumor generally shows a soft tissue cellular layer resembling a capsule. At this periphery, the cemental trabeculae are almost invariably arranged at right angles.
Treatment and Prognosis
Because of the tendency for expansion of the jaw, it is believed that extraction of the tooth is justified despite the fact that the pulp is vital. Care must be taken to distinguish this lesion from severe hypercementosis or chronic focal sclerosing osteomyelitis (i.e. condensing osteitis), both of which it may superficially resemble. The lesion does not appear to recur.
Bisphosphonate Therapy
Bisphosphonates are small inorganic molecules that bind to hydroxyapatite on the surface of damaged bones. They belong to a class of drugs that are used to prevent bone loss by demineralization of bone. At the sites of bone damage, osteoclasts are inhibited and destroyed. Since bone damage is caused by increased numbers and activity of these osteoclast bone cells, bisphosphonates reduce new bone damage and allow an opportunity for bone healing to occur. Bisphosphonates are approved for the treatment of osteoporosis, often seen in post-menopausal females, hypercalcemia of malignancy and in metastatic disease to bone (cancer spreading to bone tissue).
Bisphosphonates therefore have several beneficial effects, including:
• Preventing further bone damage
• Reducing bone pain and the need for pain relieving drugs
• Correcting and preventing hypercalcemia
• Reducing the need for radiotherapy
• Reducing pathologic fractures due to bone occupying/destroying lesions
• Improving the chances of healing and recovery of strength of the bone
Possible Side Effects of Bisphosphonates
Bisphosphonates are generally very well tolerated. The most common side effects are fever, vein irritation, general aches and pains, kidney dysfunction and osteonecrosis of the jaws (ONJ).
Osteonecrosis of the Jaws
Recently cases have been reported of individuals having difficulty in healing after undergoing tooth extraction or other invasive dental procedures, associated with a phenomenon called osteonecrosis of the jaws (ONJ). The only common factor in these patients was that they were taking bisphosphonate drugs. As a consequence, an agreement was reached that there is an association between osteonecrosis of the jaw and bisphosphonate therapy, although the drugs are not the only factor involved.
Overall, the risk is thought to be less than 1% for patients taking IV bisphosphonates, and at least ten times less likely than that for patients taking the drugs by mouth. Most reported cases occur after oral trauma (tooth extraction or oral surgical procedures). Tobacco use, treatment with corticosteroids, long term use of bisphosphonates, treatment with more than one kind of bisphosphonates, and diabetes may increase the risk of this condition developing.
Signs of Bisphosphonate-associated Osteonecrosis
The hall marks of this condition are gum wounds that heal very slowly or do not heal at all for six weeks or more after a procedure that exposes bone. Some patients report that this begins with a feeling of ‘roughness’ on the gum tissue. If these open wounds become infected, suppuration or swelling in the adjacent gum tissue develop. Many times, the condition is painless in the beginning, and pain is experienced only after the exposed bone becomes infected. If this infection lasts long enough, there may even be numbness, especially in the lower jaw.
Unfortunately at this time most reported treatments are slow to resolve osteonecrosis of the jaw, so the best treatment is prevention. Current treatment methods that are used include antiseptic rinses, systemic antibiotics, and cleaning/removal of dead bone from the affected area. Generally, therapy focuses on controlling pain and preventing infection so that the body can heal properly.
If problems persist and/or if healing is slow, consideration can be given to stopping bisphosphonate therapy for 2–4 months to facilitate recovery. There is every reason to hope that with appropriate awareness and early management, serious problems from osteonecrosis can be avoided.
Patients with documented myeloma related bone disease should not take bisphosphonates. This means that, in general, patients with monoclonal gammopathy of undetermined significance (MGUS) and smoldering (early/clinically not established) myeloma without bone disease do not need or benefit from bisphosphonates. However, this remains an area of ongoing research and clinical trials.
Bisphosphonates must be used with caution in patients with pre-existing kidney disease or known elevation in serum creatinine, especially >3.0 mg/dl but also any value above the normal range.
Patients who have allergic reactions or are intolerant to bisphosphonates should not be recruited to therapy.
Diseases of Temporomandibular Joint
The temporomandibular joints (TMJs) are one of the most commonly used joints and also one of the most complex joints. These joints play crucial roles in mastication and speech. Disorders of TMJs include congenital and developmental conditions, traumatic disturbances, arthritis, benign and malignant tumors, dysfunction of the articular disks and ligaments, disorders inside or outside the TMJ capsule, and disorders associated with mandibular or temporal bones. Disorders of TMJs are high; some studies reports as high as 86% of the population has some kind of TMJ related signs or symptoms. Progress in cross-sectional imaging, such as computed tomography (CT), magnetic resonance imaging (MRI) and cone beam computed tomography (CBCT), has allowed better evaluation of the TMJs. Plain radiographs and panoramic radiographs have limited values in diagnosing TMJ disorders. With current clinical knowledge and treatment options available, management of TMJ related conditions are difficult and not always successful.
The following sections provide a brief overview of the most common TMJ-related disorders.
Development Disturbances of Temporomandibular Joint
At the time of birth, the TMJs are incompletely formed. Therefore, developmental disturbance to the TMJs can occur either before or after birth. Kaneyama et al, have classified developmental disturbances as follows:
Condylar Hypoplasia or Aplasia
Condylar aplasia is failure of development of the mandibular condyle, while condylar hypoplasia is underdevelopment of the condyle. Aplasia or hypoplasia may be congenital or acquired, and may occur unilaterally or bilaterally (Fig 17-36A, B).
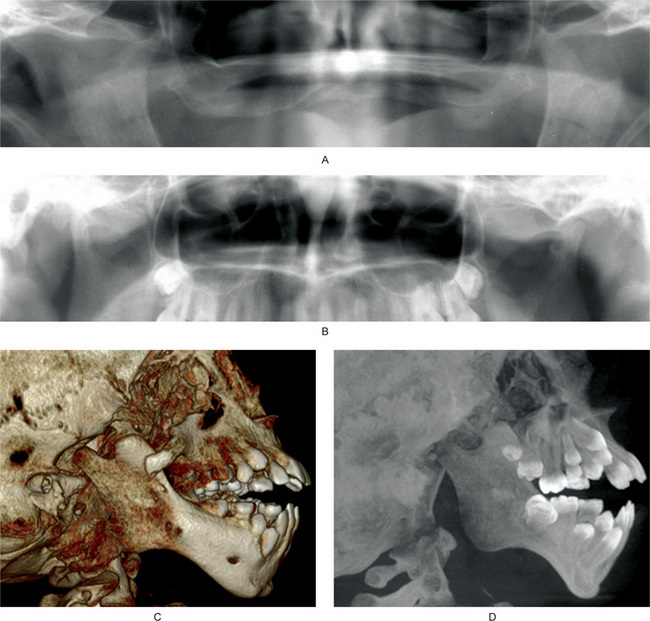
Figure 17-36 Developmental disturbances of temporomandibular joints.
Aplasia and hypoplasia of the TMJ. (A) Cropped panoramic radiograph showing bilateral aplasia of the TMJs. The finding is worse on the left side. The coronoid processes appear hyperplastic. (B) Cropped panoramic radiograph showing hypoplasia of the right condylar head. Compared to the left condylar head, the anteroposterior dimension of the right condylar head is small. The left condylar head has flattening of the lateral surface, consistent with degenerative joint disease. (C) Treacher Collins syndrome, 3-D reconstruction of a cone beam computed tomography data. (D) Same patient as in Figure C. Maximum intensity projection of cone beam computed tomography data. Figures C and D showing hypoplastic condylar head and coronoid process. Zygomatic arch is partially formed. Anterior teeth are protruded. Antegonial notch is prominent. In Treacher Collins syndrome, hypoplasia of the condylar heads is bilateral.
Congenital or primary hypoplasia or aplasia is characterized by unilateral or bilateral underdevelopment of the condyle, usually due to disturbances in the first or second branchial arches. Conditions that show congenital hypoplasia or aplasia include Treacher Collins syndrome (Fig. 17-36C, D), oculo-auriculo-vertebral syndrome, hemifacial macrosomia, Pierre Robin sequence, and Hurler syndrome. In congenital variety, both the joints are usually affected but the primary clinical findings may be unilateral.
The acquired or secondary form of hypoplasia may be due to any agent which interferes with the normal development of the condyle. Local causes that may initiate condylar hypoplasia include trauma, infection of the mandible or middle ear, and therapeutic dose of irradiation.
Clinical Features
Hypoplasia or aplasia is frequently associated with other anatomically related defects such as a defective or absent external ear, an underdeveloped mandibular ramus or macrostomia. Unilateral involvement is the most common clinical type. If the disturbance is unilateral, there is obvious facial asymmetry, and both occlusion and mastication may be altered. A shift of the mandible towards the affected side occurs during opening. A mild disturbance presents only lesser degree of these features, perhaps accompanied by a mandibular midline shift during opening and closing. The distortion of the mandible in this pathognomonic pattern results from lack of downward and forward growth of the body of the mandible due to the arrest of the chief growth center of the mandible, the condyle. Some growth continues at the outer posterior border of the angle of the mandible, resulting in thickening of the bone in this area. The older the patient at the time of the growth disturbance, the less severe will be the facial deformation.
In bilateral hypoplasia or aplasia, deviation of the jaws is not present. However, micrognathia is a prominent feature of bilateral hypoplasia or aplasia. Along with micrognathia, dental crowding and open bite may be present. The severity of the clinical findings depends on the age of the patient when the injury occurred, the duration of the injury and its severity. In aplasia or severe hypoplasia, persistent airway obstruction may occur due to posterior positioning of the tongue into the pharyngeal airway. Feeding is thus compromised as well as speech.
Treatment
Treatment of condylar aplasia or hypoplasia is difficult since there are no available means of stimulating its growth locally or compensating for its failure. Singh and Bartlett have reported different treatment modalities conducted on a series of 266 patients over a period of 27 years. Early diagnosis and intervention are important in managing the occlusion, speech and other functions. Although the condition itself is not necessarily a progressive one, the resulting disturbance may become more severe as the patient approaches puberty. Cartilage or bone transplants have been used to build-up the underdeveloped parts, preceded in some cases by unilateral or bilateral sliding osteotomy, to improve the appearance of the patient with asymmetry and retrusion. If the derangement is severe, osteoplasty may be considered as an option. If the patient exhibits little difficulty, surgical intervention is not warranted, although cosmetic surgery may aid in correcting facial deformity. Infants with condylar aplasia who exhibit respiratory difficulty may require tracheotomy.
Hyperplasia of Mandibular Condyle
Condylar hyperplasia is a rare unilateral enlargement of the condyle (Fig. 17-37A,B, C) which should not be confused with a neoplasm of this structure, although it may superficially resemble an osteoma or chondroma. The cause of this condition is obscure, but it has been suggested that mild chronic inflammation, resulting in a condition analogous to a proliferative osteomyelitis, stimulates the growth of the condyle or adjacent tissues. The unilateral occurrence strongly suggests a local phenomenon. Obewegeser and Makek have classified condylar hyperplasia into three categories. Type A is hemimandibular hyperplasia, causing asymmetry in the vertical plane. In this type, the growth is unilateral in the vertical plane, with minimal deviation of the chin. Typically, maxilla shows compensatory growth. In absence of the maxillary growth, an open bite may be present on the same side. Type B is hemimandibular elongation, causing asymmetry in the transverse plane. In this type, the chin is deviated towards the contralateral side with no vertical asymmetry. Patient may exhibit cross-bite. Type C is a combination of type A and type B, and exhibits hyperplastic features unilaterally or bilaterally.
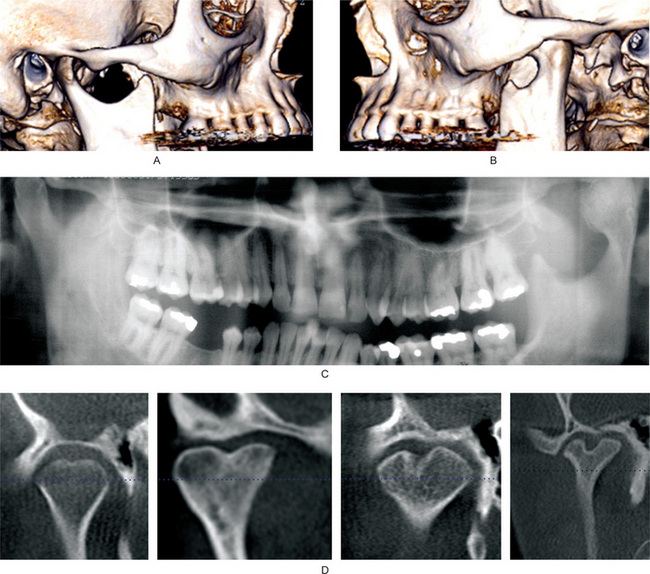
Figure 17-37 Developmental disturbances of temporomandibular joints.
Figures (A and B) are from the same patient. Three-dimensional reconstruction of a computed tomography data. The left condylar head is hyperplastic. Please note that left condylar head is too large for the articular fossa, and is permanently dislocated anterior to the articular eminence. (C) Cropped panoramic radiograph shows hyperplasia of the left condylar head. (D) Axially corrected coronal views of the temporomandibular joints from four different patients from cone beam computed tomography scans. Bifid condyles may have slight midline depression to almost a duplication of the condylar head.
Clinical Features
Condylar hyperplasia occurs more frequently in females. Patients of any age may be affected, although most common occurrence is in the third decade of life. Diagnosis of condylar hyperplasia is made by a combination of clinical and radiographic findings. The patients usually exhibit a unilateral, slowly progressive elongation of the face with deviation of the chin away from the affected side. The enlarged condyle may be clinically evident or at least palpated and presents a striking radiographic appearance in both coronal and sagittal views. The affected joint may or may not be painful. A malocclusion is a usual sequel of the condition.
Bifid Condyle
Bifid condyle is characterized by a varying depth of groove or depression around the midline of the condylar head (Fig. 17-37D). The depression may be visible on coronal or sagittal orientation. A deep groove may result into an appearance of duplicity of the condylar head. Usually, bifidity is unilateral, although bilateral bifid condyles have been reported. Bifidity is a rare condition, effecting less than 1% of the population. Even rarer are trifid condyles. The etiology of bifidity is controversial. Two etiologies of bifid condyles have been suggested. One theory speculates that bifidity may originate in embryo where blood supply to the condylar head is limited. Another theory suggests trauma being the cause of bifidity, either due to birth trauma or fracture of the condylar head. Usually patients with bifid condyles are asymptomatic and do not require any treatment. Relationship of bifid condyles to articular disks is not clearly known.
Traumatic Disturbances of Temporomandibular Joint
Dislocation of the Condyles
Condylar dislocation can primarily be of two types: anterior dislocation and cranial dislocation. Cranial dislocation, where the condylar head is dislocated into the cranial fossa due to trauma, is rare. The causative trauma is mostly motor vehicle or sports related. Trauma may also dislocate the condylar head posteriorly.
Anterior dislocation of the TMJ is more common than cranial dislocation. Anterior dislocation occurs when the head of the condyle moves anteriorly over the articular eminence into such a position that it cannot be returned voluntarily to its normal position. Some researchers believe that this inability to retrude the mandible is caused by spasm of the temporal muscle initiated by myotatic reflex. Thus, in movements of the mandible involving forward translation of the condyle, tension may be placed on the temporalis and leads to the formation of the muscle spasm.
Anterior dislocations can be ‘luxation’ or ‘subluxation’. Luxation of the joint refers to complete dislocation, which cannot be reduced by the patient (non-self-reducing). Subluxation is a partial or incomplete dislocation, which can be reduced by the patient (self-reducing). Some investigators hold the view that subluxation is actually a form of hypermobility, and should not be viewed as a form of dislocation. Anterior dislocation may occur repeatedly, and condylar reduction may become easier with successive dislocation. Repeated anterior dislocation and self-reduction is referred as habitual or recurrent dislocation.
Anterior dislocation may be acute, owing to a sudden traumatic injury resulting in fracture of the condyle (see Figure 7-42E, F for examples of traumatic dislocation), or more frequently, only in stretching of the capsule, usually at the point of attachment for the external pterygoid muscle into the capsule. There is often some tearing of the tendon at this insertion point. Most commonly, however, dislocation is a result of yawning or having the mouth opened too widely, as during extraction of teeth or during procedures such as tonsillectomy or endoscopy.
Clinical Features
The typical form of anterior dislocation is characterized by sudden locking and immobilization of the jaws when the mouth is open, accompanied by prolonged spasmodic contraction of the temporal, internal pterygoid and masseter muscles, with protrusion of the jaw. The patient experiences severe pain of the TMJs, excessive salivation and depression of the skin in the preauricular area. All activities requiring motion of the mandible, such as eating or talking, become impossible. The mouth cannot be closed, and the patients frequently become panicky, especially if it is their first experience.
Treatment
Reduction of an anteriorly dislocated condyle is accomplished by inducing relaxation of the muscles and then guiding the head of the condyle under the articular eminence into its normal position by an inferior and posterior pressure of the thumbs in the mandibular molar area. The necessary relaxation can sometimes be brought about only by means of general anesthesia or by tiring the masticatory muscles by cupping the chin in the palm of the hand and applying a posterior and superior pressure for 5–10 minutes. Treatment of recurrent dislocation may be achieved by altering the ligaments, associated musculature, and bony anatomy.
Ankylosis (Hypomobility)
Ankylosis of the TMJ is a disorder in which adhesion of joint components takes place by fibrous or bony union, resulting into loss of function (Fig. 17-38).
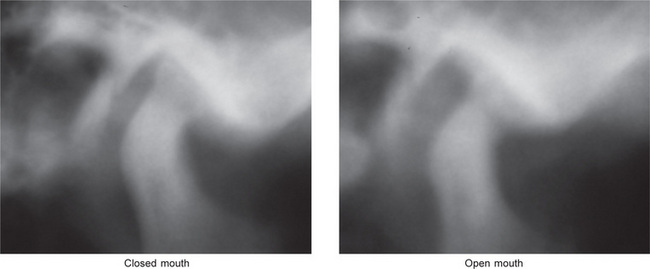
Figure 17-38 Ankylosis of the temporomandibular joint.
The images are tomography of the left temporomandibular joint in the closed and open mouth positions. A radiopaque band of tissues extends from the condylar head to the anterior slope of the articular fossa. On the open mouth view, the condylar head barely translated.
Etiology
The most frequent causes of ankylosis of the TMJ are traumatic injuries and local or systemic infections. Other causes of ankylosis include systemic diseases such as ankylosing spondylitis, rheumatoid arthritis, psoriasis, or previous TMJ surgery. Bilateral ankylosis is often a result of rheumatoid arthritis.
Classification
Sawheny in 1986 had classified ankylosis of the TMJ is into four different types. In his study of 55 patients, type I ankylosis was least common, while type IV was most common. In another study by Zhi et al, type III ankylosis was most common.
• Type I: The condylar head is flattened or deformed. Presence of fibrous adhesion makes movement impossible.
• Type II: The condylar head is deformed and a small bony adhesion exists between the condyle and articular fossa. The articular surfaces are mostly well-defined.
• Type III: A bony bridge extends from the ramus to the zygomatic arch. On the medial aspect, atrophic and displaced condylar head is still present. The articular surface of the fossa is intact. The articular disk is also probably intact.
• Type IV: The architecture of the joint is lost due to bony bridge extending from the ramus to the temporal bone.
A simpler classification of TMJ ankylosis identifies two types of ankylosis: intra-articular and extra-articular. In intra-articular ankylosis, the joint undergoes progressive destruction of the meniscus with flattening of the mandibular fossa, thickening of the head of the condyle and narrowing of the joint space. The ankylosis is basically fibrous, although ossification in the scar may result in a bony union.
Extra-articular ankylosis results in a ‘splinting’ of the TMJ by a fibrous or bony mass external to the joint proper, as in cases of infection in surrounding bone or extensive tissue destruction.
Clinical Features
Ankylosis of the joint occurs at any age, but most cases occur before the age of 10 years. Distribution is approximately equal between the genders. The patient may or may not be able to open his/her mouth to any appreciable extent, depending on the type of ankylosis. In complete ankylosis, bony fusions will absolutely limit any motion. There is usually somewhat greater motion in fibrous ankylosis than in bony ankylosis.
If the injury which brought about the ankylosis was sustained in infancy or childhood, at least before the age of 15 years, there is nearly always an associated facial deformity. The type of deformity is partially dependent upon whether the ankylosis is unilateral or bilateral. In unilateral ankylosis occurring at an early age, the chin is displaced laterally and backward on the affected side because of a failure of development of the mandible. When an attempt is made to open the mouth, the chin deviates toward, the ankylosed side, if any motion is present. Bilateral ankylosis occurring in childhood results in underdevelopment of the lower face; a receding chin and micrognathia (Fig. 17-39). The maxillary incisors often manifest overjet due to failure of this mandibular growth.
Radiographic Features
In fibrous ankylosis, the joint space is often limited. The articulating surfaces of the condyle and the fossa may be irregular. Irregular surfaces appear to interdigitate in a locking fashion. In bony ankylosis, a bony bridge exists from condylar head to the articular fossa.
Treatment
Treatment of TMJ bony ankylosis is surgical. Early surgical intervention in childhood can reduce the adverse effect on facial development. Different surgical procedures, including condylectomy, gap arthroplasty, interpositional arthroplasty, joint reconstruction have been attempted. Sometimes re-ankylosis can occur following arthroplasty. Fibrous ankylosis may be treated by functional methods.
Injuries of Articular Disk: Internal Derangement
Internal derangement of the TMJ is defined as an abnormality of the internal components of the joint, where the articular disk is displaced from its normal functional relationship with the condylar head and the articular fossa of the temporal bone (Fig. 17-40). According to Barkin and Winberg, internal derangement is considered to have four clinical stages. In stage I, the articular disk is displaced in closed mouth position and reduces to normal contact (the central part of the disk is in contact with the condylar head and articular eminence) in open mouth position. In stage II, the disk is displaced in closed mouth position, and intermittently locks in open mouth position. In stage III, the disk is displaced in closed mouth position, and does not reduce to normal contact in open mouth position (closed lock). In stage IV, the disk is displaced without reduction and with perforation of the disk or posterior attachment tissues.
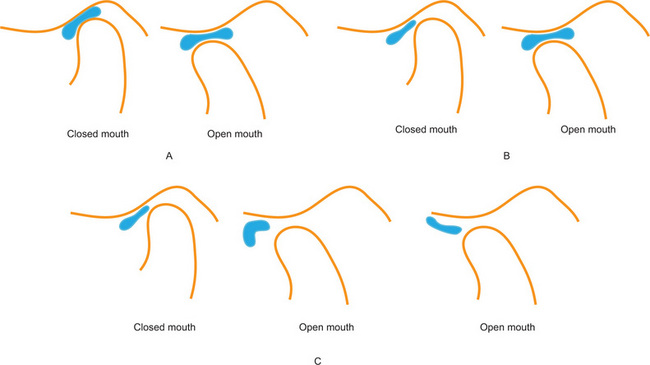
Figure 17-40 Diagrammatic representation of the relationship between bone and articular disk of the temporomandibular joint.
(A) Normal disk location in closed and open mouth position. In the closed mouth position, the posterior band of the articular disk is located between 11:30 and 12:30 of a clock face. The central narrow zone of the disk is in contact with the condylar surface as well as the articular fossa. In the open mouth position, the central narrow zone of the disk remains in contact with the condylar head and the articular eminence. (B) Disk displacement with reduction (DDWR). In the closed mouth position, the posterior band of the articular disk is displaced anterior to 11:30. The central narrow zone of the disk is not in contact with the condyle or the articular fossa. In the open mouth position, the central narrow zone of the disk is in contact with the condylar head and articular eminence. (C) Disk displacement without reduction (DDWOR). In the closed mouth position, the posterior band of the articular disk is displaced anterior to 11:30. The central narrow zone of the disk is not in contact with the condyle or the articular fossa. In the open mouth position, the disk is anteriorly displaced, and may assume normal biconcave shape or become deformed.
Etiology
The etiology of internal derangement is not known. The possible causes of internal derangement may be injury to the condylar region, variation of normal function, muscle hyperactivity or excessive opening of the mouth. The trauma causing disk displacement may be a single event (macrotrauma) or multiple excessive forces over a period of time (microtrauma), such as bruxism.
Clinical Features
Internal derangements are far more common in females than in males, the ratio being as high as 8:1. Young adults are more frequently affected than children or persons past 40 years of age.
Patients with internal derangement can be asymptomatic. Patients who have disk displacement with reduction may have normal range of jaw movement. Some patients who have disk displacement with reduction may have limitation of movement, primarily due to pain. Clinical examination may reveal clicking or joint sound during opening and closing of the mouth. Patients who have disk displacement without reduction have limited range of mouth opening. In these patients, during opening, the mouth may deviate towards the involved joint. The patient may have pain during closed mouth position as the condyle rests on the retrodiskal tissues.
Magnetic resonance imaging of the TMJ in both closed and open positions is necessary for study of the condition (Fig. 17-41). Other radiographic examinations, such as panoramic radiography or computed tomography, are not useful in determining the location of the disk. In some patients with history of trauma, fluid effusion may be present. Effusion can be identified with T2-weighted MR images, where effusion has high signal intensity.
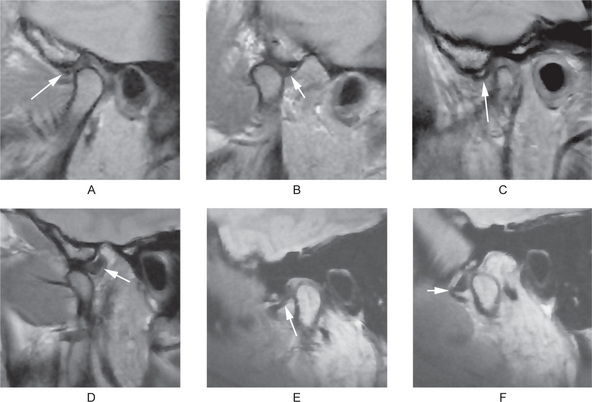
Figure 17-41 Magnetic resonance imaging of the temporomandibular joints.
(A, B) From the same patient, in closed (A) and open (B) mouth position. (A and B) show Normal disk relationship with the condylar head and articular fossa/eminence. (C, D) From the same patient, in closed (C) and open (D) mouth position. (C, D) show disc displacement with reduction (DDWR). (E, F) From the same patient, in closed (E) and open (F) mouth position. (E and F) show disc displacement without reduction. (DDWOR) The arrows point to the articular discs.
Treatment
The treatment of disk displacement can be surgical or nonsurgical. Current understanding of the function of TMJs and the roles of articular disks suggest that it may not be necessary to re-establish the position of the disk. An anterior positioning appliance may be helpful in patients who have disk displacement with reduction. The treatment for each case depends upon careful individual evaluation, and no definite guidelines have been established.
Condylar Fracture
About 17–52% mandibular fractures involve condylar fracture. Zachariades et al have provided a thorough review of condylar fractures. Condylar fractures may be classified according to the anatomic location, e.g. the condylar head (intracapsular), the condylar neck (extracapsular) and the subcondylar region. Condylar fractures may also be classified as nondisplaced, deviated, displaced (medial or lateral), and dislocated (Fig. 17-42). Another classification is according to the orientation of the fracture line, e.g. horizontal, vertical or compression type. Condylar fracture results from an acute traumatic injury to the jaw and is accompanied by limitation of motion and pain and swelling over the involved condyle. The fractured condyle fragment is frequently displaced anteriorly and medially into the infratemporal region because of the forward pull of the external pterygoid muscle, and reduction of the fracture is often difficult because of this displacement. In unilateral fracture of the condyle, the body of the contralateral mandible is also likely to fracture. In fatal automobile accidents, a fracture condyle may be dislocated into the cranium. Open reduction of the fracture fragments is indicated in case of limited range of motion or loss of function. Surgical reduction of the fracture fragments may not be necessary if the patient has adequate function.
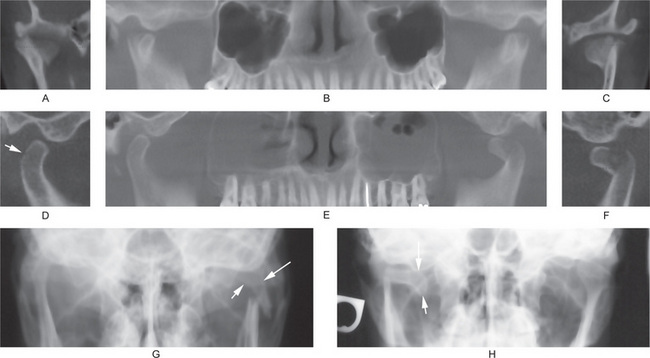
Figure 17-42 Fracture of the condylar temporomandibular joints.
(A, B, and C) From the same patient’s cone beam computed tomography scan. Both the condylar heads are vertically fractured. The reconstructed panoramic view (B) shows the fractured fragments of the condylar heads are dislocated anteriorly. (D, E, and F) From the same patient’s cone beam computed tomography scan. D, Shows a hairline fracture (arrow) of the right condylar head. Figure E shows fractured and anteriorly displaced left condylar head. Both the maxillary sinuses are cloudy. F, shows fractured left condyle, and that fracture fragment is displaced to the peak of the articular eminence. (G) Fracture of the left side at the condylar neck region. Open mouth Towne’s radiograph shows the fractured fragment (arrows) is medially inclined. (H) Fracture of the right side at the condylar neck region. Open mouth Towne’s radiograph shows the fractured fragment (arrows) is displaced medially and is horizontally oriented.
Inflammatory Disturbances of Temporomandibular Joint
Arthritis or inflammation of the joints is one of the most prevalent chronic diseases. Temporomandibular joint may suffer from any form of arthritis. Following three are the most common types of arthritis of TMJ:
Osteoarthritis (Degenerative Joint Disease)
Osteoarthritis is the most common type of arthritis and has been said to develop, at least to some degree, in all persons past 40 years of age. Although its etiology is unknown, it is a disease associated with the aging process. The joints first involved are those which bear the weight of the body and are thus subjected to continued stress and strain: the joints of the knees, hips and spine. In case of TMJ, degenerative joint disease is assumed to initiate after disk is displaced and bony contact exists between the condyle and articular fossa (Fig. 17-43).
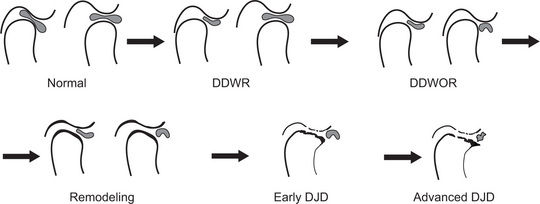
Figure 17-43 Diagrammatic representation of the progression from normal condyle to degenerative joint disease.
In normal stage, the condylar head and the articular fossa/eminence are smooth and well-defined. The posterior band of the articular disk and the central narrow zone are in normal positions. In stage II, the disc is anteriorly displaced in closed mouth position, but reduces during opening (disc displacement with reduction=DDWR). In stage III, in the open mouth position, the displaced disc does not reduce (disc displacement without reduction = DDWOR). In stage IV, the condylar head and the articular fossa/ eminence are slightly flattened and sclerosed, indicating remodeling of the joint. In stage V, the margins of the condylar head and articular fossa are irregular and slightly eroded, indicating early degenerative joint disease. In stage VI, condylar and fossa margins are eroded. A prominent osteophyte is present at the anterior margin of the condylar head. These signs indicate advanced degenerative joint disease. The disc is deformed in advance degenerative disease.
Clinical Features
Clinical signs and symptoms of osteoarthritis are often remarkably absent even in the face of severe histologic or radiographic joint changes. Since the TMJ is not a weight-bearing joint, changes here are insignificant even though arthropathy may be present in other joints. Those changes that do occur may be a result of disturbed balance of the joint due to loss of all teeth or due to external injury. Patients with osteoarthritis of other joints may complain of clicking and snapping in the TMJ, but pain is not necessarily a feature. This joint noise is probably due to atypical disk motion resulting from discordant mandibular condyle-disk function on the basis of the changes in the articular cartilage. Limitation of motion or ankylosis rarely occurs.
Histologic Features
The changes in the articular cartilage consist of surface erosions of varying degrees of severity, with the presence of vertical cracks extending often from the surface through the cartilaginous plate into the subchondral bone. The cartilage cells often exhibit degeneration, and there may be complete destruction of cartilage in localized areas.
Radiographic Features
Degenerative joint disease is diagnosed radiographically. CBCT and CT are the reliable examinations to diagnose degenerative changes (Fig. 17-44). MRI and panoramic radiographs have limited value in diagnosing early degenerative changes. Ahmad et al, have described the diagnostic criteria for degenerative joint disease. Radiographically, a joint is considered osteoarthritic if it fulfils any of the following criteria: (i) the condyle has osteophytes, (ii) condyles or fossa/eminence has subcortical erosion or (iii) condyles or fossa/eminence has subcortical pseudocysts. An osteophyte is defined as a marginal hypertrophy with sclerotic borders and exophytic angular formation of osseous tissue arising from the surface of the condyle. Subcortical erosion is defined as loss of continuity of articular cortex. A subcortical pseudocyst is defined as a cavity below the articular surface that deviates from normal marrow pattern.
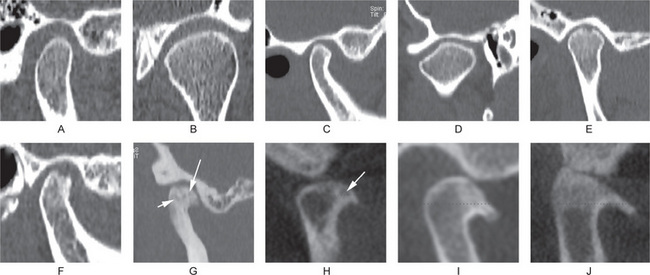
Figure 17-44 Computed tomographic findings of normal and osteoarthritic temporomandibular joints.
(A) Sagittal view of a normal temporomandibular joint, which has smooth, rounded and well-defined cortical margin of the condyle and the fossa. (B) Axially corrected coronal view of a normal temporomandibular joint which has smooth, rounded and well-defined cortical margins. (C) Sagittal view of a temporomandibular joint that shows signs of remodeling. Anterior slope of the condylar head is flat. (D) An axially corrected coronal view showing remodeling of the condylar head. The lateral slope of the condylar head is flat. (E–J) Images of joints with degenerative joint disease. (E, F) Sagittal views showing erosion of the condylar heads. The continuity of the cortical margin is lost. (G) A condyle with generalized sclerosis, osteophyte at the anterior margin, and several subcortical pseudocysts (arrows). (H–J) From cone beam computed tomography. (H) An osteophyte at the anterior margin of the condylar head and a subcortical pseudocyst (arrow). (I) A prominent osteophyte, which often appears as a bird’s beak. (J) A prominent anterior osteophyte, sclerosis of the superior part of the condylar head, and flattening of the articular eminence.
Rheumatoid Arthritis
Rheumatoid arthritis is a disease of unknown etiology which commonly begins in early adult life (35–50 years) and affects women more frequently than men, in a ratio of at least 3:1. It is a chronic, systemic, autoimmune inflammatory disorder. This is an aggressive condition that can damage a joint within 2 years. Although this disease is apparently not due to a specific bacterial infection, there is evidence to indicate that it may be a hypersensitivity reaction to bacterial toxins, specifically streptococci. The distribution of joint involvement is nearly always polyarticular and frequently symmetrically bilateral. Patients usually manifest a long series of episodic exacerbations and remissions. TMJ involvement in cases of rheumatoid arthritis is not particularly common despite the fact that this is a polyarticular disease. Treister and Glick have provided a thorough review of rheumatoid arthritis of the TMJ and dental care plan.
Clinical Features
Rheumatoid arthritis, in its early stages, may be manifested by slight fever, loss of weight and fatigability. The joints affected are swollen, and the patient complains of pain and stiffness. Involvement of the TMJ may occur concomitantly with the other joint lesions or may arise at any subsequent time. Radiographically, joints may be irregular (Fig. 17-45). Articular surfaces become flat. Subcortical pseudocysts and osteophytes may be present.
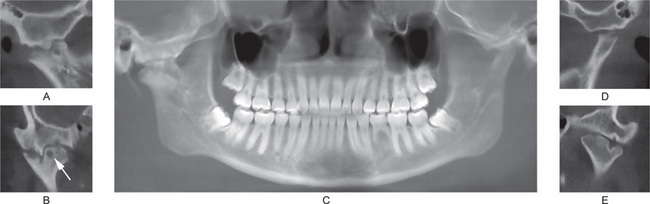
Figure 17-45 Cone beam computed tomography of a patient with bilateral involvement with rheumatoid arthritis.
(A) Sagittal view of the right joint. Superior margin of the condylar head is irregular. (B) An axially corrected coronal view of the right joint, which shows inter-digitation of bony projections that lead to ankylosis of the joint. A subcortical pseudocyst (arrow) is present in the condylar head. (C) A reconstructed panoramic radiograph from the cone beam scan. The dentition appears within normal limits, with the exception of congenitally missing mandibular left second premolar. (D, E) Sagittal and coronal sections of the left joint. Superior margin of the left condylar head is irregularly flat and has prominent notching. The articular fossa is also flat.
Movement of the jaw, as during mastication or talking, causes pain and may be limited because of the stiffness. The stiffness is commonly at its height in the morning and tends to diminish throughout the day with continued use of the jaw. Clicking and snapping of the joint are not common, but when they occur are due to alterations in the articular cartilage and meniscus. Over a period of years there may be ankylosis of the joint, but this is not inevitable.
Juvenile idiopathic arthritis (JIA) or juvenile rheumatoid arthritis or Still’s disease is a common rheumatic disease in children. TMJ involvement in JIA patients may be as high as 87%. Some studies indicate that TMJ can be the only joint involved with JIA. The patient may be asymptomatic, without any clinical signs and symptoms. The first clinical signs may be asymmetry of the mandible and class II malocclusion. When this diagnosis is made, irreversible condylar resorption has already taken place. Radiographic findings include low density of the condylar head, erosion of the surface of the condyle, eminence and fossa. In severe cases, the resorbed condylar head may assume the shape of a pencil.
Histologic Features
There has been little opportunity for microscopic examination of the TMJ in cases of rheumatoid arthritis, and findings have been reported in but few cases. There is; however, no reason to expect the histologic features to be significantly different from those in other joints. Elsewhere, the disease is characterized by the ingrowth of granulation tissue to cover the articular surfaces, the invasion of cartilage and its replacement by granulation tissue, and the ultimate destruction of the articular cartilage. Eventually fibrous adhesions occur; the articular disk may become eroded, and fibrous ankylosis results. Occasionally the connective tissue becomes ossified and a true bony ankylosis occurs.
Treatment and Prognosis
There is no specific treatment for rheumatoid arthritis, although remarkable benefit may result from the administration of adrenocorticotropic hormone (ACTH) or cortisone. Once limitation of motion and deformity have occurred, surgical intervention in the form of condylectomy may be necessary to regain movement. There is; however, a great tendency for recurrence of the ankylosis. For patients with JIA, the management includes orthodontic treatment and functional appliances. Nonsteroidal antiinflammatory drug (NSAID) therapy has shown mixed benefit. Intra-articular corticosteroid injection has shown promising results in JIA patients. Arabshahi and Cron have reported benefits of different treatment modalities for JIA.
Septic (Infectious) Arthritis
The incidence of arthritis due to a specific infection is low when compared to the occurrence of degenerative joint disease and rheumatoid arthritis. Until recently, about 40 cases have been reported in English literature. Leighty et al, have reviewed the existing literature, which showed that most common organism is Staphylococcus aureus. The spread of infection is either directly from a penetrating wound or from hematogeneous origin. Cai et al, have recently reported another 40 cases that were treated in a hospital in China. In this study, majority of the patients had hematogeneous source of infection.
Clinical Features
Patients suffering from acute infectious arthritis complain chiefly of sudden severe pain in the joint, with extreme tenderness on palpation or manipulation over the joint area. The pain is of such intensity that motion is severely limited. Healing of this form of arthritis often results in ankylosis, either osseous or fibrous. A fibrous ankylosis is more common, but in either event there is severe limitation of motion. Diagnosis of the condition is usually achieved by clinical examination, radiographic evaluation and aspiration of the fluid in the joint area.
Histologic Features
Depending upon the severity of involvement, there is a variable amount of destruction of the articular cartilage and articular disk. Osteomyelitis with destruction of bone of the condyle may develop. The joint spaces become narrower in the healing phase by the development of granulation tissue and its subsequent transformation into dense scar tissue.
Treatment
For septic arthritis, treatment of choice is antibiotics and arthrocentesis under low pressure. If treatment is instituted in the acute phase, the sequelae will be less deforming or disabling than if the disease has been allowed to enter a chronic phase. After infection subsides, physiotherapy may help in improving mobility of the joint.
Neoplastic Disturbances of Temporomandibular Joint
Neoplasms and tumor like growths, benign and malignant, may involve the TMJ, but such involvement is relatively uncommon. Such tumors may originate from the condyle, either the bone or the articular cartilage, or from the joint capsule. As might be expected, the connective tissue, cartilage, and bone give rise to the majority of these tumors. Occasionally metastatic tumors have also been reported to involve the TMJ. The rarity of these lesions precludes their discussion here, particularly since they exhibit no features significantly different from those of similar tumors occurring in other locations in and about the oral cavity which have already been described.
Loose Joint Bodies
Synovial Chondromatosis
Synovial chondromatosis is a rare benign condition where nodular cartilaginous or osteocartilaginous entities proliferate in the joint synovium (Fig. 17-46). These entities may become loose from the synovium, and continue to grow in size.
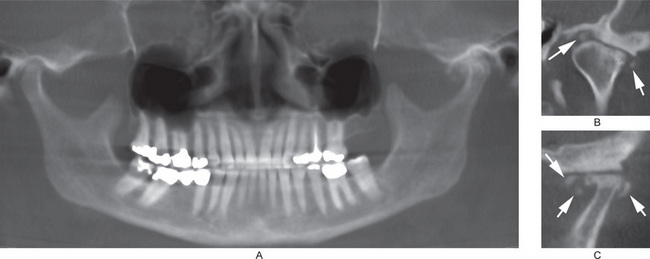
Figure 17-46 Synovial chondromatosis of the left temporomandibular joint as shown in a cone beam computed tomography.
(A) Normal right temporomandibular joint, and synovial chondromatosis associated with the left temporomandibular joint. (B) An axially corrected coronal view of the left temporomandibular joint. Loose joint bodies (arrows) are present at the superior and lateral margins of the condylar head. The condylar head is flattened and has subcortical sclerosis. (C) Sagittal view of the left temporomandibular joint. The condylar head and articular fossa are flat and sclerosed. Loose joint bodies (arrows) are present at the anterior and posterior margins of the condylar head.
Clinical Features
Mean age of patients with synovial chondromatosis is about 45 years. It is more common in females. Bilateral synovial chondromatosis is rare. Patients may be asymptomatic. However, Guarda-Nardini et al, have reported three cardinal signs and symptoms of synovial chondromatosis. These include: (i) pain in the preauricular area, (ii) swelling, facial asymmetry, and joint deformity, and (iii) limited joint function. Additional signs and symptoms include occlusal changes, headache, and joint sound. CT and MRI are useful imaging examinations for diagnosis and treatment planning.
Temporomandibular Disorders
TMJ Syndrome
Temporomandibular joint (TMJ) syndrome or temporomandibular disorder (TMD) is the most common cause of facial pain after toothache. No unequivocal definition of the disease exists; discrepancies concerning the terminology, definitions, and practical treatment methodologies exist. TMD can be classified broadly as TMD secondary to myofacial pain and dysfunction (MPD), and TMD secondary to true articular disease.
The two types can be present at the same time, making diagnosis and treatment more challenging. The MPD type forms the majority of the cases of TMD and is associated with pain without apparent destructive changes of the TMJ on radiograph. It is frequently associated with bruxism and daytime jaw clenching in a stressed and anxious person. True intra-articular disease can be grouped under disk displacement disorder, chronic recurrent dislocations, degenerative joint disorders, systemic arthritic conditions, ankylosis, infections, and neoplasia.
Etiology
The etiology of MPD is multifactorial and includes malocclusion, jaw clenching, bruxism, personality disorders, increased pain sensitivity, and stress and anxiety. The principal factors responsible for the clinical manifestations in MPD (i.e. pain, tenderness, and spasm of the masticatory muscles) are muscular hyperactivity and dysfunction due to malocclusion of variable degree and duration. The significance of psychological factors has been recognized during the past few years. Of the causes of TMD of articular origin, disk displacement is the most common. Other diseases such as degenerative joint disorders, rheumatoid arthritis, ankylosis, dislocation, infection, neoplasia, and congenital anomalies may contribute to pain. In TMD of articular origin, the spasm of the masticatory muscles is secondary in nature. One study found that in patients with chronic inflammatory connective tissue disease, the pain on mandibular movement and tenderness to palpation of TMJ is related to the level of tumor necrosis factor alpha (TNF-α) in the synovial fluid.
Clinical Features
TMD primarily affects young women aged 20–40 years. The male-to-female ratio is 1:4. A comprehensive, chronological history and physical examination of the patient, including dental history and examination, is essential to diagnose the specific condition to decide further investigations, if any, and to provide specific treatment. There are four cardinal signs and symptoms of the syndrome: (1) pain, (ii) muscle tenderness, (iii) a clicking or popping noise in the TMJ, and (iv) limitation of jaw motion, unilaterally or bilaterally in approximately an equal ratio, sometimes with deviation on opening. The pain is usually periauricular, associated with chewing, and may radiate to the head but is not like the common headache. It may be unilateral or bilateral in MPD, and usually is unilateral in TMD of articular origin, except in rheumatoid arthritis. In MPD, the pain may be associated with history of bruxism, jaw clenching, stress, and anxiety; the pain may be more severe during periods of increased stress. Clicking, popping, and snapping sounds usually are associated with pain in TMD. An isolated click is very common in the general population and is not a risk factor for development of TMD. Limited jaw opening due to pain or disk displacement may be seen. TMD may act as a trigger in patients prone to headaches, and when present in association with TMD, they tend to be severe in nature. Other symptoms associated with TMD are otalgia, neck pain and/or stiffness, shoulder pain, and dizziness. About one third of these patients have a history of psychiatric problems. History of facial trauma, systemic arthritic disease, and recurrent dislocation also should be elicited. Differential diagnoses include cluster headache, migraine headache, post herpetic neuralgia, temporal/giant cell arteritis, trigeminal neuralgia and middle ear infections.
Laboratory Findings
Blood examination is required if systemic illness is suspected to be the cause of TMD. A complete blood count is done if infection is suspected. Rheumatoid factor (RF), ESR, antinuclear antibody (ANA), and other specific antibodies are checked if rheumatoid arthritis, temporal arteritis, or a connective tissue disorder is suspected. Uric acid should be checked for gout.
Radiographic Features
Radiographic findings in TMJ correlate to the etiology of TMD; in cases of rheumatoid arthritis and seronegative spondyloarthropathies, conventional radiographs show erosions, osteophytes, subchondral bony sclerosis, and condylar-glenoid fossa remodeling. A variety of new imaging techniques are being used and perfected to study TMJ. CT scan can explore both bony structures and muscular soft tissues. It is relatively less expensive and can be done with contrasting material injected into the joint cavity. MRI, though costly, should be used as the study of choice if an articular or meniscal pathology is suspected and an endoscopic or surgical procedure is contemplated in a case of traumatic TMD. Diagnostic arthroscopy is an invasive diagnostic approach. It should be used mainly in patients suffering from internal TMJ derangements resistant to conservative treatments. A good MRI study should be obtained before contemplating arthroscopy.
Treatment and Prognosis
Most TMDs are self-limiting. Conservative treatment involving self-care practices, rehabilitation aimed at eliminating muscle spasms, and restoring correct coordination, is all that is required. NSAIDs should be used on a short-term, regular basis. On the other hand, treatment of chronic TMD can be difficult and the condition is best managed by a team approach, consisting of a primary care physician, a dentist, a physiotherapist, a psychologist, a pharmacologist, and in small number of cases, a surgeon. The different modalities include patient education and self-care practices, medication, physical therapy, splints, psychological counseling, relaxation techniques, biofeedback, hypnotherapy, acupuncture, and arthrocentesis. Most cases of TMD respond to simple treatment and the prognosis is good. Symptoms usually remit with simple care. In cases of secondary involvement of TMJ, the prognosis depends on the primary disease.
Langerhans Cell Histiocytosis
Langerhans cell histiocytosis (LCH) is a disease that primarily affects bone but occasionally may also affect other organ systems and present in a multisystemic pattern. Despite advances in understanding the clinical picture, disease course and molecular profiling, the pathogenesis of LCH is still enigmatic. The use of different terminology to describe this condition adds to this puzzle and the search for its etiology. In the past the terms that have been used to define LCH and associated conditions include histiocytosis X, self-healing histiocytosis, pure cutaneous histiocytosis, Langerhans cell granulomatosis, type II histiocytosis and non-lipid reticuloendotheliosis. The term LCH is generally preferred to the older term, histiocytosis X. This new name emphasizes the histogenesis of the condition by specifying the type of lesional cell and removes the connotation of the unknown (X) because its cellular basis has now been clarified. Unique features of Langerhans cells revealed by cytoplasmic immunostaining with S100 antigen distinguishes them from other histiocytes. The presence of HX bodies or Birbeck granules (rod-shaped with characteristic periodicity and some times a dilated terminal end called tennis racket appearance, intracellular in location, identified by electron microscopy) and the presence of CD1a antigen on the cell surface and HLA-DR positivity confirms the Langerhans cell origin of this disease.
The annual incidence of LCH is reported at 5.4 million children per year (Bhatia S et al, 1997). Males are affected to slightly greater degree than females. It is predominantly a disease of childhood, with more than 50% of cases diagnosed between the ages of one and 15 years: there is a peak in the incidence between the ages of one and four. Although diagnosis is often made in childhood, many cases progress eventually into adult life.
The term histiocytosis is a collective designation for a variety of proliferative disorders of histiocytes or macro-phages. Some such as the rare histiocytic lymphomas are clearly malignant, whereas others such as reactive histiocytic proliferation in the lymph nodes are clearly benign. Between these two extremes lies the condition called Langerhans cell histiocytosis. It is a group of idiopathic disorders characterized by the clonal proliferation of specialized bone marrow-derived, antigen presenting dendritic cells called Langerhans cells (LCs) and mature eosinophils. The clinical spectrum includes on one end, an acute fulminant, disseminated disease called Letterer-Siwe disease, and on the other end, solitary or few, indolent and chronic lesions of bone or other organs called eosinophilic granuloma. The intermediate clinical form is called Hand-Schüller-Christian disease.
Pathogenesis
The pathogenesis of LCH has remained a mystery despite intensive search in this direction. Current theories suggest a role for environmental, infectious, immunologic and genetic causes; others believe that LCH is a neoplastic process. Flowcytometric sorting coupled with polymerase chain reaction (PCR) confirmed that the CD1a + Langerhans cells in LCH lesions are clonal. However, a clonal proliferation does not necessarily mean that LCH is a neoplastic process. The favorable natural history in most cases, the high probability of survival for patients older than two years of age, and the failure to detect aneuploidy or consistent karyotypic abnormalities support the idea that LCH is not a neoplastic process or malignancy. However, the proliferation of LCs may be a physiological rather than a pathologic response.
Langerhans cell histiocytosis may represent a reaction to a virus, but there is no convincing evidence at this point. A comprehensive analysis of nine different viruses by in situ hybridization and PCR failed to prove an association among 56 specimens obtained from osseous lesions and lymph nodes (McClain K et al, 1994). A role for HHV-6 has been suggested by other studies. One report detected HHV-6 in 14 of 30 (47%) pediatric cases of LCH using PCR on archival tissues (Leahy, MA et al, 1993). The abnormal immunological response noted in this disease state may be secondary to a viral infection of LCs and the lymphocytes. This also suggests that cells other than the pathologic LCs may play a key role in the pathogenesis of LCH. In fact there are other indirect evidences supporting a viral etiology. Aberrant or uncontrolled cytokine production is known to play a role in reactive histiocytic disorders. Likewise, LCH patients may abnormally produce at least 10 different cytokines (Egeler RM et al, 1999). Most of these are of T-cell origin and may explain the recruitment of LCs, other inflammatory cells, over expression of adhesion molecules, fibrosis, bone resorbtion, and necrosis. The presence of different kinds of chromosomal alterations as well as a high variability in the number of chromosomal breaks noted in this disease is consistent with a hypothesis of a viral etiology for LCH.
Treatment
Many cases demonstrate a favorable natural history without treatment. The major difficulty in treatment decision rests with reconciling the status of the disease as either reactive or neoplastic proliferation of LCs. Because of this there have been major discrepancies in the search to find appropriate treatment for patients.
Hand-Schüller-Christian Disease (Multifocal eosinophilic granuloma)
Hand-Schüller-Christian disease is characterized by widespread skeletal and extraskeletal lesions and a chronic clinical course. It occurs primarily in early life, usually before the age of five, but it has been reported in adolescents and even in young adults. It is more common in boys than in girls, with a gender ratio of approximately two to one.
Hand-Schüller-Christian disease has certain features in common with Letterer-Siwe disease and eosinophilic granuloma, and some features which serve to distinguish it from the others. In all three diseases, the proliferative cell is the histiocyte. In Hand-Schüller-Christian disease both the skeletal system and soft tissues may be involved, while in eosinophilic granuloma, only the bone is affected, although soft-tissue extension is often observed. Letterer-Siwe disease is an acute, fulminating disease with widespread lesions of both skeletal and extraskeletal tissues, including the skin.
Clinical Features
Hand-Schüller-Christian disease is characterized by the classic triad of single or multiple areas of ‘punchedout’ bone destruction in the skull, unilateral or bilateral exophthalmos, and diabetes insipidus with or without other manifestations of dyspituitarism such as polyuria, dwarfism, or infantilism. The complete triad occurs in only about 25% of affected patients. Involvement of the facial bones, which is frequently associated with soft-tissue swelling and tenderness, causes facial asymmetry. Otitis media is also common. Other bones are frequently involved, particularly the femur, ribs, vertebrae, pelvis, humerus, and scapula. Any of the visceral organs also may be involved, and the skin sometimes exhibits papular or nodular lesions, as discussed by Winkelmann.
Oral Manifestations
Oral manifestations may be one of the earliest signs of the presence of the disease. In reported series, the frequency of oral involvement varies widely, ranging from 5% to over 75% of patients.
These oral manifestations are often nonspecific and include sore mouths, with or without ulcerative lesions; halitosis gingivitis and suppuration; and unpleasant taste; loose and sore teeth with precocious exfoliation of teeth; and failure of healing of tooth sockets following extraction (Fig. 17-43). Loss of supporting alveolar bone mimicking advanced periodontal disease is characteristic, and this finding in a child should always be viewed with suspicion. The oral findings have been discussed by Sedano and his associates and by Jones and his coworkers.
Radiographic Features
The individual lesions, particularly in the skull, are usually sharply outlined, although the lesions in the jaws may be more diffuse. The lesions in the jaws are usually manifested simply as destruction of alveolar bone with tooth displacement (Fig. 17-43).
Histologic Features
Hand-Schüller-Christian disease is usually considered as manifesting in four stages during the progression of the characteristic lesion. These are:
• A proliferative histiocytic phase with accumulation of collections of eosinophilic leukocytes scattered throughout the sheets of histiocytes.
• A vascular-granulomatous phase with persistence of histiocytes and eosinophils, sometimes with aggregation of lipidladen (cholesterol) macrophages.
• A diffuse xanthomatous phase with abundance of these ‘foam cells’.
• A fibrous or healing phase (Fig. 17-47).
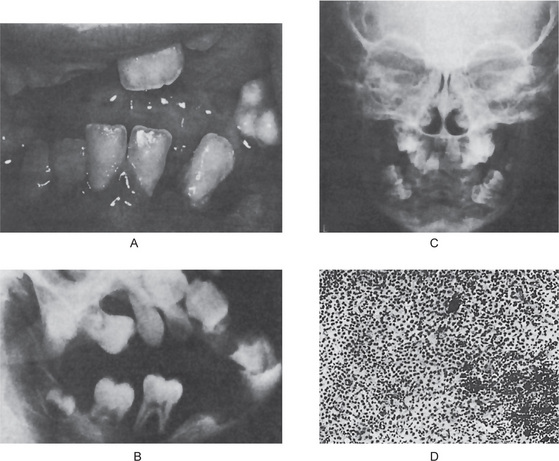
Figure 17-47 Hand-Schüller-Christian disease.
Clinically (A) the teeth are loose and have migrated. The gingiva is red, tender, swollen and hyperplastic. The radiographs (B, C) show lesions in the maxilla and mandible with severe loss of supporting bone. The microscopic appearance (D) is characteristic, showing sheets of proliferating histiocytes.
Laboratory Features
Anemia and, less frequently, leukopenia, and thrombocytopenia are occasionally found. The serum cholesterol level is nearly always normal, although the tissue cholesterol content may be elevated remarkably.
Treatment and Prognosis
The prognosis in Hand-Schüller-Christian disease is good. Approximately half of the patients undergo spontaneous remission over a period of years. The treatment of choice is curettage or excision of lesions. Inaccessible lesions may be irradiated. Some patients benefit from chemotherapeutic drugs, including prednisone, vinblastine, and cyclophosphamide. One of the most significant factors influencing the morbidity and mortality of the disease is the extent of the disease at the time of initial diagnosis and number of organ systems involved.
Eosinophilic Granuloma (Unifocal eosinophilic granuloma)
The term ‘eosinophilic granuloma of bone’ was introduced by Lichtenstein and Jaffe in 1940, although the lesion to which it referred had been described by others before them. The term is used to describe a lesion of bone which is primarily a histiocytic proliferation, with an abundance of eosinophilic leukocytes but no intracellular lipid accumulation. This disease occurs primarily in older children and young adults, and the proportion of males to females is about two to one.
Clinical Features
Clinically, the lesion may present no physical signs or symptoms and may be found only upon an incidental radiographic examination of the bones of the head or other areas. On the other hand, there may be local pain, swelling, and tenderness. The lesion may occur in the jaw and overlying soft tissues of the mouth, so that the differential diagnosis between eosinophilic granuloma and some form of dental disease becomes imperative. Although the skull and mandible are common sites of involvement, the femur, humerus, ribs, and other bones may also be affected. General malaise and fever occasionally accompany the eosinophilic granuloma of bone. The lesions are destructive and are well demarcated, roughly round or oval in shape. The area destroyed is replaced by a soft tissue, the composition of which varies, depending upon the stage at which the lesion is examined. The tissue of the early lesion is soft and brown and, since there is no necrosis, is not friable. Later the lesion becomes fibrous and grayish.
Radiographic Features
The lesions appear as irregular radiolucent areas usually involving superficial alveolar bone (Fig. 17-48). The cortex is often destroyed, and pathologic fractures may occur. If the lesions are in the jaw, they usually appear as single or multiple areas of rarefaction which may be so well circumscribed as to resemble cysts of the jaw, periapical granulomas or even periodontal disease (Fig. 17-49).
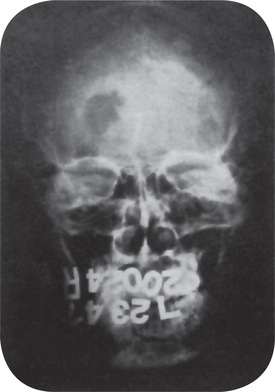
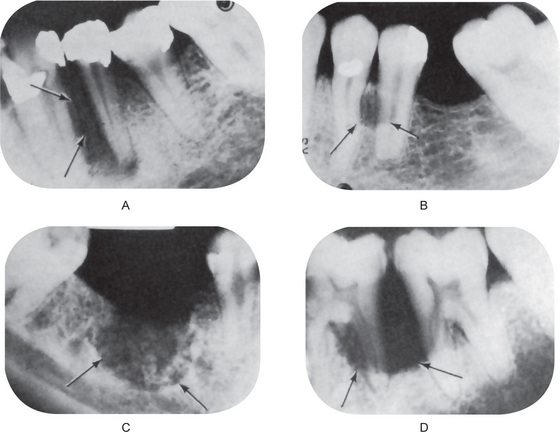
Histologic Features
Microscopically (Fig. 17-50), the primary cell is the histiocyte, which grows in sheets or sheetlike collections. The histiocytes may coalesce and form multinucleated giant cells, but this is quite uncommon. The early lesions also contain large numbers of focal collections of eosinophils. When the lesion matures, fibrosis occurs. In these older lesions, the eosinophils become less numerous, and they may even disappear so that the lesion approximates the histologic picture of Hand-Schüller-Christian disease.
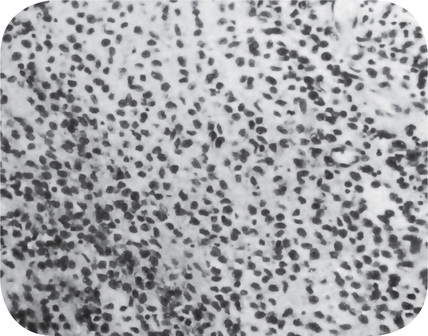
References
Ablin, D.S. Osteogenesis imperfecta: a review. Can Assoc Radiol J. 1998; 49(2):110–123. [Apr].
Afzal, A.R., Rajab, A., Fenske, C., Crosby, A., et al. Linkage of recessive Robinow syndrome to a 4 cM interval on chromosome 9q22. Human Genetics. 2000; 106:351–354.
Afzal, A.R., Rajab, A., Fenske, C., Oldridge, M., et al. Recessive Robinow syndrome: allelic to dominant brachydactyly type B is caused by mutations of ROR2. Nature Genetics. 2000; 25:419–422.
Agus, Z.S., Etiology of hypocalcemia. UpToDate CD-ROM. Wellesley, Mass: Up To Date Inc. 2000.
Ahmad, M., Hollender, L., Anderson, Q., et al. Research diagnostic criteria for temporomandibular disorders (RDC/TMD): development of image analysis criteria and examiner reliability for image analysis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009; 107(6):844–860. [Jun].
Albers-Schonberg, H. Roentgenbilder einer seltenen Knochennerkrankung. Munch Med Wochenschr. 51(365), 1904.
Albright, F., Butler, A.M., Hampton, A.D., Smith, P. Syndrome characterized by osteitis fibrosa disseminata, areas of pigmentation and endocrine dysfunction, with precocious puberty in females. New Engl J Med. 216(727), 1937.
Aldegheri, R. Distraction osteogenesis for lengthening of the tibia in patients who have limb-length discrepancy or short stature. J Bone Joint Surg Am. 1999; 81(5):624–634. [May].
Allanson, J.E. Germinal mosaicism in Apert syndrome. Clin Genet. 1986; 29(5):429–433. [May].
Altman, R.D., Brown, M., Gargano, F. Low back pain in Paget’s disease of bone. Clin Orthop. 1987; 217:152–161. [Apr].
American Academy of Pediatrics Committee on Genetics. Health supervision for children with achondroplasia. American Academy of Pediatrics Committee on Genetics. Pediatrics. 1995; 95(3):443–451. [Mar].
Anderson, D.C., Richardson, P.C., Brown, J.K. Intravenous pamidronate: evolution of an effective treatment strategy. Semin Arthritis Rheum. 1994; 23(4):273–275. [Feb].
Anderson, P.J., Netherway, D.J., Abbott, A., David, D.J. Intracranial volume measurement of metopic craniosynostosis. J Craniofac Surg, 11. 2004; 15(6):1014–1016.
Anderson, D.E., McClendon, J.L., Corneliu, E.A. Cherubism: hereditary fibrous dysplasia of the jaws I Genetic considerations II: pathologic considerations. Oral Surg. 1962; 15(Suppl 2):5.
Arabshahi, B., Cron, R.Q. Temporomandibular joint arthritis in juvenile idiopathic arthritis: the forgotten joint. Curr Opin Rheumatol. 2006; 18(5):490–495. [Sep].
Armstrong, D.G., Newfield, J.T., Gillespie, R. Orthopedic management of osteopetrosis: results of a survey and review of the literature. J Pediatr Ortho. 1999; 19(1):122–132. [Jan-Feb].
Arn, P.H., Mankinen, C., Jabs, E.W. Mild mandibulofacial dysostosis in a child with a deletion of 3p. Am J Med Genet. 1993; 46:534–536.
Arnalich, F., Plaza, I., Sobrino, J.A. Cardiac size and function in Paget’s disease of bone. Int J Cardiol. 1984; 5(4):491–505. [Apr].
Arnott, D.G. Cherubism—an initial unilateral presentation. Br J Oral Surg. 1978; 16(1):38–46.
Arthur, A. Rickets in the welfare state. N Z Med J. 2000; 113(1120):452. [Oct 27].
Aterman, K., Welch, J.P., Taylor, P.G. Presumed homozygous achondroplasia: a review and report of a further case. Pathol Res Pract. 1983; 178(1):27–39. [Aug].
Azouz, E.M., Teebi, A.S., Eydoux, P., et al. Bone dysplasias: an introduction. Can Assoc Radiol J. 1998; 49(2):105–109. [Apr].
Backstrom, M.C., Kuusala, A.L., Maki, R. Metabolic bone disease of prematurity. Ann Med. 1996; 28(4):275–282.
Baden, E., Spirgi, M. Oral manifestations of Marfan’s syndrome. Oral Surg. 19(757), 1965.
Baker, K.M., Olson, D.S., Harding, C.O., Pauli, R.M. Long-term survival in typical thanatophoric dysplasia type 1. Am J Med Genet. 1997; 70(4):427–436. [Jun 27].
Balemans, W., Patel, N., Ebeling, M., Van Hul, E., et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002; 39:91–97.
Balemans, W., Van Den, Ende J., Paes-Alves, A.F., Dikkers, F.G., et al. Localization of the gene for sclerosteosis to the van Buchem disease-gene region on chromosome 17q12-q21. Am J Hum Genet. 1999; 64:1661–1669.
Balestrazzi, P., Baeteman, M.A., Mattei, M.G., Mattei, J.F. Franceschetti syndrome in a child with a de novo balanced translocation (5, 13) (q11, p11) and significant decrease of hexosaminidase. B Hum Genet. 1983; 64:305–308.
Barcia, J.P., Strife, C.F., Langman, C.B. Infantile hypophosphatasia: treatment options to control hypercalcemia, hypercalciuria, and chronic bone demineralization. J Pediatr. 1997; 130(5):825–828. [May].
Barker, D., Welbury, R.R. Dental findings in Morquio syndrome (mucopolysaccharidoses type IVa. ASDC J Dent Child. 2000; 67(6):431–433. [407, Nov-Dec].
Barker, D.J.P., et al. Paget’s disease of bone: the Lancashire focus. Br Med J. 1956; 1:1105–1107.
Barkin, S., Weinberg, S. Internal derangements of the temporomandibular joint: the role of arthroscopic surgery and arthrocentesis. J Can Dent Assoc. Apr. 2000; 66(4):199–203.
Baroncelli, G.I., Federico, G., Bertelloni, S., et al. Assessment of bone quality by quantitative ultrasound of proximal phalanges of the hand and fracture rate in children and adolescents with bone and mineral disorders. Pediatr Res. 2003; 54(1):125–136. [Jul].
Bath, A.P., Bull, P.D. Management of upper airway obstruction in Pierre Robin sequence. J Laryngol Otol. 1997; 111(12):1155–1157. [Dec].
Bauze, R.J., Smith, R., Francis, M.J.O. A new look at osteogenesis imperfecta. J Bone Joint Surg, 57B: 1. 1975.
B Bedrune P Jammet C Chossegros. Temporomandibular joint pain- dysfunction syndrome after whiplash injury. Medico-legal problems in common law
Behrman RE, Arvin AM, Nelson WE, Kliegman RMNelson Textbook of Pediatrics (15th ed). WB Saunders, Philadelphia, 1996
Beighton, P., Hamersma, H., Cremin, B.J. Osteopetrosis in South Africa: the benign, lethal and intermediate forms. S Afr Med J, 21. 1979; 55(17):659–665. [Apr].
Bell, W.E. Clinical diagnosis of the pain-dysfunction syndrome. J Am Dent Assoc. 79(154), 1969.
Bell, W.H., Hinds, E.C. Fibrosarcoma complicating polyostotic fibrous dysplasia. Oral Surg. 23(299), 1967.
Bellus, G.A., Bamshad, M.J., Przylepa, K.A., et al. Severe achondroplasia with developmental delay and acanthosis nigricans (SAD DAN): phenotypic analysis of a new skeletal dysplasia caused by a Lys650Met mutation in fibroblast growth factor receptor 3. Am J Med Genet, 2. 1999; 85(1):53–65. [Jul].
Berg, C., Hanebuth, L., Paget’s disease. Nursing Care. Vol 10. 1977:25–26.
Bernstein, R.M., Zaleske, D.J. Familial aspects of Caffey’s disease. Am J Ortho. 1995; 24(10):777–781. [Oct].
Bhatt, S., Schreck, R., Graham, J.M., et al. Transient leukemia with trisomy. Am J Med Genet, 25 21: description of a case and review of the literature. 1995; 58(4):310–314. [Sep].
Bianco, P., Kuznetsov, S.A., Riminucci, M., Fisher, L.W., et al. Reproduction of human fibrous dysplasia of bone in immunocompromised mice by transplanted mosaics of normal and Gs-alpha-mutated skeletal progenitor cells. J Clin Invest. 1998; 101:1737–1744.
Bianco, P., Riminucci, M., Majolagbe, A., Kuznetsov, S.A., et al. Mutations of the GNAS1 gene, stromal cell dysfunction, and osteomalacic changes in non-McCune- Albright fibrous dysplasia of bone. J Bone Miner Res. 2000; 15:120–128.
Bieganski, T., Kozlowski, K. Brachytelephalangic chondrodysplasia punctata. Australas Radiol. 1998; 42(3):244–245. [Aug].
Bilezekian, J.P. Primary hyperparathyroidism. In Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, 3, Philadelphia: Williams and Wilkins; 1996:181–186.
Bisphosphonate therapy oral the cavity. The American Academy of Oral Medicine, Edmonds WA. 2008.
Bjorvatn, K., Gilhuus-Moe, O., Aarskog, D. Oral aspects of ospeopetrosis. Scand J Dent Res. 87(245), 1979.
Blackard, W.C., Robinson, R.R., White, J.E. Familial hypophosphatemia: report of a case, with observations regarding pathogenesis. New Engl J Med. 1962; 266:899–905.
Blank, E. Recurrent Caffey’s cortical hyperostosis and persistent deformity. Pediatrics. 55(856), 1975.
Bodo, M., Baroni, T., Carinci, F. Interleukin secretion, proteoglycan and procollagen alpha(1)(I) gene expression in Crouzon fibroblasts treated with basic fibroblast growth factor. Cytokine. 2000; 12(8):1280–1283. [Aug].
Boor, R., Fricke, G., Bruhl, K., Spranger, J. Abnormal subcortical somatosensory evoked potentials indicate high cervical myelopathy in achondroplasia. Eur J Pediatr. 1999; 158(8):662–667. [Aug].
Borgaonkar, D.S., Davis, M., Bolling, D.R., Herr, H.M. Evaluation of dermal patterns in Down's syndrome by predictive discrimination. I. Preliminary analysis based on frequencies of patterns. Johns Hopkins Med J. 1971; 128(3):141–152. [Mar].
Borochowitz, Z., Lachman, R., Adomian, G.E., et al. Achondrogenesis type I: delineation of further heterogeneity and identification of two distinct subgroups. J Pediatr. 1988; 112(1):23–31. [Jan].
Borochowitz, Z., Ornoy, A., Lachman, R., Rimoin, D.L. Achondrogenesis II- hypochondrogenesis: variability versus heterogeneity. Am J Med Genet. 1986; 24(2):273–288. [Jun].
Boucek, R.J., Noble, N.L., Gunja-Smith, Z., Butler, W.T. The Marfan syndrome: a deficiency in chemically stable collagen cross-links. New Eng J Med. 305(988), 1981.
Bower, C.M., Gungor, A. Pediatric obstructive sleep apnea syndrome. Otolaryngol Clin North Am. 2000; 33(1):49–75. [Feb].
Brooke, R.I., Stenn, P.G., Mothersill, K.J. The diagnosis and conservative treatment of myofascial pain dysfunction syndrome. Oral Surg. 44(844), 1977.
Brooksaler, F., Miller, J.E. Infantile cortical hyperostosis. J Pediatr. 48(739), 1956.
Brown, E.M., Harris, H.W., Vassilev, P.M. The biology of the extracellular Ca2+sensing receptor. In: Bilezikian J.P, ed. Principles of Bone Biology. San Diego Calif: Academic Press; 1996:243–262.
Brown, R.H., Cunningham, W.M. Some dental manifestations of mongolism. Oral Surg. 14(664), 1961.
Bruce, K.W., Bruwer, A., Kennedy, R.L.J. Familial intraosseous fibrous swellings of the jaws (‘cherubism’). Oral Surg. 6(995), 1953.
Brunvand, L., Brunvatne, R. Health problems among immigrant children in Norway. Tidsskr Nor Laegeforen. 2001; 121(6):715–718. [Feb 28].
Brussell, I.J. Temporomandibular joint diseases: differential diagnosis and treatment. J Am Dent Assoc. 39(532), 1949.
Bulas, D.I., Fonda, J.S. Prenatal evaluation of fetal anomalies. Pediatr Clin North Am. 1997; 44(3):537–553. [Jun].
Burbank, P.M., Lovestedt, S.A., Kennedy, R.L.J. The dental aspects of infantile cortical hyperostosis. Oral Surg. 11(1126), 1958.
Burkhart, J.M., Burke, E.C., Kelly, P.J. The chondrodystrophies. Mayo Clin Proc. 40(481), 1965.
Cabral, C.E., Guedes, P., Fonseca, T. Polyostotic fibrous dysplasia associated with intramuscular myxomas: Mazabraud’s syndrome. Skeletal Radiology. 1998; 27:278–282.
Caffey, J. Infantile Cortical Hyperostoses. J Pediatr. 1946; 29:541–559.
Caffey, J., Silverman, W.A. Infantile cortical hyperostosis: preliminary report on new syndrome. Am J Roentgenol. 54(1), 1945.
Cahn, L.R. Leontiasis ossea. Oral Surg. 6(201), 1953.
Cai, X.Y., Yang, C., Zhang, Z.Y., Qiu, W.L., Chen, M.J., Zhang, S.Y. Septic arthritis of the temporomandibular joint: a retrospective review of 40 cases. J Oral Maxillofac Surg. 2010; 68(4):731–738. [Apr].
Caillaud, C., Poenaru, L. Gene therapy in lysosomal diseases. Pharmacother Biomed. 2000; 54(10):505–512. [Oct].
Candeliere, G.A., Glorieux, F.H., Prud’Homme, J., St-Arnaud, R. Increased expression of the c-fos proto-oncogene in bone from patients with fibrous dysplasia. New Eng J Med. 1995; 332:1546–1551.
Caouette-Laberge, L., Plamondon, C., Larocque, Y. Subperiosteal release of the floor of the mouth in Pierre Robin sequence: experience with 12 cases. Cleft Palate Craniofac J. 1996; 33(6):468–472. [Nov].
Carmi, R., et al. Use of a DNA pooling strategy to identify a human obesity syndrome locus on chromosome 15. Hum Molec Genet. 1995; 4:9–13.
Carson, I.H. Polyostotic fibrous dysplasia, report of case. Oral Surg. 7(524), 1954.
Cascone P, Spallaccia F, Rivarolli A. Arthrocentesis of the temporomandibular joint. Long-term results.
Cayler, G.G., Peterson, C.A. Infantile cortical hyperostosis. Am J Dis Child. 91(119), 1956.
CDC. From the Centers for Disease Control and Prevention. Severe malnutrition among young children—Georgia, January 1997-June 1999. J Am Med Assoc. 2001; 285(20):23–30. [2573-74 May].
Chakravarty, K., Merry, P., Scott, D.G. A single infusion of bisphosphonate AHPrBP in the treatment of Paget’s disease of bone. J Rheumatol. 1994; 21(11):2118–2121. Nov
Chan, D., Cole, W.G., Chow, C.W., et al. A COL2A1 mutation in achondrogenesis type II results in the replacement of type II collagen by type I and III collagens in cartilage. J Biol Chem. 1995; 270(4):1747–1753. [Jan 27].
Chanson, P., Dib, A., Visot, A., Derome, P.J. McCune-Albright syndrome and acromegaly: clinical studies and responses to treatment in five cases. Europ J Endocr. 1994; 131:229–234.
Chapman, S., Hall, C.M. Non-accidental injury or brittle bones. Pediatr Radiol. 1997; 27(2):106–110. [Feb].
Chen, C.P., Chern, S.R., Wang, W. Second-trimester molecular diagnosis of a heterozygous 742 —> T (R248C) mutation in the FGFR3 gene in a thanatophoric dysplasia variant following suspicious ultrasound findings. Ultrasound Obstet Gynecol. 2001; 17(3):272–273. [Mar].
Chen, H., Liu, C.T., Yang, S.S. Achondrogenesis: a review with special consideration of achondrogenesis type II (Langer-Saldino. Am J Med Genet. 1981; 10(4):379–394.
Chen, J.R., Rhee, R.S., Wallach, S. Neurologic disturbances in Paget’s disease of bone: response to calcitonin. Neurology. 1979; 29(4):448–457. [Apr].
Chesney, R.W., Mazess, R.B., Rose, P. Long-term influence of calcitriol (1,25- dihydroxyvitamin D) and supplemental phosphate in X-linked hypophosphatemic rickets. Pediatrics. 1983; 71(4):559–567. [Apr].
Chitayat, D., Fernandez, B., Gardner, A., et al. Compound heterozygosity for the Achondroplasia-hypochondroplasia FGFR3 mutations: prenatal diagnosis and postnatal outcome. Am J Med Genet. 1999; 84(5):401–405. [Jun 11].
Chossegros C, Cheynet F, Blanc JL et al. Diagnostic temporomandibular arthroscopy. Principle lesion, apropos of 50 case reports.
Chowers, I, Czackes, W, Ehrenfeld, EN, Landau, S. Familial aminoaciduria in osteogenesis imperfecta. J Am Med Assoc. 181(771), 1962.
Church, L.E. Polyostotic fibrous dysplasia of bone. Oral Surg. 11(184), 1958.
Clarke, P.R., Williams, H.I. Ossification in extradural fat in Paget’s disease of the spine. Br J Surg. 1975; 62(7):571–572. [Jul].
Craniosynostosis. In: Cohen M.M., Jr., eds. Diagnosis, evaluation, and management. New York: Raven Press, 1986.
Cohen, M.M., Jr., Kreiborg, S., Lammer, E.J., Cordero, J.F., et al. Birth prevalence study of the Apert syndrome. Am J Med Genet. 1992; 42(5):655–659. [Mar 1].
Cohen, M.M., Jr., Kreiborg, S. An updated pediatric perspective on the Apert syndrome. Am J Dis Child. 1993; 147(9):989–993. [Sep].
Cohen, M.M., Jr., Kreiborg, S. Cutaneous manifestations of Apert syndrome. Am J Med Genet. 1995; 58(1):94–96. [Jul 31].
Cohen, M.M., Jr., Kreiborg, S. Hands and feet in the Apert syndrome. Am J Med Genet. 1995; 57(1):82–96. [May 22].
Cohen, M.M., Jr., Kreiborg, S. Skeletal abnormalities in the Apert syndrome. Am J Med Genet. 1993; 47(5):624–632. [Oct 1].
Cohen, M.M., Jr., Kreiborg, S. The central nervous system in the Apert syndrome. Am J Med Genet. 1990; 35(1):36–45. [Jan].
Cohen, M.M., Jr., Kreiborg, S. Visceral anomalies in the Apert syndrome. Am J Med Genet. 1993; 45(6):758–760. [Mar 15].
Cohen, M.M., Jr. Craniosynostoses: phenotypic/molecular correlations. Am J Med Genet. 1995; 56(3):334–339. [Apr 10].
Cohen, M.M., Jr. Craniosynostosis update 1987. Am J Med Genet Suppl. 1988; 4:99–148.
Cohen, M.M., Jr. Let’s call it ‘Crouzonodermoskeletal syndrome’ so we won’t be prisoners of our own conventional terminology. Am J Med Genet, 7. 1999; 84(1):74. [May].
Cohen, B.H., Lilienfeld, A.M., Sigler, A.T. Some epidemiological aspects of mongolism: a review. Am J Public Health. 54(223), 1963.
Cohen, J. Osteopetrosis. J Bone Joint Surg, 33A: 923. 1951.
Cohen, M.M., Jr. The Robin anomalad—its nonspecificity and associated syndromes. J Oral Surg. 34(587), 1976.
Cohen, M.M., Winer, R.A., Schwartz, S., Shklar, G. Oral aspects of mongolism I: periodontal disease in mongolism. Oral Surg. 14(92), 1961.
Cole DEC. Personal Communication. Toronto, Canada, 2/17/1996.
Cole, D.E.C., Fraser, F.C., Glorieux, F.H., Jequier, S., et al. Panostotic fibrous dysplasia: a congenital disorder of bone with unusual facial appearance, bone fragility, hyperphosphatasemia, and hypophosphatemia. Am J Med Genet. 1983; 14:725–735.
Comroe, B.I. Arthritis and Allied Conditions (3rd ed, 3, Philadelphia: Lea and Febiger, 1948.
Congleton, J., Burkes, E.J. Amelogenesis imperfecta with taurodontism. Oral Surg Oral Med Oral Path. 1979; 48:540–544.
Cooke, B.E.D. Paget’s disease of the jaws: 15 cases. Ann R Coll Surg Engl. 19(223), 1956.
Cooper, S.C., Flaitz, C.M., Johnston, D.A., Lee, B., et al. A natural history of cleidocranial dysplasia. Am J Med Genet. 2001; 104:1–6.
Cornelius, E.A., McClendon, J.L. Cherubism—hereditary fibrous dysplasia of the jaws: roentgenographic features. Am J Roentgenol Radium Ther Nucl Med. 106(136), 1969.
Cossio Lozano, G.E. Thesis in Medical Genetics. Hospital Infantil de Mexico. 1990.
Crawford JL. Concomitant taurodontismamelogenesis imperfecta in the American Caucasian. Dent Child J. 1970; 37:83–87.
Crawford, P.J.M., Aldred, M.J. Amelogenesis imperfecta with taurodontism and the tricho-dento-osseous syndrome: separate conditions or a spectrum of disease? Clin Genet. 1990; 38:44–50.
Crawford, P.J.M., Evans, R.D., Aldred, M.J. Amelogenesis imperfecta: autosomal dominant hypomaturation-hypoplasia type with taurodontism. Dent J Br. 1988; 164:71–73.
Crisp, A.J., Smith, M.L., Skingle, S.J. The localization of the bone lesions of Paget’s disease by radiographs, scintigraphy and thermography: pain may be related to bone blood flow. Br J Rheumatol. 1989; 28(3):266–268. [Jun].
Cros P, Freidel M, BorieJ et al. 15 years of treatment of temporomandibular joint algodysfunctional syndromes.
Cuerda, E., del Pozo, J., Rodriguez-Lozano, J., et al. Acne in Apert’s syndrome: treatment with isotretinoin. J Dermatolog Treat. 2003; 14(1):43–45. [Jan].
Dabezies, E.J., Warren, P.D. Fractures in very low birth weight infants with rickets. Clin Orthop, (335. 1997; 233–239. [Feb].
Daffner, R.H., Kirks, D.R., Gehweiler, J.A., Jr. Computed tomography of fibrous dysplasia. AJR Am J Roentgenol. 1982; 139(5):943–948. [Nov].
Davies, D.G. Paget’s disease of the temporal bone. A clinical and histopathological survey. Acta Otolaryngol, Suppl. 242(3), 1968.
Davis, J.P. A cephalometric investigation of cleidocranial dysplasia Master’s thesis. Indiana University School of Dentistry. 1974.
de Deuxchaisnes, C.N., Krane, S.M. Paget’s disease of bone: clinical and metabolic observations. Medicine. 43(233), 1964.
de Filippis C, Osti L, Osti R et al. Algodystrophic syndrome of the temporomandibular joint:a clinical experience.
De Smet, A., Travers, H., Neff, JR. Chondrosarcoma occurring in a patient with polyostotic fibrous dysplasia. Radiol Skeletal. 7(197), 1981.
Delatycki, M., Rogers, J.G. The genetics of fibrodysplasia ossificans progressiva. Clin Orthop, (346. 1998; 15–18. [Jan].
Delezoide, A.L., Lasselin-Benoist, C., Legeai-Mallet, L., et al. Abnormal FGFR 3 expression in cartilage of thanatophoric dysplasia fetuses. Hum Mol Genet. 1997; 6(11):1899–1906. [Oct].
Delmas, P.D. Biochemical markers of bone turnover in Paget’s disease of bone. J Bone Miner Res, 14 (Suppl 2. 1999; 66–69. [Oct].
Demitsu, T., Kakurai, M., Okubo, Y., et al. Skin eruption as the presenting sign of Hunter syndrome IIB. Clin Exp Dermatol. 1999; 24(3):179–182. [May].
Desmons, F., Bar, J., Brandt, A. Les signes cutanes du mongolisme (trisomie 21. Bull Soc fr Dermatol et Syphiligr. 1973; 80:233–237.
Deutsch, E.S. Tonsillectomy and adenoidectomy. Changing indications. Pediatr Clin North Am. 1996; 43(6):1319–1338. [Dec].
Dickinson, C.J. The possible role of osteoclastogenic oral bacterial products in etiology of Paget’s disease. Bone. 2000; 26(2):101–102. [Feb].
Dietz, H.C., Cutting, G.R., Pyeritz, R.E. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature, 25. 1991; 352(6333):337–339. [Jul].
Dingman, R.O. Diagnosis and treatment of lesions of the temporomandibular joint. Am J Ortho Oral Surg. 26(388), 1940.
Dionisopoulos, T., Williams, H.B. Congenital Anomalies of the Ear, Nose and Throat. Oxford University Press, New York. 1997; 243–260.
Dixon, P.H., Christie, P.T., Wooding, C. Mutational analysis of PHEX gene in X-linked hypophosphatemia. J Clin Endocrinol Metab. 1998; 83(10):3615–3623. [Oct].
Do Amaral, C.M.R, Di Domizio, G., Buzzo, C.L. Surgical treatment of Apert syndrome and Crouzon anomaly by gradual bone distraction. Online J Plast Reconstr Surg. 1, 1998.
Do, T.T. Clinical and radiographic evaluation of bowlegs. Curr Opin Pediatr. 2001; 13(1):42–46. [Feb].
Dourmishev, A., Miteva, L., Mitev, V., et al. Cutaneous aspects of Down syndrome. Cutis. 2000; 66(6):420–424. [Dec].
Dove, J. Complete fractures of the femur in Paget’s disease of bone. J Bone Joint Surg Br, 62-B(1. 1980; 12–17. [Feb].
Down, J.L. Observations on an ethnic classification of idiots. 1866. Ment Retard. 1995; 33(1):54–56. [Feb].
Drury, B.J. Paget’s disease of the skull and facial bones. J Bone Joint Surg, 44A: 174. 1962.
Dundulis, J.A., Becker, D.B., Govier, D.P., et al. Coronal ring involvement in patients treated for unilateral coronal craniosynostosis. Plast Reconstr Surg. 2004; 114(7):1695–1703. [Dec].
Dunn, F.H. Nonfamilial and nonhereditary craniofacial dysostosis: a variant of Crouzon’s disease. Am J Roentgenol Radium Ther Nucl Med. 84(472), 1960.
Duthie, R.B., Townes, P.L. The genetics of orthopaedic conditions. J Bone Joint Surg, 49B: 229. 1967.
Dyson, D.P. Osteomyelitis of the jaws in Albers-Schonberg disease. Br J Oral Surg. 7(178), 1970.
Edelson, J.G., Obad, S., Geiger, R., On, A., Artul, H.J. Pyknodysostosis: orthopedic aspects with a description of 14 new cases. Clin Ortho, 280. 1992; 273–276.
El, Deeb M, Waite, D.E., Gorlin, R.J. Congenital monostotic fibrous dysplasia—a new possibly autosomal recessive disorder. J Oral Surg. 37(520), 1979.
M., El Deeb, Waite, D.E., Jaspers, M.T. Fibrous dysplasia of the jaws report of five cases. Oral Surg. 47(312), 1967.
El Samma, M., et al. Calcitonin as treatment for hearing loss in Paget’s disease. Am J Otolaryngol. 1986; 7:241–243.
Elliott, M.A., Studen-Pavlovich, D.A., Ranalli, D.N. Prevalence of selected pediatric conditions in children with Pierre Robin sequence. Pediatr Dent. 1995; 17(2):106–111. [Mar-Apr].
Ellis, D.J., Adams, T.O. Massive osteolysis: report of a case. J Oral Surg. 29(659), 1971.
Elmore, S.M. Pycnodysostosis: a review. J Bone Joint Surg, 49A: 153. 1967.
Elmslie, F.V., W., Reardon. Craniofacial developmental abnormalities. Curr Opin Neurol. 1998; 11(2):103–108. [Apr].
el-Tawil, T., Stoker, D.J. Benign osteopetrosis: a review of 42 cases showing two different patterns. Skeletal Radiol. 1993; 22(8):587–593. [Nov].
Engel, M.B., Brodie, A.G. Condylar growth and mandibular deformities. Oral Surg. 1(790), 1948.
Ercis, M., Balci, S., Atakan, N. Dermatological manifestations of 71 Down syndrome children admitted to a clinical genetics unit. Clin Genet. 1996; 50(5):317–320. [Nov].
Eretto, P., Krohel, G.B., Shihab, Z.M. Optic neuropathy in Paget’s disease. Am J Ophthalmol. 1984; 97(4):505–510. [Apr].
Erneyi, S. Craniofacial dysplasia associated with congenital cataracta, impairment of hearing and brachydactyly. A J Ophthalmol. 1966; 62:697–702.
Esposito EJ, Panucci PJ, Farman AG. Associations in 425 patients having temporomandibular disorders.
Eversole, L.R., Sabes, W.R., Rovin, S. Fibrous dysplasia: a nosologic problem in the diagnosis of fibroosseous besions of the jaws. Oral Path J. 1(189), 1972.
Ey-Chmielewska H. An attempt to use ultrasonic technique for confirming the diagnosis, planning and observation of long term treatment results of painful temporo-mandibular joint dysfunction.
Eyre, D.R., Upton, M.P., Shapiro, F.D., et al. Nonexpression of cartilage type II collagen in a case of Langer-Saldino achondrogenesis. Am J Hum Genet. 1986; 39(1):52–67. [Jul].
Falvo, K.A., Root, L., Bullough, P.G. Osteogenesis imperfecta: clinical evaluation and management. J Bone Joint Surg, 56A: 783. 1974.
Fassauer H, Bethmann W, Begemeier I. Diseases of the temporomandibular joint-a clinical statistical study.
Feingold, M., Schneller, S. Down syndromesystemic lupus erythematosus. Genet Clin. 1995; 48(5):277. [Nov].
Felix, R., Hofstetter, W., Cecchini, M.G. Recent developments in the understanding of the pathophysiology of osteopetrosis. Eur J Endocrinol. 1996; 134(2):143–156. [Feb].
Fernandez, A.O., Ronis, M.L. The Treacher-Collins syndrome. Arch Otolaryngol. 80(505), 1964.
Fernbach, S.K. Craniosynostosis 1998: concepts and controversies. Pediatr Radiol. 1998; 28(9):722–728. [Sep].
Feshchenko, S.P., Rebrin, I.A., Sokolnik, V.P., et al. The absence of type II collagen and changes in proteoglycan structure of hyaline cartilage in a case of Langer- Saldino achondrogenesis. Hum Genet. 1989; 82(1):49–54. [Apr].
Fitzpatrick, L.A., Arnold, A. Hypoparathyroidism. In: DeGroot L.J., ed. Endocrinology. Philadelphia: WB Saunders; 1995:1123–1135.
FOA, Rickets and osteomalacia. Food and Agriculture Organization of the United Nations Web site. 2002 2002; Available at: http://www.fao.org/. Accessed April 4
Franceschetti, A., Klein, D. The mandibulo-facial dysostosis: a new hereditary syndrome Acta Ophthalmol (Kbh). 1949;27(143).
Fu, H., Samulski, R.J., McCown, T.J., et al. Neurological correction of lysosomal storage in a mucopolysaccharidosis IIIB mouse model by adeno-associated virus- mediated gene delivery. Mol Ther. 2002; 5(1):42–49. [Jan].
Gallegos-Arreola, M.P., Machorro-Lazo, M.V., Flores-Martinez, S.E., et al. Urinary glycosaminoglycan excretion in healthy subjects and in patients with mucopolysaccharidoses. Arch Med Res. 2000; 31(5):505–510. [Sep-Oct].
Gallucci M, Bozzao A, Splendiani A et al. Magnetic resonance in condylo-meniscal incoordination pathology of the temporomandibular joint. Indications, diagnostic accuracy and optimization of study techniques.
Gardner, A.F., Halpert, L. Fibrous dysplasia of the skull with special reference to the oral regions. Dent Pract Dent Rec. 13(337), 1949.
Garjian, K.V., Pretorius, D.H., Budorick, N.E., et al. Fetal skeletal dysplasia: three-dimensional US—initial experience. Radiology. 2000; 214(3):717–723. [Mar].
Genuth, S.M., Klein, L. Hypoparathyroidism and Paget’s disease: the effect of parathyroid hormone administration. J Clin Endocrinol Metab. 1972; 35(5):693–699. [Nov].
Gerry, R.G. Effects of trauma and hypermotility on the temporomandibular joint. Oral Surg. 7(876), 1954.
Gines, E., Rodriguez-Pichardo, A., Jorquera, E. Crouzon disease with acanthosis nigricans and melanocytic nevi. Pediatr Dermatol. 1996; 13(1):18–21. [Jan-Feb].
Girschick, H.J., Schneider, P., Kruse, K., Huppertz, H.I. Bone metabolism and bone mineral density in childhood hypophosphatasia. Bone. 1999; 25(3):361–367. [Sep].
Glickman, I. Fibrous dysplasia in alveolar bone. Oral Surg. 1(895), 1948.
Glorieux, F.H., Scriver, C.R., Reade, T.M. Use of phosphate and vitamin D to prevent dwarfism and rickets in X-linked hypophosphatemia. New Engl J Med, 7. 1972; 287(10):481–487. [Sep].
Golan, I., Baumert, U., Held, P., Feuerbach, S., Mubig, D. Radiological findings and molecular genetic confirmation of cleidocranial dysplasia. Clin Radiol. 2001; 56:525–529.
Gold, L. The classification and pathogenesis of fibrous dysplasia of the jaws. Oral Surg. 8: 628, 725, 856, 1955.
Goldenberg, R.R. The skull in Paget’s disease. J Bone Joint Surg, 33A: 911. 1951.
Goldman, A.B., Bullough, P., Kammerman, S. Osteitis deformans of the hip joint. AJR Am J Roentgenol. 1977; 128(4):601–606. [Apr].
Goltzman, D., Cole, D.E.C. Hypoparathyroidism. In: Favus M.J., ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Philadelphia: Lippincott- Raven; 1996:220–223.
Goodman, W.G., Coburn, J.W., Slatopolsky, E. Renal osteodystrophy in adults and children. In Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, 3, Philadelphia: Williams and Wilkins; 1996:341–360.
Gorham, L.W., Stout, A.P. Massive Osteolysis (Acute spontaneous absorption of bone, phantom bone, disappearing bone. J Bone Joint Surg, Boston, 15-A. 1955; 985–1004.
Gorham, L.W., Wright, A.W., Schultz, H.H., Maxon, F.C. Disappearing bones: a rare form of massive osteolysis, report of two cases, one with autopsy findings. Am J Med, New York. 1954; 17:674–681.
Gorlin, R.J., Jue, K.L., Jacobsen, U., Goldschmidt, E. Oculoauriculovertebral dysplasia. J Pediatr. 63(991), 1963.
Gorlin, R.J., Pindborg, J.J., Cohen, M.M., Jr. Syndromes of the Head and Neck (2nd ed. McGraw-Hill, New York. 1976.
Gott, V.L., Cameron, D.E., Pyeritz, R.E. Composite graft repair of Marfan aneurysm of the ascending aorta: results in 150 patients. J Card Surg. 1994; 9(5):482–489. [Sep].
Granat O, Pharaboz C, Gerber S et al. The diagnoastic importance of different imaging technics in temporomandibular joint dysfunction.
Greene, C.S., Laskin, D.M. Long-term evaluation of conservative treatment for myofascial pain-dysfunction syndrome. Am Dent Assoc J. 89(1365), 1974.
Greene, C.S., Lerman, M.D., Sutcher, H.D., Laskin, D.M. The TMJ pain-dysfunction syndrome: heterogeneity of the patient population. J Am Dent Assoc. 79(1168), 1969.
Grey, A., Mitnick, M.A., Masiukiecwicz, U. A role for interleukin-6 in parathyroid hormone-induced bone resorption in vivo. Endocrinology. 1999; 140:4683–4690.
Guarda-Nardini, L., Piccotti, F., Ferronato, G., Manfredini, D. Synovial chondromatosis of the temporomandibular joint: a case description with systematic literature review. Int J Oral Maxillofac Surg. 2010; 39(8):745–755. [Aug].
Gutman, A.B., Tyson, T.L., Gutman, E.B. Serum calcium, inorganic phosphorus and phosphatase activity in hyperparathyroidism, Paget’s disease, multiple myeloma and neoplastic diseases of the bone. Arch Intern Med. 57(379), 1936.
Gyapay, G., et al. The. Nature Genet. 7. 1994; 246–339. [1993-1994 Genethon human genetic linkage map.].
Haapanen, M.L., Laitinen, S., Paaso, M., Ranta, R. Quality of speech correlated to craniofacial characteristics of cleft palate patients with the Pierre Robin sequence. Folia Phoniatr Logop. 1996; 48(5):215–222.
Hammer III, J.E. The demonstration of perivascular collagen deposition in cherubism. Oral Surg. 27(129), 1969.
Handzic-Cuk, J., Cuk, V., Gluhinic, M. Mastoid pneumatization and aging in children with Pierre-Robin syndrome and in the cleft palate population out of syndrome. Eur Arch Otorhinolaryngol. 1999; 256(1):5–9.
Harris, W.H., Dudy, H.R., Jr., Barry, R.J. The natural history of fibrous dysplasia. J Bone Joint Surg Am. 44(207), 1962.
Harris, V.J., Ramilo, J. Caffey’s disease: a case originating in the first metatarsal and review of a 12-year experience. AJR. 130(335), 1978.
Harris, W.H., Dudley, H., Barry, R.J. Natural history of fibrous dysplasia An orthopedic, pathological, and roentgenographic study. J Bone Joint Surg, 44A: 207. 1962.
Hart, T.C., Bowden, D.W., Bolyard, J., Kula, K., et al. Genetic linkage of the tricho-dento- osseous syndrome to chromosome 17q21. Hum Molec Genet. 1997; 6:2279–2284.
Hasenhuttl, K. Osteopetrosis. J Bone Joint Surg, 44A: 359. 1962.
Heath, H., III., Hobbs, M.R. Familial hyperparathyroid syndromes. In Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, 3, Philadelphia: Williams and Wilkins; 1996:187–189.
Herring, J. Infantile cortical hyperostosis. In: Tachdjiana’s Pediatric Orthopaedics. Philadelphia: WB Saunders; 1996:1561–1565.
Heys, F.M., Blattner, R.J., Robinson, H.B.G. Osteogenesis imperfecta and odontogenesis imperfecta: clinical and genetic aspects in eighteen families. J Pediatr. 56(234), 1960.
Higginbottom, M.C., Jones, K.L., James, H.E. Intrauterine constraint and craniosynostosis. Neurosurgery. 1980; 6(1):39–44. [Jan].
Ho, K.L., Chang, C.H., Yang, S.S., Chason, J.L. Neuropathologic findings in thanatophoric dysplasia. Acta Neuropathol (Berl. 1984; 63(3):218–228.
Horton, C.P. Treatment of arthritic temporomandibular joint by intra-articular injection of hydrocortisone. Oral Surg. 6(826), 1953.
Hutter, R.V.P., Foote, F.W., Jr., Frazell, E.L., Francis, K.C. Giant cell tumors complicating Paget’s disease of bone. Cancer. 16(1044), 1963.
Hutton, C.E., Bixler, D., Garner, L.D. Cleidocranial dysplasia—treatment of dental problems: report of case. J Dent Child. 48(456), 1981.
Ignatowicz, R., Gdakowicz, B. Dysostoza czaszkowo-twarzowa Crouzona. Wiadomosci Lekarskie. 1971; 24:363–366.
Irving J, Wood GD, Hackett AF. Dose temporomandibular disorder pain dysfunction syndrome affect dietary intake?
Jabs, E.W. Toward understanding the pathogenesis of craniosynostosis through clinical and molecular correlates. Clin Genet. 1998; 53(2):79–86. [Feb].
Jaffe, H.L., Lichtenstein, L. Non-osteogenic fibroma of bone. Am J Pathol. 18(205), 1942.
Jaffe, H.L., Lichtenstein, L., Portis, R. Giant cell tumor of bone, its pathologic appearance, grading, supposed variants and treatment. Arch Pathol. 30(993), 1940.
Jaffe, H.L. Paget’s disease of bone. Arch Pathol. 15(83), 1933.
Jensen, B.L. Cleidocranial dysplasia: craniofacial morphology in adult patients. J Craniofac Genet Dev Biol. 1994; 14:163–176.
Johnston, C.C., Jr., Lavy, N., Lord, T., Vellios, F., et al. Osteopetrosis A clinical, genetic, metabolic and morphologic study of the dominantly inherited, benign form Medicine. 1968;47(149).
Jones, W.A. Familial multilocular cystic disease of the jaws. J Cancer Am. 1933; 17(4):946–950.
Jones, W.A., Gerrie, J., Pritchard, J. Cherubism: a familial fibrous dysplasia of the jaws. J Bone Joint Surg, 32B: 334. 1950.
Kabukcuoglu, F., Kabukcuoglu, Y., Yilmaz, B., Erdem, Y., et al. Mazabraud’s syndrome: intramuscular myxoma associated with fibrous dysplasia: Pathol Oncol Res. Epub Jun 9. 2004; 10(2):121–123. [2004].
Kainer, G., Chan, J.C. Hypocalcemic and hypercalcemic disorders in children. Curr Probl Pediatr. 1989; 19(10):489–545. [Oct].
Kalliala, E., Taskinen, P.J. Cleidocranial dysostosis. Oral Surg. 15(808), 1962.
Kaneyama, K., Segami, N., Hatta, T. Congenital deformities and developmental abnormalities of the mandibular condyle in the temporomandibular joint. Congenit Anom (Kyoto), Sep. 2008; 48(3):118–125.
Kaslick, R.S., Brustein, H.C. Clinical evaluation of osteopetrosis. Oral Surg. 15(71), 1962.
Kelln, E.E., Chaudhry, A.P., Gorlin, R.J. Oral manifestations of Crouzon’s disease. Oral Surg. 13(1245), 1960.
Kery, L., Wouters, H.W. Massive osteolysis: report of two cases. J Bone Joint Surg, 52B: 452. 1970.
Key, L., Carnes, D., Cole, S. Treatment of congenital osteopetrosis with high-dose calcitriol. New Engl J Med, 16. 1984; 310(7):409–415. [Feb].
Khosla, S., Melton, L.J., III., Wermers, R.A. Primary hyperparathyroidism and the risk of fractures: A population-based study. J Bone Miner Res. 1999; 14:1700–1707.
Kifor, O., Moore, F.D., Jr., Wang, P. Reduced immunostaining for the extracellular Ca2+- sensing receptor in primary and uremic secondary hyperparathyroidism. J Clin Endocrinol Metab. 1996; 81(4):1598–1606. [Apr].
King, M., Payne, W.S., Olfasson, S., et al. Surgical palliation of respiratory insufficiency secondary to massive exuberant polyostotic fibrous dysplasia of ribs. Ann Thoracic Surg. 39(185), 1985.
King, J.D., Bobechko, W.P. Osteogenesis imperfecta: an orthopaedic description and surgical review. J Bone Joint Surg, 53B: 72. 1971.
Kneal, K., Sante, L.R. Osteopetrosis (marble bones. Am J Dis Child. 81(693), 1951.
Kolble, N., Sobetzko, D., Ersch, J. Diagnosis of skeletal dysplasia by multidisciplinary assessment: a report of two cases of thanatophoric dysplasia. Ultrasound Obstet Gynecol. 2002; 19(1):92–98. [Jan].
Kowalski, M., Paszkowska, M., Bryjanowska, L. A case of Crouzon’s syndrome with coexistance of other congenital anomalies (author’s transl. Klin Oczna. 1977; 47(10):445–447. [Oct].
Krane, S.M., Hollick, M.F. metabolic bone disease. Harrison’s principles of internal medicine (12th ed. 1991; 1921–1931.
Kreiborg, S., Cohen, M.M., Jr. Characteristics of the infant Apert skull and its subsequent development. J Craniofac Genet Dev Biol. 1990; 10(4):399–410.
Kreiborg, S., Jensen, B.L., Larsen, P., Schleidt, D.T., et al. Anomalies of craniofacial skeleton and teeth in cleidocranial dysplasia. J Craniofac Genet Dev Biol. 1999; 19:75–79.
Kress, W., Collmann, H., Busse, M. Clustering of FGFR2 gene mutations inpatients with Pfeiffer and Crouzon syndromes (FGFR2-associated craniosynostoses. Cytogenet Cell Genet. 2000; 91(1-4):134–137.
Ladner, M.B., et al. Human CSF-1: gene structure and alternative splicing of mRNA precursors. EMBO J. 6. 1987; 2693–2698.
Laskin, D.M. Oral and maxillofacial surgery: volume II. CV Mosby, St. Louis. 1985; 585–591.
Laskin, D.M. Etiology of the pain-dysfunction syndrome. J Am Dent Assoc. 79(147), 1969.
Lathrop, G.M., Lalouel, J.M., Julier, C., Ott, J. Multilocus linkage analysis in humans: detection of linkage and estimation of recombination. Proc Natn Acad Sci USA, 81. 1984; 3443–3446.
Leighty, S.M., Spach, D.H., Myall, R.W., Burns, J.L. Septic arthritis of the temporomandibular joint: review of the literature and report of two cases in children. Int J Oral Maxillofac Surg, Oct. 1993; 22(5):292–297.
Lejeune, J., Gautier, M., Turpin, R. Study of somatic chromosomes from 9 mongoloid children. C R Hebd Seances Acad Sci, 16. 1959; 248(11):1721–1722. [Mar].
Lerner, L.H., Wiss, K., Gellis, S., Barnhill, R. An unusual pustular eruption in an infant with Down syndrome and a congenital leukemoid reaction. J Am Acad Dermatol, 35(2 Pt 2. 1996; 330–333. [Aug].
Lichenstein, L., Jaffe, H.L. Fibrous dysplasia of bone: a condition affecting one, several or many bones, the graver cases of which may present abnormal pigmentation of skin, premature sexual development, hyperthyroidism or still other extraskeletal abnormalities. Arch Pathol. 33(777), 1942.
Lichtenstein, L. Polyostotic fibrous dysplasia. Arch Surg. 36(874), 1938.
Liptak, G.S., Serletti, J.M. Pediatric approach to craniosynostosis (published erratum appears in Pediatr Rev, 20(1):20, Jan, 1999. Pediatr Rev. 1998; 19(10):352. [quiz 359, Oct].
Lomri, A., Lemonnier, J., Hott, M., de Parseval, N., et al. Increased calvaria cell differentiation and bone matrix formation induced by fibroblast growth factor receptor 2 mutations in Apert syndrome. J Clin Invest. 1998; 101(6):1310–1317. [Mar 15].
Losapio PL, Amaddeo P. A case of true congenital temporomandibular ankylosis.
Losee, J.E. Corde McCune-Albright syndrome on a deformational plagiocephaly: diagnosis, prevention,treatment. Plast Surg Clin. 2005; 32(1):53–64. [Jan].
Manusov, E.G., Douville, D.R. Page LV Osteopetrosis (‘marble bone’ disease. Am Fam Physician. 1993; 47(1):175–180. [Jan].
Marbach, J.J. Arthritis of the temporomandibular joints. Radiogr Photogr Dent. 42(51), 1969.
Maroteaux, P., Lamy, M. LaPyknodysostose. Presse Med, 70. 1962; 999–1002.
Maroteaux, P., Lamy, M. The malady of Toulouse-Lautrec. J Am Med Assoc, 191. 1965; 715–717.
Marques, I.L., Barbieri, M.A., Bettiol, H. Etiopathogenesis of isolated Robin sequence. Cleft Palate Craniofac J. 1998; 35(6):517–525. [Nov].
Marx, S.J. Causes of Hypocalcemia or Osteomalacia: a review of endocrinology diagnosis and treatment. NIH syllabus. 1999; 506–513. Oct
Marx, S.J. Familial hypocalciuric hypercalcemia. In Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, 3, Philadelphia: Williams and Wilkins; 1996:190–192.
Marx, S.J. Therapy of PTH or Calciferol Deficiency States: A Review ofEndocrinology Diagnosis and Treatment. NIH syllabus. Oct. 1999; 6–10. [515-25].
Maumenee, I.H. The eye in the Marfan syndrome. Trans Am Ophthalmol Soc. 1981; 79:684–733.
McClendon, J.L., Anderson, D.E., Cornelius, E.A. Cherubism: hereditary fibrous dysplasia of the jaws II Pathologic considerations. Oral Surg. 15(Suppl 2), 1962. [17].
McDonald, R.E., Shafer, W.G. Disseminated juvenile fibrous dysplasia of the jaws. Am J Dis Child. 89(354), 1955.
McKusick, V.A. The cardiovascular aspects of Marfan’s syndrome: a heritable disorder of connective tissue. Circulation. 1955; 11:321–342.
McKusick, V.A., Scott, C.I. A nomenclature for constitutional disorders of bone. J Bone Joint Surg, 53A: 978. 1971.
McKusick, V.A. Heritable Disorders of Connective Tissue (4th ed. CV Mosby, St Louis. 1972.
Meijer, R., Walker, J.C. Waardenburg’s syndrome. Plast Reconstr Surg. 34(363), 1964.
Miklaszewska, M., Racawska, A., Ziarkiewicz, M. Syndroma Crouzon and syndromal acanthosis nigricans. Demonstration of case on the meeting of Polish Dermatological Society February (oral presentation. 1990.
Miklaszewska, M., Dysostosis cranio-facialis hederitariaMiklaszewska M., ed. Wasik FDermatologia pediatryczna. Volumed Wrocaw. 2000;2:650–654.
Miller, J.R. Dermatoglyphics. J Invest Dermatol. 1973; 60(6):435–442. [Jun].
Miller, A.S., Cuttino, C.L., Elzay, R.P., Levy, W.M., et al. Giant cell tumor of the jaws associated with Paget’s disease of bone Report of two cases and review of the literature. Arch Otolaryngol, 100, 233. 1974.
Minch, C.M., Kruse, R.W. Osteogenesis imperfecta: a review of basic science and diagnosis (published erratum appears in orthopedics 1998 Aug, 21(8):842. Orthopedics. 1998; 21(5):558–567. [quiz 568-69, May].
Modica R, Mongini F. Limitations in the opening of the mouth of an arthrogenic nature.
Moloney, D.M., Slaney, S.F., Oldridge, M., Wall, S.A., et al. Exclusive paternal origin of new mutations in Apert syndrome. Nat Genet. 1996; 13(1):48–53. [May].
Montgomery, A.H. Ossifying fibroma of the jaw. Arch Surg. 15(30), 1927.
Moorman, W.C. Diseases of the temporomandibular joint. Thesis, Indiana University. 1950.
Morgan, D.H. Mandibular joint pathology Importance of radiographs. Dent Radiogr Photgr. 43(3), 1970.
Morony, S., Capparelli, C., Lee, R. A chimeric form of osteoprotegerin inhibits hypercalcemia and bone resorption induced by IL-1 beta, TNF-alpha, PTH, PTHrP, and 1,25 (OH)2D3. J Bone Miner Res. 1999; 14:1478–1485.
Morovic, C.G., Monasterio, L. Distraction osteogenesis for obstructive apneas in patients with congenital craniofacial malformations. Plast Reconstr Surg. 2000; 105(7):2324–2330. [Jun].
Murdoch, J.L., Walker, B.A., Halpern, B.L., et al. Life expectancy and causes of death in the Marfan syndrome. New Engl J Med, 13. 1972; 286(15):804–808. [Apr].
Murphy, J.B., Doku, H.C., Carter, B.L. Massive osteolysis: phantom bone disease. J Oral Surg. 36(318), 1978.
Myer, C.M., 3rd., Reed, J.M., Cotton, R.T., et al. Airway management in Pierre Robin sequence. Otolaryngol Head Neck Surg. 1998; 118(5):630–635. [May].
Nagle, R.J. Temporomandibular function. Oral Surg. 8(500), 1978.
Nelson, C.L., Hutton, C.E. Condylectomy for temporomandibular joint dysfunction A survey of seventeen postoperative patients. Oral Surg. 51(351), 1981.
Newberg, A.H., Tampas, J.P. Familial infantile cortical hyperostosis: an update. Am J Res. 137(93), 1981.
Nicholas, J.A., Saville, P.D., Bronner, F. Osteoporosis, osteomalacia, and the skeletal system. J Bone Joint Surg, 45A: 391. 1963.
NIH, Osteoporosis and Related Bone Disorders-National Resource Center Website. Fast Facts on Fibrous Dysplasia page. National Institutes of Health, Washington, DC, 2001. Available at:http://www.osteo.org/default.asp.
O’Riordan, M.L., Robinson, J.A., Buckton, K.E., Evans, H.J.. Distinguishing between the chromosomes involved in Down’s syndrome (trisomy 21) and chronic myeloid leukaemia (Ph1) by flourescence Nature. 1971;230(167).
Oatis, G.W., Jr., Burch, M.S., Samuels, H.S. Marfan’s syndrome with multiple maxillarymandibular cysts: report of case. Oral Surg J. 29(515), 1971.
Obwegeser, H.L., Makek, M.S. Hemimandibular hyperplasia--hemimandibular elongation. J Maxillofac Surg. 1986; 14(4):183–208. [Aug].
Oldridge, M., Zackai, E.H., McDonald-McGinn, D.M., Iseki, S., et al. De novo alu- element insertions in FGFR2 identify a distinct pathological basis for Apert syndrome. Am J Hum Genet. 1999; 64(2):446–461. [Feb].
Oshrain, H.I., Sackler, A. Involvement of the temporomandibular joint in a case of rheumatoid arthritis. Oral Surg. 8(1039), 1955.
Osteopetrosis acro-osteolytica:. syndrome of osteopetrosis. a acro-osteolysis sutures of the skull. open Acta Chir Scand. 1962; 124:496–507.
Otto F, Thornell AP, Crompton T et al. CBFA 1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development Cell 997, 89: 765-71.
Ozonoff, M.B., Steinbach, H.L., Mamunes, P. The trisomy 18 syndrome. J Roentgenol Am Ther Radium Nucl, Med. 91(618), 1964.
Pal, B.R., Shaw, N.J. Rickets resurgence in the United Kingdom: improving antenatal management in Asians. J Pediatr. 2001; 139(2):337–338. [Aug].
Park, W.J., Theda, C., Maestri, N.E., Meyers, G.A., et al. Analysis of phenotypic features and FGFR2 mutations in Apert syndrome. Am J Hum Genet. 1995; 57(2):321–328. [Aug].
Pavsek, E.J. Mandibulofacial dysostosis (Treacher-Collins syndrome. Am J Roentgenol, Radium Ther, Nucl Med. 79(598), 1958.
Petersen, D.J., Boniface, A.M., Schranck, F.W. X-linked hypophosphatemic rickets: a study (with literature review) of linear growth response to calcitriol and phosphate therapy. J Bone Miner Res. 1992; 7(6):583–597. [Jun].
Phemister, D.B., Grimson, K.S. Fibrous osteoma of the jaws. Ann Surg. 105(564), 1937.
Pike, M.M. Paget’s disease with associated osteogenic sarcoma: report of three cases. Arch Surg. 46(750), 1943.
Pindborg, J.J., Kramer, I.R.H., Torloni, H. Histological typing of odontogenic tumors, jaw cysts and allied lesions. In: International Histological Classification of Tumors. Geneva: World Health Organization; 1971:18–19.
Pitt, M.J., Rickets and osteomalaciaResnick D., ed. Bralow LBone and Joint Imaging. 2, 1996:511–524.
Porretta, C.A., Dahlin, D.C., Janes, J.M. Sarcoma in Paget’s disease of bone. J Bone Joint Surg, 39A: 1314. 1957.
Posnick, J.C., Ruiz, R.L. The craniofacial dysostosis syndromes: current surgical thinking and future directions. Cleft Palate Craniofac J. 2000; 37(5):433. [Sep].
Poswillo, D. The pathogenesis of the first and second branchial arch syndrome. Oral Surg. 35(302), 1973.
Prowler, J.R., Glassman, S. Agenesis of the mandibular condyles. Oral Surg. 7(133), 1954.
Pugh, D.G. Fibrous dysplasia of the skull, a probable explanation for leontiasis ossea Radiology. 1945;44(458).
Purdue, P.E., Skoneczny, M., Yang, X., Zhang, J.W., et al. Rhizomelic chondrodysplasia punctata, a peroxisomal biogenesis disorder caused by defects in Pex7p, a peroxisomal protein import receptor: a minireview. Neurochem Res. 1999; 24(4):581–586. [Apr].
Pycnodysostosis. a lysosomal disease caused by cathepsin K deficiency. Science. 1996; 273:1236–1238.
Pycnodysostosis: a clinical, pathological, and ultramicroscopic study of a case. J BoneJoint Surg. 60A: 1122-28, 1978.
Ravikiran, Ongole, Pillai, Rejeev S., Keerthilatha, M. Pai. Cherubism in Siblings: a case report. J Can Dent Assoc. 2003; 69(3):150–154.
Rebel, A., Basle, M., Pouplard, A., Kouyoumdjian, S., et al. Viral antigens in osteoclasts from Paget’s disease of bone. Lancet. 2(344), 1980.
Rebel, A., Malkani, K., Basle, M., Bregeon, Ch. Is Paget’s disease of bone a viral infection? Calcif Tissue Res. 22(Suppl: 283), 1977.
Reed, T.E., Borgaonkar, D.S., Conneally, P.M., et al. Dermatoglyphic nomogram for the diagnosis of Down’s syndrome. J Pediatr. 1970; 77(6):1024–1032. [Dec].
Reed, R.J. Fibrous dysplasia of bone: a review of 25 cases. Arch Pathol. 75(480), 1963.
Renier, D., Arnaud, E., Cinalli, G., Sebag, G., et al. Prognosis for mental function in Apert’s syndrome. J Neurosurg. 1996; 85(1):66–72. [Jul].
Renton, P. Radiology of rickets, osteomalacia and hyperparathyroidism. Hosp Med. 1998; 59(5):399–403. [May].
Resnick, D., Niwayama, G. Diagnosis of Bone and Joint Disorders, 2, Philadelphia: WB Saunders, 1988. [4057-4070].
Rex, A.P., Preus, M. A diagnostic index for Down syndrome. J Pediatr. 1982; 100(6):903–906. [Jun].
Robin, N.H. Molecular genetic advances in understanding craniosynostosis. Plast Reconstr Surg. 1999; 103(3):1060–1070. [Mar].
Robins, P.R., Moe, J.H., Winter, R.B. Scoliosis in Marfan’s syndrome: its characteristics and results of treatment in thirty-five patients. J Bone Joint Surg Am. 1975; 57(3):358–368. [Apr].
Robinson, HBG. Osseous dysplasia, reaction of bone to injury. J Oral Surg. 14(3), 1956.
Robinson, M. Polyostotic fibrous dysplasia of bone. J Am Dent, Assoc. 42(47), 1951.
Roizen, N.J. Down syndrome: progress in research. Ment Retard Dev Disabil Res Rev. 2001; 7(1):38–44.
Roughley, P.J., Rauch, F., Glorieux, F.H. Osteogenesis imperfecta—clinical and molecular diversity. Eur Cell Mater, 30. 2003; 5:41–47. [discussion 47, Jun].
Rovin, S., Dachi, S.F., Borenstein, D.B., Cotter, W.B. Mandibulofacial dysostosis, a familial study of five generations. J Pediatr. 64(215), 1964.
Rowe, P.M. Why is rickets resurgent in the USA? Lancet. 2001; 357(9262):1100. [Apr 7].
Rowley, J.D. Down syndromeacute leukaemia: Increased risk may be due to trisomy 21. Lancet. 2(1020), 1981.
Rushton, M.A. An anomaly of cementum in cleidocranial dysostosis. Br Dent J. 100(81), 1956.
Satge, D., Sommelet, D., Geneix, A., et al. A tumor profile in Down syndrome. Am J Med Genet. 1998; 78(3):207–216. [Jul 7].
Saul, R.A., Lee, W.H., Stevenson, R.E. Caffey’s disease revisited: further evidence for autosomal dominant inheritance with incomplete penetrance. Am J Dis Child. 1982; 136(1):55–60. [Jan].
Sawhney, C.P. Bony ankylosis of the temporomandibular joint: follow-up of 70 patients treated with arthroplasty and acrylic spacer interposition. Plast Reconstr Surg. 1986; 77(1):29–40. [Jan].
Schaefer, G.B., Sheth, R.D., Bodensteiner, J.B. Cerebral dysgenesis: an overview. Neurol Clin. 1994; 12(4):773–788. [Nov].
Scherbenske, J.M., Benson, P.M., Rotchford, J.P., James, W.D. Cutaneous and ocular manifestations of Down syndrome. J Am Acad Dermatol. 1990; 933–938. [May, 22(5 Pt 2)].
Schlumberger, H.G. Fibrous dysplasia (ossifying fibroma) of the maxilla and mandible. Am J Orthod Oral Surg. 32(579), 1946.
Schmorl, G. Uber Osteitis deformans Paget Virchows Arch. [Pathol Anat]. 283(694), 1932.
Schreiber, H.R. An anatomicphysiological approach to treatment of temporomandibular joint disturbances. Am Dent Assoc J. 48(261), 1954.
Schwartz, B.T., Alpert, M. The malignant transformation of fibrous dysplasia. Am J Med Sci. 241(35), 1964.
Schwartz, L.Disorders of the Temporomandibular Joint. Philadelphia: WB Saunders, 1959.
Schwindinger, W.F., Francomano, C.A., Levine, M.A. Identification of a mutation in the gene encoding the alpha subunit of the stimulatory G-protein of adenylyl cyclase in McCune-Albright syndrome. Proc Nat Acad Sc. 1992; 89:5152–5166.
Scott, J.A., Wenger, S.L., Steele, M.W., Chakravarti, A. Down syndrome consequent to a cryptic maternal 12p, 21q chromosome translocation. Am J Med Genet. 1995; 56(1):67–71. [Mar 13].
Scutellari PN, Orzincolo C, Ceruti S. The temporomandibular joint in pathologic conditions: rheumatoid arthritis and seronegative spondyloarthritis.
Sedano, H.O., Gorlin, R.J., Anderson, V.E. Pyknodysostosis: clinicalgenetic considerations. J Dis Child Am. 116(70), 1968.
Sedano, H.O., Sauk, J.J., Jr., Gorlin, R.J. Oral Manifestations of Inherited Disorders Woburn, Mass. Butterworth Publishers Inc. 1977.
Shane, E. Hypercalcemia: Pathogenesis, manifestations clinical diagnosis differential management. In Primer on the Metabolic Bone DiseasesDisorders of Mineral Metabolism, 3, Philadelphia: Lippincott, WilliamsWilkins; 1996:177–181.
Shapiro, H.H., Truex, R.C. The temporomandibular joint and the auditory function. J Am Dent Assoc. 30(1147), 1943.
Shapiro, H.H. The anatomy of the temporomandibular joint. Oral Surg. 3(1521), 1950.
Sheth, R.D., Mullett, M.D., Bodensteiner, J.B., Hobbs, G.R. Longitudinal head growth in developmentally normal preterm infants. Arch Pediatr Adolesc Med. 1995; 149(12):1358–1361. [Dec].
Sheth, R.D., Schaefer, G.B., Keller, G.M., et al. Size of the corpus callosum in cerebral palsy. J Neuroimaging. 1996; 6(3):180–183. [Jul].
Shoenfeld, Y., Fried, A., Ehrenfeld, N.E. Osteogenesis imperfecta. Am J Dis Child. 129(679), 1975.
Shores, J., Berger, K.R., Murphy, E.A., Pyeritz, R.E. Progression of aortic dilatation and the benefit of long-term beta-adrenergic blockade in Marfan’s syndrome. New Engl J Med. 1994; 330(19):1335–1341. [May 12].
Sillence, D.O., Senn, A., Danks, D.M. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979; 16(2):101–116. [Apr].
Silverberg, S.J., Shane, E., Jacobs, T.P. A 10-year prospective study of primary hyperparathyroidism with or without parathyroid surgery. New Engl J Med. 1999; 341:1249–1255.
Silverman, F.N., Kuhn, J.P., Metabolic abnormalities of the skeleton. Caffey’s Pediatric X-Ray Diagnosis: An Integrated Imaging Approach (9th ed). Year Book, Mosby. 1993:666–674.
Singer, F.R. Paget’s Disease of Bone. Plenum Medical Book Company, New York. 1977.
Singer, F.R. Paget’s disease of bone: a slow virus infection? Calcif Tissue Int. 31(185), 1980.
Slaney, S.F., Oldridge, M., Hurst, J.A., Moriss-Kay, G.M., et al. Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. Am J Hum Genet. 1996; 58(5):923–932. [May].
Smith, B.J., Eveson, J.W. Paget’s disease of bone with particular reference to dintistry. J Oral Pathol. 10(233), 1981.
Smith, N.H.H. A histologic study of cementum in a case of cleidocranial dysostosis. Oral Surg. 25(470), 1968.
Smyth, F.S., Potter, A., Silverman, W. Periosteal reaction, fever and irritability in young infants: new syndrome? Am J Dis Child. 71(333), 1946.
Soares, S.R., Templado, C., Blanco, J., et al. Numerical chromosome abnormalities in the spermatozoa of the fathers of children with trisomy 21 of paternal origIn: generalised tendency to meiotic non-disjunction. Hum Genet. 2001; 108(2):134–139. [Feb].
Soudant J, Lamas G. Surgical treatment of temporomandibular joint dysfunctions by Myrhaug's technic. Apropos 60b interventions.
Speculaand B, Goss AN, Huges A et al. Temporomandibular joint dysfunction: pain and illness behaviour.
Spilka, C.J., Callahan, K.R. A review of the differential diagnosis of oral manifestations of early osteitis deformans. Surg Oral. 11(809), 1958.
Spitzer, R. A case of unilateral ankylosis of the temporomandibular joint with malposed unerupted mandibular molar. Oral Surg. 6(588), 1953.
Sponseller, P.D., Hobbs Riley LH 3rd, W., Pyeritz, R.E. The thoracolumbar spine in Marfan syndrome. J Bone Joint Surg Am. 1995; 77(6):867–876. [Jun].
Stafne, E.C., Austin, L.T. A study of dental roentgenograms in cases of Paget’s disease (osteitis deformans), fibrosa cystica osteitis osteoma. Am Dent Assoc J. 25(1202), 1938.
Stam HJ, McGrath PA, Brooke RI. The effects of a cognitive-behavioral treatment program on temporo-mandibular pain and dysfunction syndrome.
Stark, R.B., Saunders, D.E. The first branchial syndrome The oral-mandibular- auricular syndrome. Reconstr Surg Plast. 29(229), 1962.
Stein, I., Stein, R.O., Beller, M.L. Living Bone in Health and Disease. JB Lippincott, Philadelphia. 1955.
Stickler, G.B., Morgenstern, B.Z. Hypophosphataemic rickets: final height and clinical symptoms in adults. Lancet. 1989; 2(8668):902–905. [Oct 14].
Stout, A.P. Fibrous and granulomatous lesions of the jaws. New York Dent J. 13(127), 1947.
Sugiura, Y., Yama, Y., Koh, J. Pyknodysostosis in Japan: report of six cases and review of Japanese literature. in Birth Defects, Original Article Series, Vol. 10 (Alan R. Liss, New York. 1974.
Tachdjian, M.O. Marfan’s syndrome. In: Herring J.A., ed. Tachdjian’s Pediatric Orthopaedics. Philadelphia: WB Saunders; 1990:829–837.
Taitz, L.S. Child abuse and osteogenesis imperfecta. Br Med J (Clin Res Ed. 1987; 295(6606):1082–1083. [Oct 31].
Tampas, J.P., Van Buskirk, F.W., Peterson, O.S., Jr., Soule, A.B. Infantile cortical hyperostosis. J Am Med Assoc. 175(491), 1961.
Tanner, H.C., Jr., Dahlin, D.C., Childs, D.S., Jr. Sarcoma complicating fibrous dysplasia Probable role of radiation therapy. Oral Surg. 14(837), 1961.
Tewfik, T.L., Teebi, A.S., Der Kaloustian, V.M. Selected syndromes and conditions. In: Tewfik T.L., Der Kaloustian V.M., eds. Congenital Anomalies of the Ear, Nose and Throat. New York: Oxford University Press; 1997:516–517.
Thakker, R.V. Molecular basis of PTH underexpression. In: Bilezikian J.P, et al, eds. Principles of Bone Biology. San Diego, Calif: Academic Press, 1996. [837-851].
ThoMcCune-Albright syndrome, L., Augey, F., Chamchikh, N., et al. [Cutaneous signs of trisomy 21]. Ann Dermatol Venereol. 1994; 121(4):346–350.
Thompson, D.N., Slaney, S.F., Hall, C.M., Shaw, D., et al. Congenital cervical spinal fusion: a study in Apert syndrome. Pediatr Neurosurg. 1996; 25(1):20–27. [Jul].
Tiley, F., Albright, J.A. Osteogenesis imperfecta: treatment by multiple osteotomy and intramedullary rod insertion: report on thirteen patients. J Bone Joint Surg Am. 1973; 55(4):701–713. [Jun].
Tillman, H.H. Paget’s disease of bone: a clinical, radiographic, and histopathologic study of twenty-four cases involving the jaws. Oral Surg. 15(1225), 1962.
Treister, N., Glick, M. Rheumatoid arthritis: a review and suggested dental care considerations. Am Dent Assoc. 1999; 130(5):689–698.
Tomashek, K.M., Nesby, S., Scanlon, K.S., et al. Nutritional rickets in Georgia. Pediatrics, 107(4): E45, Apr. 2001.
Tossier, P. The definitive plastic surgical treatment of cranio-facial-dysostosis. Plast Reconstr Surg. 1971; 48:419–442.
Tschopp K, Bachmann R. Temporomandibular myoarthropathy syndrome-a frequent cause of facial pain.
Tsuyama M, Kondoh T, Seto K et al. Complications of temporomandibular joint arthroscopy: a retrospective analysis of 301 lysis and lavage procedures performed using the triangulation technique.
Ulmansky, M., Hjorting-Hansen, E., Andreasen, J.O. Paget’s disease of bone: report of a case. Sart Odontol Tidskr. 72(204), 1964.
“Understanding bisphosphonate therapy”. International Myeloma Foundation, 2006. North Hollywood, Calif.
Unger, S., Mornet, E., Mundlos, S., Blaser, S., et al. Severe cleidocranial dysplasia can mimic hypophosphatasia. Euro J Paed. 2002; 161:623–626.
United States Pharmacopeia. USP Dispensing Information: Advice to the Health Care Professional. 1999; 1:2966–2971.
Van Buchem, F.S.P., Hadders, H.N, Ubbens, R. An uncommon familial systemic disease of the skeleton: hyperostosis corticalis generalisata familialis. Acta Radio. 44(109), 1955.
Van Buskirk, F.W., Tampas, J.P., Peterson, D.S., Jr. Infantile cortical hyperostosis An inquiry into its familial aspects. Am J Roentgenol Radium Ther Nucl Med. 85(613), 1961.
Van, Horn P.E., Jr., Dahlin, D.C., Bickel, W.H. Fibrous dysplasia: a clinical pathologic study of orthopedic surgical cases. Mayo Clin Proc. 38(175), 1963.
Vegter, F., Hage, J.J., Mulder, J.W. Pierre Robin syndrome: mandibular growth during the first year of life. Ann Plast Surg. 1999; 42(2):154–157. [Feb].
Viner, R.M., Shimura, N., Brown, B.D., et al. Down syndrome in association with features of the androgen insensitivity syndrome. J Med Genet. 1996; 33(7):574–577. [Jul].
Vintzileos, A.M., Egan, J.F. Adjusting the risk for trisomy 21 on the basis of second- trimester ultrasonography. Am J Obstet Gynecol. 1995; 172(3):837–844. [Mar].
Von, Wowern N. Cherubism. Int J Oral Surg. 1(240), 1972.
Wada, M., Nagano, N., Nemeth, E.F. The calcium receptor and calcimimetics. Curr Opin Nephrol Hypertens. 1999; 8:429–433.
Waldron, C.A., Giansanti, J.S. Benign fibro-osseous lesions of the jaws: a clinical- radiologic-histologic review of sixty-five cases. Oral Surg. 35: 190, 340, 1973.
Waldron, C.A. Giant cell tumors of the jawbones. Oral Surg. 6(1055), 1953.
Wauters, I.M., van Soesbergen, R.M. [Disease caused by lack of sunlight: rickets and osteomalacia]. Ned Tijdschr Geneeskd. 1999; 143(12):593–597. [Mar 20].
Weinstein, L.S., Shenker, A., Gejman, P.V., et al. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. New Eng J Med. 1991; 325:1688–1695.
Welch, T.R., Bergstrom, W.H., Tsang, R.C. Vitamin D-deficient rickets: the reemergence of a once-conquered disease. J Pediatr. 2000; 137(2):143–145. [Aug].
Whyte, M.P., Kurtzberg, J., McAlister, W.H., et al. Marrow cell transplantation for infantile hypophosphatasia. J Bone Miner Res. 2003; 18(4):624–636. [Apr].
Whyte, M.P. Hypophosphatasia. In: Scriver C.R., Beaud, Sly W.L., et al, eds. The Metabolic and Molecular Basis of Inherited Disease. 7. New York: McGraw-Hill; 1995:4095–4111.
Whyte, M.P., Sclerosing Bone Dysplasias. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 4. 1999:367–383.
Wick, M.R., Siegal, G.P., Unni, K.K., McLeon, R.A., Greditzer III, H.G. Sarcomas of bone complicating osteitis deformans (Paget’s disease) Fifty years’ experience. Am J Surg, Pathol. 5(47), 1981.
Wilkes, D., Rutland, P., Pulleyn, L.J. A recurrent mutation, ala391glu, in the transmembrane region of FGFR3 causes Crouzon syndrome and acanthosis nigricans. J Med Genet. 1996; 33(9):744–748. [Sep].
Wilkie, A.O., Slaney, S.F., Oldridge, M., Poole, M.D., et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet. 1995; 9(2):165–172. [Feb].
Wilms, A., Dummer, R. Elastosis perforans serpiginosa in Down syndrome. Hautarzt. 1997; 48(12):923–925. [Dec].
Wilner, D., Sherman, R.S. Roentgen diagnosis of Paget’s disease (osteitis deformans. Dent Radiogr Photogr, 43 : 47. 1970.
Winter, G.R., Maiocco, P.D. Osteogenesis imperfecta and odontogenesis imperfecta. Oral Surg. 2(782), 1949.
Winters, R.W., Graham, J.B., Williams, T.F., McFalls, V.W., et al. A genetic study of familial hypophosphatemia and vitamin D-resistant rickets with a review of the literature. Medicine. 1958; 37:97–142.
Wirth, W.A., Leavitt, D., Enzinger, F.M. Multiple intramusculr myxomas: another extraskeletal manifestation of fibrous dysplasia. Cancer. 27(1167), 1971.
Woolf, R.M., Georgiade, N., Pickrell, K. Micrognathia and associated cleft palate (Pierre-Robin syndrome. Plast Reconstr Surg. 26(199), 1960.
Yamane, G.M., Fleuchaus, P.T. Paget’s disease (osteitis deformans. Oral Surg. 7(939), 1954.
Yoshida, H., et al. The murine mutation osteopetrosis is in the coding region of the macrophage clony stimulating factor gene. Nature, 345. 1990; 442–443.
Zachariades, N., Mezitis, M., Mourouzis, C., Papadakis, D., Spanou, A. Fractures of the mandibular condyle: a review of 466 cases. Literature review, reflections on treatment and proposals. J Craniomaxillofac Surg. 2006; 34(7):421–432. [Oct].
Zhi, K., Ren, W., Zhou, H., et al. Management of temporomandibular joint ankylosis: 11 years’ clinical experience. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009; 108(5):687–692. [Nov].
Zimmerman, D.C., Dahlin, D.C., Stafne, E.C. Fibrous dysplasia of the maxilla and mandible. Oral Surg. 11(55), 1958.
Zunin, C. Two cases of Pierre-Robin’s syndrome (micrognathia, cleft palate and glossoptosis. Pediatrics. 63: 95, 1955.
Zurutuza, L., Muller, F., Gibrat, J.F., et al. Correlations of genotype and phenotype in hypophosphatasia. Hum Mol Genet. 1999; 8(6):1039–1046. [45].