Diseases of the Blood and Blood-forming Organs
 . Diseases Involving Red Blood Cells
. Diseases Involving Red Blood CellsHematologic abnormalities are varied in nature, in its causation as well as in its clinical manifestations, most of which manifest in the oral cavity. In fact the oral site in many instances could act as the forerunner of its manifestations before overt systemic signs and symptoms express. The need of a thorough oral examination is a requisite in any attempt to look into manifestations of hematologic abnormalities and therefore its role as a clinical adjunct cannot be overemphasized.
The symptoms of hematologic disorders are so varied and nonspecific that in themselves they may not suggest a hematologic problem. Thus, unexplained fever, extreme fatigability, or recurrent infections may or may not be caused by a hematologic condition. Likewise, physical examinations may or may not direct attention to the hematopoietic system. Certain details of history must receive special enquiry. These include exposure to physical or chemical agents that may have caused injury and to drugs prescribed or self-medicated. Also deserving special enquiry is the diet, the degree and frequency of chronic blood loss, and the presence or absence of fever. In addition, family history is important in the differential diagnosis of hematologic disorders. Knowledge of ethnic origin or a history of jaundice, anemia, or bleeding in male rather than in female members of the family, for example, may offer useful clues. Furthermore, history alone may be insufficient. Although symptoms may be denied, a palpable spleen on physical examination or morphologic changes seen in blood smear, may direct attention to a hitherto unsuspected hereditary disorder. A family history is only as good as the thoroughness of the enquiry (Wintrobe, 1998).
The formed elements of the blood, as well as its liquid portion, play extraordinary roles in many physiologic mechanisms and processes in the human body. When a disturbance of one of these constituents occurs, severe clinical manifestations result. In some cases, the alteration of cells, serum, or other components is a result of a hereditary diathesis, nutritional deficiency, or exposure to certain chemicals. Other times, a focal or disseminated infection or a defect in one of the elements associated with the clotting mechanism causes the disturbance. A neoplastic overproduction of white cells is recognized as one of the most dreaded of blood dyscrasias.
The various blood diseases present polymorphic clinical expressions, one of which is the relatively constant involvement of oral structures. The dentist is often consulted by the patient suffering from one of the hematologic disorders who, unaware of his condition, only seeks relief of his harassing physical discomforts. Oral manifestations of many diseases of the blood are clinically similar to those lesions which occur in the oral cavity as a result of some local phenomenon, usually irritation or infection. For this reason a specific diagnosis of blood dyscrasia is difficult, if not impossible, to establish on the basis of the oral findings alone.
The hematologic disorders discussed in the following section are grouped, for ease of consideration, according to the cell type involved. No attempt is made to describe every known blood disease or even all the common ones. The sole criterion for inclusion in this section is the occurrence of oral manifestations and their obvious dental implications.
Diseases Involving Red Blood Cells
Anemia
Anemia is defined as an abnormal reduction in the number of circulating red blood cells, the quantity of hemoglobin and the volume of packed red cells in a given unit of blood. The etiologies of the condition are extremely varied, and the classification presented in Table 18-1 based upon causes has been offered by Wintrobe.
Table 18-1
Etiologic classification of the anemia

Modified from MM Wintrobe: Clinical Hematology. 8th ed. Lea and Febiger, Philadelphia, 1981.
In addition to this etiologic classification, a morphologic classification (Table 18-2) has been found of great value. It expresses the characteristic changes in the size and hemoglobin content of the red blood cell and thus acts as a guide to treatment. A recent classification based on cellular kinetic parameters was suggested (Table 18-3).
Table 18-2
Morphologic classification of the anemia
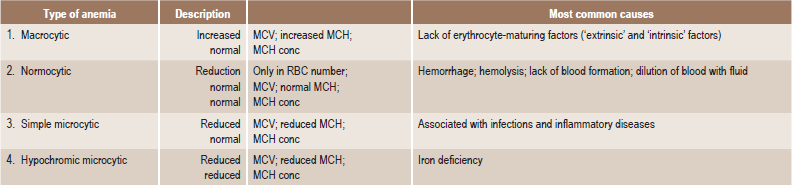
MCV = mean corpuscular volume (Volume/RBC).
MCH = mean corpuscular hemoglobin (Hb/RBC).
MCH conc = mean corpuscular hemoglobin concentration (Hb/Vol).
Table 18-3
Kinetic classification of anemia

Adopted from Wintrobe’s Clinical Haematology. 10th ed. Lippincott Williams and Wilkins, Philadelphia, 1998.
A number of different types of anemia may exhibit oral manifestations. These may be unusually varied, but often are so characteristic that the dentist should at least strongly suspect, if not actually confirm, the diagnosis of the anemia. In the discussion to follow, only those forms of anemia which are known to exhibit specific oral signs and symptoms will be considered.
Pernicious Anemia: (Vitamin B12 deficiency, Addisonian anemia, Biermer anemia, Hunter-Addison anemia, Lederer anemia, Biermer-Ehrlich anemia, Addison-Biermer disease)
Pernicious anemia is a relatively common chronic hematologic disease. It is an adult form of anemia that is associated with gastric atrophy and a loss of intrinsic factor production in gastric secretions and a rare congenital autosomal recessive form in which intrinsic factor (IF) production is lacking without gastric atrophy. The term pernicious anemia is reserved for patients with vitamin B12 deficiency due to a lack of production of IF in the stomach. Intrinsic factor in gastric secretions is necessary for the absorption of dietary vitamin B12. Vitamin B12, a substance now thought to be synonymous with the ‘erythrocyte-maturing factor’ or ‘hemopoietic principle’ and present in many foods, particularly liver, beef, milk and dairy products. Body stores of the vitamin usually exceed 1000 mcg and the daily requirement is about 1 mcg.
Pernicious anemia probably is an autoimmune disorder with a genetic predisposition and the disease is associated with human leucocyte antigen (HLA) types A2, A3, and B7 and A blood group. Antiparietal cell antibodies occur in 90% of patients with pernicious anemia but in only 5% of healthy adults. Similarly, binding and blocking antibodies to IF are found in most patients with pernicious anemia. A greater association than anticipated exists between pernicious anemia and other autoimmune diseases, which include thyroid disorders, type I diabetes mellitus, ulcerative colitis, Addison disease, and acquired agammaglobulinemia. An association between pernicious anemia and Helicobacter pylori infections has been postulated but not clearly proven.
Clinical Features
Pernicious anemia is rare before the age of 30 years and increases in frequency with advancing age. In the United States, males are affected more commonly than females; in other countries, notably Scandinavia, females are more commonly affected. No apparent racial predilection is noticed.
The disease is often characterized by the presence of a triad of symptoms: generalized weakness, a sore, painful tongue, and numbness or tingling of the extremities. In some cases the lingual manifestations are the first sign of the disease. Other typical complaints are easy fatigability, headache, dizziness, nausea, vomiting, diarrhea, loss of appetite, shortness of breath, loss of weight, pallor and abdominal pain.
Patients with severe anemia exhibit a yellowish tinge of the skin and sometimes of the sclerae. The skin is usually smooth and dry. Nervous system involvement is present in over 75% of the cases of pernicious anemia, and this consists of sensory disturbances including the paresthetic sensations of the extremities described above, weakness, stiffness and difficulty in walking, general irritability, depression or drowsiness as well as incoordination and loss of vibratory sensation. These nervous aberrations are referable to the degeneration of posterior and lateral tracts of the spinal cord with loss of nerve fibers and degeneration of myelin sheaths. Degeneration of the peripheral nerves also occurs.
Oral Manifestations
Glossitis is one of the more common symptoms of pernicious anemia. The patients complain of painful and burning lingual sensations which may be so annoying that the dentist is often consulted first for local relief.
The tongue is generally inflamed, often described as ‘beefy red’ in color, either in entirety or in patches scattered over the dorsum and lateral borders (Fig. 18-1). In some cases, small and shallow ulcers — resembling aphthous ulcers — occur on the tongue. Characteristically, with the glossitis, glossodynia and glossopyrosis, there is gradual atrophy of the papillae of the tongue that eventuate in a smooth or ‘bald’ tongue which is often referred to as Hunter’s glossitis or Moeller’s glossitis and is similar to the ‘bald tongue of Sandwith’ seen in pellagra. Loss or distortion of taste is sometimes reported accompanying these changes. The fiery red appearance of the tongue may undergo periods of remission, but recurrent attacks are common. On occasion, the inflammation and burning sensation extend to involve the entire oral mucosa but, more frequently, the rest of the oral mucosa exhibits only the pale yellowish tinge noted on the skin. Millard and Gobetti have emphasized that a nonspecific persistent or recurring stomatitis of unexplained local origin may be an early clinical manifestation of pernicious anemia. Not uncommonly the oral mucous membranes in patients with this disease become intolerant to dentures.
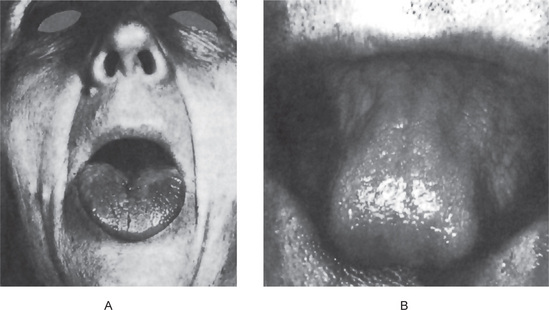
Figure 18-1 Pernicious anemia. The tongue is inflamed and painful in each case, and there is beginning of atrophy of the papillae in (A) and advanced atrophy in (B) (Courtesy of Dr Boynton H Booth and Dr Stephen F Dachi).
Farrant and Boen and Boddington have reported that cells from buccal scrapings of patients with pernicious anemia presented nuclear abnormalities consisting of enlargement, irregularity in shape and asymmetry. These were postulated to be due to a reduced rate of nucleic acid synthesis with a reduced rate of cell division. These epithelial cell alterations are rapidly reversible after administration of vitamin B12.
Laboratory Findings
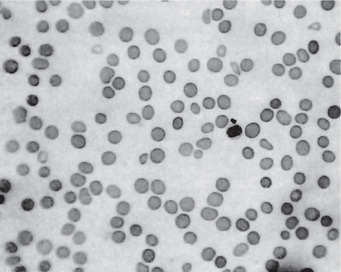
This chronic disease often exhibits periods of remission and exacerbation, and the blood changes generally parallel these clinical states. The red blood cell count is seriously decreased, often to 1,000,000 or less per cubic millimeter. Many of the cells exhibit macrocytosis; this, in fact, is one of the chief characteristics of the blood in this disease, although poikilocytosis, or variation in shape of cells, is also present (Fig. 18-2). The hemoglobin content of the red cells is increased, but this is only proportional to their increased size, since the mean corpuscular hemoglobin concentration is normal. A great many other red blood cell abnormalities have been described, particularly in advanced cases of anemia, including polychromatophilic cells, stippled cells, nucleated cells, Howell-Jolly bodies and Cabot’s rings punctate basophilia. Leukocytes are also often remarkably reduced in number, but are increased in average size, in number of lobes to the nucleus (becoming the so-called macropolycytes) and anisopoikilocytosis. Mild to moderate thrombocytopenia is noticed. Coexistent iron deficiency is common because achlorhydria prevents solubilization of dietary ferric iron from foodstuffs. Striking reticulocyte response and improvement in hematocrit values after parenteral administration of cobalamin is characteristic.
The indirect bilirubin may be elevated because pernicious anemia is a hemolytic disorder associated with increased turnover of bilirubin. The serum lactic dehydrogenase usually is markedly increased. The serum potassium, cholesterol, and skeletal alkaline phosphatase often are decreased. Serum antibodies for IF are highly specific.
Total gastric secretions are decreased to about 10% of the reference range. Most patients with pernicious anemia are achlorhydric, even with histamine stimulation. IF is either absent or is markedly decreased.
The bone marrow biopsy and aspirate usually are hypercellular and show trilineage differentiation. Erythroid precursors are large and often oval. The nucleus is large and contains coarse motley chromatin clumps, providing a checkerboard appearance. Nucleoli are visible in the more immature erythroid precursors. Imbalanced growth of megakaryocytes is evidenced by hyperdiploidy of the nucleus and the presence of giant platelets in the smear. Lymphocytes and plasma cells are spared from the cellular gigantism and cytoplasmic asynchrony observed in other cell lineages. The bone marrow histology is similar in both folic acid and cobalamin deficiency.
The treatment of pernicious anemia consists of the administration of vitamin B12 and folic acid. Early recognition and treatment of pernicious anemia provides a normal, and usually uncomplicated, lifespan. Delayed treatment permits progression of the anemia and neurological complications. The mental and neurological damage can become irreversible without therapy.
Celiac Sprue: (Celiac disease, nontropical sprue, gluten-sensitive enteropathy, Gee-Herter disease)
Sprue is one disease of a large group which constitutes the ‘malabsorption syndrome’. It is a chronic disease of the digestive tract that interferes with the absorption of nutrients from food. Sprue is not basically an anemic disorder. It is considered here; however, because it presents so many signs and symptoms in common with pernicious anemia that the differentiation is often difficult. This disease, also called ‘idiopathic steatorrhea’ to distinguish it from steatorrhea resulting from fibrocystic disease of the pancreas with resultant decrease in pancreatic enzyme secretion.
People with celiac sprue cannot tolerate gluten, a protein commonly found in wheat, rye, barley, and sometimes, oats. When affected individuals ingest gluten, the mucosa of their small intestine is damaged by an immunologically mediated inflammatory response, resulting in maldigestion and malabsorption. Genetics play an important role in celiac sprue. The incidence of disease in relatives of celiac sprue patients is significantly higher than in the general population. The prevalence in first-degree relatives of celiac sprue patients is approximately 10%. Concordance for the disease in HLA identical siblings is about 30% and that for identical twins approaches 70%. Strong association exists between the disease and two human leukocyte antigen (HLA) haplotypes, DR3 and DQw2.
Clinical Features
Sprue occurs both in tropical countries and in temperate zones in persons of all ages, including infants. For example, the frequency of the disease is between 1 in 250 persons and 1 in 300 persons in Italian and Irish populations. In comparison, the disease is rare in Africans or Asians. The symptoms of untreated celiac sprue are divided into gastrointestinal and extraintestinal symptoms.
Gastrointestinal symptoms include diarrhea, which is the most common symptom in untreated celiac sprue due to maldigestion and malabsorption of nutrients. Malabsorption of ingested fat (steatorrhea) resulting in excessive amount of fat passed in the stools. Flatulence results from the release of intestinal gas by the bacterial flora flourishing on undigested and unabsorbed food materials. In infants and young children with untreated celiac sprue, failure to gain weight and growth retardation is common. Other symptoms include weakness and fatigue, severe abdominal pain and excessive malodorous flatus. Occasionally, severe hypokalemia due to the loss of potassium in the stool can cause muscle weakness.
Extraintestinal symptoms include anemia which is usually due to impaired absorption of iron or folate from the proximal small intestine. In severe disease with ileal involvement, absorption of vitamin B12 might be impaired. A bleeding diathesis caused by prothrombin deficiency due to impaired absorption of fat-soluble vitamin K. Excessive amounts of fat are passed in the stools, inducing a concomitant excessive loss of calcium, which in turn causes a calcium deficiency with ensuing low blood calcium levels and occasional tetany. Bone pain occurs due to osteoporosis as a result of calcium and vitamin D deficiency. Nervous irritability as well as numbness and tingling of the extremities occurs, but seldom is there spinal cord involvement as in pernicious anemia. Malaise and generalized weakness are also common. The skin changes are often identical with those of pernicious anemia, but also include irregular brownish pigmentation, particularly on the face, neck, arms and legs, and drying of the skin with a scaly eruption.
Oral Manifestations
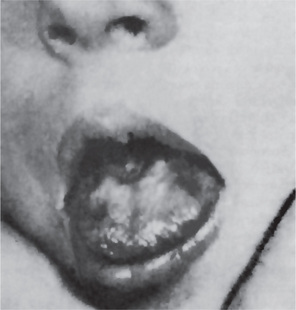
The oral changes in sprue are similar to those of pernicious anemia and have been described by Adlersberg from observation of 40 cases. There may be a severe glossitis with atrophy of the filiform papillae, although the fungiform papillae often persist for some time on the atrophic surface. Painful, burning sensations of the tongue and oral mucosa are common, and small, painful erosions may occur. These severe oral manifestations are seldom absent in cases of sprue (Fig. 18-3). Tyldesley has reviewed this problem recently and concluded that there is an association between recurrent oral ulceration, or recurrent aphthous ulcers, and celiac disease and that proper dietary treatment leads to remission of the oral lesions.
Laboratory Findings
The blood and bone marrow changes are often identical with those of pernicious anemia and include a macrocytic anemia and leukopenia. Hypochromic microcytic anemia occasionally occurs. A low serum iron level is common. The prothrombin time (PT) might be prolonged because of malabsorption of vitamin K. The patients do not usually exhibit achlorhydria, nor is the ‘intrinsic’ factor absent.
Small intestinal biopsy, along with appropriate serum antibodies, usually will establish the diagnosis.
Histologic Findings
Celiac sprue primarily involves the mucosa of the small intestine. The submucosa, muscularis, and serosa usually are not involved. The villi are atrophic or absent, and crypts are elongated. The cellularity of the lamina propria is increased with a proliferation of plasma cells and lymphocytes. The number of intraepithelial lymphocytes per unit length of absorptive epithelium is increased.
Treatment
Sprue responds well in most cases to the administration of vitamin B12 and folic acid, although the diet must be carefully supervised and supplemented with vitamins and minerals. Use of food grains containing gluten should be avoided. A small percentage of celiac sprue patients fail to respond to a gluten free diet. In some patients who are refractory, corticosteroids might be helpful. The patients who fail to respond to corticosteroids, other conditions such as lymphomas of the small intestine should be suspected.
Aplastic Anemia
Aplastic anemia is a bone marrow failure syndrome characterized by peripheral pancytopenia and general lack of bone marrow activity. It may affect not only the red blood cells but also the white cells and platelets, resulting in a pancytopenia. The clinical manifestations of the disease vary according to the type of cell chiefly affected. Paul Ehrlich, introduced the concept of aplastic anemia in 1888 when he studied the case of a pregnant woman who died of bone marrow failure. However, it was not until 1904 when this disorder was termed aplastic anemia by Chauffard.
It is common to recognize two chief forms of aplastic anemia, primary and secondary. Primary aplastic anemia is a disease of unknown etiology which occurs most frequently in young adults, develops rapidly and usually terminates fatally. A disease known as Fanconi’s syndrome consists of congenital, and sometimes familial, aplastic anemia associated with a variety of other congenital defects including bone abnormalities, microcephaly, hypogenitalism and a generalized olive-brown pigmentation of the skin.
Secondary aplastic anemia, on the other hand, is of known etiology, occurs at any age and presents a better prognosis, particularly if the cause is removed. The etiology of this secondary anemia is the exposure of the patient to various drugs or chemical substances or to radiant energy in the form of X-rays, radium or radioactive isotopes. In many cases the development of aplastic anemia after exposure to the drug or chemical seems to be an allergic phenomenon, since the amount of the substance absorbed is too small to result in an actual poisoning or intoxication. The chemicals which have been found most frequently to cause the development of this condition are acetophenetidine, amidopyrine, organic arsenicals, particularly sulfarsphenamine, benzol, chloramphenicol, quinacrine hydrochloride (Atabrine), trinitrotoluene, dinitrophenol, colloidal silver, bismuth, mercury, sulfonamides and penicillin, although many others have also produced the disease. On few occasions aplastic anemia is preceded by infection by hepatitis viruses, Epstein-Barr virus (EBV), HIV, parvovirus, and mycobacterial infections.
The role of an immune dysfunction was suggested in 1970, when autologous recovery was documented in a patient with aplastic anemia who had failed to engraft after marrow transplantation. It was proposed that the immunosuppressive regimen used for conditioning promoted the return of normal marrow function. Subsequently, numerous studies have shown that, in approximately 70% of patients with acquired aplastic anemia, immunosuppressive therapy improves marrow function. Although the inciting antigens that breach immune tolerance with subsequent autoimmunity are unknown, HLA-DR2 is over represented among European and American patients with aplastic anemia.
Suppression of hematopoiesis likely is mediated by an expanded population of the cytotoxic T lymphocytes (CD8 and HLA-DR+), which are detectable in both the blood and bone marrow of patients with aplastic anemia. These cells produce inhibitory cytokines, such as gamma interferon and tumor necrosis factor, which are capable of suppressing progenitor cell growth. These cytokines suppress hematopoiesis by affecting the mitotic cycle and cell killing through induction of Fas-mediated apoptosis. It also has been shown that these cytokines induce nitric oxide synthase and nitric oxide production by marrow cells, which contributes to immune-mediated cytotoxicity and elimination of hematopoietic cells.
The effect of irradiation is usually more pronounced on the white blood cell series, although the development of aplastic anemia after exposure to X-ray radiation is well recognized.
Clinical Features
The clinical manifestations of aplastic anemia are referable not only to the anemia, but also to the leukopenia and thrombocytopenia which are variably present. There are few differences in the clinical features of the primary and secondary forms of the disease except in the ultimate prognosis. The onset is insidious, with the initial symptom relating to anemia or bleeding, but fever or infections often are noted at presentation.
The patients usually complain of severe weakness with dyspnea following even slight physical exertion and exhibit pallor of the skin. Numbness and tingling of the extremities and edema are also encountered due to anemia. Petechiae in the skin and mucous membranes occur, owing to the platelet deficiency, while the neutropenia leads to a decreased resistance to infection.
Oral Manifestations
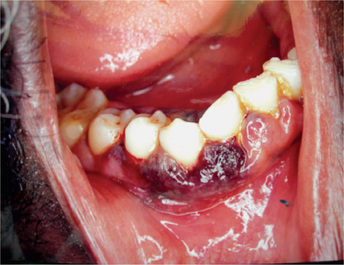
Petechiae purpuric spots or frank hematomas of the oral mucosa may occur at any site, while hemorrhage into the oral cavity, especially spontaneous gingival hemorrhage, is present in some cases. Such findings are related to the blood platelet deficiency (Fig. 18-4). As a result of the neutropenia there is a generalized lack of resistance to infection, and this is manifested by the development of ulcerative lesions of the oral mucosa or pharynx. These may be extremely severe and may result in a condition resembling gangrene because of the lack of inflammatory cell response.
Laboratory Findings
The red blood cell count is remarkably diminished, often to as low as 1,000,000 cells per cubic millimeter, with a corresponding reduction in the hematocrit and hemoglobin levels. A paucity of granulocytes, monocytes, and reticulocytes is found. The thrombocytopenia results in a prolonged bleeding time; the clotting time remains normal. Clot retraction is poor and the tourniquet test is positive. The degree of cytopenia is useful in assessing the severity of aplastic anemia. The presence of teardrop poikilocytes and leucoerythroblastic changes suggest marrow aplasia from infiltrative and dysplastic causes.
Bone marrow smears exhibit variable findings depending on the extent of the anemia and/or pancytopenia. If only an anemia exists, there is erythropoietic depression. Occasionally, however, the marrow appears normal or even hyperplastic. In pancytopenia there is hypoplasia of all marrow elements, and only occasional cells of any type may be found. In cases of less severe damage, moderate numbers of primitive cells persist. In severe cases, hypocellular bone marrow with fatty replacement and relatively increased nonhematopoietic elements such as plasma cells and mast cells may be found. Hemoglobin electrophoresis and blood group testing may show elevated fetal hemoglobin and red cell 1 antigen suggesting stress erythropoiesis, which is observed in both aplastic anemia and myelodysplastic syndromes.
Treatment
Patients with aplastic anemia require transfusion support until the diagnosis is established and specific therapy can be instituted. Infections should be treated appropriately as it is the major cause of mortality. Other treatment options are bone marrow transplantation and immunosuppressive therapy.
Thalassemia: (Cooley’s anemia, Mediterranean anemia, erythroblastic anemia)
Thalassemic syndromes are genetically determined disorders of hemoglobin synthesis with decreased production of either alpha or beta polypeptide chains of hemoglobin molecules, which results from markedly decreased amounts of globin messenger ribonucleic acid. Features first described by Thomas B Cooley in 1925 are seen primarily in Mediterranean populations, in races bordering the Eastern Mediterranean sea or in families originating from these areas (thalassa means ‘sea’ in Greek).
Normal adult hemoglobin is a large complex molecule in which an iron-containing pigment (heme) is conjugated to a complex protein (globin). The globin component consists two pairs of unlike polypeptide chains, alpha and nonalpha chains (e.g. beta, gamma, delta). In the normal adult hemoglobin (HbA), which constitutes over 95% of the hemoglobin in normal persons older than one year, the globin component consists of two alpha and two beta chains. The thalassemia group of anemias is a heterogeneous group characterized by diminished synthesis of the alpha (α)-or beta (β)-globin chain of hemoglobin A. The disease is termed α thalassemia when there is deficient synthesis of the α-chain and β-thalassemia when the β-chain is deficient. Thus, in β-thalassemia there is an excess of α-chains, producing ‘unstable hemoglobins’ that damage the erythrocytes and increase their vulnerability to destruction. In heterozygotes, the disease is mild and is called thalassemia minor or thalassemia trait. It represents both α-and β-thalassemia. Homozygotes may exhibit a severe form of the disease that is called thalassemia major or homozygous β-thalassemia, in which the production of β-chains is markedly decreased or absent, and a consequent decrease in synthesis of total hemoglobin occurs. This results in severe hypochromic anemia. Furthermore, excess α-chains, which synthesize at the normal rate, precipitate as insoluble inclusion bodies within the erythrocytes and their precursors. The presence of such intracellular inclusion bodies (Fessas bodies) leads to increased erythrocyte hemolysis and severe ineffective hematopoiesis. Approximately 70–85% of marrow normoblasts are destroyed in severely affected patients. These processes result in profound anemia and an associated increase in marrow activity, which is estimated to increase 5- to 30-fold.
Two other forms of thalassemia major that represent α-thalassemia also exist. These are:
• Hemoglobin H disease, which is a very mild form of the disease in which the patient may live a relatively normal life.
• Hemoglobin Bart’s disease, with hydrops fetalis, in which the infants are stillborn or die shortly after birth.
Clinical Features
In high-risk areas (i.e. Greek and Italian islands), 10% of the population may have homozygous β-thalassemia; 5% in Southeast Asian populations; and 1.5% in African and American black populations. The onset of the severe form of the disease (homozygous β-thalassemia) occurs within the first two years of life, often in the first few months. Siblings are commonly affected. The child has a yellowish pallor of the skin and exhibits fever, chills, malaise and a generalized weakness. Splenomegaly and hepatomegaly may cause protrusion of the abdomen. The face often develops mongoloid features due to prominence of the cheek bones, protrusion or flaring of the maxillary anterior teeth, and the depression of the bridge of the nose which gives rise to the characteristic rodent facies. The child does not appear acutely ill, but the disease follows an ingravescent course which is often aggravated by intercurrent infection. Some patients; however, die within a few months, especially when the disease is manifested at a very early age. Logothetis and his associates have shown that the degree of cephalofacial deformities in this disease (including prominent frontal and parietal bones, sunken nose bridge, protruding zygomas and mongoloid slanting eyes) is closely related to the severity of the disease and the time of institution of treatment.
Thalassemia minor (thalassemia trait) is generally without clinical manifestations.
Oral Manifestations
An unusual prominence of the premaxilla has been described in cases of erythroblastic anemia, such as that reported by Novak, and this results in an obvious malocclusion. The oral mucosa may exhibit the characteristic anemic pallor observed on the skin.
Laboratory Findings
The pronounced anemia is of a hypochromic microcytic type, the red cells exhibiting a poikilocytosis and anisocytosis. These cells are extremely pale, but in some instances appear as ‘target’ cells with a condensation of coloring matter in the center of the cell. The presence of typical safety-pin cells and of normoblasts or nucleated red blood cells in the circulating blood is also a characteristic feature. The white blood cell count is frequently elevated, often as high as 10,000–25,000 or more per cubic millimeter. Supravital staining (methyl violet) of peripheral blood can demonstrate inclusion bodies. Bone marrow smears show cellular hyperplasia with large numbers of immature, primitive and stem forms of red blood cells, all indicating maturation arrest. The serum bilirubin in these patients is also elevated, indicative of the severe hemosiderosis which is almost invariably present. This systemic hemosiderosis has suggested a possible block in iron utilization with accumulation of iron pigment and subsequent inadequate formation of hemoglobin.
Radiographic Features
The skeletal changes in thalassemia are most striking and have been thoroughly described by Caffey. A frequent finding in rib has been referred to as the rib-within-a-rib appearance and is noted particularly in the middle and anterior portions of the ribs. The finding consists of a long linear density within or overlapping the medullary space of the rib and running parallel to its long axis. In the skull, there is extreme thickening of the diploe (medulla), the inner and outer plates (cortices) become poorly defined, and the trabeculae between the plates become elongated, producing a bristle like crew-cut or hair-on-end appearance of the surface of the skull (Fig. 18-5). Because of the lack of hematopoietic marrow, the occipital bone usually is not involved.
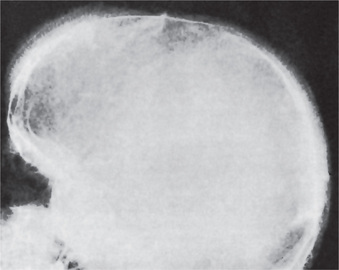
Figure 18-5 Thalassemia. The ‘hair-on-end’ effect is well demonstrated in the radiograph (Courtesy of Dr Robert J Gorlin).
Both the skull and long bones exhibit some degree of osteoporosis, but spontaneous fracture of bones is not common. There is typically a widening of the medulla with thinning of the cortices of the long bones. The bony changes may occur early in life and tend to persist, particularly those in the skull.
Proliferation of marrow within the frontal and facial bones impedes pneumatization of the paranasal sinuses. This results in hypertrophy of osseous structures and a consequent prominence of the lateral margins of the malar eminences, together with anterior and medial displacement of developing teeth. Characteristically, ethmoidal sinuses are not involved, a factor attributable to the absence of red marrow in the sinus walls.
Intraoral radiographs in some cases reveal a peculiar trabecular pattern of the maxilla and mandible, characterized by an apparent coarsening of some trabeculae and the blurring and disappearance of others, resulting in a salt and pepper effect. In general, thinning of the lamina dura and circular radiolucencies in the alveolar bone are also found (Dewey et al).
Gall bladder imaging and ultrasound evaluation may reveal pigment stones. Splenic ultrasound may reveal splenomegaly.
Treatment
There is no treatment for this form of anemia. The administration of liver extract, iron or vitamin B6 is fruitless. Blood transfusions do provide temporary remissions. Bone marrow transplantation may be a definitive treatment option, but long-term results from transplants already performed are not available. The disease is usually fatal, although mild forms which are compatible with life apparently exist. Generally, the earlier in infancy the disease occurs, the more rapidly it proves fatal. Death is generally due to intercurrent infection, cardiac damage as a result of anoxia, or liver failure.
Sickle Cell Anemia: (Sickle cell disease)
Sickle cell anemia is a hereditary type of chronic hemolytic anemia transmitted as a mendelian dominant, nongenderlinked characteristic, which occurs almost exclusively in blacks, and in whites of Mediterranean origin. Malaria is possibly the selecting agent for sickle cell disease because a concordance exists between the prevalence of malaria and HbS. The name is derived from the peculiar microscopic appearance of sickle-or crescent-shaped erythrocytes found in the circulating blood. Normal adult hemoglobin (HbA) is genetically altered to produce sickle hemoglobin (HbS) by the substitution of valine for glutamine at the sixth position of the β-globin chain. In the heterozygote, only about 40% of the hemoglobin is HbS, so that the individual has only the sickle-cell trait and manifests clinical evidence of sickling only under conditions of severe hypoxia. About 8% of American blacks are heterozygous for hemoglobin S. In the homozygote, nearly all hemoglobin is HbS, and the individual suffers from sickle cell anemia. This occurs in about 1 in 600 American blacks.
Deoxygenation of the heme moiety of HbS leads to hydrophobic interactions between adjacent HbS molecules, which then aggregate into larger polymers, distorting the red blood cell (RBC) into the classic sickle shape. The RBCs with sickle shape become much less deformable; therefore, obstructing the microcirculation and thus caused tissue hypoxia, further promotes sickling. Sickle-shaped RBCs are rapidly hemolyzed and have a life span of only about 10–20 days.
The clinical manifestations of sickle cell anemia are diverse, and any organ system may be affected. These manifestations commonly are divided into vaso-occlusive, hematologic, and infectious crises.
Clinical Features
Sickle cell anemia is more common in females and usually becomes clinically manifest before the age of 30 years. Patients manifest a variety of features related to the anemia per se. Thus the patient is weak, short of breath and easily fatigued. Pain in the joints, limbs and abdomen, as well as nausea and vomiting, is common. Systolic murmur and cardiomegaly also occur. One additional feature characteristically seen is packing of red blood cells in peripheral vessels with erythrostasis and subsequent local tissue anoxia. An infarct of the mandible on this basis has been reported by Walker and Schenck. Sickle cell crises (vaso-occlusive, hematologic, and infectious crises) may occur under a variety of situations, including the administration of a general anesthetic, probably as a result of decreased oxygenation of the blood. Other triggering causes of deoxygenation may include exercise or exertion, infections, pregnancy or even sleep.
Oral Manifestations
According to the studies of Robinson and Sarnat, a majority of patients with sickle cell anemia exhibit significant bone changes in the dental radiographs. These alterations consist of a mild to severe generalized osteoporosis and a loss of trabeculation of the jaw bones with the appearance of large, irregular marrow spaces. The trabecular change is prominent in the alveolar bone. There are no alterations in the lamina dura or periodontal ligament. Similar findings were reported by Morris and Stahl and by Prowler and Smith, not only in patients with sickle cell anemia but also in many with only the sickling trait. However, in a study of 80 patients with sickle cell anemia who were compared with an apparently normal group of patients, Mourshed and Tuckson stated that these two radiographic features of the jaws—increased radiolucency and coarse trabeculation—cannot be considered reliable diagnostic criteria for the disease.
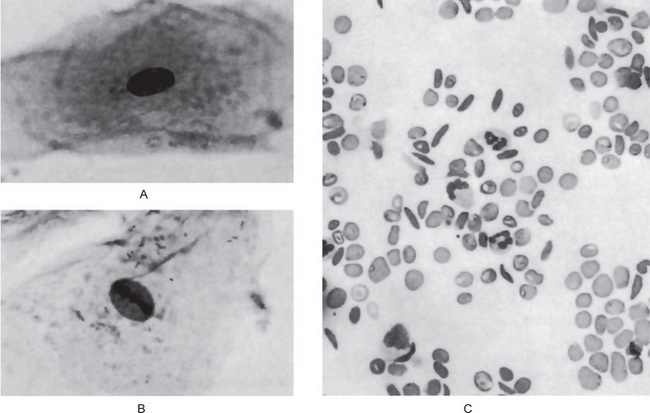
Goldsby and Staats have reported morphologic alterations in the nuclei of epithelial cells in scrapings of the oral mucosa in 90% of all studied cases of patients with homozygous sickle cell disease. These changes were chiefly nuclear enlargement, binucleation and an atypical chromatin distribution. These changes are similar to those that have been reported occurring in pernicious anemia and sprue (Fig. 18-6A, B).
Radiographic Features
Radiographs of the skull exhibit an unusual appearance, with perpendicular trabeculations radiating outward from the inner table producing a ‘hair-onend’ pattern, identical to that seen in thalassemia, congenital hemolytic jaundice and sometimes in chronic iron deficiency anemia and secondary polycythemia of cyanotic congenital heart disease. The outer table of bone may appear absent and the diploe thickened. Generalized osteoporosis may be present. The long bones of children may exhibit enlarged medullary cavities with thin cortices, while the same bones in adults become sclerotic with cortical thickening due to fibrosis of the marrow.
Laboratory Findings
The red blood cell count may reach a level of 1,000,000 cells or less per cubic millimeter with a decreased hemoglobin level. High reticulocyte count indicates anemia and increased marrow response. A major drop in hemoglobin (i.e. more than 2 gm/dl) from previous values indicates a hematological crisis. If the reticulocyte count is low, an aplastic crisis is the probable cause.
On the blood smear, typical sickle-shaped red blood cells are commonly seen, although they are present also in cases of the sickle trait without clinical evidence of the disease (Fig. 18-6C). Elevated levels of lactate dehydrogenase and decreased levels of haptoglobin confirm the presence of hemolysis. Hemoglobin electrophoresis can be done to differentiate homozygous from heterozygous.
Treatment
Treatment strategies include the following five goals:
• Management of vaso-occlusive crisis
• Management of chronic pain syndromes
• Management of the chronic hemolytic anemia
• Prevention and treatment of infections
• Management of the complications and the various organ damage syndromes associated with the disease.
Because this is a lifelong disease, prognosis is not good. The goal is to achieve a normal lifespan with minimal morbidity.
Erythroblastosis Fetalis
Congenital hemolytic anemia due to Rh incompatibility results from the destruction of fetal blood brought about by a reaction between maternal and fetal blood factors.
The Rh factor, named after the rhesus monkey, was discovered by Landsteiner and Wiener in 1940 as a factor in human red blood cells that would react with rabbit antiserum produced by administration of red blood cells from the rhesus monkey. The Rh factor, a dominant hereditary characteristic, is present in the red blood cells of approximately 85% of the Caucasian population of the United States.
Pathogenesis
Erythroblastosis fetalis is essentially due to the inheritance by the fetus of a blood factor from the father that acts as a foreign antigen to the mother. The transplacental transfer of this antigen, actually transplacental leaks of red cells, from the fetus to the mother results in immunization of the mother and formation of antibodies which, when transferred back to the fetus by the same route, produce fetal hemolysis. Occasionally, the ABO system may produce a similar type of immunization and hemolysis.
The basic inheritance of the Rh factor is relatively simple. If both parents are homozygously Rh-positive (have the Rh factor), the infant will be Rh-positive, but maternal immunization cannot occur, since both mother and fetus have the same antigen. If the mother is homozygously positive, but the father Rh-negative, the same situation actually exists, since both the mother and the fetus have the same antigen and no immunization can occur. If the father is Rh-positive and the mother Rh-negative; however, the fetus inherits the paternal factor, which may then act as an antigen to the mother and immunize her with resultant antibody formation.
The problem is complicated; however, by the occurrence of numerous immunologically distinct Rh antigens. The strongest of these antigens have been termed C, D and E, and the presence of any one of them constitutes an Rh-positive person. Each of these antigens is normally present in a specific gene, but, if absent, their place is taken by less potent Hr antigens, known as c, d and e. Thus, three Rh or Hr genes are inherited from each parent, constituting three pairs of factors. Any combination of C, D, E and c, d, e is therefore possible, but the only combination producing an Rh-negative person is cde-cde. The D antigen, by far the strongest, is most frequently responsible for the clinical manifestations of erythroblastosis fetalis, and the 85% of the population generally considered Rh-positive actually have the D antigen homozygously (DD) or heterozygously (D-d). The 15% who are Rh-negative have the d antigen homozygously (d-d). Mathematically, according to the laws of random mating, there should be 10 cases of erythroblastosis fetalis in every 100 pregnancies. Clinically, it has been found that only one case in every 200 pregnancies occurs. There are several possible explanations for this discrepancy:
• In some cases the mother may be unable to form antibodies even though immunized by the Rh-positive fetus.
• Even though the fetus is Rh-positive, transplacental transfer of the antigen does not occur, so that there is no maternal immunization.
• Immunization may occur, but its level is so low as to be clinically insignificant. Recent evidence has shown that, in general, women have a reduced immunologic responsiveness during pregnancy.
Subsequent pregnancies might cause further immunization with increased antibody formation, so that in ensuing pregnancies clinical hemolysis does occur. This latter explanation is plausible since it explains adequately why the first pregnancy is often uneventful, while erythroblastosis frequently occurs in succeeding pregnancies.
It is of great interest to note that the frequency of erythroblastosis fetalis of Rh incompatibility has shown a dramatic decrease in the past few years and that the eventual elimination of the disease through immunization prevention techniques is a probability. At present, Rh-negative mothers are being given anti-D gamma globulin to prevent immunization, since it binds to antigenic receptor sites on fetal red cells, making them nonimmunogenic.
Clinical Features
The manifestations of the disease depend upon the severity of the hemolysis. Some infants are stillborn. Those that are born alive characteristically suffer from anemia with pallor, jaundice, compensatory erythropoiesis, both medullary and extramedullary, and edema resulting in fetal hydrops. It is of considerable interest that the severe anemia and jaundice do not begin to develop until at least several hours after birth and frequently not for several days. The most important aid in diagnosis of the disease is a positive direct Coombs test on cord blood.
Oral Manifestations
Erythroblastosis fetalis may be manifested in the teeth by the deposition of blood pigment in the enamel and dentin of the developing teeth, giving them a green, brown or blue hue (Fig. 18-7). Ground sections of these teeth give a positive test for bilirubin. The stain is intrinsic and does not involve teeth or portions of teeth developing after cessation of hemolysis shortly after birth.
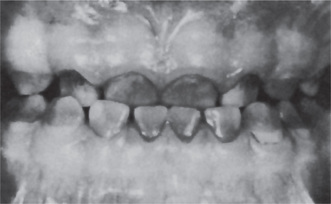
Figure 18-7 Pigmentation of teeth in erythroblastosis fetalis. The teeth had a definite blue cast. The sharp line of separation between affected and unaffected tooth substance is seen near the cervical area of the mandibular cuspids and first molars (Courtesy of Dr Ralph E McDonald).
Enamel hypoplasia is also reported occurring in some cases of erythroblastosis fetalis. This usually involves the incisal edges of the anterior teeth and the middle portion of the deciduous cuspid and first molar crown. Here a characteristic ring-like defect occurs which has been termed the Rh hump by Watson.
Many infants with this disease are stillborn, but an increasing number of those born alive have survived after a total replacement of their blood by transfusion at birth. Thus the dentist may expect to see more children with the peculiar pigmentations of teeth characteristic of the condition, and should be aware of its nature.
Laboratory Findings
The red blood count at birth may vary from less than 1,000,000 cells per cubic millimeter to near a normal level. There are characteristically large numbers of normoblasts, or nucleated red cells, in the circulating blood. Ultimately, severe anemia usually develops within a few days. The icterus index is invariably high and may reach a level of 100 units.
Iron Deficiency Anemia and Plummer-Vinson Syndrome: (Paterson-Brown-Kelly syndrome, Paterson-Kelly syndrome, sideropenic dysphagia)
Iron deficiency is an exceedingly prevalent form of anemia, particularly in females. Iron deficiency is the most prevalent single deficiency state on a worldwide basis. It has been estimated that between 5 and 30% of women in the United States are iron deficient, while in some parts of the world, this may reach 50%. Men are only rarely affected. In healthy people, the body concentration of iron (approximately 60 parts per million) is regulated carefully by absorptive cells in the proximal small intestine, which alter iron absorption to match body losses of iron. Persistent errors in iron balance lead to either iron deficiency anemia or hemosiderosis.
The iron deficiency leading to this anemia usually arises through:
• Chronic blood loss (as in patients with a history of profuse menstruation
• Increased requirements for iron, as during infancy, childhood and adolescence and during pregnancy.
An adult male absorbs and loses about 1 mg of iron from a diet containing 10–20 mg of iron daily. During childbearing years, an adult female loses an average of 2 mg of iron daily (extra 500 mg of iron with each pregnancy, menstrual losses are highly variable is about 4–100 mg of iron) and must absorb a similar quantity of iron in order to maintain equilibrium. Growing children must obtain approximately 0.5 mg more iron daily.
The Plummer-Vinson syndrome is one manifestations of iron-deficiency anemia and was first described by Plummer in 1914 and by Vinson in 1922 under the term ‘hysterical dysphagia’. Not until 1936; however, was the full clinical significance of the condition recognized. Ahlbom then defined it as a predisposition for the development of carcinoma in the upper alimentary tract. It is, in fact, one of the few known predisposing factors in oral cancer. It is thought that the depletion of iron-dependent oxidative enzymes may produce myasthenic changes in muscles involved in the swallowing mechanism, atrophy of the esophageal mucosa, and formation of webs as mucosal complications. It is also thought to be an autoimmune phenomenon as the syndrome is seen in association with autoimmune conditions such as rheumatoid arthritis, pernicious anemia, celiac disease, and thyroiditis. Other factors such as nutritional deficiencies, genetic predisposition are thought to play roles in the causation of this disease.
Clinical Features
While an iron-deficiency anemia may occur at any age, the Plummer-Vinson syndrome occurs chiefly in women in the fourth and fifth decades of life. Presenting symptoms of the anemia and the syndrome are cracks or fissures at the corners of the mouth (angular cheilitis), a lemon-tinted pallor of the skin, a smooth, red, painful tongue (glossitis) with atrophy of the filiform and later the fungiform papillae, and dysphagia limited to solid food resulting from an esophageal stricture or web. These oral findings are reminiscent of those seen in pernicious anemia. The mucous membranes of the oral cavity and esophagus are atrophic and show loss of normal keratinization. Koilonychia (spoon-shaped fingernails) or nails that are brittle and break easily have been reported in many patients; splenomegaly has also been reported in 20–30% of the cases.
The depletion of iron stores in the body, manifested as iron-deficiency anemia, may be the direct cause of the mucous membrane atrophy, since the integrity of epithelium is dependent upon adequate serum iron levels. The atrophy of the mucous membranes of the upper alimentary tract predisposes to the development of carcinoma in these tissues. This relationship was first noted by Ahlbom, who reported that half of all women with carcinoma of the hypopharynx and upper part of the esophagus seen at Radiumhemmet in Stockholm suffered from Plummer-Vinson syndrome. Subsequently the predisposition to the development of oral carcinoma was also established.
Laboratory Findings
Blood examination reveals a hypochromic microcytic anemic of varying degree, while sternal marrow examination shows no megaloblasts typical of pernicious anemia. The red blood cell count is generally between 3,000,000 and 4,000,000 cells per cubic millimeter, and the hemoglobin is invariably low. That the anemia is of an irondeficiency type can be confirmed by lack of a reticulocyte response following administration of vitamin B12. A low serum iron and ferritin with an elevated total iron binding capacity (TIBC) are diagnostic of iron deficiency. There is an absence of free hydrochloric acid in the stomach. The achlorhydria is generally the cause of the faulty absorption of iron, since the absence of hydrochloric acid prevents the conversion of unabsorbable dietary ferric iron to the absorbable ferrous state. The absence of stainable iron in a bone marrow aspirate is further diagnostic of iron deficiency. Unusual alterations in exfoliated squamous epithelial cells of the tongue in cases of severe iron-deficiency anemia have been reported by Monto and his associates. These changes consisted of a deficiency of keratinized cells, a reduced cytoplasmic diameter of cells with a paradoxical enlargement of the nucleus, and abnormal cellular maturation characterized by a disturbed nuclear pattern, an increase in nucleoli, presence of double nuclei and karyorrhexis. Testing stool for the presence of hemoglobin is useful in establishing gastrointestinal bleeding as the etiology of iron deficiency anemia; however, they produce a high incidence of false-positive results in people who eat meat.
Treatment and Prognosis
The anemia responds well to iron therapy and a high-protein diet. Because of the predisposition to the development of carcinoma of oral mucous membranes, it is essential that the diagnosis be established early so that treatment may be instituted as soon as possible. Dysphagia may improve with iron replacement alone, particularly in patients whose webs are not substantially obstructive. Dysphagia caused by more advanced webs is unlikely to respond to iron replacement alone, and thus is managed with mechanical dilation.
Polycythemia
Polycythemia is defined as an abnormal increase in the number of red blood cells in the peripheral blood, usually with an increased hemoglobin level. Three forms of the disease are recognized: relative polycythemia; primary polycythemia or erythremia (polycythemia rubra vera) of unknown etiology; and secondary polycythemia or erythrocytosis, due to some known stimulus.
Relative polycythemia is an apparent increase in the number of circulating red blood cells that occurs as a result of loss of blood fluid with hemoconcentration of cells, and is seen in cases of excessive loss of body fluids such as chronic vomiting, diarrhea, or loss of electrolytes with accompanying loss of water. This increase in the number of red blood cells is only relative to the total blood volume, and therefore, is not a true polycythemia.
Primary polycythemia, or polycythemia rubra vera, is characterized by a true idiopathic increase in the number of circulating red blood cells and of the hemoglobin level. It is characterized by bone marrow with an inherent increased proliferative activity.
Secondary polycythemia is similar to primary polycythemia except that the etiology is known. Secondary polycythemia is caused due to absolute increase in red blood cell mass resultant to enhanced stimulation of red blood cell production. In general, the stimulus responsible for producing a secondary polycythemia is either bone marrow anoxia or production of an erythropoietic stimulating factor. Bone marrow anoxia may occur in numerous situations such as pulmonary dysfunction, heart disease, habitation at high altitudes or chronic carbon monoxide poisoning. Erythropoietic stimulatory factors include a variety of drugs and chemicals such as coal-tar derivatives, gum shellac, phosphorus, and various metals such as manganese, mercury, iron, bismuth, arsenic and cobalt. Some types of tumors such as certain brain tumors, liver and kidney carcinomas and the uterine myoma have also been reported associated with polycythemia. The mechanism for increased production of the red blood cells by these tumors is unknown, but has been postulated as due to elaboration of a specific factor which stimulates erythropoiesis.
Polycythemia Vera: (Polycythemia rubra vera, erythremia, Vaquez’s disease, Osler’s disease)
Polycythemia vera (PV) is a chronic stem cell disorder with an insidious onset characterized as a panhyperplastic, malignant, and neoplastic marrow disorder. The most prominent feature is an absolute increase in the number of circulating red blood cells and in the total blood volume because of uncontrolled red blood cell production. This is accompanied by increased white blood cell (myeloid) and platelet (megakaryocytic) production, which is due to an abnormal clone of the hematopoietic stem cells with increased sensitivity to the different growth factors for maturation.
The bone marrow of patients with polycythemia vera shows normal and abnormal stem cells (Figs. 18-8, 18-9, and 18-10). The clonal proliferation of abnormal stem cells interfere with or suppress normal stem cell growth and maturation. Evidence indicates that the etiology of this panmyelosis is unregulated neoplastic proliferation. The cause of the stem cell transformation remains unknown.
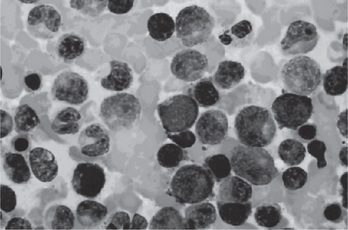
Figure 18-8 Polycythemia vera, bone marrow aspirate. Increased number of both erythroid and myeloid precursors are seen. PV results in a panhyperplasia of marrow cell elements (Wright Giemsa, Oil).
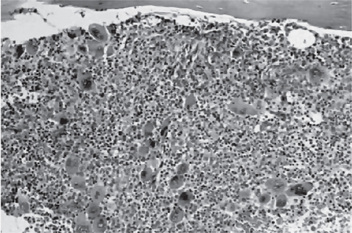
Figure 18-9 Polycythemia vera, bone marrow core biopsy. Hypercellular, megakaryocytes are increased.
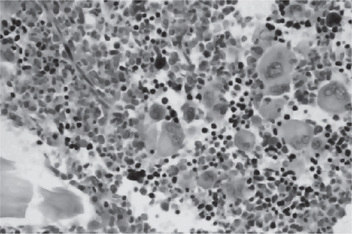
Figure 18-10 Polycythemia vera, megakaryocytes proliferation. This biopsy illustrates the proliferation of megakaryocytes in PV.
All clinical manifestations of this disease are identical with those of secondary polycythemia, so the two conditions are considered together here.
Clinical Features
Polycythemia vera often manifests itself primarily by headache or dizziness, weakness and lassitude, tinnitus, visual disturbances, mental confusion, slurring of the speech and inability to concentrate. The skin is flushed or diffusely reddened, as a result of capillary engorgement and high red cell mass, as though the patient were continuously blushing. This condition is most obvious on the head, neck and extremities, although the digits may be cyanotic. Increased red blood cell mass increases blood viscosity and decreases tissue perfusion, and also predisposes for thrombosis. If secondary polycythemia is secondary to hypoxia, patients can also appear cyanotic or may have acrocyanosis which is caused by sluggish blood flow through small blood vessels. The skin of the trunk is seldom involved. Splenomegaly is one of the most constant features of polycythemia vera, and the spleen is sometimes painful. Gastric complaints such as gas pains, belching and peptic ulcers are common, and hemorrhage from varices in the gastrointestinal tract may occur. Pruritus results from increased histamine levels released from increased basophils and mast cells and can be exacerbated by a warm bath or shower in up to 40% of patients. The disease is more common in men and usually occurs in middle age or later.
Cytogenetic studies show the presence of an abnormal karyotype in the hematopoietic progenitor cells in approximately 34% of patients with PV, depending on the stage of the disease. Approximately 20% of patients have cytogenetic abnormalities at diagnosis, increasing to more than 80% for those with more than 10 years of follow-up care.
The chromosomal abnormalities observed in patients with PV are deletion of 20q (8.4%), deletion of 13q (3%), trisomy 8 (7%), trisomy 9 (7%), trisomy of 1q (4%), deletion of 5q or monosomy 5 (3%), deletion of 7q or monosomy 7 (1%). These are similar to the abnormal karyotypes observed in patients with myelodysplastic syndromes and other myeloproliferative disorders.
Oral Manifestations
The oral mucous membranes appear deep purplish red, the gingiva and tongue being most prominently affected. The cyanosis is due to the presence of reduced hemoglobin in amounts exceeding 5 gm/dl. The gingivae are often engorged and swollen and bleed upon the slightest provocation. Submucosal petechiae are also common, as well as ecchymoses and hematomas. Intercurrent infection may occur, but this is not related directly to the disease.
Laboratory Findings
Red blood cell mass and plasma volume can be measured directly using radiochromiumlabeled red blood cells which show an increase in mass with a normal or slightly decreased plasma volume. The red blood cell count is elevated and may even exceed 10,000,000 cells per cubic millimeter (Figs. 18-11). The red blood cells in patients with PV are usually normochromic normocytic. The hemoglobin content of the blood is also increased, often as high as 20 gm/dl, although the color index is less than 1.0. Because of the great number of cells present, both the specific gravity and the viscosity of the blood are increased.
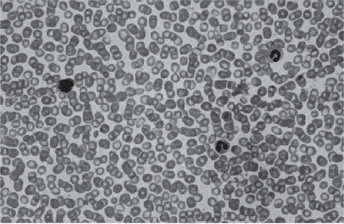
Figure 18-11 Polycythemia vera, peripheral blood. It is often difficult to prepare a good peripheral blood smear in PV due to the increased viscosity of the blood. The red cells are crowded together (Wright Giemsa).
Leukocytosis is usual, as is a great increase in the number of platelets (400,000–800,000/dl) (Figs. 18-12, 18-13); in addition, the total blood volume is elevated through distention of even the smallest blood vessels of the body. The leukocyte alkaline phosphatase score is elevated (>100 U/L) in 70% of patients. There is usually hyperplasia of all elements of the bone marrow. Bleeding and clotting times are normal.
Treatment
No specific treatment for polycythemia is known, although several methods are used for relieving its symptoms. The patient may be periodically bled, or substances may be administered either to destroy blood cells (phenylhydrazine) or to interfere with its formation (nitrogen mustard or even X-ray radiation). In recent years, the radioactive isotope of phosphorus, P32, has been used. Any such treatment; however, produces only a remission of the disease; it does not affect a cure. The course of the disease may be protracted over many years.
Diseases Involving White Blood Cells
Leukopenia
Leukopenia is an abnormal reduction in the number of white blood cells in the peripheral blood stream. This decrease involves predominantly the granulocytes, although any of the cell types may be affected. The etiology of this particular sign of disease is extremely varied, but the classification shown in Table 18-4 has been devised by Wintrobe.
Table 18-4
Modified from MM Wintrobe: Clinical Hematology, 8th ed. Lea and Febiger, Philadelphia, 1981.
Oral lesions are present in certain diseases that are characterized by a reduction in the number of white cells. These lesions are related to the inability of the tissues to react in the usual manner to infection or trauma. Because of the dangerous sequelae which may result if the disease is not recognized, the dentist must be fully acquainted with each disorder and its serious consequences.
Agranulocytosis: (Granulocytopenia, agranulocytic angina, malignant leukopenia or neutropenia)
Agranulocytosis is a serious disease involving the white blood cells. It is characterized by decreased number of circulating granulocytes. It is often classified with reference to etiology as primary or secondary in type, primary agranulocytosis being that form of the disease in which the etiology is unknown, and secondary agranulocytosis being that form in which the cause is recognized. Since the clinical and laboratory findings in both forms are identical, the disease will be discussed here as a single entity.
Etiology
The most common known cause of agranulocytosis is the ingestion of any one of a considerable variety of drugs (Table 18-5) and infections. Those compounds chiefly responsible for the disease are also those to which patients commonly manifest idiosyncrasy in the form of urticaria, cutaneous rashes and edema. For this reason and because often only small amounts of these drugs are necessary to produce the disease, it appears that the reaction may be an allergic phenomenon, although attempts to demonstrate antibodies in affected patients have not been successful. Moreover, in the case of some of the drugs, the disease occurs only after continued administration.
Table 18-5
Risks of agranulocytosis associated with select drugs
| Drug | RR | Excess risk |
| Antithyroid drugs | 97 | 5.3 |
| Macrolides | 54 | 6.7 |
| Procainamide | 50 | 3.1 |
| Aprindine | 49 | 2.7 |
| Dipyrone | 16 | 0.6 |
| Trimethoprim-sulfamethoxazole | 16 | 2.4 |
| Thenalidine | 16 | 2.4 |
| Carbamazepine | 11 | 0.6 |
| Digitalis | 2.5–9.9 | 0.1–0.3 |
| Indomethacin | 6.6 | 0.4 |
| Sulfonylureas | 4.5 | 0.2 |
| Corticosteroids | 4.1 | |
| Butazones | 3.9 | 0.2 |
| Dipyridamole | 3.8 | 0.2 |
| β-Lactams | 2.8 | 0.2 |
| Propranolol | 2.5 | 0.1 |
| Salicylates | 2.0 | 0.0006 |
From the International Aplastic Anemia and Agranulocytosis Study.
RR = multivariant relative risk estimate; excess risk is expressed as number of cases per 1 million users in 1 week. Reproduced from Young NS. Agranulocytosis. JAMA 271: 935–938, 1995.
Kracke, in 1931, was one of the first to point out that a rapid increase in the number of cases of agranulocytosis occurred at the time of the introduction of certain coal-tar derivatives for use in therapy. The following drugs and compounds are some of those which have been reported to produce agranulocytosis in some persons:
Barbiturates (including amobarbital and phenobarbital)
Phenothiazines and related compounds (including chlorpromazine promazine, mepazine, prochlorperazine and imipramine)
Sulfonamides (including sulfanilamide, sulfapyridine, sulfathiazole and sulfadiazine)
Most persons can be exposed to these drugs with near impunity; the hematologic reaction to the compounds is actually an uncommon one.
The mechanism that causes agranulocytosis is not understood completely. In drug-induced agranulocytosis, the drug may act as a hapten and induce antibody formation. Thus produced antibodies destroy the granulocytes or may form immune complexes which bind to the neutrophils and destroy them. Autoimmune neutropenia due to antineutrophil antibodies is seen in few cases.
Other uncommon causes of agranulocytosis include Kostmann syndrome (severe congenital neutropenia) which is most often inherited in autosomal recessive pattern. Autosomal dominant and sporadic cases have also been reported, most often due to mutations in the granulocyte colony-stimulating factor (G-CSF) receptor.
Chronic severe neutropenia has an underlying unknown cause. Myelodysplasia occurs in early infancy and is associated with recurrent infections. The condition is due to accelerated apoptosis and decreased expression of bcl-x in neutrophil precursors.
Clinical Features
Agranulocytosis can occur at any age, but is somewhat more common in adults, particularly women. The disease frequently affects workers in the health professions and in hospitals (e.g. physicians, dentists, nurses, hospital orderlies, and pharmacists), probably because they have easy access to the offending drugs and often use drug samples injudiciously.
The disease commences with a high fever, accompanied by chills and sore throat. The patient suffers malaise, weakness and prostration. The skin appears pale and anemic, or in some cases, jaundiced. The most characteristic feature of the disease is the presence of infection, particularly in the oral cavity, but also throughout the gastrointestinal tract, genitourinary tract, respiratory tract and skin. Regional lymphadenitis accompanies the infection in any of these locations. If treatment is not promptly instituted, the infection progresses to generalized sepsis, which may be life threatening.
The clinical signs and symptoms develop rapidly in the majority of cases, usually within a few days, and death may occur within a week.
Oral Manifestations
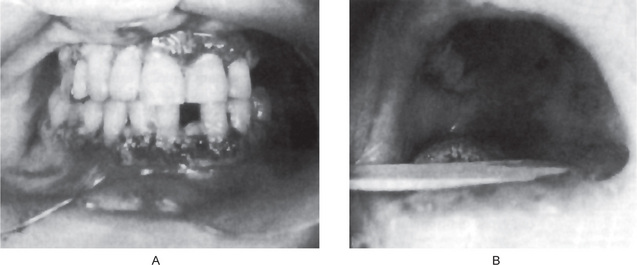
The oral lesions constitute an important phase of the clinical aspects of agranulocytosis. These appear as necrotizing ulcerations of the oral mucosa, tonsils and pharynx. Particularly involved are the gingiva and palate. The lesions appear as ragged necrotic ulcers covered by a gray or even black membrane (Fig. 18-12). Usually no purulent discharge is noticed. Significantly, there is little or no apparent inflammatory cell infiltration around the periphery of the lesions, although hemorrhage does occur, especially from the gingiva. In addition, the patients often manifest excessive salivation.
It is obvious that all oral surgical procedures, particularly tooth extraction, are contraindicated in cases of agranulocytosis.
Histologic Features
The microscopic appearance of sections through the ulcerated oral lesions is a pathognomonic one and accounts for certain clinical features of the disease. Since the essential fault is the lack of development of normal granular leukocytes, the ulcerated areas exhibit no polymorphonuclear reaction to the bacteria in the tissues, and rampant necrosis ensues.
Bauer studied the microscopic appearance of the jaws in agranulocytosis and reported necrosis of the gingiva, beginning adjacent to the sulcus and spreading into the free gingiva, periodontal ligament and even alveolar bone. Rapid destruction of the supporting tissues of the teeth follows.
Laboratory Findings
The white blood cell count in agranulocytosis is often below 2000 cells per cubic millimeter with an almost complete absence of granulocytes or polymorphonuclear cells. The red blood cell count and platelet count are usually normal, although occasionally anemia is present.
The bone marrow is relatively normal except for the absence of granulocytes, metamyelocytes and myelocytes. Promyelocytes and myeloblasts are usually present in near normal numbers; however, and for this reason it appears that the basic defect is an arrest in cell maturation.
Treatment and Prognosis
The treatment of agranulocytosis is not specific, but should consist principally in recognition and withdrawal of the causative drug and in administration of antibiotic drugs to control the infection.
Death is usually related to massive infection, and for this reason the disease carried a high mortality before the advent of the antibiotics. Today, although it is still a serious disease, agranulocytosis has a good prognosis if the responsible agent is discovered. Agranulocytosis secondary to viral infections is usually self-limited, and patients with such conditions have a good prognosis.
Cyclic Neutropenia: (Periodic neutropenia, cyclic agranulocytic angina, periodic agranulocytosis)
Cyclic neutropenia is an unusual form of agranulocytosis characterized by a periodic or cyclic diminution in circulating polymorphonuclear neutrophilic leukocytes as a result of bone marrow maturation arrest, accompanied by mild clinical manifestations, which spontaneously regresses only to recur subsequently in a rhythmic pattern. The etiology of this disease is unknown. Excellent reviews of cyclic neutropenia with its oral manifestations have been published by Page and Good, Becker and his coworkers, and Gorlin and Chaudhry. Although the role of hormonal and allergic factors in the etiology of the disease has been suggested by some workers, there is no sound evidence to indicate that this is the case. There appear to exist at least two additional rare hereditary forms of the disease, one cyclic and the other noncyclic. In addition, a chronic idiopathic neutropenia, noncyclic and nonfamilial, associated with severe persistent gingivitis has been reported by Kyle and Linman.
Clinical Features
This type of agranulocytosis may occur at any age, although the majority of cases have been reported in infants or young children. The symptoms are similar to those of typical agranulocytosis except that they are usually milder. The patients manifest fever, malaise, sore throat, stomatitis and regional lymphadenopathy, as well as headache, arthritis, cutaneous infection and conjunctivitis. In contrast to other types of primary agranulocytosis, rampant bacterial infection is not a significant feature (Table 18-6), presumably because the neutrophil count is low for such a short time. Entities closely mimic the clinical characteristics of cyclic neutropenia are variable, encompassing a wider spectra (Table 18-7).
Table 18-6
Infection associated with neutropenia
| Viruses and viral illness | Bacterial |
| Colorado tick fever | Brucellosis |
| Cytomegalovirus | Gram-negative septicemia |
| Dengue fever | Paratyphoid fever |
| Epstein-Barr virus | Tuberculosis |
| Hepatitis virus | Tularemia |
| Herpes simplex virus | Typhoid fever |
| Human immunodeficiency virus type A and B | Fungal |
| Influenza | Histoplasmosis |
| Measles | Protozoal |
| Mumps | Leishmaniasis |
| Parvovirus | Malaria |
| Poliomyelitis | Rickettsial |
| Psittacosis | Rickettsial pox |
| Respiratory syncytial virus | Rocky Mountain |
| Roseola | spotted fever |
| Rubella | Typhus fever |
| Sandfly fever | |
| Smallpox | |
| Varicella | |
| Yellow fever |
Adapted from Wintrobe’s Clinical Hematology. 10th ed. Williams and Wilkins, 1998.

Oral Manifestations
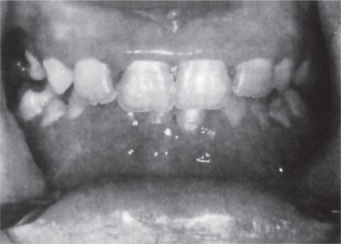
Patients with this disease typically exhibit a severe gingivitis, sometimes a stomatitis with ulceration, which corresponds to the period of the neutropenia and is due to bacterial invasion, chiefly from the gingival sulcus, in the absence of a defense mechanism (Fig. 18-13). With return of the neutrophil count to normal, the gingiva assumes a nearly normal clinical appearance. In children, the repeated insult of infection often leads to considerable loss of supporting bone around the teeth (Fig. 18-14). The widespread severe ulceration usually seen in agranulocytosis does not often occur. However, isolated painful ulcers may occur which persist for 10–14 days and heal with scarring. On this basis, it has been suggested by Gorlin and Chaudhry that some cases diagnosed clinically as periadenitis mucosa necrotica recurrens may actually be cyclic neutropenia.
Radiographic Features
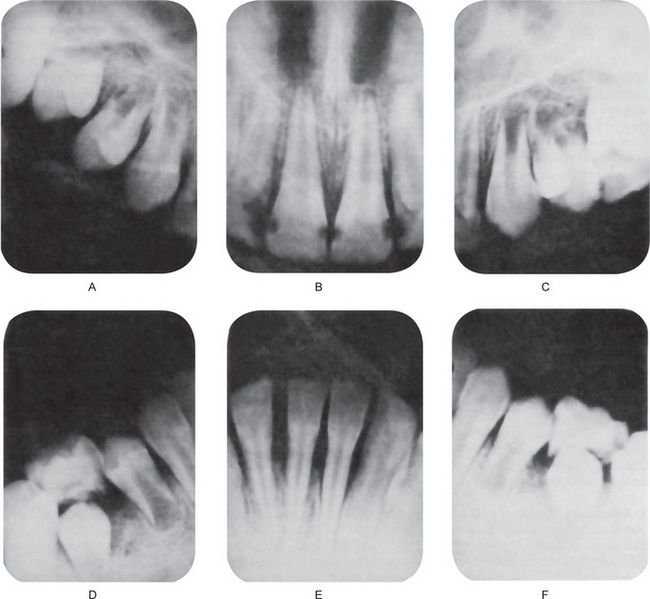
The intraoral radiographs typically exhibit mild to severe loss of superficial alveolar bone, even in children, as a result of the repeated cyclic gingivitis, advancing to periodontitis. In children, this loss of bone around multiple teeth has sometimes been termed ‘prepubertal periodontitis’, and it is frequently indicative of a serious systemic disease. Cohen and Morris have discussed the periodontal manifestations of cyclic neutropenia.
Laboratory Findings
Cyclic neutropenia is an unusual disease which manifests the clinical signs and symptoms and blood changes in a periodic fashion. The cycle commonly occurs every three weeks, although in some cases it may be several months or even longer in duration.
The patient may exhibit a normal blood count which, over a period of four to five days, begins to show a precipitous decline in the neutrophil count compensated by an increase in monocytes and lymphocytes. At the height of the disease, the neutrophils may completely disappear for a period of one or two days. Soon; however, the cells begin to reappear, and within four to five days the blood cell count and differential count are essentially normal.
Treatment and Prognosis
There is no specific treatment for the disease, although in some instances splenectomy has proved beneficial. Death occasionally results, usually from intercurrent infection, but the prognosis is generally far better than in typical agranulocytosis. The patients may suffer from their periodic disease for years.
Chediak-Higashi Syndrome: (Béguez César syndrome, Chédiak-Steinbrinck-Higashi syndrome)
Chédiak-Higashi syndrome (CHS) was described by Béguez Cesar in 1943, Steinbrinck in 1948, Chédiak in 1952, and Higashi in 1954. Chédiak-Higashi syndrome is an autosomal recessive immunodeficiency disorder characterized by abnormal intracellular protein transport.
Clinical Features
Chédiak-Higashi syndrome affects all races and usually appears soon after birth or in children younger than five years. This disease is characterized by immune deficiency; partial oculocutaneous albinism; easy bruisability and bleeding as a result of deficient platelet dense bodies; recurrent infections with neutropenia, impaired chemotaxis, and bactericidal activity; and abnormal natural killer (NK) cell function.
The Chédiak-Higashi syndrome gene was characterized in 1996 as the LYST or CHS1 gene and is localized to bands 1q42–43 which encodes a lysosomal trafficking regulator. The CHS gene affects the synthesis and/or maintenance of storage or secretory granules in these cells, e.g. lysosomes of leukocytes and fibroblasts, dense bodies of platelets, azurophilic granules of neutrophils, and melanosomes of melanocytes. The impaired function in the polymorphonuclear leukocytes may be due to abnormal microtubular assembly. Defective melanization of melanosomes, i.e. autophagocytosis of melanosomes results in oculocutaneous albinism in CHS.
The disease is often fatal in childhood as a result of terminal phase characterized by nonmalignant lymphohistiocytic lymphoma like infiltration of multiple organs that occurs in more than 80% of patients. This stage is precipitated by virus infection, particularly by the Epstein-Barr virus. It is associated with anemia, bleeding episodes, and overwhelming infections leading to death. Infections secondary to abnormal functioning of polymorphonuclear leukocytes commonly involve the skin, the lungs, and the respiratory tract. Infections are usually caused by Staphylococcus aureus, Streptococcus pyogenes, and Pneumococcus species. Very few patients live to adulthood and in these patients, a progressive neurologic dysfunction may be the dominant feature. Neurologic involvement is variable but often includes peripheral neuropathy.
Oral Manifestations
Ulcerations of the oral mucosa, severe gingivitis, and glossitis are the commonly described oral lesions, as in the case report of Gillig and Caldwell. Hamilton and Giansanti have pointed out that periodontal breakdown, probably related to defective leukocyte function, may also be a common oral feature.
Laboratory Findings
Hematologic studies show that the patients classically exhibit giant abnormal granules in the peripheral circulating leukocytes, in their marrow precursors, and in many other cells of the body as well. These granules are the hallmark of the syndrome and are invariably present. They are thought to represent abnormal lysosomes and bear resemblance to toxic granulations and Dohle bodies. Pancytopenia is sometimes present. Ultrastructurally viable dividing bacteria, along with abnormal granules, are found in the cytoplasm of periodontal polymorphonuclear leukocytes.
Leukocytosis
Leukocytosis is defined as an abnormal increase in the number of circulating white blood cells. This condition is usually considered to be a manifestation of the reaction of the body to a pathologic situation. Any increase in the number of circulating white blood cells, particularly when involving only one type of cell, should prompt suspicion of and investigation for a particular disease, especially when the laboratory findings are correlated with the clinical findings in the patient. Care must be exercised in separating an absolute from a relative leukocytosis. But this should offer little difficulty.
A tabulation of the various conditions in which a pathologic increase in the number of each form of white blood cell is found has been compiled by Wintrobe. This classification is presented in Table 18-8. In addition, a transient peripheral plasmacytosis, a cell not normally seen in circulating blood, may be found occasionally in a variety of pathologic situations or conditions listed in Table 18-9.
Table 18-8
Causes of neutrophilia, eosinophilia, basophilia, lymphocytosis, and monocytosis
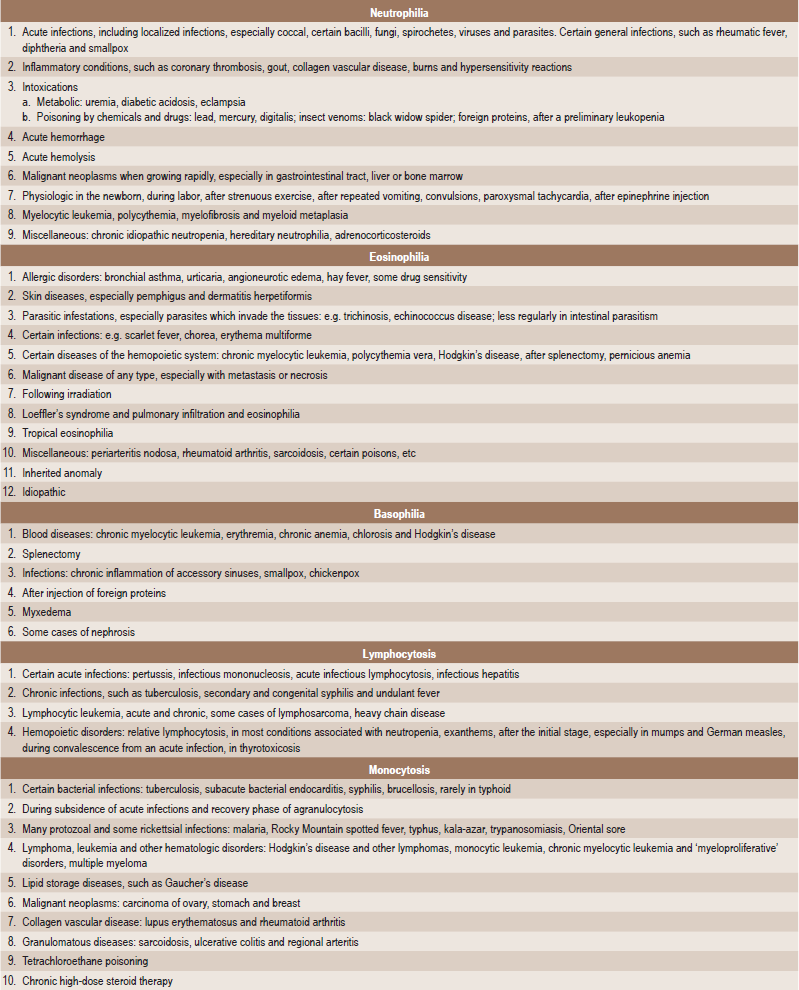
Modified from MM Wintrobe: Clinical Hematology, 8th ed. Lea and Febiger, Philadelphia, 1981.

Infectious Mononucleosis: (Glandular fever)
Infectious mononucleosis was first described by Sprunt and Evans in the Johns Hopkins Medical Bulletin in 1920. These authors described the clinical characteristics of Epstein-Barr virus (EBV) infectious mononucleosis, and, at the time, their paper was entitled ‘Mononuclear leukocytosis in reaction to acute infection (infectious mononucleosis)’. Since the 1800s, infectious mononucleosis has been recognized as a clinical syndrome consisting of fever, pharyngitis, and adenopathy. The term glandular fever was first used in 1889 by German physicians and was termed Drusenfieber.
The disease occurs chiefly in children and young adults. It has been transmitted experimentally to monkeys by the administration either of emulsified material from lymph nodes or of Seitz filtrate of the blood from affected human beings. EB virus is transmitted via intimate contact with body secretions, primarily oropharyngeal secretions and one important means is thought to be through ‘deep kissing’ or intimate oral exchange of saliva. For this reason, the condition has sometimes been called the ‘kissing disease’. It is known that oral excretion of the EB virus may continue for as long as 18 months following onset of the disease, although this excretion may be either constant or intermittent.
Clinical Features
Frequently seen in epidemic form, infectious mononucleosis is characterized by fever, sore throat, headache, chills, cough, nausea or vomiting and lymphadenopathy (bilateral and symmetrical). Splenomegaly and hepatitis also occur with considerable frequency. Most patients with EBV infectious mononucleosis can be asymptomatic.
The cervical lymph nodes are usually the first to exhibit enlargement, followed by the nodes of the axilla and groin (Fig. 18-15A). Pharyngitis and tonsillitis are common, but not invariably present, and skin rash has occasionally been reported.
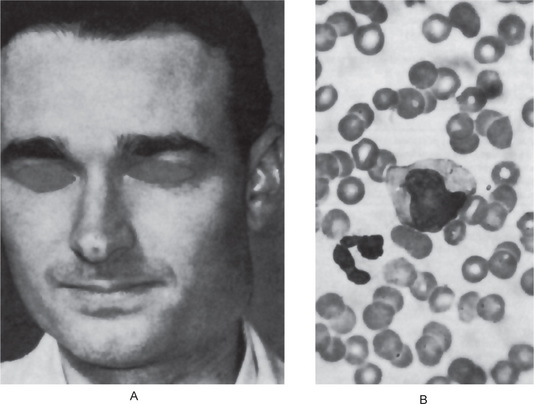
Figure 18-15 Infectious mononucleosis. Severe cervical lymphadenopathy is present (A), while the peripheral blood smear exhibits numerous atypical lymphocytes with indented nuclei such as that illustrated in (B).
The majority of cases in children appear to be asymptomatic. However, the peak incidence of the disease occurs in the 15 to 20-year-old age group. There does not appear to be a sex or seasonal predilection for occurrence.
Oral Manifestations
There are apparently no specific oral manifestations of infectious mononucleosis, although secondary lesions do occur. An excellent review of the literature and study of the oral lesions occurring in 140 patients with infectious mononucleosis was reported by Fraser-Moodie. The oral manifestations consisted chiefly of acute gingivitis and stomatitis, the appearance of a white or gray membrane in various areas, palatal petechiae and occasional oral ulcers. Of his entire series of 140 patients, 32% exhibited oral manifestations, and interestingly, in 50% of the patients with stomatitis the oral lesions were the first sign of the disease. Edema of the soft palate and uvula has also been reported in some cases.
Reports by Shiver and his coworkers and by Courant and Sobkov emphasized the finding of petechial hemorrhages of the soft palate near the junction with the hard palate as an early manifestation of infectious mononucleosis (Fig. 18-16). These have been described as pinpoint petechiae, numbering from a dozen to several hundred, which appeared a few days after other symptoms in 50–65% of the patients in these series. The lesions persisted for 3–11 days and then gradually faded. They must be differentiated from areas of increased vascularity and pigmented areas. This occurrence of palatine petechiae as an early clinical diagnostic sign of infectious mononucleosis has also been confirmed by Schumacher and Barcay. They reported that approximately 7% of a series of 452 patients with this disease had hemorrhagic manifestations, nearly half of these presenting with palatine petechiae. About one-third of the patients with the hemorrhagic tendency exhibited oronasopharyngeal bleeding, including bleeding from the gingiva.
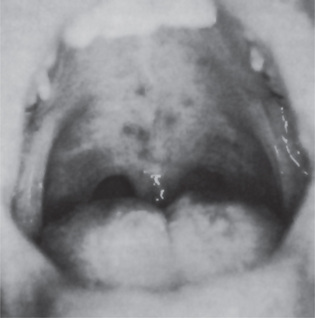
Laboratory Findings
The patient exhibits atypical lymphocytes in the circulating blood, as well as antibodies to the EB virus and an increased heterophil antibody titer (Fig. 18-15B). However, the increased heterophil is present only in a small minority of children with the disease. The normal titer of agglutinins and hemolysins in human blood against sheep red blood cells does not exceed 1 : 8. In infectious mononucleosis; however, the titer may rise to 1 : 4096. This is referred to as a positive Paul-Bunnell test and is both characteristic and pathognomonic of the disease. Agglutination of horse RBCs on exposure to EB virus heterophile antibodies (Monospot test) is a highly specific test. In acute infection, the viral core antigen antibody of IgM class titer against EBV is increased. Later in the course of infection, the increase in IgM viral core-antigen (VCA) antibodies may be accompanied by an increase in IgG VCA antibodies and an increase in IgG EBNA (EBV nuclear antigen antibodies). EBNA appears after one to two months and persists throughout life. Thus the presence of this antibody suggests previous exposure to the antigen. The erythrocyte sedimentation rate is elevated in most patients with EBV infectious mononucleosis. An increase in the white blood cell count is also common, and this is almost invariably a lymphocytosis. In fact, infectious mononucleosis is defined partly on the basis that the patient has more than a 50% lymphocytosis, of which 10% or more are the ‘atypical’ forms. These ‘atypical’ forms consist of either oval, horseshoe-shaped or indented nuclei with dense, irregular nuclear chromatin and a basophilic, foamy or vacuolated cytoplasm. A thrombocytopenia is also present in some patients. It is an interesting finding that during the acute phase of infectious mononucleosis, patients frequently have a normal sedimentation rate.
Treatment
There is no specific treatment for this disease. The various antibiotics have been used without great success. Bed rest and adequate diet are probably of as great a benefit as any other form of therapy. Short-term steroid therapy has occasionally been used, but the results have been somewhat inconsistent. The disease generally runs its course in two to four weeks, and there seldom are complications.
Leukemia
Leukemia is a disease characterized by the progressive overproduction of white blood cells which usually appear in the circulating blood in an immature form. This proliferation of white blood cells or their precursors occurs in such an uncoordinated and independent fashion that leukemia is generally considered a true malignant neoplasm, particularly since the disease is so often fatal. Any of the white blood cells may be involved by this disorder, and for this reason the disease is often classified according to the following types:
1. Lymphoid (lymphoblastic, lymphocytic) leukemia—involving the lymphocytic series.
2. Myeloid (myelogenous) leukemia—involving progenitor cell that gives rise to terminally differentiated cells of the myeloid series (erythrocytes, granulocytes, monocytes and platelets), e.g. acute myelogenous leukemia, acute promyelocytic leukemia, acute monocytic leukemia, acute erythroleukemia, acute megakaryocytic leukemia (Cotran et al, 2001).
This classification may be modified to indicate the course of the disease by application of the terms ‘acute’, ‘subacute’, and ‘chronic’. An acute form of leukemia is one in which survival is less than six months; chronic leukemia implies a survival of over one year, and the subacute form lies between these two. In general, the course of the disease closely parallels the degree of anaplasia of the malignant cells; thus the more undifferentiated the cell, the more acute is the course. The relation of the leukemias to other malignant diseases of the lymphoid tissues is discussed under the section dealing with the malignant lymphomas.
Etiology
The etiology of leukemia is unknown. Certain aspects of the disease have suggested an infectious origin to some investigators, but a specific causative organism has never been isolated. Of these, viruses have been suspected for many years of being most closely related to this disease. It has been recognized for many years that a variety of animal leukemias were almost certainly of viral origin. It has been shown by Stewart and Eddy that the ‘polyoma’ virus is capable of producing numerous different types of neoplasms in a variety of animals, one of these neoplasms being leukemia.
It is rather well accepted by most workers in the field of viral oncology today that avian, feline and murine leukemia are caused by leukemogenic viruses. However, the animals must be rendered immunologically vulnerable and it is possible that, in the human as well as in the experimental animal, radiation and a variety of chemicals, both of which have been closely associated with leukemia for many years, may be at least one key to this immunologic susceptibility. Not only is the incidence of leukemia among radiologists approximately 10 times higher than among general practitioners of medicine, but also the data indicate a general rise in the incidence of this disease among the Japanese exposed to the atomic bomb blasts at Hiroshima and Nagasaki. In addition, chronic exposure to benzol, aniline dyes and related chemicals has been recognized for many years as being associated with the development of leukemia.
The Epstein-Barr (EB) virus, a herpes like virus, has been implicated as being the most likely leukemogenic virus in humans because of the high antibody titer against this virus in leukemic patients, as well as the finding in leukemic cells of viruses with a morphologic similarity to the EB virus. Human T-cell leukemia virus-1 (HTLV-1) is known to be associated with a form of T-cell leukemia/lymphoma that is endemic in certain parts of Japan and the Caribbean basin.
It is also recognized that chromosomal abnormalities commonly occur in leukemic patients. One such abnormality is the finding of the Philadelphia chromosome in between 85 and 95% of patients with chronic myeloid leukemia. This Philadelphia chromosome, at one time thought to be a partial deletion of the long arm of chromosome 22, is now recognized as a translocation of chromosomal material from chromosome 22 to chromosome 9. In about 5% of cases, the translocation occurs to other chromosomes. It is interesting to note that this chromosome disappears from the circulation during remission of the disease in many cases but will reappear when there is a relapse. In addition, a variety of other chromosomal abnormalities also have been recognized as occurring in over 50% of patients with different forms of poorly differentiated leukemia.
It should be remembered that mongolism or Down syndrome is due to a defect or trisomy of chromosome 21. Interestingly, it has been found that the incidence of leukemia in mongoloids is between three and 15–20 times that of the general population. However, this type of leukemia in mongoloids is generally an acute form of leukemia in contrast to the chronic leukemia associated with the Philadelphia chromosome.
The importance of various cofactors or predisposing characteristics, such as genetics, age, hormones, immune competence and stress, must all be considered in determining the susceptibility to tumor development of an individual infected with an oncogenic virus. Only when this has been accomplished can there be any attempt at specific cure or even prevention of the disease.
Clinical Features
The age of the patients affected by leukemia varies remarkably, but generally may be correlated with the course of the disease. Thus acute leukemia occurs more commonly in children and young adults, while chronic leukemias are most frequently seen in adults of middle age or older. There are; however, many exceptions to this general rule. There is some difference in the gender predilection, males being affected more often than females. No notable differences exist in the clinical manifestations of the morphologic forms of leukemia except that most cases of acute leukemia in adults are of the monocytic variety; thus all types of acute leukemia present a similar clinical picture and cannot be differentiated without recourse to laboratory studies. The same is true for chronic leukemia. For this reason the clinical features of leukemia can be discussed under the general categories of acute and chronic forms of the disease.
The development of acute leukemia is sudden, characterized by weakness, fever, headache, generalized swelling of lymph nodes, petechial or ecchymotic hemorrhages in the skin and mucous membranes and evidence of anemia. The lymphadenopathy is often the first sign of the disease, although many cases are recorded in which the oral lesions were the initial manifestation. In a survey of children with acute lymphoblastic leukemia, White has shown that in at least two-thirds of the cases, cervical lymph nodes are palpable before diagnosis and treatment of the disease have been established.
Numerous organs, such as the spleen, liver and kidney, become enlarged, owing to leukemic infiltration, especially in cases of long duration. In the fulminating variety of the disease there is not time for gross pathologic changes to develop. Hemorrhages are common due to the decrease in platelets incident to involvement of the bone marrow and decrease in megakaryocytes. Terminal infection is frequent and may be related to the crowding out of myeloid tissue which ordinarily produces granulocytes.
In contrast to acute leukemia, chronic leukemia develops so insidiously that the disease may be present for months or even several years before the symptoms lead to discovery. It is not unusual for this form of leukemia to be found by a routine hematologic examination in which an unexplained leukocytosis is noted.
The patient may appear in excellent health or exhibit features such as an anemic pallor and emaciation suggestive of a chronic debilitating disease. Lymph node enlargement is common in chronic lymphatic leukemia, but uncommon in myeloid leukemia, as might be expected, particularly in the early stages of the disease. The protracted course of the disease allows sufficient time for full development of splenomegaly and hepatomegaly. Enlargement of the salivary glands and tonsils also may occur, owing to leukemic infiltration, and this results in xerostomia.
The skin is frequently involved in chronic leukemia and may manifest petechiae or ecchymoses. In other instances there may be leukemids: papules, pustules, bullae, areas of pigmentation, herpes zoster, itching and burning sensations or a variety of other disturbances. Finally, nodular lesions composed of leukemic cells may occur on the skin.
Destructive lesions of bone are reported in some cases of chronic leukemia, and these may result in pathologic fracture or osteomyelitis.
Laboratory Findings
Hematologic examination constitutes the basis for the final diagnosis of any type of leukemia. It is recognized; however, that ‘subleukemic’ or ‘aleukemic’ forms of the disease exist in which the white blood cell count of the peripheral blood is normal or even subnormal and in which these are or are not abnormal or immature leukocytes present.
Anemia and thrombocytopenia are both characteristic of acute leukemia. As a result, in some instances, both bleeding time and coagulation time are prolonged. The tourniquet test is usually positive.
The leukocyte count may be subnormal, particularly in the early stages of the disease, but it usually rises in the terminal stages to 100,000 or more cells per cubic millimeter, and there is a corresponding increase in the proportion of the involved cell in the differential count. This increase in cells is due to a single cell type, usually very immature. In myeloid leukemia the predominant cell often resembles the myeloblast, or undifferentiated myelocyte. The cells of lymphoid leukemia may exhibit considerable variation in degree of differentiation. Monocytic leukemia also manifests poorly differentiated cells (Fig. 18-17).
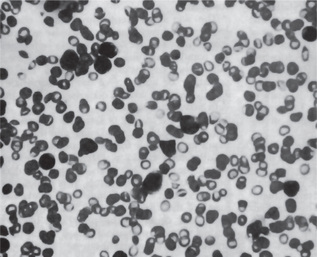
Figure 18-17 Monocytic leukemia, acute. Vast numbers of atypical, pleomorphic monocytes are present in the peripheral blood smear.
In many instances, it is difficult if not impossible for even an experienced hematologist to distinguish the exact type of acute leukemia. The term ‘stem cell leukemia’ is sometimes applied to those types in which the leukemic cells are highly undifferentiated. Such cases are most difficult to diagnose.
Anemia and thrombocytopenia are also common in the chronic form of leukemia. The leukocytosis may be great, and white blood cell counts of over 500,000 cells per cubic millimeter are not uncommon. On the other hand, very low white blood cell counts also occur. In all forms of the chronic dyscrasia the differential count is elevated in the cell type involved, and often over 95% of the total number of cells are leukemic cells.
Oral Manifestations
Oral lesions occur in both acute and chronic forms of all types of leukemia: myeloid, lymphoid and monocytic. These manifestations are far more common; however, in the acute stage of the disease, and according to Burket, are most common in monocytic leukemia. In a series of cases he reported oral lesions in 87% of patients with monocytic leukemia, in 40% of patients with myeloid leukemia, and in 23% of those with lymphoid leukemia. Osgood found a similar high incidence of oral manifestations in monocytic leukemia, reporting that 80% of affected patients exhibited gingival hyperplasia. An 80% incidence of positive oral findings was reported in a series of 38 leukemic patients by Duffy and Driscoll. Interestingly, those patients not manifesting oral lesions were either very young children or edentulous persons. In a study of 292 children with leukemia of different types, Curtis found that only slightly less than 30% had oral findings suggestive of leukemia. He pointed out that this infrequency of oral manifestations in childhood leukemia is due primarily to the high incidence of acute lymphocytic leukemia in this age group, since this type is least likely to produce oral lesions.
Often a patient with leukemia presents himself to his/her dentist for treatment of oral lesions, not suspecting that they are more than local in nature. These primary clinical manifestations of the disease may consist of gingivitis, gingival hyperplasia, hemorrhage, petechiae and ulceration of the mucosa.
The gingival hyperplasia, which may be one of the most constant features of the disease except in edentulous patients, is usually generalized and varies in severity. In severe cases the teeth may be almost completely hidden (Fig. 18-18). The gingivae are boggy, edematous and deep red. They bleed easily. The gingival swelling is due to the leukemic infiltration in areas of mild chronic irritation (Fig. 18-19). Purpuric lesions of the oral mucosa analogous to the cutaneous ecchymoses may also be seen.
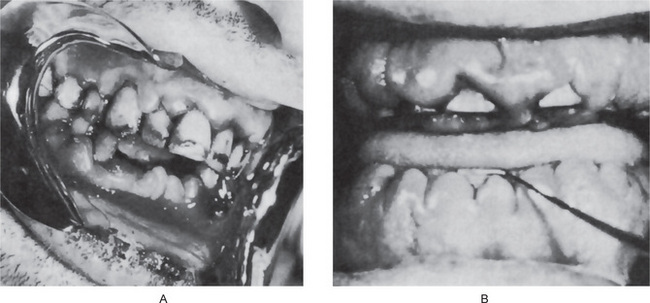
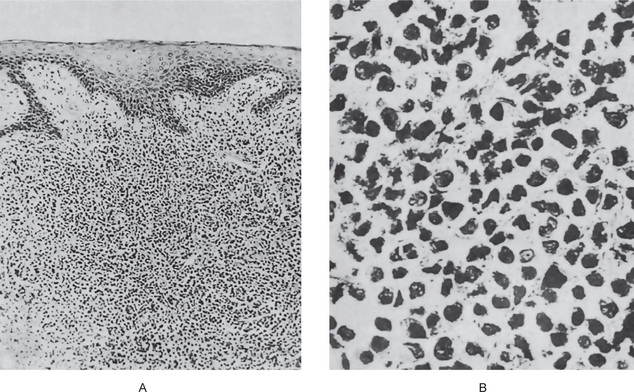
Figure 18-19 Monocytic leukemia.
The gingival tissue is densely infiltrated by atypical blood cells (A) with a mononuclear configuration (B).
The gingival hemorrhage which commonly occurs is due to ulceration of the sulcus epithelium and necrosis of underlying tissue. Since the normal white blood cells distribution is greatly disturbed, a normal inflammatory response to even a mild infection is impossible. For this reason severe ulceration of the oral mucosa and even the development of a nomalike condition is not unusual. Thrombosis of gingival vessels appears to contribute to this phenomenon.
Rapid loosening of the teeth due to necrosis of the periodontal ligament has been reported, and destruction of alveolar bone also occurs in some cases. The use of panoramic radiographs in a study of 214 children with acute leukemia has been reported by Curtis to be useful in demonstrating previously overlooked changes in the jaws. Of this group, approximately 63% exhibited osseous changes in the jaws, including alterations in developing tooth crypts, destruction of lamina dura, displacement of teeth and poor radiographic definition of bone, sometimes extending to the crest of alveolar bones, with destruction of the bone in this area.
It is imperative that the dentist maintain a high index of suspicion in cases of periodontal lesions with a somewhat unusual appearance. The complaint of a patient that he/she has experienced sudden gingival bleeding or gingival hyperplasia should suggest the possibility of leukemia. As Michaud and her coworkers have indicated in a study of 77 children with the disease, the oral manifestations of acute leukemia may be varied; they are not pathognomonic. Any disease that causes immunosuppression, bone marrow suppression, and disease of the blood-forming organs may have one or more of the oral findings of acute leukemia at the time of its initial diagnosis.
Treatment
Spectacular advances have been made in the treatment of the leukemias over the past few years. At one time, the prognosis for this disease was almost hopeless. Today, a wide array of chemotherapeutic drugs, radiation therapy and corticosteroids under certain circumstances offer prolonged remissions and apparent cures in at least some forms of the disease. For example, the most common form of leukemia in children, acute lymphocytic leukemia, once almost always fatal within a few months, now has a prolonged remission and a probable cure rate approaching 50%. Because this area of treatment is changing so rapidly with the introduction of new drugs and new techniques, to cite data on therapeutic responses would not be meaningful. It is sufficient to note that while leukemia is still a serious disease, the outlook for the leukemic patient today is far more promising than it was only a few decades ago and will probably continue to improve.
Diseases Involving Blood Platelets
Blood platelets have a variety of unique and very necessary functions which include:
• Adhesion to a variety of substances, primarily collagen fibrils in the damaged vessel wall, which initiates a secretory process in which these are extruded from the cell (release reaction) granules including serotonin, adenosine triphosphate (ATP) and adenosine diphosphate (ADP). ADP can directly aggregate platelets, thus accounting for the primary and temporary arrest of bleeding after vascular wall disruption.
• Participation in the blood-clotting mechanism by providing a lipid or lipoprotein surface that may catalyze on or more reactions in the conversion of prothrombin to thrombin. This thrombin, in addition to converting fibrinogen to fibrin, can also aggregate platelets. An additional function, recently discovered, is their synthesis of certain prostaglandins which act as potent inhibitors of platelet aggregation in normal blood flow.
There has been very extensive research within the past two decades to clarify our understanding of platelet function, and particularly, our understanding of the exact mechanisms involved in some of the bleeding disorders which may be encountered clinically. Thus, the finding of a prolonged bleeding time in a patient with a normal platelet count would suggest some disturbance in platelet function. This could be a result of an inherent defect of the platelets, which is the usual case, or a deficiency of a plasma factor necessary for some certain aspects of platelet function.
Platelet physiology and abnormalities in their function have been thoroughly reviewed by Weiss and by Zieve and Levin, and the reader is referred to these sources for a discussion of hemostasis. Only the more common and well-recognized diseases involving blood platelets can be discussed in this section, but it should be emphasized that new pathologic entities in this area are being reported frequently.
Purpura
Purpura is defined as a purplish discoloration of the skin and mucous membranes due to the spontaneous extravasation of blood, and in itself, is a symptom rather than a disease entity. There are many causes of purpura, and the clinical manifestations of the condition are widely diversified.
Blood platelets play an obviously important role in the clotting mechanism, and if the platelets are defective or deficient, purpura may result (Fig. 18-20, Table 18-10). On the other hand, many times purpura will occur even though there are adequate numbers of thrombocytes in the circulating blood; in such cases the purpura is due to an unexplained increase in capillary fragility. This variability in presence or absence of blood platelet deficiency in cases of purpura has formed the basis of the following classification:
Table 18-10
International nomenclature of blood clotting factors
| Factor | Preferred synonyms |
| I | Fibrinogen |
| II | Prothrombin |
| III | Tissue thromboplastin |
| IV | Ionized calcium |
| V | Accelerator globulin |
| Proaccelerin | |
| Labile factor | |
| VI | Term no longer used; factor VI-activated factor V |
| VII | Serum prothrombin conversion accelerator (SPCA) |
| Convertin | |
| Stable factor | |
| VIII | Antihemophilic globulin (AHG) |
| IX | Plasma thromboplastin component (PTC) |
| Christmas factor | |
| X | Stuart factor |
| XI | Plasma thromboplastin antecedent (PTA) |
| XII | Hageman factor |
| XIII | Fibrin stabilizing factor |
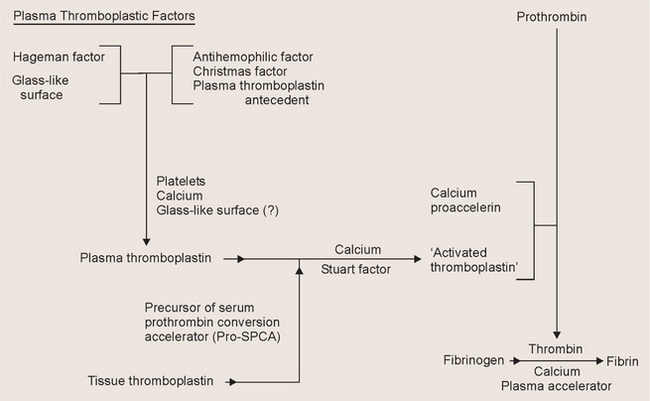
Nonthrombocytopenic Purpura
Nonthrombocytopenic purpura constitutes a heterogeneous group of diseases which have in common only the fact that they may cause purpura. As the name indicates, this type of purpura is not mediated through changes in the blood platelets, but rather through alteration in the capillaries themselves that result in many instances in increased permeability. The most common causes of nonthrombocytopenic purpura or conditions with which this form of purpura is associated are shown in Table 18-11.
Table 18-11
Bleeding disorders mainly due to vascular abnormalities (nonthrombocytopenic purpura)
Modified from MM Wintrobe: Clinical Hematology, 8th ed. Lea and Febiger, Philadelphia, 1981.
The oral manifestations of this form of purpura vary considerably both in incidence of occurrence and in nature, and many of these will be discussed in other sections dealing with the specific etiologic agents. In general terms, the oral purpuric lesions resemble those to be described under thrombocytopenic purpura.
Thrombocytopenic Purpura
Thrombocytopenia is a disease in which there is an abnormal reduction in the number of circulating blood platelets. When this occurs, the patient develops focal hemorrhages into various tissues and organs, including the skin and mucous membranes.
Two basic forms of thrombocytopenia are recognized: primary, which is of unknown etiology; and secondary, which may be due to a wide variety of situations listed in Table 18-12. One subtype, thrombotic thrombocytopenic purpura, will be discussed separately because of its unusual clinical and histologic features.

Primary Thrombocytopenia: (Werlhof 's disease, purpura hemorrhagica and idiopathic purpura)
Primary thrombocytopenia is thought by some investigators to be an autoimmune disorder in which a person becomes immunized and develops antibodies against his/her own platelets. The discovery in the serum of thrombocytopenic patient of an antiplatelet globulin which results in a decrease in the number of circulating platelets when administered to normal patients has given credence to this theory. However, some cases appear due to the absence of a platelet-stimulating or megakaryocyteripening factor. The acute form of the primary type of disease commonly occurs in children, often following certain viral infections, while the chronic type occurs most frequently in adults, especially women of childbearing age.
The various manifestations of primary and secondary thrombocytopenic purpura are nearly identical, and for this reason, may be described together.
Clinical Features
Thrombocytopenic purpura is characterized by the spontaneous appearance of purpuric or hemorrhagic lesions of the skin which vary in size from tiny, red pinpoint petechiae to large purplish ecchymoses and even massive hematomas. The patient also exhibits a bruising tendency.
Epistaxis, or bleeding from the nose, is a common manifestation of the disease, as are bleeding in the urinary tract, resulting in hematuria, and bleeding in the gastrointestinal tract, producing melena or hematemesis. A possible complication is intracranial hemorrhage, which may result in hemiplegia. The spleen is usually not palpable. If it is palpable, leukemia should be suspected instead of thrombocytopenic purpura.
According to Wintrobe and his associates, over 80% of cases of primary thrombocytopenic purpura occur before the age of 30 years, with the greatest incidence before 10 years. Many patients present a familial history of purpura. Secondary thrombocytopenia has no particular age predilection.
Oral Manifestations
One of the prominent manifestations of thrombocytopenic purpura is the severe and often profuse gingival hemorrhage which occurs in the majority of cases (Fig. 18-21). This hemorrhage may be spontaneous and often arises in the absence of skin lesions.
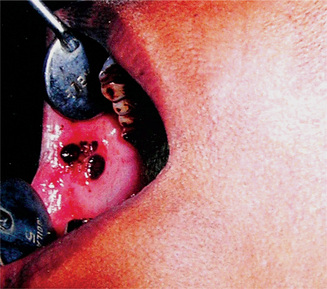
Petechiae also occur on the oral mucosa, commonly on the palate, and appear as numerous tiny, grouped clusters of reddish spots only a millimeter or less in diameter. Actual ecchymoses do occur occasionally.
The tendency for excessive bleeding contraindicates any oral surgical procedure, particularly tooth extraction, until the deficiency has been compensated.
Laboratory Findings
The thrombocytopenia may be exceptionally severe, and the platelet count is usually below 60,000 platelets per cubic millimeter. As a consequence, the bleeding time is prolonged, often to one hour or more. The coagulation time is normal, although the clot does show failure of retraction. As might be expected from the clinical findings, the capillary fragility is increased and the tourniquet test is strongly positive. The red and white blood cell counts are normal unless secondarily disturbed by frequent episodes of hemorrhage or drug or X-ray-induced pancytopenia. Giant platelets on peripheral smear suggest congenital thrombocytopenia.
It is important to understand the basic mechanisms underlying the determination of bleeding and clotting times. Cessation of bleeding as measured by the bleeding time, depends upon the physical blockade of severed capillaries by platelets; as long as the number of platelets present in the blood stream is normal and the platelets aggregate properly, there is no alteration in bleeding time. But if the number of circulating platelets is decreased, the normal platelet plugging of the capillaries occurs more slowly and the bleeding time is consequently prolonged. On the other hand, the role of the platelets in the blood clotting mechanism is through release of a thromboplastic factor from agglutinated platelets. This is present in sufficiently large quantities so that, even when there is a reduction in the number of circulating platelets, sufficient thromboplastic substance is released to maintain normal coagulation. Therefore, in thrombocytopenia, the coagulation time remains normal.
The blood platelets are probably also related to capillary fragility, although the exact mechanism is unknown. It has been suggested that all capillaries undergo 'daily wear and tear' with minor injuries to their walls which are normally plugged by the platelets. If the platelets are diminished; however, there is failure of this maintenance of capillary integrity, resulting in an apparent increase in the capillary fragility.
Treatment and Prognosis
There is no specific treatment for this disease, although splenectomy probably has proved more beneficial than any other form of therapy aside from symptomatic relief such as transfusions and bed rest. Corticosteroids have been used in many cases with excellent results, although remissions may be temporary. The prognosis for patients with this disease is fairly good, since remissions are common. Unfortunately, exacerbations are also common. When death ensues, it is usually from sudden severe hemorrhage.
In secondary thrombocytopenia, correction or removal of the etiologic factor is essential.
Thrombotic Thrombocytopenic Purpura: (Moschcowitz disease)
Thrombotic thrombocytopenic purpura (TTP), an uncommon form of thrombocytopenic purpura, is a life-threatening multisystem disorder of an obscure nature but may be immunologically mediated. It was first described by Eli Moschcowitz in 1924. TTP and hemolytic uremic syndrome (HUS) are thrombotic microangiopathies characterized by microvascular lesions with platelet aggregation.
Thrombotic thrombocytopenic purpura and hemolytic uremic syndrome share the same pathophysiological etiology and may be varied expressions of the same underlying disease process. TTP is more common in adults and is associated with pregnancy; diseases such as HIV, cancer, bacterial infection, and vasculitis; bone marrow transplantation; and drugs.
The TTP syndrome is characterized by microangiopathic hemolysis and platelet aggregation/hyaline thrombi in microcirculation, whose formation is unrelated to coagulation system activity. The thrombi partially occlude the vascular lumina with overlying proliferative endothelial cells. The endothelia of the kidneys and brain are particularly vulnerable to TTP. No inflammatory changes are seen, but the partial occlusion causes fragmentation of erythrocytes and hemolysis.
Clinical Features
The disease generally occurs in young adults and is more common in females than in males. It is characterized by thrombocytopenia, hemolytic anemia, fever, transitory neurologic dysfunction and renal failure.
Histologic Features
The major findings in this disease are the widespread microthrombi in the arterioles, venules, and capillaries in all tissues and organs throughout the body. These intravascular thrombi are composed of loose aggregates of platelets that become organized into amorphous plugs, which are then replaced by fibrin. All of the clinical features can be traced to the thrombosed microcirculation.
It has been reported by Goldenfarb and Finch that biopsy of gingival tissue in patients suspected of having this disease will frequently confirm the diagnosis. Although tissue from many other sites may be used, they believe that gingival tissue is preferable because of its accessibility to rapid hemostasis. The characteristic microscopic gingival changes are described as occlusive subintimal deposits of PAS (periodic acid-Schiff)-positive material at arteriolocapillary junctions.
Laboratory Findings
On blood examination thrombocytopenia and anemia can be noted. Fragmented RBCs (schistocytes) consistent with hemolysis are noticed in peripheral smear. Reticulocyte count is also elevated in few cases. Prothrombin time (PT) and activated partial thromboplastin time (aPTT) are within normal limits. LDH levels are increased. Indirect bilirubin is elevated due to extensive hemolysis. Urinalysis shows proteinuria and microscopic hematuria.
Wiskott-Aldrich Syndrome: (Hypogammaglobulinemia M)
Wiskott-Aldrich syndrome (WAS) is an X-linked recessive genetic condition with variable expression, commonly includes immunoglobulin M (IgM) deficiency. This disorder is a severe congenital immunodeficiency, found almost exclusively in boys. This syndrome results from an X-linked genetic defect in a protein now termed Wiskott-Aldrich syndrome protein (WASp). The gene resides on Xp11. 22–23, and its expression is limited to cells of hematopoietic lineage. The exact function of WASp is not fully elucidated, but it seems to function as a bridge between signaling and actin polymerization in the cytoskeleton.
Clinical Features
The disease is characterized by thrombocytopenic purpura, eczema, usually beginning on the face, and a markedly increased susceptibility to infection due to cellular and humoral immunodeficiency and an increased risk of autoimmune disease and hematologic malignancy. Petechiae and a purpuric rash or ecchymoses of the skin may be early signs of the disease. The eczema has been thought to be allergic in nature. These patients commonly manifest boils, otitis media, bloody diarrhea, and respiratory infection. The increased susceptibility to infection appears related to an antibody deficiency—in particular, a poor antibody response to protein antigens. Serum IgM levels are low, IgG levels are relatively normal, but IgA and IgE levels may be normal or elevated. Thus, patients are unable to form antibody against polysaccharide-containing organisms such as pneumococci, Hemophilus influenzae and coliform bacilli. It is generally agreed that these patients have T- and B-cell abnormalities, although these cells may be relatively normal during early infancy.
One of the important features of the disease is the occurrence of a lymphoreticular malignant neoplasm, commonly a malignant lymphoma, which is often discovered incidentally at autopsy, although it is the specific cause of death in about 10% of cases.
Oral Manifestations
Spontaneous bleeding of the gingiva is frequently seen as well as bleeding from the gastrointestinal tract and nose. Palatal petechiae may also be present.
Laboratory Findings
One of the basic defects appears to be both a qualitative and quantitative abnormality of the platelets. Because of the thrombocytopenia, generally between 18,000 and 80,000 per cubic millimeter, these patients have a prolonged bleeding time. In addition, there is considerable anisocytosis—alterations in the size and shape of platelets, with most platelets being smaller than normal. At the electron microscope level there are alterations in the cell membrane, while biochemically there is a deficiency of the adenosine diphosphate nucleotide storage pool, although platelets can aggregate. Quantitatively, there appears to be decreased production and defective maturation of platelets since normal megakaryocytes may be seen in the marrow, but little platelet formation. There also seems to be accelerated platelet clearance from peripheral blood.
Treatment and Prognosis
There is no specific treatment for the disease, and death usually occurs within the first five years of life as a result of secondary infection or hemorrhage. Some patients have been treated with antibiotics and platelet transfusions, bone marrow transplantation, and even transfer factor. The eventual prognosis, however, is poor.
Thrombocytasthenia
‘Thrombocytasthenia’ is the term used to designate a variety of diseases characterized by a qualitative defect in blood platelets. Some forms are congenital and/or familial, while others are acquired.
Familial Thrombasthenia: (Glanzmann thrombasthenia or disease)
Familial thrombasthenia is a hereditary, chronic hemorrhagic disease transmitted as an autosomal recessive trait. There appears to be at least several varieties or forms of Glanzmann disease, thus accounting for the heterogeneous nature of various descriptions of the condition and the bewildering array of biochemical alterations cited.
Clinical Features
Patients with this disease exhibit the usual characteristics of excessive bleeding, either spontaneous or following minor traumatic injury. Both genders may be affected, and in females, the onset of menarche may be a critical event. Purpuric hemorrhages of skin are common, as are epistaxis and gastrointestinal bleeding. Hemarthrosis has also been reported.
Oral Manifestations
Spontaneous bleeding from the oral cavity, particularly gingival bleeding, is often seen in these patients as are palatal petechiae.
Laboratory Findings
The bleeding time is prolonged in familial thrombasthenia, while clot retraction characteristically is impaired. However, the platelet count is normal, as is the clotting time. The aggregation of platelets by epinephrine, ADP and thrombin is defective. In addition, it is now recognized that there are reduced amounts of certain membrane glycoproteins on the surface of platelets in this disease. This membrane abnormality may be at least partly responsible for the hemostatic defect.
Treatment
There is no specific treatment. However, Perking and his coworkers have discussed this disease and reported two cases of patients requiring oral surgery who were treated with a microfibrillar collagen preparation and with a fibrinolytic inhibitor, c-aminocaproic acid, to control postoperative hemorrhage.
Thrombocytopathic Purpura: (Thrombocytopathia)
Thrombocytopathic purpura is a group of rare diseases of unknown etiology in which the patient manifests a bleeding tendency referable to qualitative defects in the blood platelets. It is not related to thrombocytopenic purpura, since the platelet count is usually normal, although the two diseases have been reported to occur simultaneously. It is clinically indistinguishable from thrombasthenia. An acquired form is also recognized associated with a variety of disease conditions such as uremia.
Clinical Features
Patients with thrombocytopathic purpura have a severe bleeding tendency and bruise easily after only minor trauma. Spontaneous ecchymoses are common, although petechial hemorrhages are rare. Epistaxis and bleeding into the gastrointestinal tract are frequent clinical findings. In some cases, menstrual bleeding has been so severe as to require blood transfusions.
Oral Manifestations
The oral manifestations are those that might be expected in such a hemorrhagic disorder. Spontaneous gingival bleeding is common, while mucosal ecchymoses occasionally occur. Excessive and prolonged bleeding from dental extractions may be a serious management problem.
Laboratory Findings
The platelet count is nearly always normal, but the bleeding time is either normal or prolonged. This is generally due to defective platelet aggregation, even though there is normal release of ADP and ATP, so that normal capillary plugging is impaired. This failure of normal aggregation can often be seen in routine blood smears. Since there are a number of forms of this disease, a variety of different platelet defects seem to exist. For example, in one type called 'storage pool disease', there is a deficiency in the nonmetabolic storage pool of platelet adenine nucleotides. Another form is the 'Portsmouth syndrome', discussed by Roser and his associates, in which there is normal ADPinduced platelet aggregation but abnormal or absent collageninduced aggregation. In the Bernard-Soulier syndrome, there is normal platelet aggregation to collagen and ADP but an abnormal response to fibrinogen.
Thrombocythemia: (Thrombocytosis)
Thrombocythemia is a condition characterized by an increase in the number of circulating blood platelets. As in thrombocytopenia, two forms are recognized: primary (or ‘essential’) and secondary. The etiology of primary thrombocythemia is unknown. Secondary thrombocythemia may occur after traumatic injury, inflammatory conditions, surgical procedures or parturition. In addition, a number of cases have been reported to occur in association with polycythemia and myeloid leukemia, anemia, tuberculosis and sarcoidosis, hyperadrenalism, rheumatoid arthritis and bronchial carcinoma with osseous metastases. Secondary thrombocytosis may be due to the overproduction of proinflammatory cytokines, such as IL-1, IL-6, and IL-11, that occurs in chronic inflammatory, infective, and malignant states. The presence of elevated IL-1, IL-6, C-reactive protein, granulocyte colony-stimulating factor (G-CSF), and granulocyte-macrophage colony-stimulating factor (GM-CSF) in individuals with this condition suggests that these cytokines may be involved in reactive thrombocytosis.
Clinical Features
No gender or age predilection is seen. Patients with thrombocythemia almost invariably show a bleeding tendency in spite of the fact that their platelet count is elevated. Epistaxis and bleeding into the gastrointestinal tract as well as bleeding into the genitourinary tract and central nervous system are common. Hemorrhage into the skin is also found. Few patients can be asymptomatic and are identified on routine blood counts.
Oral Manifestations
Spontaneous gingival bleeding is one of the more commonly reported findings in cases of thrombocythemia, but petechiae are rare. Excessive and prolonged bleeding also frequently occurs after dental extractions. Pogrel has discussed this disease as a cause of oral hemorrhage.
Laboratory Findings
The platelet count in thrombocythemia is greatly increased, and it has been suggested that this high concentration interferes with the formation of thromboplastin. One case reported in the literature showed 14,000,000 platelets per cubic millimeter by a method whereby the normal value was approximately 250,000. In addition, there is abnormal platelet aggregation in response to several aggregating agents. The clotting time, prothrombin time, clot retraction and tourniquet test are all normal, although the bleeding time is frequently prolonged. In primary thrombocythemia, both the red and white blood cell counts are normal. But in secondary thrombocythemia, there may be alterations in the red and white cell counts, depending upon the associated condition.
Diseases Involving Specific Blood Factors
Hemophilia: (Bleeder's disease, disease of the Hapsburg, the disease of Kings)
Hemophilia is a blood disease with a long and interesting history. It is characterized by a prolonged coagulation time and hemorrhagic tendencies. The disease is hereditary, the defect being carried by the X chromosome, and is transmitted as a gender-linked Mendelian recessive trait; thus hemophilia occurs only in males, but is transmitted through an unaffected daughter to a grandson. The sons of a hemophiliac are normal and are not carriers of the trait; the heterozygous daughters carry the defect to half of their sons and as a recessive trait to half of their daughters. The occurrence of hemophilia is theoretically possible in a homozygous female, and occasional rare cases have been recorded.
Etiology
There are a number of different types of hemophilia, and there has been extensive investigation and clarification of this disease in recent years. In light of our present knowledge, three chief forms of hemophilia may be described: hemophilia A (classic hemophilia), B, and C. Each of these differs from the others only in the particular deficiency of the blood clotting factor involved:
| Type | Clotting factor deficiency |
| Hemophilia A | Plasma thromboplastinogen (antihemophilic globulin, AHG, factor VIII) |
| Hemophilia B | Plasma thromboplastin component (PTC, factor IX) |
| Hemophilia C | Plasma thromboplastin antecedent (PTA, factor XI) |
The genes for factor VIII and factor IX are located on the long arm of the X chromosome in bands q28 and q27, respectively. Genetic abnormalities include deletions of variable size, abnormalities with stop codons, and frame-shift defects. Recent data suggest that 45% cases of severe hemophilia A result from an inversion mutation. In hemophilia B several mutations such as partial and total deletions, missense mutations that result in the decrease or absence of factor IX or the production of an abnormal molecule. The factor XI gene is located on chromosome 4. Mutations of factor XI gene cause failure, reduced production of the active protein, and rarely production of an abnormal molecule result in factor XI deficiency.
A deficiency of AHG (factor VIII) results in the occurrence of hemophilia A, which is the most common type of hemophilia. However, recent studies now show that factor VIII is a glycoprotein which contains three distinct components:
• A clot-promoting factor that corrects the coagulation defect in patients with classic hemophilia.
• A factor VIII antigen that is present in patients with classic hemophilia but deficient in those with von Willebrand's disease (q.v.).
• A component called the von Willebrand factor that is synthesized by endothelial cells that will correct the platelet adherence defect in von Willebrand's disease.
Therefore, in hemophilia A (classic hemophilia), there is only an absence of the clot-promoting factor. Hemophilia B, due to a PTC deficiency, is also known as Christmas disease (named after the first patient in whom it was described). Apparently two forms of hemophilia B exist: one in which there are apparently normal levels of the inactive protein, another in which there are deficient levels of the coagulant factor. A PTA deficiency is the cause of hemophilia C. Despite the fact that different blood components are involved in each of these diseases, their clinical and oral manifestations are identical. They will therefore be described together as a single disease. In addition, some of the characteristics of the various hemophilioid disorders are shown in Table 18-13.
Table 18-13
Characteristics of the hemophilioid disorders
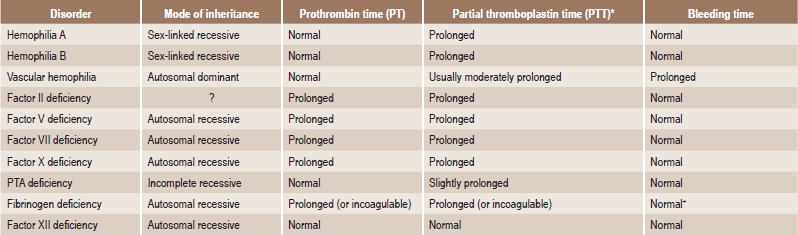
*Either original test or activated test (kaolin, etc). may be used.
+May occasionally be prolonged.
Courtesy of Dr Harold R Roberts. Modified from HR Roberts and KM Brinkhous: Blood coagulation and hemophilioid disorders. Postgrad Med, 43: 114, 1968.
Clinical Features
Patients with hemophilia exhibit persistent bleeding, either spontaneous or following even slight trauma that produces the mildest of abrasions or cuts. Hemorrhage into the subcutaneous tissues, internal organs, and joints is also a common feature and may result in massive hematomas. It is of interest, though still unexplained, that there is a wide range in the degree of severity of factor VIII deficiencies, with some patients showing only rare and mild bleeding, and others frequent and severe bleeding.
The disease is usually present from birth, but may not become clinically apparent for several years. Approximately, 30–50% of patients with severe hemophilia present with manifestations of neonatal bleeding such as prolonged bleeding from the umbilical cord. Spontaneous cyclic remissions and exacerbations of hemophilia are common.
Petechiae usually do not occur in patients with hemophilia because they are manifestations of capillary blood leaking, which typically is the result of vasculitis or abnormalities in the number or function of platelets.
Hemophilia is classified according to the clinical severity as mild, moderate, or severe (Table 18-14).
Table 18-14
| Classification | Factor activity (percentage) | Cause of hemorrhage |
| Mild | >5 | Major trauma or surgery |
| Moderate | 1–5 | Mild-to-moderate trauma |
| Severe | <1 | Spontaneous, hemarthrosis, soft tissue bleeding |
Hemophilia C can be distinguished from hemophilias A and B by the absence of bleeding into joints and muscles and by its occurrence in individuals of either genders.
Oral Manifestations
Hemorrhage from many sites in the oral cavity is a common finding in hemophilia, and gingival hemorrhage may be massive and prolonged. Even the physiologic processes of tooth eruption and exfoliation may be attended with severe prolonged hemorrhage. The oral manifestations of the various forms of hemophilia have been discussed by Spiegel and by Steg and his coworkers. In addition, mandibular 'pseudotumor' of hemophilia has been reported by Stoneman and Beierl, a condition in which there is subperiosteal bleeding, with reactive new bone formation causing tumor like expansion of the bone.
The problem of dental extractions is a difficult one in hemophiliacs. Without proper premedication, even a minor surgical procedure may result in death from exsanguination. Tooth extraction by means of rubber bands has often been used successfully, the rubber band being placed around the cervix of the tooth and allowed to migrate apically, causing exfoliation of the tooth through pressure necrosis of the periodontal ligament.
Laboratory Findings
The characteristic defect of hemophilia is a prolonged coagulation time. The bleeding time is normal, as is the prothrombin time and platelet aggregation. Usually, the activated partial thromboplastin time (aPTT) is prolonged; however, normal aPTT does not exclude mild or even moderate disease. Functional assay of factors is useful in diagnosing hemophilia caused due to dysfunction of coagulation factors. The combination of low factor VIII and low von Willebrand's factor indicate von Willebrand's disease. In vitro, the deficiency of the clot-promoting factor in the plasma of hemophiliacs impairs clotting because it appears to retard development of the substance responsible for conversion of prothrombin to thrombin. Separation of the various forms of hemophilia and proper diagnosis depends upon demonstration that the plasma of a patient with a known form of hemophilia does not correct the plasma clotting defect in the patient under observation.
Treatment and Prognosis
There is no known cure for hemophilia. The affected persons should be protected from traumatic injuries.
If a surgical procedure such as tooth extraction must be carried out, the operation should be considered a major one, to be performed only in a hospital.
The greatest number of fatalities in hemophiliacs have resulted from surgical procedures, including tooth extraction. Preoperative transfusion of whole blood and the administration of antihemophilic factor concentrate are recommended. Nevertheless, oral surgery is a dangerous procedure and should be avoided whenever possible. Unfortunately, a small percentage of hemophiliacs have circulating anticoagulant, probably an antibody, which specifically inactivates antihemophilic factor, negating the effects of transfusion.
The prognosis is variable, and many affected persons die during childhood.
von Willebrand's Disease: (Pseudohemophilia, vascular hemophilia, vascular purpura)
von Willebrand's disease, or pseudohemophilia, is a disease characterized by the tendency to excessive bleeding in patients who have a normal platelet count, normal clotting time, normal serum fibrinogen and normal prothrombin time. Only the bleeding time is prolonged. Therefore, other diseases characterized by an abnormal bleeding time must be ruled out before the diagnosis is established. It is the most common hereditary bleeding disorder first described by Erik Adolf von Willebrand in 1926. It is now accepted to be a hereditary disease, inherited as an autosomal dominant trait transmitted by and manifested in both males and females but detected more often in females. von Willebrand disease (vWD) is a family of bleeding disorders caused by an abnormality of the von Willebrand factor (vWF).
von Willebrand disease is caused by an inherited defect in the amount and/or quality of vWF. A gene on chromosome 12p (vWF gene) codes for the synthesis of this macromolecule. A variety of point mutations, insertions, and deletions at the vWF locus have been described. Acquired forms of vWD can be observed in the conditions such as Wilms tumor, systemic lupus erythematosus, etc.
Clinical Features
Prevalence worldwide is estimated at 0.9–1.3%. Many children with vWD are asymptomatic and are diagnosed as a result of a positive family history or during routine preoperative screening. Excessive bleeding, either spontaneously or following even minor trauma, is the chief feature of the disease. The most common sites of bleeding are nose, skin, and gingiva. Spontaneous nosebleeds and spontaneous cutaneous ecchymoses can be seen in these cases. Bleeding into the gastrointestinal tract and severe menorrhagia are also common, although hemarthrosis is rare. However, a wide variation in the clinical manifestations exists, even for the members of the same family. This bleeding tendency is often cyclic or sporadic.
Oral Manifestations
Gingival bleeding occurred in this same series in 39% of the patients. In some instances this was spontaneous bleeding; in other cases bleeding occurred only after brushing of the teeth.
The disease may be discovered after dental extractions because of the prolonged and excessive bleeding. The profuse bleeding may commence at the time of the extraction and continue indefinitely, or it may begin several hours subsequent to surgery and result in an almost unmanageable flow.
Laboratory Findings
The bleeding time of patients with this disease is increased to an extremely variable degree. Bleeding times of over 300 minutes have been recorded, but more often they range between several minutes and one hour. The bleeding time also shows wide variation in the same patient at different times. Prothrombin time (PT) is normal and aPTT increased in approximately 25% of type 1 vWD. The clotting time of the blood is usually normal, but may be slightly prolonged, while capillary fragility is reportedly increased with a positive tourniquet test in about 50% of cases. The clot retraction is normal. Characteristically, poor platelet adherence is also demonstrable.
Treatment
Bleeding episodes are best treated by transfusions of plasma and/or antihemophilic factor and by local control of hemostasis. Unfortunately, some patients become refractory to this treatment after repeated transfusions, and occasionally patients develop antibodies against antihemophilic factor.
Death from bleeding in pseudohemophilia is reportedly rare despite what appears to be excessive loss of blood. Nevertheless inherent dangers of tooth extraction should be recognized by the dentist so that if such a procedure is absolutely necessary, he/she may be on guard to institute prompt measures to control bleeding, should it occur. In general, all surgical procedures of an unessential nature should be avoided.
Parahemophilia
Parahemophilia is a rare hemorrhagic disorder, clinically similar to hemophilia, but caused by a deficiency of an unrelated blood factor, proaccelerin (factor V), which is one of the substances responsible for the conversion of prothrombin to thrombin.
Clinical Features
Parahemophilia is generally thought to be inherited as an autosomal recessive trait. Both genders are affected. Patients with parahemophilia exhibit a severe bleeding tendency. Spontaneous epistaxis, bleeding into the gastrointestinal tract and menorrhagia are common. Cutaneous ecchymoses and hematomas are frequently seen, although petechiae are rare. Intraocular hemorrhage and hemorrhage into the central nervous system have been reported in some patients, but hemarthrosis is seldom seen.
Oral Manifestations
Spontaneous gingival bleeding occurs in some cases of parahemophilia. Petechiae of the oral mucosa are rare. Prolonged bleeding following dental extractions is common, and this may terminate fatally.
Afibrinogenemia and Hypofibrinogenemia: (Hypofibrinogenopenia)
Afibrinogenemia is an uncommon disease in which the patient has little or no fibrinogen present in either his/her plasma or tissues. For this reason the blood cannot clot, even after the addition of thrombin.
A fibrinogen deficiency may be either congenital or acquired. Congenital afibrinogenemia is a rare hereditary disease, probably an autosomal recessive trait, occurring in both genders, but with some predilection for males. It is present from the time of birth and appears to be due to an inability of the patient to synthesize fibrinogen rather than any excessive destruction of fibrinogen.
Acquired hypofibrinogenemia generally occurs secondary to defective fibrinogen formation, to an increase in fibrinogen consumption during intravascular clotting, or to destruction or digestion of fibrinogen by fibrinolytic or proteolytic enzymes circulating in the blood stream. It may also occur in extravascular sequestration of the protein, in loss of blood through hemorrhage, in a transfusion reaction and in association with other conditions, including amyloidosis, polycythemia, certain neoplasms, and pregnancy.
There is generally not a complete absence of fibrinogen in the acquired form of the disease as there is in the congenital type, and this accounts for the difference in use of the terms 'afibrinogenemia' and 'hypofibrinogenemia'. But since the clinical features in both forms of the disease are almost identical, they will be described together.
Clinical Features
Patients with hypofibrinogenemia or afibrinogenemia exhibit severe bleeding episodes, throughout their lives in the congenital type, and the disease is clinically indistinguishable from hemophilia. However, characteristically in the congenital type, the patients may have long periods of freedom from bleeding. Epistaxis, bleeding into the gastrointestinal tract and central nervous system, and cutaneous ecchymoses and hematomas are all common. Hemarthrosis is not as prominent as in hemophilia. In affected females, menstrual bleeding is usually normal.
Oral Manifestations
The oral manifestations of congenital afibrinogenemia have been reviewed by Kranz and Ruff, and those of the acquired type by Rose. These consist of spontaneous gingival bleeding and prolonged and excessive bleeding following dental extractions. Petechiae of the oral mucosa are rare.
Laboratory Findings
Patients with congenital afibrinogenemia have normal red blood cell, white blood cell and platelet counts, although thrombocytopenia has been occasionally reported. The bleeding time may be normal or slightly prolonged. The most dramatic feature is that the clotting time and prothrombin time are infinite, although this is not necessarily the case in hypofibrinogenemia. The peripheral blood fails to clot even after the addition of thrombin. The tourniquet test in these patients is normal. Finally, the erythrocyte sedimentation rate is zero, the cells remain suspended even after 24 hours.
Treatment
There is no specific treatment for the disease except for transfusions, particularly of concentrated fibrinogen, during bleeding episodes. Occasional patients develop antibodies against the administered fibrinogen, thus disrupting therapy. Unfortunately, the prognosis is poor, and many patients die of hemorrhage during infancy or early childhood. Some patients do reach adult life. The acquired form of the disease is less serious if recognized in time.
Dysfibrinogenemia
Dysfibriongenemia is a congenital disease probably transmitted as an autosomal dominant characteristic which appears to represent a group of familial disorders rather than a single entity. For example, there may be impairment of the rate at which thrombin cleaves fibrinopeptides from fibrinogen. There may be replacement of one amino acid residue by another in the NH2 terminal part of the A<chain of fibrinogen, as in fibrinogen detroit, in which arginine replaces serine. In fibrinogen Philadelphia, the abnormal protein is catabolized at an accelerated rate.
Fibrinogen is usually present in normal amounts in this disease, but is defective in its structure and coagulability so that the aggregation of fibrin monomers is impeded. In one variant, the abnormality of fibrinogen appears to be one manifesting defective cross-linking between fibrin strands after clotting has occurred.
Acquired dysfibrinogenemias, often called dysfibrinogenemia of liver disease, commonly due to severe liver disease secondary to cirrhosis, hepatoma, or hepatitis exhibit bleeding complications.
The disease manifests itself clinically by a mild to severe bleeding tendency although, interestingly, paradoxical thrombosis has also been reported. (Fibrinogen Oslo I is an abnormal fibrinogen that is associated with thromboembolic complications. The abnormal fibrinogen in these patients forms a fibrin clot that is resistant to fibrinolysis by plasmin).
Fibrin-stabilizing Factor Deficiency: (Factor XIII deficiency)
Congenital factor XIII or fibrin-stabilizing factor (factor XIII) deficiency, originally recognized by Duckert in 1960, is a rare autosomal recessive disease, with a high incidence of consanguinity. Acquired factor XIII deficiency has been described in association with hepatic failure, inflammatory bowel disease, and myeloid leukemia.
Inherited factor XIII deficiency is usually due to mutations in the gene encoding the catalytic a subunit, located on chromosome 6. More than 40 different mutations have been identified, half of which are missense mutations. In patients homozygous for this defect, the a subunit is absent in plasma, platelets, and monocytes, resulting in a severe bleeding diathesis; the concentration of ß subunits is relatively normal. Biochemically, thrombin appears to activate factor XIII from an inactive precursor form. This activated factor XIII then crosslinks fibrin or stabilizes it by transamidation. In the absence of this factor, there is failure of permanent peptide bonds between fibrin molecules so that the fibrinogen monomer aggregates (fibrins) break up under certain conditions. Factor XIII covalently binds fibronectin, 2-plasmin inhibitor, and other molecules to the fibrin plug; this enhances adherence to the wound site, resistance to fibrinolysis, and wound healing.
Clinical Features
Patients with this deficiency have severe postsurgical bleeding episodes which are typically delayed for 24–36 hours, hemarthrosis and defective wound healing. Bleeding and clotting times are both normal. Bleeding from the stump of the umbilical cord within the first days to weeks of life is a characteristic sign that occurs in 80% of affected individuals. Soft tissue bleeding and bruising are very common, as is bleeding into the mouth and gums during teething.
Laboratory Findings
Measurement of clot stability is the most commonly used screening test for factor XIII deficiency. In the absence of factor XIII, the clot dissolves in minutes to hours. Factor XIII a and factor XIII ß antigen levels can be quantified by means of enzyme-linked immunosorbent assay (ELISA) techniques.
Macroglobulinemia: (Waldenstrom hypergammaglobulinemia, macroglobulinemia of Waldenstrom)
Macroglobulinemia is not specifically a blood 'factor' disease, but is included here because of the hemorrhagic tendency of the disease, thus mimicking the other hemorrhagic diatheses previously described. The condition was first described as an entity by Waldenstrom in 1948, and, by 1958, he found over 100 cases in the literature in his classic review of the disease. Since then many more cases have been discovered, and this disease should not be considered rare.
Etiology
The etiology of macroglobulinemia is unknown. It has been suggested to be related to:
• A variant of multiple myeloma, IgM monoclonal gammopathies of undetermined significance (MGUS) are considered a precursor of Waldenstrom macroglobulinemia.
• The Bing-Neel syndrome (hyperglobulinemia with central nervous system involvement on a toxi-infectious basis).
• An altered immunologic reaction.
• Reports of familial cases suggest a genetic predisposition.
It is now generally classified as a plasma cell dyscrasia (monoclonal gammopathies) so that the excessive proliferation of B-lymphocytes, the precursor of the plasma cell, results in the production of large amounts of electrophoretically homogeneous M-type IgM globulins which characterize the disease.
The clinical presentation of WM is similar to that of multiple myeloma (MM) except that organomegaly is common in WM and is uncommon in MM, and lytic bony disease and renal disease are uncommon in WM but are common in MM.
Clinical Features
The protean clinical manifestations of this rare disorder result from two important components of the disorder. First, secretion of the IgM paraprotein leads to hyperviscosity and consequent vascular complications because of certain physical, chemical, and immunological properties of this paraprotein. The monoclonal IgM causes hyperviscosity syndrome; cryoglobulinemia types 1 and 2; coagulation abnormalities; polyneuropathies; cold agglutinin disease and anemia; primary amyloidosis; and tissue deposition of amorphous IgM in skin, the GI tract, kidneys, and other organs.
Second, neoplastic lymphoplasmacytic cells infiltrate tissue. The abnormal clone of lymphoplasmacytic cells infiltrate the bone marrow, spleen, lymph nodes, liver, lungs, GI tract, kidneys, skin, eyes, and CNS. The infiltration of these organs causes numerous organ specific clinical symptoms and signs. Occasionally, IgM paraprotein has rheumatoid factor activity, antimyelin activity that can contribute to peripheral neuropathy, and immunologically related lupus anticoagulant activity.
Macroglobulinemia occurs most frequently in persons over the age of 50 years, seldom under 40 years. Males and females are about equally affected.
The chief clinical signs are pallor, weakness and weight loss, lymphadenopathy and hepatomegaly occurring commonly. Hemorrhages from the nasal and oral cavity are characteristic of the disease, and subarachnoid and ocular hemorrhages are also frequently seen, according to Voight and Frick. Bone lesions such as those that occur in myeloma are exceedingly rare.
Oral Manifestations
Oral lesions are common in macroglobulinemia, and these have been reviewed by Gamble and Driscoll. They consist of spontaneous gingival hemorrhage, often with continued oozing of blood; bleeding oral ulcers on the tongue, palate, buccal mucosa or gingiva; and focal areas of hyperemia which appear edematous and are painful. Severe and prolonged bleeding following dental extractions is common. Salivary gland involvement with xerostomia has also been reported.
These bleeding diatheses appear to be related to proteinprotein interactions, with formation of complexes between IgM globulins and coagulation factors such as fibrinogen, thrombin and factors V and VII, as well as interference with platelet agglutination and capillary damage.
Laboratory Findings
Waldenstrom was the first to demonstrate by ultracentrifugation technique that the serum of patients with this disease contained a fraction in the serum proteins, presumably globulins, with molecular weights near 1,000,000 as contrasted to the highest normal globulin molecular weight of 150,000. In addition to the macroglobulinemia and hyperglobulinemia, these patients generally manifested severe anemia with hemoglobin levels near 4–6 gm/dl, an extremely elevated sedimentation rate, demonstrable euglobulins and frequent gelling of the serum upon cooling to room temperature or lower. The viscosity of the blood serum was usually extremely high.
The white blood cell and platelet counts, as well as the bleeding, clotting, and prothrombin times, are usually within normal limits, although lymphocytosis, neutropenia, and thrombocytopenia are occasionally observed.
Bone marrow smears are generally confusing, since they show an increase in mononuclear cells that have been interpreted variously as plasma cells, lymphoid cells, or lymphoid reticulosis.
Bence Jones proteinuria is present in a limited number of patients with macroglobulinemia. Serum protein electrophoresis results indicate evidence of a monoclonal spike.
Cryoglobulinemia: (Cryoproteinemia)
Cryoglobulinemia is a disease characterized by the presence of cryoglobulins in varying amounts in the blood. These cryoglobulins are globulins which have the ability to precipitate on exposure to cold and redissolve upon return to body temperature. This condition has been discussed in detail by Brouet and his associates.
A mild cryoglobulinemia has been found to occur in a large variety of diseases including some of the collagen diseases such as rheumatoid arthritis, periarteritis nodosa and systemic lupus erythematosus, as well as in certain of the malignant lymphomas including lymphosarcoma, Hodgkin's disease and lymphatic leukemia. It is also sometimes found in polycythemia, heart disease and cirrhosis. In multiple myeloma, it has occasionally been found in large quantities. In some cases there is no apparent associated disease, and in these instances, it has been referred to as ‘essential cryoglobulinemia’, or mixed IgG-IgM cryoglobulinemia. The etiology of this disease is not known.
The current system of classification of the types of cryoglobulinemia (by composition) includes the following:
Type I cryoglobulinemia, or simple cryoglobulinemia, is the result of a monoclonal immunoglobulin, usually immunoglobulin M (IgM) or immunoglobulin G (IgG) observed in lymphoproliferative disorders (e.g. multiple myeloma, Waldenström macroglobulinemia).
Type II and type III cryoglobulinemia (mixed cryoglobulinemia) contain rheumatoid factors (RF), which usually are IgM. These RFs form complexes with the fragment crystallizable (Fc) portion of polyclonal IgG. The actual RF may be monoclonal (in type II cryoglobulinemia) or polyclonal (in type III cryoglobulinemia) immunoglobulin. This type is observed in lymphoproliferative disorders such as chronic lymphocytic leukemia (CLL), chronic liver disease, infections, and coexistent connective tissue diseases.
Cryoglobulinemia does not usually produce clinical manifestations. On occasion, however, spontaneous bleeding from the nose and mouth with purpuric hemorrhages into the skin and retina may be found. Other sequelae are related directly to cryoprecipitation in vivo, including plugging and thrombosis of small arteries and capillaries in the extremities (gangrene) and glomeruli (acute renal failure). Circulating large-molecular weight cryoprotein complexes, even when unprecipitated in vivo, can lead to clinical hyperviscosity syndrome.
Treatment
The goal of therapy is the limitation of precipitant cryoglobulin and the resultant inflammatory effects; however, asymptomatic cryoglobulinemia does not require treatment. In case of secondary cryoglobulinemia underlying malignancy or associated disease should be treated. Otherwise, cryoglobulinemia is treated simply by suppression of the immune response.
Hematopoietic Stem Cells (HSCs)
Hematopoietic stem cells (HSCs) are multipotent stem cells that give rise to all the blood cell types from the myeloid (monocytes and macrophages, neutrophils, basophils, eosinophils, erythrocytes, megakaryocytes/platelets, dendritic cells), and lymphoid lineages (T cells, B cells, NK cells). HSCs are a heterogeneous population. Three classes of stem cells exist, distinguished by their ratio of lymphoid to myeloid progeny (L/M) in blood. Myeloid-biased (My-bi) HSC have low L/M ratio (>0, <3), whereas lymphoid biased (Ly-bi) HSC show a large ratio (>10). The third category consists of the balanced (Bala) HSC for which 3< L/M<10. Much work is currently being undertaken to investigate the properties of these different classes of HSCs, but it appears that only the myeloid–biased and balanced HSCs have durable self-renewal properties.
HSCs are found in the bone marrow of adults, which includes femurs, hip, ribs, sternum, and other bones. Cells can be obtained directly by removal from the hip using a needle and syringe, or from the blood following pre-treatment with cytokines, such as G-CSF (granulocyte colony-stimulating factors), that induce cells to be released from the bone marrow compartment. Other sources for clinical and scientific use include umbilical cord blood, peripheral blood in which a small number of stem and progenitor cells circulate in the blood stream. These cells can migrate from marrow to blood in greater numbers by injecting the donor with a cytokine, such as G-CSF. As stem cells, HSCs are defined by their ability to replenish all blood cell types (multipotency) and their ability to self-renew. It is known that a small number of HSCs can expand to generate a very large number of daughter HSCs. This phenomenon is used in bone marrow transplantation, when a small number of HSCs reconstitute the hematopoietic system. This indicates that, subsequent to bone marrow transplantation, symmetrical cell divisions in-to two daughter HSCs must occur.
Stem cell self-renewal is thought to occur in the stem cell niche in the bone marrow, and it is reasonable to assume that key signals present in this niche will be important in self-renewal. There is much interest in the environmental and molecular requirements for HSC self-renewal, as understanding the ability of HSC to replenish themselves will eventually allow the generation of expanded populations of HSC in vitro that can be used therapeutically.
HSCs have a higher potential than other immature blood cells to pass the bone marrow barrier, and thus, may travel in the blood from the bone marrow in one bone to another bone. If they settle in the thymus, they will develop into T cells. In the case of fetuses and other extramedullary hematopoiesis, HSCs may also settle in the liver or spleen and develop. This ability is the reason why HSCs may be harvested directly from the blood.
With regard to morphology, HSCs resemble lymphocytes. They are non-adherent, and rounded, with a rounded nucleus and low cytoplasm-to-nucleus ratio. Since peripheral HSC cannot be isolated as a pure population, it is not possible to identify them under a microscope. The above description is based on the morphological characteristics of a heterogeneous population, of which PHSC are a component.
Hematopoietic stem cells are identified by their surface markers, many of these markers belong to the cluster of differentiation (CD) series, like: CD34, CD38, CD90, CD133, CD105, CD45, and also c-kit, the receptor for stem cell factor. There are many differences between the human and mice hematopoietic cell markers for the commonly accepted type of HSCs.
Hematopoetic stem cells cannot be easily observed directly and, therefore, their behaviors need to be inferred indirectly. Clonal studies are likely the closest technique for single cell in vivo studies of HSC. Here, sophisticated experimental and statistical methods are used to ascertain that, with a high probability, a single HSC is contained in a transplant administered to a lethally irradiated host. The clonal expansion of this stem cell can then be observed over time by monitoring the percent donor-type cells in blood as the host is reconstituted. The resulting time series is defined as the repopulation kinetic of the HSC.
References
Aalto, S.M., Linnavuori, K., Peltola, H., et al. Immunoreactivation of Epstein-Barr virus due to cytomegalovirus primary infection. J Med Virol. 1998; 56(3):186–191. [Nov].
Adlersberg, D. Newer advances in sprue. Oral Surg. 1(1109), 1948.
Ahlbom, H.E. Simple achlorhydric anemia, Plummer Vinson syndrome, and carcinoma of the mouth, pharynx and oesophagus in women. Br Med J. 2(231), 1936.
Akashi, K., Eizuru, Y., Sumiyoshi, Y., et al. Brief report: severe infectious mononucleosis-like syndrome and primary human herpesvirus 6 infection in an adult. New Engl J Med. 1993; 329(3):168–171. [Jul 15].
Akman, I.O., Ostrov, B.E., Neudorf, S. Autoimmune manifestations of the Wiskott- Aldrich syndrome. Semin Arthritis Rheum. 1998; 27(4):218–225. [Feb].
Andersson, J.P. Clinical aspects on Epstein-Barr virus infection. Scand J Infect Dis Suppl. 1991; 80:94–104.
Andiman, W.A., Miller, G. Antibody responses to Epstein-Barr virus. In: Ros N.R., Friedman H., eds. Manual of Clinical Immunology. Washington DC: American Society for Microbiology; 1980:628–633. [(2nd ed)].
Anwar, R., Miloszewski, K.J. Factor XIII deficiency. Br J Haematol. 1999; 107(3):468–484. [Dec].
Anwar, R., Minford, A., Gallivan, L., et al. Delayed umbilical bleeding—a presenting feature for factor XIII deficiency: clinical features, genetics, and management. Pediatrics. 109: E32, 2002.
Araneda, M., Krishnan, V., Hall, K., et al. Reactive and clonal thrombocytosis: proinflammatory and hematopoietic cytokines and acute phase proteins. South Med J. 2001; 94(4):417–420. [Apr].
Ariens, R.A., Lai, T.S., Weisel, J.W., et al. Role of factor XIII in fibrin clot formation and effects of genetic polymorphisms. Blood. 2002; 100(3):743–754. [Aug 1].
Asakai, R., Chung, D.W., Ratnoff, O.D. Factor XI (plasma thromboplastin antecedent) deficiency in Ashkenazi Jews is a bleeding disorder that can result from three types of point mutations. Proc Natl Acad Sci USA. 1989; 86(20):7667–7671. [Oct].
Aslan, Y., Erduran, E., Gedik, Y., et al. The role of high dose methylprednisolone and splenectomy in the accelerated phase of Chediak-Higashi syndrome. Acta Haematol. 1996; 96(2):105–107.
Atmatzidis, K., Papaziogas, B., Pavlidis, T., et al. Plummer-Vinson syndrome. Dis Esophagus. 2003; 16(2):154–157.
Bacigalupo, A., Brand, R., Oneto, R., et al. Treatment of acquired severe aplastic anemia: bone marrow transplantation compared with immunosuppressive therapy—the European group for blood and marrow transplantation experience. Semin Hematol. 2000; 37(1):69–80. [Jan].
Bacigalupo, A., Broccia, G., Corda, G., et al. Antilymphocyte globulin, cyclosporin, and granulocyte colony-stimulating factor in patients with acquired severe aplastic anemia (SAA): a pilot study of the EBMT SAA Working Party. Blood. 1995; 85(5):1348–1353. [Mar 1].
Baldus, M., Zunftmeister, V., Geibel-Werle, G., et al. Chediak-Higashi-Steinbrinck syndrome (CHS) in a 27-year-old woman—effects of G-CSF treatment. Ann Hematol. 1999; 78(7):321–327. [Jul].
Banks, P. Infectious mononucleosis: a problem of differential diagnosis to the oral surgeon. Br J Oral Surg. 4(227), 1967.
Barak, Y., Nir, E. Chediak-Higashi syndrome. Am J Pediatr Hematol Oncol Spring. 1987; 9(1):42–55.
Barbosa, M.D., Barrat, F.J., Tchernev, V.T., et al. Identification of mutations in two major mRNA isoforms of the Chediak-Higashi syndrome gene in human and mouse. Hum Mol Genet. 1997; 6(7):1091–1098. [Jul].
Barnfield, W.F. Leukemia and dental procedures. Am J Orthod Oral Surg. 31(329), 1945.
Barton, G.M.G. Recurrent agranulocytosis. Lancet. (103):1948.
Batlle, J., Torea, J., Rendal, E., Fernandez, M.F. The problem of diagnosing von Willebrand’s disease. Intern Med Suppl J. 1997; 740:121–128.
Bauer, W.H. The supporting tissues of the tooth in acute secondary agranulocytosis (arsphenamine neutropenia. J Dent Res. 25(501), 1946.
Bazzan, M., Tamponi, G., Vaccarino, A. Natural and acquired inhibitors of hemostasis in selected symptomatic outpatients with venous thromboembolic disease. Haematologica. 1997; 82(4):420–422. [Jul-Aug].
Beauchesne, M.F., Shalansky, S.J. Nonchemotherapy drug-induced agranulocytosis: a review of 118 patients treated with colony-stimulating factors. ALYSIS. 1999; 19(3):299–305. [Mar].
Becker, F.T., Coventry, W.D., Tuura, J.L. Recurrent oral and cutaneous infections associated with cyclic neutropenia. AMA Arch Dermatol. 80(731), 1959.
Bell, W.R., Braine, H.G., Ness, P.M., Kickler, T.S. Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: clinical experience in 108 patients. New Engl J Med. 1991; 325(6):398–403. [Aug 8].
Bell, W.R. Thrombotic thrombocytopenic purpura/hemolytic uremic syndrome relapse: frequency, pathogenesis, and meaning. Semin Hematol. 1997; 34(2):134–139. [Apr].
Bender, I.B. Bone changes in leucemia. Am J Orthod Oral Surg. 30(556), 1944.
Berk, P.D., Goldberg, J.D., Donovan, P.B., et al. Therapeutic recommendations in polycythemia vera based on Polycythemia Vera Study Group protocols. Semin Hematol. 1986; 23(2):132–133. [Apr].
Berlin, N.I. Diagnosis and classification of the polycythemia. Semin Hematol. 1975; 12(4):339–351. [Oct].
Berliner, D. Spontaneous gingival bleeding in aplastic anemia. J Maine Med Assoc. 41(200), 1950.
Berntropp, E., Astermark, J., Bjorkman, S. Consensus perspectives on prophylactic therapy for hemophilia: summary statement. Hemophilia, 9(Supp 1. 2003; 1–4.
Bethell, F.H. Leukemia: the relative incidence of its various forms and their response to radiation therapy. Ann Intern Med. 18(757), 1943.
Betticher, D.C., Hsu Schmitz, S.F., Ratschiller, D., et alSwiss Group for Clinical Cancer Research (SAKK). Cladribine (2-CDA) given as subcutaneous bolus injections is active in pretreated Waldenstrom’s macroglobulinaemia. Br J Haematol. 1997; 99(2):358–363. [Nov].
Beutler, E., Lichtman, M.A., Coller, B.S., Williams Hematology. McGraw-Hill, New York, 2001:295–304. [6th ed 447-70].
Birch, C.L., Snider, F.F. Tooth extraction in hemophilia. J Am Dent Assoc. 26(1933), 1939.
Bizzozero, O.J., Jr., Johnson, K.G., Ciocco, A. Radiation-related leukemia in Hiroshima and Nagasaki. New Engl J Med. 1966; 274(1095):1946–1964. [Distribution, incidence and appearance time].
Boddington, M.M. Changes in buccal cells in the anaemias. J Clin Pathol. 12(222), 1959.
Boen, S.T. Changes in the nuclei of squamous epithelial cells in pernicious anaemia. Acta Med Scand. 159(425), 1957.
Bolton-Maggs, P.H., Hill, F.G. The rarer inherited coagulation disorders: a review. Blood Rev. 1995; 9(2):65–76. [Jun].
Bolton-Maggs, P.H., Patterson, D.A., Wensley, R.T. Definition of the bleeding tendency in factor XI-deficient kindreds—a clinical and laboratory study. Thromb Haemost. 1995; 73(2):194–202. Feb
Bolton-Maggs, P.H. Factor XI deficiency. Baillieres Clin Haematol. 1996; 9(2):355–368. [Jun].
Bolton-Maggs, P.H. The management of factor XI deficiency. Haemophilia. 1998; 4(4):683–688. [Jul].
Borgna-Pignatti, C., De Stefano, P., Pajno, D. Cholelithiasis in children with thalassemia major: an ultrasonographic study. J Pediatr. 1981; 99(2):243–244. [Aug].
Bothwell, T.H., Charlton, R.W., Cook, J.D. Iron Metabolis in. Man. 1979; 1–77.
Bowie, E.J.W., Jr., Thompson, J.H., Owen, C.A., Jr. The blood platelet (including a discussion of the qualitative platelet diseases). Clin Proc Mayo. 40(625), 1965.
Boyd, W.C. Rh blood factors; an orientation review. Arch Pathol. 40(114), 1945.
Boyle, P.E., Dinnerman, M. Natural vital staining of the teeth of infants and children. Am J Orthod Oral Surg. 27(377), 1941.
Braun-Falco, O., Plewig, G., Wolff, H.H., Burgdorf, W.H.C. Chediak-higashi syndrome. In: Dermatology. Berlin: Springer-Verlag; 2000. [(2nd ed). 1029].
Bredenkamp, J.K., Castro, D.J., Mickel, R.A. Importance of iron repletion in the management of Plummer-Vinson syndrome. Ann Otol Rhinol Laryngol. 1990; 99(1):51–54. [Jan].
Brenner, B., Laor, A., Lupo, H. Bleeding predictors in factor-XI-deficient patients. Blood Coagul Fibrinolysis. 1997; 8(8):511–515. [Nov].
Brettler, D.B., Levine, P.H. Clinical manifestations and therapy of inherited coagulation factor deficiencies. In RW Colman, J Hirsh, VJ Marder et alHemostasis and Thrombosis: Basic Principles and Clinical Practice (3rd ed). JB Lippincott, Philadelphia. 1994; 169–183.
Bridgman, J., Witting, M. Thrombotic thrombocytopenic purpura presenting as a sudden headache with focal neurological findings. Annals of emergency medicine. 1996; 27:95–97.
Brinkhous, K.M., Denicola, P. Hemophilia and other hemorrhagic states. Chapel Hill NC: University of North Carolina Press; 1959. [(Ed Assoc)].
Brouet, J.C., Clauvel, J.P., Danon, F. Biologic and clinical significance of cryoglobulins: a report of 86 cases. Am J Med. 1974; 57(5):775–788. [Nov].
Brown, Kelly A. Spasm at the entrance of the oesophagus. J Laryngol Rhinol Otol. 1919; 34:285–289.
Brunning, R.D. Philadelphia chromosome positive. leukemia hum pathol. 11(307), 1980.
Buchwald, D.S., Rea, T.D., Katon, W.J., et al. Acute infectious mononucleosis: characteristics of patients who report failure to recover. Am J Med. 2000; 109(7):531–537. [Nov].
Buckley, R.H. Advances in the correction of immunodeficiency by bone marrow transplantation. Pediatr Ann. 1987; 16(5):412–413. [416-21, May].
Buckley, R.H. Primary immunodeficiency diseases. In: E. Middleton, Jr., et al, eds. Allergy: Principles and Practice. St. Louis: Mosby-Year Book; 1998:713–734. [Vol 2 (5th ed)].
Bunn, H.F. Pathogenesis and treatment of sickle cell disease. New Engl J Med. 1997; 337(11):762–769. [Sep 11].
Burgio, G.R., Monafo, V. Infectious mononucleosis fifty years after the discovery of the Paul- Bunnell test. Infection. 1983; 11(1):1–5. [Jan-Feb].
Burket, L.W. A histopathologic explanation for the oral lesions in the acute leucemias. Am J Orthod Oral Surg. 30(516), 1944.
Buse, P.E., Zuckerman, G.R., DM, Balfe. Cervical esophageal web associated with a patch of heterotopic gastric mucosa. Imaging Abdom. 1993; 18(3):227–228.
Caffey, J. Cooley’s anemia: a review of the roentgenographic findings in the skeleton. Am J Roentgenol, Radium Ther, Nucl Med. 78(381), 1957.
Carcao, M.D., Blanchette, V.S., Dean, J.A., et al. The platelet function analyzer (PFA-100): a novel in-vitro system for evaluation of primary haemostasis in children. Br J Haematol. 1998; 101(1):70–73. [Apr].
Carnide, E.M., Jacob, C.M., Pastorino, A.C., et al. Chediak-Higashi syndrome: presentation of seven cases. Rev Paul Med. 1998; 116(6):1873–1878. [Nov-Dec].
Carulli, G., Sbrana, S., Azzara, A. Reversal of autoimmune phenomena in autoimmune neutropenia after treatment with rhG-CSF: two additional cases [letter; comment]. Br J Haematol. 1997; 96(4):877–878. [Mar].
Chan, L. A case of Plummer-Vinson syndrome. Oral Surg. 5(325), 1952.
Chanet, V., Tournilhac, O., Dieu-Bellamy, V., et al. Isolated spleen agenesis: a rare cause of thrombocytosis mimicking essential thrombocythemia. Haematologica. 2000; 85(11):1211–1213. [Nov].
Chin-Yee, I., Bezchlibnyk-Butler, K., Wong, L. Use of cytokines in clozapine-induced agranulocytosis. Can J Psychiatry. 1996; 41(5):280–284. [Jun].
Chisholm, M. The association between webs, iron and post-cricoid carcinoma. Postgrad Med J. 1974; 50(582):215–219. [Apr].
Cimino, R., Rametta, V., Matera, C., et al. Recombinant interferon alpha-2b in the treatment of polycythemia vera. Am J Hematol. 1993; 44(3):155–157. [Nov].
Cohen, J.I. Epstein-Barr virus infection. New Engl J Med. 2000; 343(7):481–492. [Aug 17].
Cohen, D.W., Morris, A.L. Periodontal manifestations of cyclic neutropenia. J Periodontol. 32(159), 1961.
Conley, M.E., Notarangelo, L.D., Etzioni, A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999; 93(3):190–197. [Dec].
Conrad, M.E., Umbreit, J.N. Iron absorption and transport—an update. Am J Hematol. 2000; 64(4):287–298. [Aug].
Cook, T.J. Blood dyscrasias as related to periodontal disease: with special reference to leukemia. J Periodontol. 18(159), 1947.
Cooley, T.B., Witwer, E.R., Lee, P. Anemia in children with splenomegaly and peculiar changes in the bones. Am J Dis Child. 34(347), 1927.
Cooley, T.B., Lee OF!. Erythroblastic anemia. Am J Dis Child. 43(705), 1932.
Cotran, R.S., Kumar, V., T, Collins. Robbins pathologic basis of disease. India Private Limited. Harcourt. 2001.
Courant, P., Sobkov, T. Oral manifestation of infectious mononucleosis. J Periodontol. 40(279), 1969.
Cunha, B.A. False positive heterophile tests for Epstein-Barr virus infectious mononucleosis. Infect Dis Pract. 2001; 25:7–9.
Currarino, G., Erlandson, M.E. Premature fusion of the epiphyses in Cooley’s anemia. Radiology. 83(656), 1964.
Curtis, A.B. Childhood leukemias: initial oral manifestations. J Am Dent Assoc. 83(159), 1971.
Custer, R.P.An Atlas of the Blood and Bone Marrow. WB Saunders: Philadelphia, 1974. [(2nd ed)].
Daniels, J.C., Ritzmann, S.E., Levin, W.C. Lymphocytes: morphological, development, and functional characteristics in health, disease, and experimental study—an analytical review. Tex Rep Biol Med. 26(5), 1968.
de Planque, M.M., Bacigalupo, A., Wursch, A., et al. Long-term follow-up of severe aplastic anaemia patients treated with antithymocyte globulin. Severe aplastic anaemia working party of the European Cooperative group for Bone Marrow Transplantation (EBMT). Br J Haematol. 1989; 73(1):121–126. [Sep].
Delannoy, A., Van Den, Neste E., Michaux, J.L., et al. Cladribine for Waldenstrom’s macroglobulinaemia. Br J Haematol. 1999; 104(4):933–934. [Mar].
Delcourt-Debruyne, E.M., Boutigny, H.R., Hildebrand, H.F. Features of severe periodontal disease in a teenager with Chediak-Higashi syndrome. J Periodontol. 2000; 71(5):816–824. [May].
Desikan, R., Dhodapkar, M., Siegel, D., et al. High-dose therapy with autologous haemopoietic stem cell support for Waldenstrom’s macroglobulinaemia. Br J Haematol. 1999; 105(4):993–996. [Jun].
Dewey, K.W., Grossman, H., Canale, V.C. Cholelithiasis in thalassemia major. Radiology. 1970; 96(2):385–388. [Aug].
Di, Paola J., Nugent, D., Young, G. Current therapy for rare factor deficiencies. Haemophilia, 7 Suppl. 2001; 1:16–22. [Jan].
Dieterich, W., Ehnis, T., Bauer, M. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med. 1997; 3(7):797–801. [Jul].
Dimopoulos, M.A., Alexanian, R. Waldenstrom’s macroglobulinemia. Blood. 1994; 83(6):1452–1459. [Mar 15].
Dimopoulos, M.A., Galani, E., Matsouka, C. Waldenstrom’s macroglobulinemia. Hematol Oncol Clin North Am. 1999; 13(6):1351–1366. [Dec].
Dimopoulos, M.A., O’Brien, S., Kantarjian, H., et al. Fludarabine therapy in Waldenstrom’s macroglobulinemia. Am J Med. 1993; 95(1):49–52. [Jul].
Dimopoulos, M.A., Panayiotidis, P., Moulopoulos, L.A., et al. Waldenstrom’s macroglobulinemia: clinical features, complications, and management. J Clin Oncol. 2000; 18(1):214–226. [Jan].
Dimopoulos, M.A., Weber, D.M., Kantarjian, H., et al. Chlorodeoxyadenosine therapy of patients with Waldenstrom macroglobulinemia previously treated with fludarabine. Ann Oncol. 1994; 5(3):288–289. [Mar].
Dimopoulos, M.A. Waldenstrom’s macroglobulinemia-therapy. American Society of Hematology, Washington DC. 1999.
Doi, H., Inaba Mm yamamoto, Y., et al. Pluripotent hemopoietic stem cells are c-kit. Proc Natl Acad Sci USA, 18. 1997; 94(6):2513–2517.
Duffy, J.H., Driscoll, E.J. Oral manifestations of leukemia. Oral Surg. 11(484), 1958.
Dunnet, W.N. Infectious mononucleosis. Br Med J. 1(1187), 1963.
Eberst, M., Hereditary hemolytic anemias. Tintinalli JE, Ruiz E, Krome RL,Emergency medicine: A Comprehensive Study Guide (4th ed). McGraw-Hill, New York. 1996:987–990.
Egbring, R., Kroniger, A., Seitz, R. Factor XIII deficiency: pathogenic mechanisms and clinical significance. Semin Thromb Hemost. 1996; 22(5):419–425.
Elkins, S.L., Wilson, P.P., Jr., Files, J.C. Thrombotic thrombocytopenic purpura: evolution across 15 years. University of Mississippi Medical Center. 1997.
Elwood, P.C., Jacobs, A., Pitman, R.G. Epidemiology of the Paterson-Kelly syndrome. Lancet. 1964; 2:716–720.
Epstein, M.A., Achong, B.C. The Epstein-Barr Virus. Springer-Verlag, New York. 1979.
Erlandson, M.E., Hilgartner, M. Hemolytic disease in the neonatal period and early infancy. J Pediatr. 54(566), 1959.
Estren, S., Medal, L.S., Dameshek, W. Pseudohemophilia blood 1946;1(504).
Fallaux, F.J., Hoeben RC. Gene therapy for the hemophilias. Opin Hematol Curr. 1996; 3(5):385–389. [Sep].
Farrant, P.C. Nuclear changes in squamous cells from buccal mucosa in pernicious anaemia. B Med J. 1(1694), 1960.
Ferri, C., Moriconi, L., Gremignai, G. Treatment of the renal involvement in mixed cryoglobulinemia with prolonged plasma exchange. Nephron. 1986; 43(4):246–253.
Ferri, C., Pietrogrande, M., Cecchetti, R. Low-antigen-content diet in the treatment of patients with mixed cryoglobulinemia. Am J Med. 1989; 87(5):519–524. [Nov].
Filipovich, A.H., Stone, J.V., Tomany, S.C., et al. Impact of donor type on outcome of bone marrow transplantation for Wiskott-Aldrich syndrome: collaborative study of the International Bone Marrow Transplant Registry and the National Marrow Donor Program. Blood. 2001; 97(6):1598–1603. [Mar 15].
Foerster, J. Waldenstrom’s macroglobulinemia. In Lee G.R., ed.: Wintrobe’s Clinical Hematology, 10th, Balttimore: Williams and Wilkins, 1999. [2681].
Foran, J.M., Rohatiner, A.Z., Coiffier, B., et al. Multicenter phase II: study of fludarabine phosphate for patients with newly diagnosed lymphoplasmacytoid lymphoma, Waldenstrom’s macroglobulinemia, and mantle-cell lymphoma. J Clin Oncol. 1999; 17(2):546–553. [Feb].
Formiga, F., Mitjavila, F., Pac, M. Effective splenectomy in agranulocytosis associated with systemic lupus erythematosus [letter]. J Rheumatol. 1997; 24(1):234–235. [Jan,].
Fraser-Moodie, W. Oral lesions in infectious mononucleosis. Oral Surg. 12(685), 1959.
Friedman, L.L., Bowie, E.J.W., Thompson, J.H., Jr., Brown, A.L., Jr., Owen, C.A., Jr. Familial Glanzmann’s thrombasthenia. Mayo Clin Proc. 39(908), 1964.
Fruchtman, S.M., Mack, K., Kaplan, M.E., et al. From efficacy to safety: a polycythemia vera study group report on hydroxyurea in patients with polycythemia vera. Semin Hematol. 1997; 34(1):17–23. [Jan].
Fruchtman, S.M., Pettit, R.M., Gilbert, H.S., et al. Anagrelide. Analysis of long-term safety and leukemogenic potential in myeloproliferative diseases (MPDs) [abstract]. Blood. 100(70), 2002.
Frye, J.L., Thompson, D.F. Drug-induced thrombocytosis. J Clin Pharm Ther. 1993; 18(1):45–48. [Feb].
Furlan, M., Robles, R., Galbusera, M., et al. von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. New Engl J Med. 1998; 339(22):1578–1584. [Nov 26].
Gaffey, M.J., Weiss, L.M. Association of Epstein-Barr virus with human neoplasia. Pathol Annu, 27 Pt. 1992; 1:55–74.
Galanakis, D.K. Inherited dysfibrinogenemia: emerging abnormal structure associations with pathologic and nonpathologic dysfunctions. Semin Thromb Hemost. 1993; 19(4):386–395.
Gamble, J.W., Driscoll, E.J. Oral Manifestations of macroglobulinemia of Waldenstron. Oral Surg. 13(104), 1960.
Geerlings, S.E., Statius, van Eps L.W. Pathogenesis and consequences of Plummer- Vinson syndrome. Clin Investig. 1992; 70(8):629–630. [Aug].
Genvresse, I., Spath-Schwalbe, E., Lukowsky, A. Delayed response to granulocyte colony-stimulating factor (G-CSF) in a case of severe neutropenia associated with large granular lymphocyte (LGL) leukemia [letter]. Eur J Haematol. 1998; 60(2):133–134. [Feb].
Gillig, J.L., Caldwell, C.H. The Chediak-Higashi syndrome: case report. J Dent Child. 37(527), 1970.
Godkin, A., Jewell, D. The pathogenesis of celiac disease. Gastroenterology. 1998; 115(1):206–210. [Jul].
Goldenfarb, P.B., Finch, S.C. Thrombotic thrombocytopenic purpura: a ten-year survey. J Am Med Assoc. 226(644), 1973.
Goldman, A.M. Leukemia—importance of recognition by the dentist. New York Dent J. 15(329), 1949.
Goldsby, J.W., Staats, O.J. Characteristic cellular changes in oral epithelial cells in sickle cell diseases. Cent Afr J Med. 10(336), 1964.
Goodheart, C.R. Herpesviruses and cancer. J Am Med Assoc. 211(91), 1970.
Gootenberg, J.E. Factor concentrates for the treatment of factor XIII deficiency. Curr Opin Hematol. 1998; 5(6):372–375. [Nov].
Gordon, L.I., Kwaan, H.C. Cancer- and drug-associated thrombotic thrombocytopenic purpura and hemolytic uremic syndrome. Semin Hematol. 1997; 34(2):140–147. [Apr].
Gorevic, P.D., Kassab, H.J., Levo, Y. Mixed cryoglobulinemia: clinical aspects and long-term follow-up of 40 patients. Am J Med. 1980; 69(2):287–308. [Aug].
Gorevic, P.D. Connective tissue disease associated with other immunologic disorders. cryoglobulinemia. In: Koopman WJArthritis and Allied Conditions: A Textbook of Rheumatology (13th ed). Lippincott, Williams and Wilkins, Philadelphia. 1997; 1572–1578.
Gorevic, P.D., Cryopathies: Cryoglobulins and Cryofibrinogenemia. Samter’s Immunologic Disease (5th ed). Little Brown, Boston. 1995:951–974.
Gorlin, R.J., Chaudhry, A.P. The oral manifestations of cyclic (periodic) neutropenia. Arch Dermatol. 82(344), 1960.
Grabenstein, J.D., Immune globulin intravenous (human. Immunofacts: Vaccines and Immunologic Drugs. Facts and comparisons, CV Mosby, St Louis. 1999:212–224.
Gross, L. Mouse leukemia: an egg-borne virus disease (with a note on mouse salivary gland carcinoma. Acta Haematol (Basel. 13(13), 1955.
Gross, L. Viral etiology of cancer and leukemia: a look into the past, present, and future—GHA clowes memorial lecture. Cancer Res. 38(485), 1978.
Guill, M.F., Brown, D.A., Ochs, H.D., et al. IgM deficiency: clinical spectrum and immunologic assessment. Ann Allergy. 1989; 62(6):547–552. [Jun].
Haddy, T.B., Rana, S.R., Castro, O. Benign ethnic neutropenia: what is a normal absolute neutrophil count? J Lab Clin Med. 1999; 133(1):15–22. [Jan].
Halliwell, H.L., L, Brigham. Pseudohemophilia. Intern Med Ann. 29(803), 1948.
Hamilton, R.E., Jr., Giansanti, J.S. The Chediak Higashi syndrome. Oral. 37(754), 1974.
Hann, I., Viscoli, C., Paesmans, M. A comparison of outcome from febrile neutropenic episodes in children compared with adults: results from four EORTC studies. International Antimicrobial Therapy Cooperative Group (IATCG) of the European Organization for Research and Treatment of Cancer. ALYSIS. 1997; 99(3):580–588. [Dec].
Hanzlik, P.J. Agranulocytosis: a critical review of causes and treatment. J Am Dent Assoc. 22(487), 1935.
Hastrup, J., Grahl-Madsen, R. Wiskott-Aldrich’s syndrome; thrombocytopenia, eczema and recurrent infection. Danish Med Bull. 12(99), 1965.
Haverkate, F., Samama, M. Familial dysfibrinogenemia and thrombophilia: report on a study of the SSC Subcommittee on Fibrinogen. Thromb Haemost. 1995; 73(1):151–161. [Jan].
Hayward, J.R., Capodanno, J.A. Trigeminal neurologic signs in leukemia. J Oral Surg, Anesth Hosp Dent Serv. 21(499), 1963.
Henle, G., Henle, W., Diehl, V. Relation of Burkitt’s tumor-associated herpes-type virus to infectious mononucleosis. Proc Nat Acad Sci USA. 59(94), 1968.
Henle, W., Henle, G., Ho, H.C., Burtin, P., et al. Antibodies to Epstein-Barr virus in nasopharyngeal carcinoma, other head and neck neoplasms, and control groups. J Natl Cancer Inst. 44(225), 1970.
Henshaw, P.S., Hawkins, J.W. Incidence of leukemia in physicians. J Natl Cancer Inst. 4(339), 1944.
Hirsch, D., Luboshitz, J., Blum, I. Treatment of antithyroid drug-induced agranulocytosis by granulocyte colony-stimulating factor: a case of primum non nocere. Thyroid. 1999; 9(10):1033–1035. [Oct].
Hjorting-Hansen, E., Philipsen, H.P., Drivsholm, A. Oral manifestations of Waldenstrom’s macroglobulinemia. Ugeskr Laeger. 124(133), 1962.
Hobbs, J.R., Milner, R.D., Watt, P.J. Gamma-M deficiency predisposing to meningococcal septicaemia. Br Med J. 1967; 4(579):583–586. [Dec 9].
Hoffman, R., Benz, E.J., Jr., Shattil, S.J. Hematology: Basic Principles and Practice (3rd ed. Churchill Livingstone, New York. 2000; 446–484.
Hoffman, R. Primary thrombocythemia. In: Hoffman R., et al, eds. Hematology: Basic Principles and Practice. 3rd. Philadelphia: Churchill Livingstone; 2000:1188–1204.
Hoffman, R.M., Jaffe, P.E. Plummer-Vinson syndrome. a case report and literature review. Arch Intern Med. 1995; 155(18):2008–2011. [Oct 9].
Hoffmann, R.M., Ott, S., Parhofer, K.G. Interferon-alpha-induced agranulocytosis in a patient on long-term clozapine therapy [letter]. J Hepatol. 1998; 29(1):170. [Jul].
Horowitz, M.M. Current status of allogeneic bone marrow transplantation in acquired aplastic anemia. Semin Hematol. 2000; 37(1):30–42. [Jan].
Huynh, P.T., de Lange, E.E., Shaffer, H.A. Symptomatic webs of the upper esophagus: treatment with fluoroscopically guided balloon dilation. Radiology. 1995; 196(3):789–792. Sep
Hymes, K.B., Karpatkin, S. Human immunodeficiency virus infection and thrombotic microangiopathy. Semin Hematol. 1997; 34(2):117–125. [Apr].
Ichinose, A. Physiopathology and regulation of factor XIII. Thromb Haemost. 2001; 86(1):57–65. [Jul].
Introne, W., Boissy, R.E., Gahl, W.A. Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome. Mol Genet Metab. 1999; 68(2):283–303. [Oct].
Israels, L.G., Israels, E.D., Fibrinogen, factor XIII, and fibrinolysis. Mechanisms in Hematology (3rd ed). Charlottesville Va. Core Health Services Inc. 2002:355–367.
Jacobs, A., Kilpatrick, G.S. The Paterson-Kelly syndrome. Br Med J. 1964; 2:79–82.
Jandl, J.H. Blood: Textbook of Hematology (2nd ed. Little Brown, Boston. 1996; 251–288.
Jessner, W., Vogelsang, H., Puspok, A., et al. Plummer-Vinson syndrome associated with celiac disease and complicated by postcricoid carcinoma and carcinoma of the tongue. Am J Gastroenterol. 2003; 98(5):1208–1209. [May].
Kaito, K., Kobayashi, M., Katayama, T., et al. Long-term administration of G-CSF for aplastic anaemia is closely related to the early evolution of monosomy 7 MDS in adults. Br J Haematol. 1998; 103(2):297–330. [Nov].
Kanfer, J.N., Blume, R.S., Yankee, R.A., Wolff, S.M. Alteration of sphingolipid metabolism in leukocytes from patients with the Chediak-Higashi syndrome. New Engl J Med. 279(410), 1968.
Kapoor, A., Munjal, S., Arya, R. Chediak-Higashi syndrome—a case report. Indian J Pathol Microbiol. 2000; 43(3):373–375. [Jul].
Kasper, B., Herbst, A., Pilz, C. Severe congenital neutropenia patients with point mutations in the granulocyte colony-stimulating factor (G-CSF) receptor mRNA express a normal G-CSF receptor protein [letter; comment]. Blood. 1997; 90(7):2839–2841. [Oct 1].
Kauder, E., Mauer, A.M. Neutropenias of childhood. J Pediatr. 69(147), 1966.
Kenney, D.M. Wiskott-Aldrich syndrome and related X-linked thrombocytopenia. Curr Opin Pediatr. 1990; 2:931–944.
Keusch, G.T., Acheson, D.W. Thrombotic thrombocytopenic purpura associated with Shiga toxins. Semin Hematol. 1997; 34(2):106–116. [Apr].
Kirschbaum, J.D., Preuss, F.S. Leukemia; a clinical and pathologic study of one hundred and twenty-three fatal cases in a series of 14,000 necropsies. Arch Intern Med. 71(777), 1943.
Kitchin, P.C. Oral observations in the case of periodic agranulocytosis. J Dent Res. 14(315), 1934.
Klein, A.S., Sitzmann, J.V., Coleman, J., et al. Current management of the Budd-Chiari syndrome. Ann Surg. 1990; 212(2):144–149. [Aug].
Kolmeier, K.H., Bayrd, E.D. Familial leukemia: report of instance and review of the literature. Proc Mayo Clin. 38(523), 1963.
Korsten, J., Grossman, H., Winchester, P.H. Extramedullary hematopoiesis in patients with thalassemia anemia. Radiology. 1970; 95(2):257–263. [May].
Kracke, R.R. Recurrent agranulocytosis. Am J Clin Pathol. 1(385), 1931.
Kranz, W.C., RuffJD. Congenital absence of fibrinogen: a rare cause of oral bleeding. Oral Surg. 12(88), 1959.
Krevsky, B., Pusateri, J.P. Laser lysis of an esophageal web. Gastrointest Endosc. 1989; 35(5):451–453. [Sep-Oct].
Kurtz, J.E., Andres, E., Maloisel, F. Drug-induced agranulocytosis in older people: a case series of 25 patients [letter]. Age Ageing. 1999; 28(3):325–326. [May].
Kutti, J., Wadenvik, H. Diagnostic and differential criteria of essential thrombocythemia and reactive thrombocytosis. Leuk Lymphoma, 22 Suppl. 1996; 1:41–45. [Sep].
Kwaan, H.C., Ganguly, P. Introduction: Thrombolytic thrombocytopenia purpura and the hemolytic uremic syndrome. Semin Hematol. 1997; 81–82.
Kwaan, H.C., Soff, G.A. Management of thrombotic thrombocytopenic purpura and hemolytic uremic syndrome. Semin Hematol. 1997; 34(2):159–166. [Apr].
Kwan, S.P., Hagemann, T.L., Radtke, B.E., et al. Identification of mutations in the Wiskott- Aldrich syndrome gene and characterization of a polymorphic dinucleotide repeat at DXS6940, adjacent to the disease gene. Proc Natl Acad Sci USA. 1995; 92(10):4706–4710. [May 9].
Kyle, R.A., Linman, J.W. Gingivitis and chronic idiopathic neutropenia: report of two cases. Mayo Clin Proc. 45(494), 1970.
Landolfi, R., Marchioli, R., Kutti, J., et al. Efficacy and safety of low-dose aspirin in polycythemia vera. New Engl J Med. 2004; 350(2):114–124. [Jan 8].
Landolfi, R. Bleeding and thrombosis in myeloproliferative disorders. Curr Opin Hematol. 1998; 5(5):327–331. [Sep].
Landsteiner, K., Wiener, A.S. Studies on an agglutinogen (Rh) in human blood reacting with anti-rhesus sera and with human isoantibodies. J Exp Med. 74(309), 1941.
Lange, R.D., Moloney, W.C., Yamawaki, T. Leukemia in atomic bomb survivors I general observations blood. 1954;9(574).
Larsson, L.G., Sandstrom, A., P, Westling. Relationship of Plummer-Vinson disease to cancer of the upper alimentary tract in Sweden. Res Cancer. 1975; 35(11 Pt. 2):3308–3316. [Nov].
Laurence, J., Mitra, D. Apoptosis of microvascular endothelial cells in the pathophysiology of thrombotic thrombocytopenic purpura/sporadic hemolytic uremic syndrome. Semin Hematol. 1997; 34(2):98–105. [Apr].
Lawrence, J.S. Irradiation leukemogenesis. J Am Med Assoc. 190(1049), 1964.
Lawson, J.P., Ablow, R.C., Pearson, H.A. Calvarial and phalangeal vascular impressions in thalassemia. AJR Am J Roentgenol. 1984; 143(3):641–645. [Sep].
Lawson, J.P., Ablow, R.C., Pearson, H.A. Premature fusion of the proximal humeral epiphyses in thalassemia. AJR Am J Roentgenol. 1983; 140(2):239–244. [Feb].
Lawson, J.P., Ablow, R.C., Pearson, H.A. The ribs in thalassemia I: the relationship to therapy. Radiology. 1981; 140(3):663–672. [Sep].
Lawson, J.P., Ablow, R.C., Pearson, H.A. The ribs in thalassemia II: the pathogenesis of the changes. Radiology. 1981; 140(3):673–679. [Sep].
Lee, C. Recombinant clotting factors in the treatment of hemophilia. Thromb Haemost. 1999; 82(2):516–524. [Aug].
Lee, C.A., Brettler, D.B. Guidelines for the diagnosis and management of von Willebrand disease. Haemophilia. 1997; 3:1–25.
Lee, G.R., Foerster, J., Lukens, J. Wintrobe’s Clinical Hematology (10th ed. Williams and Wilkins, Baltimore. 1999; 941–978.
Lee, G.R., Foerster, J., Lukens, J. Wintrobe’s Clinical Hematology, Vol 10 (10th ed. Williams and Wilkins, Baltimore. 1999; 979–1011.
Lewis, E.B. Leukemia and ionizing radiation. Science. 125(965), 1957.
Lewis, H.B. Leukemia, multiple myeloma, and aplastic anemia in American radiologists. Science. 142(1492), 1963.
Lewis, J.H. Coagulation defects. J Am Med Assoc. 178(1014), 1961.
Lilleyman, J., Hann, I., Blanchette, V., Hemophilia. Pediatric Hematology. 1999:585–598. [2nd ed].
Liu, H., Mihara, K., Kimura, A., et al. Induction of apoptosis in CD34+ cells by sera from patients with aplastic anemia. Hiroshima J Med Sci. 1999; 48(2):57–63. [Jun].
Livingston, R.J., White, N.S., Catone, G.A., Hartsock, R.J. Diagnosis and treatment of von Willebrand’s disease. J Oral Surg. 32(65), 1974.
Ljung, R.C. Prophylactic infusion regimens in the management of hemophilia. Thromb Haemost. 1999; 82(2):525–530. [Aug].
Logothetis, J., Economidou, J., Constantoulakis, M., Augoustaki, O., et al. Cephalofacial deformities in thalassemia major (Cooley’s anemia. Am J Dis Child. 121(300), 1971.
Long, J.A., Jr., Doppman, J.L., Nienhuis, A.W. Computed tomographic studies of thoracic extramedullary hematopoiesis. J Comput Assist Tomogr. 1980; 4(1):67–70. [Feb].
Long, J.A., Jr., Doppman, J.L., Nienhus, A.W. Computed tomographic analysis of beta- thalassemic syndromes with hemochromatosis: pathologic findings with clinical and laboratory correlations. J Comput Assist Tomogr. 1980; 4(2):159–165. [Apr].
Lunel, F., Musset, L., Cacoub, P. Cryoglobulinemia in chronic liver diseases: role of hepatitis C virus and liver damage. Gastroenterology. 1994; 106(5):1291–1300. [May].
Mac, Manus M., Lamborn, K., Khan, W. Radiotherapy-associated neutropenia and thrombocytopenia: analysis of risk factors and development of a predictive model. Blood. 1997; 89(7):2303–2310. [Apr 1].
Maleki, D., Cameron, A.J. Plummer-Vinson syndrome associated with chronic blood loss anemia and large diaphragmatic hernia. Am J Gastroenterol. 2002; 97(1):190–193. [Jan].
Mannucci, P.M. Treatment of von Willebrand’s disease. J Intern Med Suppl, Therapeutic Use. 1997; 129–132.
Marsland, E.A., Gerrard, J.W. Intrinsic staining of teeth following icterus gravis. Br Dent J. 94(305), 1953.
Martinez, J. Congenital dysfibrinogenemia. Curr Opin Hematol. 1997; 4(5):357–365. [Sep].
Martinez, J. Quantitative and qualitative disorders of fibrinogen. In Hoffman et alHematology: Basic Principles and Procedures, 2nd, Philadelphia: Churchill Livingstone; 1995:1703–1713. [2011 13].
Martino, R., Muniz-Diaz, E., Arilla, M. Combined autoimmune cytopenias. Haematologica. 1995; 80(4):305–310. [Jul-Aug].
McCarthy, F.P., Karcher, P.H. The oral lesions of monocytic leukemia. New Engl J Med. 234(787), 1946.
Meltzer, M., Franklin, E.C., Elias, K. Cryoglobulinemia—a clinical and laboratory study II: cryoglobulins with rheumatoid factor activity. Am J Med. 1966; 40(6):837–856. [Jun].
Meltzer, M., Franklin, E.C. Cryoglobulinemia—a study of twenty-nine patients I: IgG and IgM cryoglobulins and factors affecting cryoprecipitability. Am J Med. 1966; 40(6):828–836. Jun
Messinezy, M., Westwood, N., Sawyer, B., et al. Primary thrombocythaemia: a composite approach to diagnosis. Clin Lab Haematol. 1994; 16(2):139–148. [Jun].
MGH. Case records ofthe Massachusetts general hospital. Weekly clinicopathological exercises: case. 1968; 278(14):3–1968. [New Engl J Med 782-91 Apr 4].
Michaud, M., Baehner, R.L., Bixler, D., Kafrawy, A.H. Oral manifestations of acute leukemia in children. J Am Dent Assoc. 95(1145), 1977.
Miescher, P.A., Huang, Y.P., Izui, S. Type II cryoglobulinemia. Semin Hematol. 1995; 32(1):80–85. [Jan].
Millard, H.D., Gobetti, J.P. Nonspecific stomatitis—a presenting sign in pernicious anemia. Oral Surg. 39(562), 1975.
Miller, R. Counselling about diagnosis and inheritance of genetic bleeding disorders: haemophilia A and B. Haemophilia. 1999; 5(2):77–83. [Mar].
Miller, J. Pigmentation of teeth due to rhesus factor. Br Dent J. 9(121), 1951.
Miners, A.H., Sabin, C.A., Tolley, K.H., Lee, C.A. Assessing the effectiveness and cost-effectiveness of prophylaxis against bleeding in patients with severe haemophilia and severe von Willebrand’s disease. J Intern Med. 1998; 244(6):515–522. [Dec].
Minot, G.R., Murphy, E.J. Treatment of pernicious anemia by a special diet. J Am Med Assoc. 87(470), 1926.
Moake, J.L., Landry, P.R., Oren, M.E., Sayer, B.L., Heffner, L.T. Transient peripheral plasmacytosis. Am J Clin Pathol. 62(8), 1974.
Monti, G., Galli, M., Invernizzi, F. Cryoglobulinaemias: a multi-centre study of the early clinical and laboratory manifestations of primary and secondary disease. GISC. Italian Group for the Study of Cryoglobulinaemias. QJM. 1995; 88(2):115–126. [Feb].
Monto, R.W., Rizek, R.A., Fine, G. Observations on the exfoliative cytology and histology of the oral mucous membranes in iron deficiency. Oral Surg. 14(965), 1961.
Mori, T., Ikeda, Y. [Acquired dysfibrinogenemia]. Ryoikibetsu Shokogun Shirizu. 1998; 529–531. [21 Pt 2].
Morris, A.L., Stahl, S.S. Intraoral roentgenographic changes in sickle-cell anemia. Oral Surg. 7(787), 1954.
Moseley, J.E. Bone Changes in Hematologic Disorders (Roentgen Aspects). Grune and Stratton, New York. 1963. [26].
Mosesson, M.W. Dysfibrinogenemia and thrombosis. Semin Thromb Hemost. 1999; 25(3):311–319.
Mourshed, F., Tuckson, C.R. A study of the radiographic features of the jaws in sickle cell anemia. Oral Surg. 37(812), 1974.
Murphy, P.T., Casey, M.C. Unusual presentation of primary autoimmune neutropenia [letter]. Br J Haematol. 1999; 107(1):214–215. [Oct].
Nakao, S. Immune mechanism of aplastic anemia. Int J Hematol. 1997; 66(2):127–134. [Aug].
Nathan, D.G., Oski, F.A., Hemophilia. Hematology of Infancy and Childhood. 5th. 1998:1631–1645.
Nichols, W.C., Ginsburg, D. von Willebrand disease. Medicine (Baltimore. 1997; 76(1):1–20. [Jan].
Niederman, J.C., Miller, G., Pearson, H.A., Pagano, J.S., et al. Infectious mononucleosis Epstein-Barr-virus shedding in saliva and the oropharynx. New Eng J Med. 294(1355), 1976.
Nosher, J.L., Campbel, W.L., Seaman, W.B. The clinical significance of cervical esophageal and hypopharyngeal webs. Radiology. 1975; 117(1):45–47. [Oct].
Novak, A.J. The oral manifestations of erythroblastic (Cooley’s) anemia. Am J Orthod Oral Surg. 30(539), 1944.
Okamura, H., Tsutsumi, S., Inaki, S. Esophageal web in Plummer-Vinson syndrome. Laryngoscope 1988 Sep. 1919; 98(9):289–291. [994-98 Paterson DR: a clinical type of dysphagia. J Laryngol Rhinol Otol, 34:].
Okano, M., Gross, T.G. Epstein-Barr virus-associated hemophagocytic syndrome and fatal infectious mononucleosis. Am J Hematol. 1996; 53(2):111–115. [Oct].
Okano, M., Thiele, G.M., Davis, J.R., et al. Epstein-Barr virus and human diseases: recent advances in diagnosis. Clin Microbiol Rev. 1988; 1(3):300–312. [Jul].
Okpala, I. The management of crisis in sickle cell disease. Eur J Haematol. 1998; 60(1):1–6. [Jan].
Osgood, E.E. Monocytic leukemia. Arch Int Med. 59(931), 1931.
Page, A.R., Good, R.A. Studies on cyclic neutropenia. Am Med Assoc J Dis Child. 94(623), 1957.
Paul, J.R., Brunnell, W.W. The presence of heterophile antibodies in infectious mononucleosis. Am J Med Sci. 1932; 183:90–104.
Pearson, T.C. Diagnosis and classification of erythrocytoses and thrombocytoses. Baillieres Clin Haematol. 1998; 11(4):695–720. [Dec].
Perinol, K.E., James, R.B. Wiskott-Aldrich syndrome: review of literature and report of case. J Oral Surg. 38(297), 1980.
Perkin, R.F., White, G.C., Webster, W.P. Glanzmann’s thrombasthenia. Oral Surg. 47(36), 1979.
Piaggio, G., Podesta, M., Pitto, A., et al. Coexistence of normal and clonal haemopoiesis in aplastic anaemia patients treated with immunosuppressive therapy. Br J Haematol. 1999; 107(3):505–511. [Dec].
Pogrel, M.A. Thrombocythemia as a cause of oral hemorrhage. Oral Surg. 44(535), 1977.
Pollack, C.V., Jr. Emergencies in sickle cell disease. Emerg Med Clin North Am. 1993; 11(2):365–378. [May].
Porter, D.D., Wimberly, I., Benyesh-Melnick, M. Prevalence of antibodies to EPSTEIN-BARR virus and other herpes viruses. J Am Med Assoc. 208(1675), 1969.
Powell, J.W., Weens, H.S., Wenger, N.K. The skull roentgenogram in iron deficiency anemia and in secondary polycythemia. Am J Roentgenol Radium Ther Nucl Med. 95(143), 1965.
Poyton, H.G., Davey, K.W. Thalassemia Changes visible in radiographs used in dentistry. Oral Surg. 25(564), 1968.
Price, F.V., Legro, R.S., Watt-Morse, M., Kaplan, S.S. Chediak-Higashi syndrome in pregnancy. Obstet Gynecol. 1992; 79:804–806. [(5 (Pt 2) May].
Prowler, J.R., Smith, E.W. Dental bone changes occurringin sickle-cell diseases and abnormal hemoglobin traits. Radiology. 65(762), 1955.
Quick, A.J., Adlam, R.T. Coexistence of von Willebrand’s disease and hemophilia in a family. J Am Med Assoc. 185(635), 1963.
Ragab, A.H., Vietti, T.J. Infectious mononucleosis, lymphoblastic leukemia and the Epstein-Barr virus. Cancer. 24(261), 1969.
Rapaport, S.I. Infectious mononucleosis. Ann West Med Surg. 2(261), 1969.
Rapp, C.E., Jr., Hewetson, J.F. Infectious mononucleosis and the Epstein-Barr virus. Am J Dis Child. 132(78), 1978.
Rapp, F., Reed, C.L. The viral etiology of cancer: a realistic approach. Cancer. 40(419), 1977.
Ratnoff, O.D. Treatment of Hemorrhagic Disorder. 1968; [Harper and Row, New York].
Ratnoff, O.D., Thomas, C.C. Bleeding syndromes. a clinical manual springfield III. 1960.
Resch, C.A. Oral manifestations of leucemia. Am J Orthod, Oral Surg. 26(901), 1940.
Rettberg, W.A.H. Symptoms and signs referable to the oral cavity in blood dyscrasias. Oral Surg. 6(614), 1953.
Rister, M., Haneke, C. [Therapy of the Steinbrinck-Chediak-Higashi-Syndrome (author’s transl)]. Klin Padiatr. 1980; 192(1):19–24. [Jan].
Robbins, S.L., Cotran, R.S. Pathologic Basis of Disease. WB Saunders, Philadelphia. 1979. [2nd ed].
Robinson, I.B., Sarnat, B.G. Roentgen studies of the maxillae and mandible in sickle-cell anemia. Radiology. 58(517), 1952.
Rodgers, G.M., Greenberg, C.S., et al. Wintrobe’s Clinical Hematolog (10th ed). Inherited coagulation disorders. In: Lee GR. Baltimore: Williams and Wilkins; 1999:1702–1703.
Rodriguez, A., Yood, R.A., Condon, T.J. Recurrent uveitis in a patient with adult onset cyclic neutropenia associated with increased large granular lymphocytes [letter]. Br J Ophthalmol. 1997; 81(5):415. [May].
Rose, S.A. Hypofibrinogenopenia. Oral Surg. 11(966), 1958.
Roser, S.M., Gracia, R., Guralnick, W.C. Portsmouth syndrome: review of the literature and clinicopathological correlation. J Oral Surg. 33(668), 1975.
Rydell, R.O. Blood factor XIII deficiency: review of literature and report of case. J Oral Surg. 29(628), 1971.
Sadler, J.E. A revised classification of von Willebrand disease. For the subcommittee on von Willebrand factor of the scientific and standardization Committee of the international society on thrombosis and haemostasis. Thromb Haemost. 1994; 71(4):520–525. [Apr].
Sandberg, A.A. Chromosomes and leukemia. CA. 15(2), 1965.
Santagostino, E., Mannucci, P.M., Bianchi, Bonomi A. Guidelines on replacement therapy for haemophilia and inherited coagulation disorders in Italy. Haemophilia. 2000; 6(1):1–10. [Jan].
Schaar, F.E. Familial idiopathic thrombocytopenic purpura. J Pediatr. 62(546), 1963.
Schorer, A.E., Singh, J., Basara, M.L. Dysfibrinogenemia: a case with thrombosis (fibrinogen Richfield) and an overview of the clinical and laboratory spectrum. Am J Hematol. 1995; 50(3):200–208. [Nov].
Schroeder, T. Haematopoietic stem cell heterogeneity: subtypes, not unpredictable behavior. Cell Stem Cell, 6(3):203-7. 2010.
Schumacher, H.R., Barcay, S.J. Hemorrhagic phenomena in infectious mononucleosis. Am J Med Sci. 234(175), 1962.
Scopes, J., Daly, S., Atkinson, R., et al. Aplastic anemia: evidence for dysfunctional bone marrow progenitor cells and the corrective effect of granulocyte colony- stimulating factor in vitro. Blood. 1996; 87(8):3179–3185. [Apr 15].
Scriver, C.R., Beaudet, A.L., Sly, W.S. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 1995. [2nd ed 3129–49].
Segal, N.A. Idiopathic thrombocytopenic purpura. Oral Surg. 6(631), 1953.
Shiver, C.B., Jr., Berg, P., Frenke, E.P. Palatine petechiae, an early sign in infectious mononucleosis. J Am Med Assoc. 161(592), 1956.
Smith, P.F., Taylor, C.T. Vancomycin-induced neutropenia associated with fever: similarities between two immune-mediated drug reactions. Pharmacotherapy. 1999; 19(2):240–244. [Feb].
Smith, H.W. Oral manifestations and systemic blood diseases. WV Med, J. 43(236), 1947.
Socie, G., Rosenfeld, S., Frickhofen, N., et al. Late clonal diseases of treated aplastic anemia. Semin Hematol. 2000; 37(1):91–101. [Jan].
Sodeman, W.A., Jr., Sodeman, T.M. Sodeman’s Pathologic Physiology: Mechanisms of Disease (6th ed). Philadelphia: WB Saunders, 1979.
Soucie, J.M., Nuss, R., Evatt, B., et al. Mortality among males with hemophilia: relations with source of medical care. The hemophilia surveillance system project investigators. Blood. 2000; 96(2):437–442. [Jul 15].
Southam, C.M., Craver, L.F., Dargeon, H.W., Burchenal, J.H. A study of the natural history of acute leukemia, with special reference to the duration of the disease and the occurrence of remissions. Cancer. 4(39), 1951.
Spiegel, L.H. Christmas disease. Oral Surg. 11(376), 1958.
Stanbury, J.B., Wyngaarden, J.B., Fredrickson, D.S. McGraw-Hill, New York, 1978. [The Metabolic Basis of Inherited Sisease (4th ed).].
Steen, R.G., Emudianughe, T., Hankins, G.M., et al. Brain imaging findings in pediatric patients with sickle cell disease. Radiology. 2003; 228(1):216–225. [Jul].
Steg, R.F., Gores, R.J., Thompson, J.H., Jr., Owens, C.A., Jr. Bleeding due to deficiency of plasma thromboplastin antecedent (PTA) and plasma thromboplastin compenent (PTC. Oral Surg. 13(671), 1960.
Stephens, C.R. Sickle cell disease: a review of state-of-the-art emergency management and outcome-effective therapy. Emerg Med Reports. 1999; 20:183–192.
Stephens, D.J., Lawrence, J.S. Cyclical agranulocytic angina. Ann Intern Med. 9(31), 1935.
Stoneman, D.W., Deierl, C.D. Pseudotumor of hemophilia in the mandible. Oral Surg. 40(811), 1975.
Stoy, P.J. Three cases of acute monocytic leukaemia, with special reference to their oral condition. Br Dent J. 92(144), 1952.
Straus, S.E., Cohen, J.I., Tosato, G., Meier, J. NIH conference. Epstein-Barr virus infections: biology, pathogenesis, and management. Ann Intern Med. 1993; 118(1):45–58. [Jan 1].
Streiff, M.B., Smith, B., Spivak, J.L. The diagnosis and management of polycythemia vera in the era since the Polycythemia Vera Study Group: a survey of American society of hematology members’ practice patterns. Blood. 2002; 99(4):1144–1149. [Feb 15].
Tank, G. Two cases of green pigmentation of the deciduous teeth associated with hemolytic disease of the newborn. J Am Dent Assoc. 42(302), 1951.
Tefferi, A., Ho, T.C., Ahmann, G.J., et al. Plasma interleukin-6 and C-reactive protein levels in reactive versus clonal thrombocytosis. Am J Med. 1994; 97(4):374–378. [Oct].
Thoma, K.H., Cascario, N., Jr., Bacevicz, F.J. Erythroblastic anemia. Am J Orthod Oral Surg. 30(643), 1944.
Trier, J.S. Celiac sprue. New Engl J Med. 1991; 325(24):1709–1719. [Dec 12].
Trier, J.S. Diagnosis of celiac sprue. Gastroenterology. 1998; 115(1):211–216. [Jul].
Tyldesley, W.R. Recurrent oral ulceration and coeliac disease: a review. Br Dent J. 151(81), 1981.
Van, Creveld S. Coagulation disorders in the newborn period. J Pediatr. 54(633), 1959.
Vichinsky, E., Sickle cell disease. Schwarz GR, Cayton CG et alPrinciples and Practice of Emergency Medicine (3rd ed) Vol 2. Lea and Febiger, Philadelphia. 1992:2019–2024.
Vinson, P.P. Hysterical dysphagia. Minn Med. 1922; 5:107–108.
Voight, A.E., Frick, P.G. macroglobulinemia of Waldenstrom: a review of the literature and presentation of a case. Ann Intern Med. 44(419), 1956.
Vora, A.J., Lilleyman, J.S. Secondary thrombocytosis. Arch Dis Child. 1993; 68(1):88–90. [Jan].
Waldenstrom, J., Kjellberg, S.R. The roentgenological diagnosis of sideropenic dysphagia (Plummer-Vinson’s syndrome. Acta Radiol. 1939; 20:618–638.
Waldenstrom, J. Die Makroglobulinamie; in L Heilmeyer (ed): Ergebnisse der inneren Medizin und Kinderheilkunde. Springer-Verlag, Berlin. 1958.
Waldenstrom, J. Two interesting syndromes with hyperglobulinemia. Schweiz Med Wochenschr. 78(927), 1948.
Walker, R.D., Schenck, K.L., Jr. Infarct of the mandible in sickle cell anemia: report of a case. J Am Dent Assoc. 87(661), 1973.
Watson, A.O. Infantile cerebral palsy: a survey of dental conditions and treatment emphasizing the effect of parental Rh incompatibility on the deciduous teeth. Dent J Aust. 27: 6, 72, 1955.
Weinfeld, A., Swolin, B., Westin, J. Acute leukaemia after hydroxyurea therapy in polycythaemia vera and allied disorders: prospective study of efficacy and leukaemogenicity with therapeutic implications. Eur J Haematol. 1994; 52(3):134–139. [Mar].
Weiss, H.J., Chervenick, P.A., Zalusky, R., Factor, A. A familial defect in platelet function associated with impaired release of adenosine diphosphate. New Engl J Med. 281(1264), 1969.
Weiss, H.J. Platelet physiology and abnormalities of platelet function. New Engl J Med. 239(531), 1975.
Wentz, F.M., Anday, G., Orban, B. Histopathologic changes in the gingiva in leukemia. J Periodontol. 20(119), 1949.
Werner, E.J., Abshire, T.C., Giroux, D.S., et al. Relative value of diagnostic studies for von Willebrand disease. J Pediatr, analysis(1. 1992; 34–38. [Jul].
Werner, E.J. von Willebrand disease in children and adolescents. Pediatr Clin North Am. 1996; 43(3):683–707. [Jun].
Whipple, G.H., Robscheit-Robbins, F.S. Blood regeneration in severe anemia. Am J Physiol. 72(395), 1925.
White, L.R., Karofsky, P.S. Review of the clinical manifestations, laboratory findings, and complications of infectious mononucleosis. Wis Med J. 1985; 84(12):19–25. [Dec].
White, G.E. Oral manifestations of leukemia in children, Oral Surg 1970;29(420).
Windhorst, D.B., Zelickson, A.S., RA, Good. Chediak-Higashi syndrome. gigantism of cytoplasmic organelles. Science hereditary. 151(81), 1966.
Wintrobe, M.M. Clinical Hematology (10th ed. Williams and Wilkins, Baltimore. 1999; 2:1862–1882.
Wintrobe, M.M. Clinical Hematology (8th ed. Lea and Febiger, Philadelphia. 1981.
Wintroble, M.M., Hanrahan, E.M., Jr., Thomas, C.B. Purpura hemorrhagica, with special reference to course and treatment. J Am Med Assoc. 109(1170), 1937.
Wolff, J.A., Ignatov, V.G. Heterogeneity of thalasemia major. Am J Dis Child. 105(234), 1963.
Wyngaarden, J.B., Smith, L.H. Cecil Textbook of Medicine (16th ed. WB Saunders, Philadelphia. 1982.
Zhang, Z., Blomback, M., Anvret, M. Understanding von Willebrand’s disease from gene defects to the patients. J Intern Med Suppl. 1997; 740:115–119.
Zhao, H., Boissy, Y.L., Abdel-Malek, Z., et al. On the analysis of the pathophysiology of Chediak-Higashi syndrome: defects expressed by cultured melanocytes. Lab Invest. 1994; 71(1):25–34. [Jul].
Zhong, F., McCombs, C.C., Olson, J.M. An autosomal screen for genes that predispose to celiac disease in the western counties of Ireland. Nat Genet. 1996; 14(3):329–333. [Nov].
Zieve, P.D., Levin, J. Disorders of hemostasis Vol 10 in major problems in internal medicine. WB Saunders, Philadelphia. 1976.
Zurcher, R. Sickle cell anemia. In: Hamilto G.C., ed. Presenting Signs and Symptoms in the Emergency Department. Philadelphia: Lippincott, Williams and Wilkins; 1993:328–336.