Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by autoantibodies, immune complex formation, and immune dysregulation resulting in damage to essentially any organ, including the kidney, skin, blood cells, and the CNS. The natural history of this illness is unpredictable; patients may present with many years of symptoms or with acute life-threatening disease. Because of its protean manifestations, lupus must be considered in the differential diagnosis of many problems, including fevers of unknown origin, arthralgia, anemia, nephritis, psychosis, and fatigue. Early diagnosis and careful treatment tailored to individual patient symptoms has improved the prognosis from what was once perceived as an often fatal disease.
Etiology
The specific cause of SLE remain undefined. Research suggests that many factors, including genetics, hormones, and the environment (e.g. sunlight, drugs), contribute to the immune dysregulation observed in lupus. In lupus, greater production of autoantibodies leads to immune complex formation and tissue damage due to direct binding and/or immune complex deposition in tissues. Whether these antibodies are produced in reaction to exposure of normally nonexposed self-antigens or as a consequence of a broad spectrum of immune dysregulation resulting in excessive production of many antibodies without regard to prior stimulation is unclear. Patients with SLE produce autoantibodies against DNA, other nuclear antigens, ribosomes, platelets, erythrocytes, leukocytes, and other tissue-specific antigens. Thus, the resulting immune complexes result in widespread tissue damage. Cell-mediated autoimmune responses also play a pathophysiologic role.
Clinical Features
Systemic lupus erythematosus is a serious cutaneous-systemic disorder which characteristically manifests repeated remissions and exacerbations. This disease has its peak age of onset at about 30 years in females but about 40 years in males. The disease may occur in childhood as reported by Jacobs. Prevalence rates are higher in females than in males. A female-to-male ratio of approximately 2 : 1 occurs before puberty, and a ratio of 4 : 1 occurs after puberty.
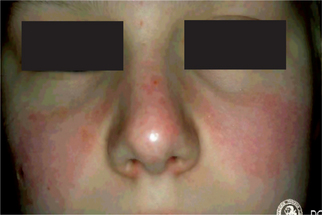
The cutaneous lesions consist of erythematous patches on the face which coalesce to form a roughly symmetrical pattern over the cheeks and across the bridge of the nose in a so-called butterfly distribution (Fig. 19-31). Also involved are the neck, upper arms, shoulders and fingers. These lesions may present itching or burning sensations as well as areas of hyperpigmentation. The acute erythematous patches either arise on previously uninvolved skin or develop in old chronic lesions. Their severity is intensified by exposure to sunlight.
The generalized manifestations of the systemic disease are referable to involvement of various organs, including the kidney and heart. In the kidney, fibrinoid thickening of glomerular capillaries occurs, producing the characteristic ‘wire loops,’ which may be sufficient to result in renal insufficiency. The heart may suffer from an atypical endocarditis involving the valves, as well as fibrinoid degeneration of the epicardium and myocardium. The widespread tissue involvement and the nature of the lesions have led to the inclusion of this disease in that group known as the ‘collagen diseases,’ which also includes rheumatic fever, rheumatoid arthritis, polyarteritis nodosa, scleroderma and dermatomyositis.
Oral Manifestations
Oral mucous membrane involvement is reported in 20–50% of cases of discoid lupus erythematosus, and slightly more frequently in the systemic form of the disease, according to Andreasen. With the oral mucous membranes affected in such a high percentage of cases, the dentist must be aware of this problem. The oral mucosa reportedly may be involved either prior to or following the development of skin lesions or even in the absence of skin manifestations.
Oral lesions are found in systemic lupus erythematosus. These are very similar to those found in discoid lupus except that hyperemia, edema and extension of the lesions is sometimes more pronounced, and there may be a greater tendency for bleeding, petechiae and superficial ulcerations which are surrounded by a red halo as a result of localized telangiectasis. Superimposed oral moniliasis as well as xerostomia have also been reported.
Sugarman pointed out that great variation in the oral lesions exists and that these frequently simulate other diseases, chiefly leukoplakia and lichen planus. For this reason, diagnosis based upon the clinical appearance of the oral lesions should not be encouraged. In fact, it has been stressed by Schiodt and his coworkers in a long-term follow-up study of 52 patients with discoid lupus erythematosus that those oral lesions which had undergone a transition into the leukoplakia-like lesions over a period of time even showed histopathologic and immunopathologic features similar to leukoplakia not preceded by lupus erythematosus.
Histologic Features
The histologic appearance of systemic lupus erythematosus and discoid lupus erythematosus is similar, differing only in the degree of certain of the findings. According to Lever, discoid lupus erythematosus of the skin is characterized by hyperkeratosis with keratotic plugging, atrophy of the rete pegs, liquefaction, degeneration of the basal layer of cells, perivascular infiltration of lymphocytes and their collection about dermal appendages, and basophilic degeneration of collagen and elastic fibers, with hyalinization, edema and fibrinoid change, particularly prominent immediately beneath the epithelium. Not all features are invariably present in each case, however. In the systemic form of the disease the cutaneous lesions are similar in appearance, although the degenerative features and collagen disturbance are usually more prominent and the inflammatory features less severe. The histologic appearance of the tissue from a cutaneous lesion of any type of lupus erythematosus is not pathognomonic of the disease, but is certainly suggestive.
The histologic findings in oral lesions of both discoid and systemic lupus erythematosus have been described in detail by Andreasen and Poulsen. Shklar and McCarthy have also discussed the histopathology of the oral lesions in 25 cases and concluded that it is sufficiently characteristic that a definitive diagnosis can be made. In the discoid form, the lesions exhibit hyperorthokeratosis and/or hyperparakeratosis alternating with areas of epithelial atrophy. In the majority of cases, keratotic plugging down into the spinous layer, acanthosis and pseudoepitheliomatous hyperplasia are present. Hydropic degeneration and liquefaction necrosis of the basal cell layer also invariably occur as well as subepithelial vesiculation or ulceration (Fig. 19-32). In most cases, a thickening of the basement membrane can be demonstrated as a homogeneous, broad, eosinophilic and PAS-positive acellular band. Finally, there is a diffuse infiltrate of lymphocytes with smaller numbers of plasma cells and occasional polymorphonuclear leukocytes in superficial and deep connective tissue. This is quite reminiscent of that seen in lichen planus. Small focal perivascular collections of lymphocytes are found also, as well as degeneration and disintegration of collagen.
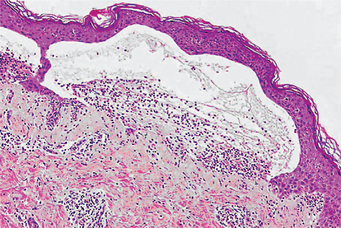
The systemic form of lupus exhibits histologic changes in the oral lesions that are virtually identical to those in the discoid type with the possible exception of the absence of keratinization.
Direct immunofluorescent testing is often used to confirm a suspected diagnosis of lupus erythematosus. It is basically a test used to detect the presence of immunoglobulins (IgG, IgM and IgA) at the epidermal-dermal junction or basement membrane zone of skin or oral mucosa of patients with the disease by incubating a biopsy specimen (either frozen section or one specially fixed in Michel solution) with a fluorescein-conjugated antiglobulin. The appearance of the immunoglobulins deposited in this location in discoid lupus generally is the ‘particulate’ (or ‘speckled’) pattern. These immunoglobulins were present at this specific histologic location in oral lesions in all patients with the systemic form and in nearly 75% of patients with the discoid form in a series of 52 patients with lupus erythematosus reported by Schiodt and his associates in 1981. They also found a high incidence of complement C3 and of fibrinogen at this same zone utilizing appropriate conjugates. Interestingly, these immunoglobulins may also be demonstrated in the uninvolved skin and mucosa of a significant percentage of patients with systemic lupus, as well as in lesional skin or mucosa, but almost never in normal or uninvolved skin or mucosa of discoid patients. In a retrospective study of 130 cases of oral mucosal disease by direct immunofluorescence technique, Daniels and Quadra-White have concluded that this can provide a valuable criterion in diagnosing chronic ulcerative or erosive disease of the oral mucosa if the biopsy specimens are taken from appropriate sites and have attached epithelium.
Laboratory Findings*
Routine clinical tests which suggest that the person has an active systemic disease include:
1. Sedimentation rate (ESR) and CRP (C-reative protein) binding, both of which are frequently elevated in inflammation from any cause.
2. Serum protein electrophoresis which may reveal increased gammaglobulin and decreased albumin.
3. Routine blood counts which may reveal anemia and low platelet and white cell counts.
These abnormalities alert to the presence of a systemic disease with multiple organ involvement.
Commonly used blood tests in the diagnosis of SLE are:
1. Antinuclear antibody test (ANA) to determine if autoantibodies to cell nuclei are present in the blood
2. Anti-DNA antibody test to determine if there are antibodies to the genetic material in the cell
3. Anti-Sm antibody test to determine if there are antibodies to Sm, which is a ribonucleoprotein found in the cell nucleus
4. Serum (blood) complement test to examine the total level of a group of proteins which can be consumed in immune reactions
5. Complement proteins C3 and C4 test to examine specific levels.
The immunofluorescent antinuclear antibody (ANA, or FANA) test is positive in almost all individuals with SLE (97%), and is the most sensitive diagnostic test currently available for confirming the diagnosis of SLE when accompanied by typical clinical findings. However, a positive ANA test, by itself, is not proof of lupus since the test may also be positive in other connective tissue diseases, such as, scleroderma, Sjogren's syndrome, rheumatoid arthritis, thyroid diseases, liver diseases, juvenile arthritis and in individuals being treated with certain drugs like procainamide, hydralazine, isoniazid, chlorpromazine, etc. in viral illnesses, such as, infectious mononucleosis, in other chronic infections such as, hepatitis, lepromatous leprosy, subacute bacterial endocarditis, malaria, etc. in other autoimmune diseases, including thyroiditis, multiple sclerosis, etc. and in as many as 30–40% of asymptomatic first-degree relatives of people with lupus.
Other Autoantibodies
• Antibodies to DNA are found primarily in SLE.
• Antibodies to histones (DNA packaging proteins) are usually found in people with drug induced lupus (DIL), but may also be found in those with SLE.
• Antibodies to the Sm antigen are found almost exclusively in lupus, and often help to confirm the diagnosis of SLE.
• Antobodies to RNP (ribonucleoprotein) are found in a number of connective tissue diseases. When present in very high levels, RNP antibodies are suggestive of mixed connective tissue disease (MCTD), a condition with symptoms like those of SLE, polymyositis, and scleroderma.
• Antibodies to Ro/SS-A are found in people with either lupus or Sjogren's syndrome, and are almost always found in babies who are born with neonatal lupus.
• Antibodies to Jo-1 are associated with polymyositis.
• Antibodies to PM-Scl are associated with certain cases of polymyositis that also have features of scleroderma.
• Antobodies to centromere (structure involved in cell division) are found in people with a limited form of scleroderma which tends to have a chronic course.
Complement
Laboratory tests which measure complement levels in the blood may also be helpful in making a diagnosis of SLE. The most common complement tests are C3, C4, and CH50 (total hemolytic complement). If the total blood complement level is low, or the C3 or C4 complement values are low and the person also has a positive ANA, the findings are more suggestive of lupus. Low C3 or C4 complement levels with a positive ANA may signify the presence of active disease.
Treatment
The most important tool in the medical care of the patient with SLE is careful and frequent clinical and laboratory evaluation to tailor the medical regimen and to provide prompt recognition and treatment of disease flare, which is the cornerstone of successful intervention. Lupus is a lifelong illness, and patients must be monitored indefinitely. SLE is a high-risk disease with the possibility of end-organ damage to any organ, vital or otherwise. This damage can severely affect organ function and can lead to decreased quality of life.
Discoid Lupus Erythematosus
Discoid lupus erythematosus (DLE) is a chronic, scarring, atrophy producing, photosensitive dermatosis. DLE may occur in patients with systemic lupus erythematosus (SLE), and some patients (<5%) with DLE progress to SLE. Serologic abnormalities are uncommon. Patients with DLE rarely have clinically significant systemic disease. Lesions may produce scarring or atrophy. Scarring alopecia is particularly disturbing.
Etiology
DLE probably occurs in genetically predisposed individuals, but the exact genetic connection has not been determined. The pathophysiology of DLE is not well understood. It has been suggested that a heat shock protein is induced in the keratinocyte following ultraviolet (UV) light exposure or stress, and this protein may act as a target for T-cell-mediated epidermal cell cytotoxicity.
Clinical Features
Discoid lupus erythematosus is a relatively common disease which, like the systemic form, occurs predominantly in the third and fourth decades. It is also considerably more common in women than in men. Although any skin area may be involved by the discoid form of lupus erythematosus, the most common sites are the face (Fig. 19-33), oral mucous membranes, chest, back and extremities.
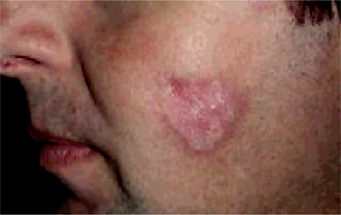
The typical cutaneous lesions are slightly elevated red or purple macules that are often covered by gray or yellow adherent scales. Forceful removal of the scale reveals numerous ‘carpet tack’ extensions which had dipped into enlarged pilosebaceous canals. The lesions increase in size by peripheral growth, this feature partially characterizing the disease. The periphery of the lesion appears pink or red, while the center exhibits an atrophic, scarred appearance indicative of the long-standing nature of the disease with characteristic central healing. The discoid form of the disease may also assume a typical ‘butterfly’ distribution on the malar regions and across the bridge of the nose. Since this is not a constant feature of the disease and since a similar distribution of lesions may occur in certain other diseases, its diagnostic significance should not be overemphasized.
Epidermoid carcinoma, and less commonly, basal cell carcinoma have been reported developing in healed scars of discoid lupus. This is only an occasional finding, and is thought to occur only in cases of 20 years duration or more.
Oral Manifestations
Oral mucous membrane involvement is reported in 20–50% of cases of discoid lupus erythematosus, and slightly more frequently in the systemic form of the disease, according to Andreasen. With the oral mucous membranes affected in such a high percentage of cases, the dentist must be aware of this problem. The oral mucosa reportedly may be involved either prior to or following the development of skin lesions or even in the absence of skin manifestations.
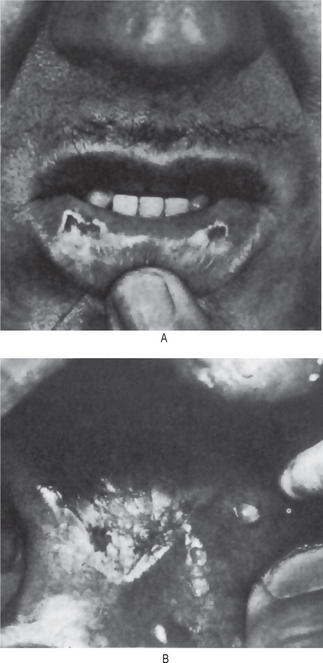
The oral lesions in the discoid form begin as erythematous areas, sometimes slightly elevated but more often depressed, usually without induration and typically with white spots. Occasionally, superficial, painful ulceration may occur with crusting or bleeding but no actual scale formation as is seen on the skin (Figs. 19-34, 19-35). The margins of the lesions are not sharply demarcated but frequently show the formation of a narrow zone of keratinization. Often, fine white striae radiate out from the margins. Central healing may result in depressed scarring. These lesions, which were symptomatic in 75% of a group of 32 patients with oral manifestation described by Schiodt and his associates, are most common on the buccal mucosa, palate and tongue. In the case of the tongue, atrophy of the papillae and severe fissuring are also seen. The vermilion border of the lips, particularly the lower, is a very common site for these lesions. The erythematous, atrophic plaques, surrounded by a keratotic border, may involve the entire lip and extend onto the skin surface. Malignant transformation of these lip lesions occurs with some frequency, and the reported cases have been reviewed by Andreasen.
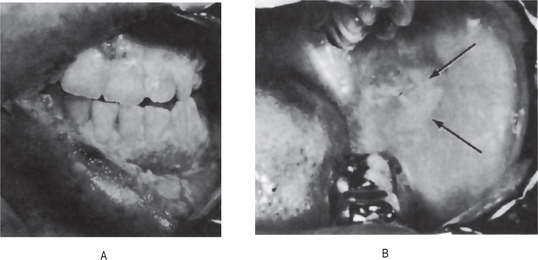
Treatment
The goals of management are to improve the patient's appearance, to control existing lesions and limit scarring, and to prevent the development of further lesions. The prognosis of patients with chronic DLE is favorable regarding mortality; however, many patients continue to experience pain in their lesions or may experience disfigurement from the scars or atrophy that can develop.
Systemic Sclerosis: (Scleroderma, dermatosclerosis, hidebound disease)
Systemic sclerosis (SSc) is a systemic connective tissue disease, characterized by vasomotor disturbances; fibrosis; subsequent atrophy of the skin, subcutaneous tissue, muscles, and internal organs (e.g. alimentary tract, lungs, heart, kidney, CNS); with associated immunologic disturbances.
Etiology
Systemic sclerosis is an autoimmune disease of unclear etiology; however, different factors, including genetic, environmental, and vascular factors are involved in SSc pathogenesis. One theory states that antigens from the human leukocyte antigen (HLA) histocompatability complex, including HLA-B8, HLA-DR5, HLA-DR3, HLA-DR52, and HLA-DQB2, are involved in SSc. Some data suggest that apoptosis and the generation of free radicals may be involved in the pathogenesis of SSc. Increased collagen deposition in tissues is a characteristic feature of SSc.
Clinical Features
Progressive systemic sclerosis is a disease characterized by the ultimate induration of the skin and fixation of the epidermis to the deeper subcutaneous tissues. It may begin in children or young adults, although the greatest incidence is between 30 and 50 years of age, and exhibits a definite gender predilection, females being affected more frequently then males (3-6:1). Systemic sclerosis usually begins on the face, hands or trunk. Simultaneously with the development of the early typical indurated edema of the skin, neuralgia and paresthesia may occur as well as arthritis or simply vague joint pain. Erythema usually accompanies this cutaneous change. The disease progresses at a variable rate, but eventually much, if not all, of the body surface becomes involved. The skin takes on a yellow, gray or ivory-white waxy appearance. Brown pigmentation of the skin may also occur, but this is usually a late manifestation of the disease. Sometimes deposition of calcium in affected areas is also found. The skin becomes hardened and atrophic and cannot be wrinkled or picked up because of its firm fixation to the deep connective tissue. This contracture of the skin gives a mask-like appearance to the face (Fig. 19-36) and a claw-like appearance to the hands (Figs 19-37, 19-38).
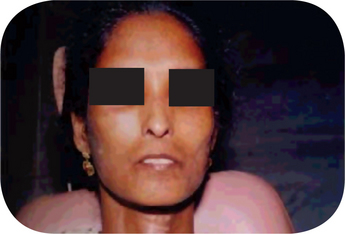
Figure 19-36 Progressive systemic sclerosis.
Characterized by induration of the skin and fixation of the epidermis with the deeper subcutaneous connective tissue.
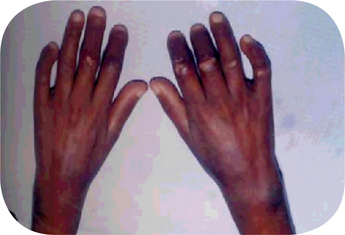
Progressive diffuse systemic sclerosis may ultimately involve many internal organs by fibrosis, loss of smooth muscle and loss of visceral function. Those organs most frequently involved are the gastrointestinal tract, lungs cardiovascular-renal system, musculoskeletal system, and central nervous system.
One variant of systemic sclerosis is the CREST syndrome, an acronym of the five major findings: calcinosis cutis, Raynaud's phenomenon, esophageal dysfunction, sclerodactyly and telangiectasia. This form of the disease is sometimes not as severe as the usual systemic type.
Circumscribed scleroderma, commonly termed morphea, is manifested by the appearance of one or more well-defined, slightly elevated or depressed cutaneous patches, which are white or yellowish and are surrounded by a violaceous halo. The plaques are varied in both size and shape. The lesions commonly occur on the sides of the chest and the thighs.
Occasionally the lesions occur as linear bands or ribbons on the face, particularly the forehead, on the chest and trunk or on an extremity. This has been termed linear scleroderma. Such a band, made up of a furrow with an elevated ridge on one side, is often termed a coup de sabre, since it resembles the mark produced by the blow of a saber. The circumscribed lesions eventually become stiff and hard. It has been reported that facial hemiatrophy is associated with this form of the disease occurring in children. The lesions are generally asymptomatic, although prickling, tingling and itching sensations have been described. The disease may persist for several months to many years, but, in this form, causes no deaths.
Oral Manifestations
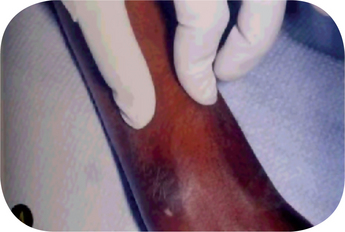
The tongue, soft palate and larynx are the intraoral structures usually involved in progressive systemic sclerosis. Early mild edema of these structures is gradually followed by atrophy and induration of mucosal and muscular tissues. The tongue often becomes stiff and board like, causing the patient difficulty in eating and speaking (Fig. 19-39). The gingival tissues are pale and unusually firm.
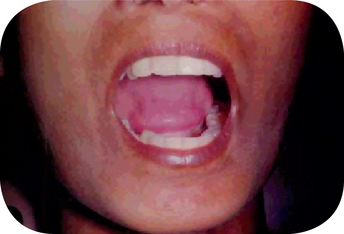
The lips become thin, rigid and partially fixed, producing microstomia. Dysphagia, a choking sensation, inability to open and close the mouth and difficulty in breathing also occur. The reduced opening of the mouth and fixation of the jaw are a result of involvement of the peritemporomandibular joint tissues, and make dental care very difficult. Limitation of mouth opening was found in 80% of a series of these patients by Marmary and his coworkers. Both Smith and Wade also have reviewed this disease with particular emphasis on the oral manifestations.
In addition, Alarcon-Segovia and his coworkers, studying 25 patients with progressive systemic sclerosis, found that all had pathologic changes in the minor salivary glands characteristic of Sjögren's disease: lymphocyte infiltration, duct cell proliferation and collagen infiltration. Weisman and Calcaterra also reported evidence of alterations of salivary gland function characteristic of the sicca syndrome in 12% of 71 patients with scleroderma.
Radiographic Features
Extreme widening of the periodontal ligament, two to four times normal thickness, has been reported originally by Stafne and Austin as characteristic of scleroderma (Fig. 19-40). This may be so striking that, once the association is recognized, the occurrence of the periodontal disturbance as found on routine dental radiographs may be sufficient to establish a tentative diagnosis of systemic sclerosis. This has been confirmed by many studies such as those of White and his coworkers and of Marmary and his associates.
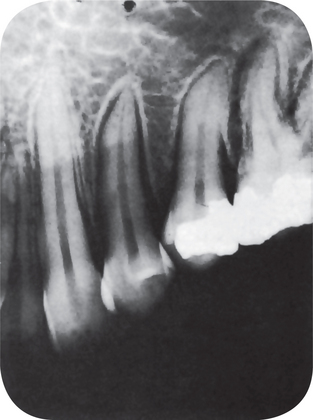
Figure 19-40 Systemic sclerosis.
The extreme widening of the periodontal ligament is obvious in the dental radiograph Courtesy of Dr Edward C Stafne.
Bone resorption of the angle of the mandibular ramus, usually bilaterally, has also been reported by numerous investigators as occurring frequently in this disease. One additional radiographic feature reported has been partial or complete resorption of condyles and/or coronoid processes of the mandible.
Histologic Features
Diffuse systemic sclerosis is characterized microscopically by thickening and hyalinization of the collagen fibers in the skin, the loss of dermal appendages, particularly the sweat glands, and atrophy of the epithelium with loss of rete pegs and increased melanin pigmentation. There is an increase in PAS-positive, diastase-resistant material present in the areas of the homogeneous collagen. Subcutaneous fat disappears, and the walls of the blood vessels become sclerotic. Mucous membrane changes are similar to those occurring in the skin.
The microscopic changes in the periodontal ligament consist of a widening due to an increase of collagen and oxytalan fibers as well as an appearance of hyalinization and sclerosis of collagen with a diminution in the number of connective tissue cells usually found. These changes have been described by Fullmer and Witte.
Ehlers-Danlos Syndrome: (Tenascin-X deficiency syndrome, lysyl hydroxylase deficiency syndrome, cutis hyperelastica)
Ehlers-Danlos syndrome (EDS) is the name given to a group of more than 10 different inherited disorders; all involving a genetic defect in collagen and connective-tissue synthesis and structure. EDS can affect the skin, joints, and blood vessels. This syndrome is clinically heterogeneous; the underlying collagen abnormality is different for each type. Clinical recognition of the types of EDS is important. One type, type IV, is associated with arterial rupture and visceral perforation, with possible life-threatening consequences.
Etiology
Ehlers-Danlos syndrome is a heterogeneous group of inherited connective-tissue disorders characterized by joint hypermobility, cutaneous fragility, and hyperextensibility. The collagen defect has been identified in only six of the 11 types of EDS.
Clinical Features
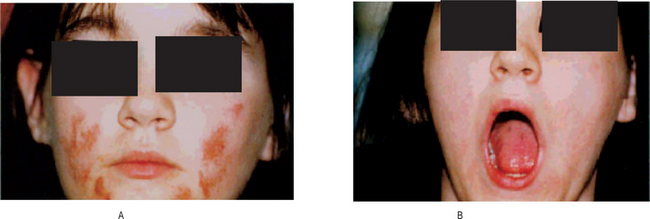
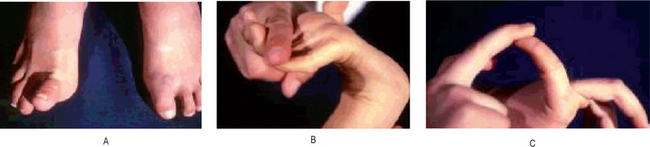
The characteristic clinical features of this disease are the hyperelasticity of skin, hyperextensibility of the joints, and fragility of the skin and blood vessels resulting in excessive bruising as well as defective healing of skin wounds (Fig. 19-41). However, there may be considerable variation in the clinical manifestations depending upon the type of the syndrome present in the patient (Table 19.1). For example, hyperextensibility of skin and joints is striking in EDS I and EDS III but very limited in EDS II and EDS IV. EDS IV is often called the ecchymotic type, since rupture of even large arteries as well as the intestine often occurs, producing a life-threatening situation. In instances in which the skin extensibility is pronounced, the patient has become known as the circus rubber man (Fig. 19-42). The facies are frequently distinctive with hypertelorism, a wide nasal bridge and epicanthic folds being common features. Protruding ears and frontal bossing are often present. Freely movable subcutaneous nodules are frequently found, and these appear to represent fibrosed lobules of fat. The scarring of the skin following wound healing in these patients is unusual inasmuch as the scars tend to spread rather than contract in time.
Table 19-1
Classification of Ehlers-Danlos syndrom
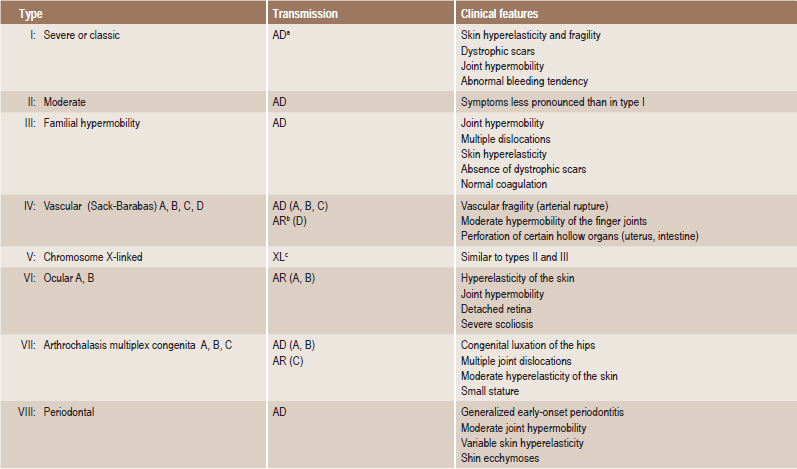
aAD Autosomal dominant;
bAR Autosomal recessive;
cXL Chromosome X-linked
Approximate percentage including all other types (V-XI, Hernandez, Friedman-Harrod, Beasley-Cohen, Viljoen.
Oral Manifestations
The oral manifestations of this disease have been described in detail by Barabas and Barabas. In their series of cases, they found that the oral mucosa was of normal color but was excessively fragile and bruised easily. Although the mucosa did not hold sutures satisfactorily, healing was only slightly retarded and there was no defective scar formation. No remarkable hyperextensibility of mucous membrane could be demonstrated, and the patients had no difficulty in wearing dentures. The gingival tissues appeared fragile and bled after toothbrushing, gingival hyperplasia and fibrous nodules were also noted. Tooth mobility was not increased.
Hypermobility of the temporomandibular joint, resulting in repeated dislocations of the jaw, has been reported.
Alterations in the structure of the teeth have also been reported by Barabas and these consist of a lack of normal scalloping of the dentinoenamel junction, the passage of many dentinal tubules into the enamel, the formation of much irregular dentin and an increased tendency to form pulp stones. Hypoplastic changes in the enamel have also been reported, the teeth have been reported to be fragile and have a tendency for fracture.
Several families have also been reported with apparent Ehlers-Danlos syndrome with extensive periodontal destruction. This has been reviewed by Linch and Action, who described an additional case.
Histologic Features
Histologic study of skin and connective tissues by routine techniques generally fails to reveal any characteristic or diagnostic abnormality. Ultrastructural changes in collagen have been reported in some forms of the disease, according to Byers and his associates.
Treatment
There is no known treatment for the disease. Surgical procedures should be carried out with care because difficulty in suturing and healing problems may exist. With the exception of EDS type IV, all the other variants of this syndrome are not too dangerous, and affected individuals can live a normal life with a few restrictions.
Focal Dermal Hypoplasia Syndrome: (Goltz's syndrome)
Focal dermal hypoplasia (FDH) is an uncommon genetic disorder characterized by distinctive skin abnormalities and a wide variety of defects that affect the eyes, teeth, and skeletal, urinary, gastrointestinal, cardiovascular, and central nervous system. The name is somewhat of a misnomer because the skin lesions appear to evolve as accumulations of fat rather than hypoplasia of the dermis. The mnemonic FOCAL can be used to remember some of the key features of this syndrome: Female sex; Osteopathia striata; Coloboma; Absent ectodermis-, mesodermis-, and neurodermis-derived elements; and Lobster claw deformity. Affected individuals are recognized at birth or prenatally, but cases involving a minor expression of the syndrome may be diagnosed later. Focal dermal hypoplasia is also known as Goltz syndrome; this is not to be confused with Gorlin or Goltz-Gorlin syndrome, which is also known as basal cell nevus syndrome.
Etiology
Focal dermal hypoplasia has an X-linked dominant inheritance pattern and is usually lethal in males. The underlying molecular defect in FDH is not clear. On the basis of common findings of syndactyly, oligodactyly, and polydactyly, the fetal expression of FDH is postulated to occur before the eighth week of gestation; by the eighth week, the hands and feet have differentiated and developed separate and elongated digits.
Clinical Features
The syndrome is characterized by relative focal absence of the dermis associated with herniation of the subcutaneous fat into the defects; skin atrophy, streaky pigmentation and telangiectasia; multiple papillomas of the mucosa and/or skin; anomalies of the extremities including syndactyly, polydactyly and adactyly; an asymmetrical face with pointed chin and notched nasal alae, asymmetrical ears; sunken eyes with sparse eyebrows and scalp hair; eye anomalies, most frequently iris and choroid colobomata and strabismus; and dental and oral anomalies. Mental retardation is often present as is some retardation of physical growth. In addition, many other anomalies have also been reported with varying frequency.
Oral Manifestations
Papillomas of the lips have been a striking feature in a number of these patients as well as papillomas of the buccal mucosa or gingiva. In addition, the teeth are commonly defective in size, shape or structure. Microdontia is a common finding as is enamel hypoplasia. Cleft lips/cleft palate has also been described in several cases. Details of the disease have been discussed by Gorlin and his associates.
Histologic Features
The atrophic reticulated patches of skin reveal attenuation of dermal collagen fibers with partialto-complete absence of significant portions of the dermis. An accompanying change is the appearance of adipose cells in the dermis. In mild cases, adipocytes may be noted only around blood vessels; in severe cases, they may replace all or part of the dermal connective tissue. A layered effect sometimes occurs, with attenuated collagenous connective tissue lying both above and beneath an adipose layer. If the accumulation of adipose tissue is pronounced, it may cause the apparent herniation of subcutaneous tissue through the thinned skin. The papillomatous lesions typically consist of a fibrovascular stalk composed of loose connective tissue with dilated vessels and a variable perivascular admixture of inflammatory cells.
Solar Elastosis: (Senile elastosis, actinic elastosis)
Solar elastosis is a dermatologic disease which is essentially a degenerative condition of skin associated with the general process of aging which itself may be influenced by hereditary factors including skin coloration or pigmentation or its absence, and exposure to the elements, especially sunlight and wind. Such skin, damaged by prolonged exposure to elements of the weather, has often been termed sailor's skin or farmer's skin. It is interesting that this disease, though common, has not been widely reported.
Clinical Features
This disturbance seldom occurs on the oral mucous membranes, but does involve the lip with considerable frequency. Although not confined to elderly patients, it is most common in this age group. The affected skin is wrinkled and appears dry, atrophic and flaccid. On the lip there may be mild keratosis and subtle blending of the vermilion with the skin surface.
Histologic Features
The chief microscopic characteristic is the apparent increase in the amount of elastic connective tissue fibers, a phenomenon that is best observed by special stains. In routine hematoxylin and eosin stained sections, the connective tissue may appear hyalinized, but it stains with hematoxylin rather than with eosin, and this has been termed basophilic degeneration.
References
Abbey, L., Shklar, G. A histochemical study of oral lichen planus. Oral Surg. 1971; 31:226.
Abe, J., Kotzin, B.L., Jujo, K., et al. Selective expansion of T cells expressing T-cell receptor variable regions V beta 2 and V beta 8 in Kawasaki disease. Proc Natl Acad Sci USA. 1992; 89(9):4066–4070. [May 1].
Aberer, W., Wolff-Schreiner, E.C., Stingl, G., Wolff, K. Azathioprine in the treatment of pemphigus vulgaris: a long-term follow-up. J Am Acad Dermatol. 1987; 16(3 Pt 1):527–533. [Mar].
Abreu-Velez, A.M., Beutner, E.H., Montoya, F., et al. Analyses of autoantigens in a new form of endemic pemphigus foliaceus in Colombia. J Am Acad Dermatol. 2003; 49(4):609–614. [Oct].
Abreu-Velez, A.M., Hashimoto, T., Bollag, W.B., et al. A unique form of endemic pemphigus in northern Colombia. J Am Acad Dermatol. 2003; 49(4):599–608. [Oct].
Accili, D., Barbetti, F., Cama, A., et al. Mutations in the insulin receptor gene in patients with genetic syndromes of insulin resistance and acanthosis nigricans. J Invest Dermatol. 1992; 98(6 Suppl):77S–81S. [Jun].
Ackerman, A.B., Pityriasis rosea. Histiologic Diagnosis of Inflammatory Skin Disease: A Method by Pattern Analysis. 1978.
Ackerman, A.B. Focal acantholytic dyskeratosis. Arch Dermatol. 1972; 106:702.
Acosta, A.E., Hietanen, J., Ivanyi, L. Direct immunofluorescence on cytological smears in oral pemphigus. Br J Dermatol. 1981; 105:645.
Aggett, P.J., Atherton, D.J., More, J., et al. Symptomatic zinc deficiency in a breast-fed preterm infant. Arch Dis Child. 1980; 55(7):547–550. [Jul].
Agnello, V. Complement deficiency states. Medicine, Baltimore. 1978; 57(1):1–23. [Jan].
Ahmed, A.R., Moy, R. Death in pemphigus. J Am Acad Dermatol. 1982; 7(2):221–228. [Aug].
Ahmed, A.R., Wagner, R., Khatri, K., et al. Major histocompatibility complex haplotypes and class II genes in non-Jewish patients with pemphigus vulgaris. Proc Natl Acad Sci USA. 1991; 88(11):5056–5060. [Jun 1].
Alarcon-Segovia, D., Ibanez, G., Hernandez-Ortiz, J., Velasquez-Forero, F., et al. Sjogren’s syndrome in progressive systemic sclerosis (scleroderma. Am J Med. 1974; 57:78.
Alkadhi, H., Wildermuth, S., Desbiolles, L., et al. Vascular emergencies of the thorax after blunt and iatrogenic trauma: multi-detector row CT and three-dimensional imaging. Radiographics. 2004; 24(5):1239–1255. [Sep-Oct].
Allen, A.C. The Skin: A Clinocopathologic Treatise, 2nd. New York: Grune and Stratton, 1967.
Allman, S., Haynes, L., MacKinnon, P., Atherton, D.J. Nutrition in dystrophic epidermolysis bullosa. Pediatr Dermatol. 1992; 9(3):231–238. [Sep].
Almeyda, J., Levantine, A. Drug reactions. XVI. Lichenoid drug eruptions. Br J Dermatol. 1971; 85(6):604–607.
Alster, T.S., Wilson, F. Focal dermal hypoplasia (Goltz’s syndrome): treatment of cutaneous lesions with the 585-nm flashlamp-pumped pulsed dye laser. Arch Dermatol. 1995; 131(2):143–144. [Feb].
Altman, J., Perry, H.O. The variations and course of lichen planus. Arch Dermatol. 1961; 84:179.
Amagai, M., Hashimoto, T., Green, K.J., et al. Antigen-specific immunoadsorption of pathogenic autoantibodies in pemphigus foliaceus. J Invest Dermatol. 1995; 104(6):895–901. [Jun].
Amagai, M., Klaus-Kovtun, V., Stanley, J.R. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell. 1991; 67(5):869–877. [Nov 29].
Ames, W.A., Mayou, B.J., Williams, K.N., Williams, K. Anaesthetic management of epidermolysis bullosa. Br J Anaesth. 1999; 82(5):746–751. [May].
Anderson, V.M., Bauer, H.M., Kelly, A.P. Mucocutaneous lymph node syndrome in an adult receiving diphenyl hydantoin. Cutis. 1979; 23(4):493–498. [Apr].
Andreasen, J.O., Poulsen, H.E. Oral manifestations in discoid and systemic lupus erythematosus II: histologic investigation. Acta Odontol, Scand. 1964; 22:389.
Andreasen, J.O., Hjorting-Hansen, E., Ulmansky, M. Milia formation in oral lesions in epidermolysis bullosa. Acta Pathol Microbiod Scand [A]. 1965; 63:37.
Andreasen, J.O. Oral lichen planus I: a clinical evaluation of 115 cases. Oral Surg. 1968; 25:31.
Andreasen, J.O. Oral lichen planus II: a histologic evaluation of ninety-seven cases. Oral Surg. 1968; 25:158.
Andreasen, J.O. Oral manifestations in discoid and systemic lupus erythematosus I: clinical investigation. Acta Odontol Scand. 1964; 22:295.
Anhalt, G.J., Kim, S.C., Stanley, J.R., et al. Paraneoplastic pemphigus: an autoimmune mucocutaneous disease associated with neoplasia. New Engl J Med. 1990; 323(25):1729–1735. [Dec, 20].
Anonymous. [Assessment of a complex of external phenotypic signs for the detection of minor cardiac anomalies]. Klin Med (Mosk). 2004; 82(7):30–33.
Aractingi, S., Morinet, F., Mokni, M., et al. Absence of picornavirus genome in pityriasis rosea. Arch Dermatol Res. 1996; 289(1):60–61. [Dec].
Aradhya, S., Courtois, G., Rajkovic, A., et al. Atypical forms of incontinentia pigmenti in male individuals result from mutations of a cytosine tract in exon 10 of NEMO (IKK-gamma. Am J Hum Genet. 2001; 68(3):765–771. Mar
Archard, H.O., Roebuck, N.F., Stanley, H.R., Jr. Oral manifestations of chronic discoid lupus erythematosus: report of a case. Oral Surg. 1963; 16:696.
Aringer, M., Wintersberger, W., Steiner, C.W. High levels of bcl-2 protein in circulating T lymphocytes, but not B lymphocytes, of patients with systemic lupus erythematosus. Arthritis Rheum. 1994; 37(10):1423–1430. [Oct].
Arndt, K.A., Paul, B.S., Stern, R.S., Parrish, J.A. Treatment of pityriasis rosea with UV radiation. Arch Dermatol. 1983; 119(5):381–382. [May].
Arslanian, S.A. Type 2 diabetes mellitus in children: pathophysiology and risk factors. J Pediatr Endocrinol Metab. 2000; 13(Suppl 6):1385–1394.
Artlett, C.M., Smith, J.B., Jimenez, S.A. New perspectives on the etiology of systemic sclerosis. Mol Med Today. 1999; 5(2):74–78. [Feb].
Arwill, T., Bergenholtz, A., Olsson, O. Epidermolysis bullosa hereditaria III: a histologic study of changes in teeth in the polydysplastic dystrophic and lethal forms. Oral Surg. 1965; 19:723.
Assier, H., Bastuji-Garin, S., Revuz, J., Roujeau, J.C. Erythema multiforme with mucous membrane involvement and Stevens-Johnson syndrome are clinically different disorders with distinct causes. Arch Dermatol. 1995; 131(5):539–543. [May].
Athreya, B.H., Rafferty, J.H., Sehgal, G.S. Adenohypophyseal and sex hormones in pediatric rheumatic diseases. J Rheumatol. 1993; 20(4):725–730. [Apr].
Atra, E., Sato, E.I. Treatment of the cutaneous lesions of systemic lupus erythematosus with thalidomide. Clin Exp Rheumatol. 1993; 11(5):487–493. [Sep-Oct].
Axell, T., Rundquist, L. Oral lichen planus—a demographic study. Community Dent Oral Epidemiol. 1987; 15(1):52–56. [Feb].
Aydingoz, U., Midia, M. Central nervous system involvement in incontinentia pigmenti: cranial MRI of two siblings. Neuroradiology. 1998; 40(6):364–366. [Jun].
Baden, H.P. Familial Schamberg’s disease. Arch Dermatol. 1964; 90:400.
Ballabio, A. MLS, Aicardi and Goltz syndromes: how many genes involved? Am J Med Genet. 1995; 59(1):100. [Oct, 23].
Balow, J.E., Austin, H.A., 3d., Tsokos, G.C. NIH conference: lupus nephritis. Ann Intern Med. 1987; 106(1):79–94. [Jan].
Bang, G. Acanthosis nigricans maligna. Oral Surg. 1970; 29:370.
Banjar, H.H. Cystic fibrosis: presentation with other diseases, the experience in Saudi Arabia. J Cyst Fibros. 2003; 2(3):155–159. [Sep].
Banoczy, J., Sugar, L., Frithiof, L. White sponge nevus—leukoedema exfoliativum mucosae oris: a report on forty-five cases. Swed Dent J. 1973; 66:481.
Barabas, G.M., Barabas, A.P. The Ehlers-Danlos syndrome: a report of the oral and haematological findings in nine cases. Br Dent J. 1967; 123:473.
Barabas, G.M. The Ehlers-Danlos syndrome abnormalities of the enamel, dentine, cementum and the dental pulp: an histological examination of 13 teeth from 6 patients. Br Dent J. 1969; 126:509.
Baroni, A., Perfetto, B., Ruocco, E. Cytokine pattern in blister fluid and sera of patients with pemphigus. Dermatology. 2002; 205(2):116–121.
Basarab, T., Dunnill, M.G., Munn, S.E., Russell-Jones, R. Incontinentia pigmenti: variable disease expression within an affected family. J Eur Acad Dermatol Venereol. 1998; 11(2):173–176. [Sep].
Bastuji-Garin, S., Rzany, B., Stern, R.S., et al. Clinical classification of cases of toxic epidermal necrolysis, Stevens-Johnson syndrome, and erythema multiforme. Arch Dermatol. 1993; 129(1):92–96. [Jan].
Bastuji-Garin, S., Souissi, R., Blum, L., et al. Comparative epidemiology of pemphigus in Tunisia and France: unusual incidence of pemphigus foliaceus in young Tunisian women. J Invest Dermatol. 1995; 104(2):302–305. [Feb].
Bauer, E.A., Herron, G.S., Marinkovich, M.P., et al. Gene therapy for a lethal genetic blistering disease: a status report. Trans Am Clin Climatol Assoc. 1999; 110:86–92.
Baykal, C., Okan, G., Sarica, R. Childhood bullous pemphigoid developed after the first vaccination. J Am Acad Dermatol. 2001; 44(2 Suppl):348–350. [Feb].
Bean, S.F., Alt, T.H., Katz, H.I. Oral pemphigus and bullous pemphigoid. J Am Med Assoc. 1971; 216:673.
Belmont, H.M., Storch, M., Buyon, J. New York University/Hospital for Joint Diseases experience with intravenous cyclophosphamide treatment: efficacy in steroid unresponsive lupus nephritis. Lupus. 1995; 4(2):104–108. [Apr].
Bennett, C.G., Shulman, St, Baughman, R.A. Prepubertal oral pemphigus vulgaris. J Am Dent Assoc. 1980; 100:64.
Berg, D., Weingold, D.H., Abson, K.G., Olsen, E.A. Sweating in ectodermal dysplasia syndromes: a review. Arch Dermatol. 1990; 126(8):1075–1079. [Aug].
Bernard, P., Bedane, C., Bonnetblanc, J.M. Anti-BP180 autoantibodies as a marker of poor prognosis in bullous pemphigoid: a cohort analysis of 94 elderly patients. Br J Dermatol. 1997; 136(5):694–698. [May].
Bernard, P., Prost, C., Durepaire, N., et al. The major cicatricial pemphigoid antigen is a 180-kD protein that shows immunologic cross-reactivities with the bullous pemphigoid antigen. J Invest Dermatol. 1992; 99(2):174–179. [Aug].
Bernard, P., Vaillant, L., Labeille, B., et al. Incidence and distribution of subepidermal autoimmune bullous skin diseases in three French regions. Bullous Diseases French Study Group. Arch Dermatol. 1995; 131(1):48–52. [Jan].
Bernier, J.L., Reynolds, M.C. The relationship of senile elastosis to actinic radiation and to squamous cell carcinoma of the lip. Milit Med. 1955; 117:209.
Bernier, J.L., Tiecke, R.W. Pemphigus, J Oral Surg. 1951; 9:253.
Berth-Jones, J., Smith, S.G., Graham-Brown, R.A. Benign familial chronic pemphigus (Hailey-Hailey disease) responds to cyclosporin. Clin Exp Dermatol. 1995; 20(1):70–72. [Jan].
Besserman-Nielsen, M. Hypohidrotisk ektodermal dysplasi Tandlaegebladet. 1971; 75:1057.
Bethea, B.T., Fitton, T.P., Alejo, D.E., et al. Results of aortic valve-sparing operations: experience with remodeling and reimplantation procedures in 65 patients. Ann Thorac Surg. 2004; 78(3):767–772. [discussion 767-72, Sep].
Bhonsle, R.B., Murti, P.R., Daftary, D.K., Mehta, F.S. An oral lesion in tobacco-lime users in Maharashtra, India. J Oral Pathol. 1979; 8:47–52.
Bielschowsky, M., Heyer, B.J., Howie, J.B. Spontaneous anemia in mice of the NZB/B1 strain. Proc Univ Otago Med School. 1959; 37:9.
Bilinski, D.L., Ehrenkranz, R.A., Cooley-Jacobs, J., McGuire, J. Symptomatic zinc deficiency in a breast-fed, premature infant. Arch Dermatol. 1987; 123(9):1221–1224. [Sep].
Bjornberg, A., Hellgren, L. Pityriasis rosea: a statistical, clinical and laboratory investigation of 826 patients and matched healthy controls. Acta Derm Venereol. 1962; 42(Suppl 50):1–68.
Bjornberg, A., Tegner, E. Pityriasis rosea. In: Freedberg I.M., Eisen A.Z., Wolff K., eds. Dermatology in General Medicine. 5th. New York: McGraw-Hill; 1999:541–546.
Black, C.M. The aetiopathogenesis of systemic sclerosis. J Intern Med. 1993; 234(1):3–8. [Jul].
Blaszczyk, M., Jablonska, S. Linear scleroderma en Coup de Sabre: relationship with progressive facial hemiatrophy (PFH. Adv Exp Med Biol. 1999; 455:101–104.
Blaszczyk, M., Jarzabek-Chorzelska, M., Jablonska, S. Autoantibodies to nucleolar antigens in systemic scleroderma: clinical correlations. Br J Dermatol. 1990; 123(4):421–430. [Oct].
Blaszczyk, M., Krysicka-Janiger, K., Jablonska, S. Primary atrophic profound linear scleroderma. Report of three cases. Dermatology. 2000; 200(1):63–66.
Bolton-Maggs, P.H., Perry, D.J., Chalmers, E.A., et al. The rare coagulation disorders— review with guidelines for management from the United Kingdom Haemophilia Centre Doctors’ Organisation. Haemophilia. 2004; 10(5):593–628. [Sep].
Bonfa, E., Golombek, S.J., Kaufman, L.D. Association between lupus psychosis and anti-ribosomal P protein antibodies. New Engl J Med. 1987; 317(5):265–271. [Jul, 30].
Bouma, P., Cabral, W.A., Cole, W.G., Marini, J.C. COL5A1 exon 14 splice acceptor mutation causes a functional null allele, haploinsufficiency of alpha 1(V) and abnormal heterotypic interstitial fibrils in Ehlers-Danlos syndrome II. J Biol Chem. 2001; 276(16):1356–1364. [Apr 20].
Boumpas, D.T., Austin, H.A., 3d., Vaughn, E.M. Controlled trial of pulse methylprednisolone versus two regimens of pulse cyclophosphamide in severe lupus nephritis. Lancet. 1992; 340(8822):741–745. [Sep 26].
Bowcock, A.M. Genetic locus for psoriasis identified. Ann Med. 1995; 27(2):183–186. [Apr].
Bowden, P.E., Haley, J.L., Kansky, A., et al. Mutation of a type II: keratin gene (K6a) in pachyonychia congenita. Nat Genet. 1995; 10(3):363–365. [Jul].
Bowness, P., Davies, K.A., Norsworthy, P.J. Hereditary C1q deficiency and systemic lupus erythematosus. QJM. 1994; 87(8):455–464. [Aug].
Brayshaw, H.A., Orban, B. Psoriasis gingivae. J Periodontol. 1953; 24:156.
Brice, S.L. Erythema multiforme. In: Weston W.L., ed. Current Problem in Dermatology. Chicago: Year Book; 1990:4.
Brinciotti, M., Ferrucci, G., Trasatti, G. Reflex seizures as initial manifestations of systemic lupus erythematosus in childhood. Lupus. 1993; 2(4):281–283. [Aug].
Brodie, A.G., Sarnat, B.G. Ectodermal dysplasia (anhidrotic type) with complete anodontia. Am J Dis Child. 1942; 64:1046.
Brown, J., Winkelmann, R.K. Acanthosis nigricans: a study of 90 case. Medicine, Baltimore. 1968; 47:33.
Bruns, M., Herrmann, K., Haustein, U.F. Immunologic parameters in systemic sclerosis. Int J Dermatol. 1994; 33(1):25–32. [Jan].
Buchner, S.A., Itin, P. Focal dermal hypoplasia syndrome in a male patient: report of a case and histologic and immunohistochemical studies. Arch Dermatol. 1992; 128(8):1078–1082. Aug
Buchner, A., Begleiter, A. Oral lesions in psoriatic patients. Oral Surg. 1976; 41:327.
Buchner, A., Lozada, F., Silverman, S., Jr. Histopathologic spectrum of oral erythema multiforme. Oral Surg. 1980; 49:221.
Buckley, C. Pityriasis rosea-like eruption in a patient receiving omeprazole. Br J Dermatol. 1996; 135(4):660–701. [Oct].
Budinger, L., Borradori, L., Yee, C., et al. Identification and characterization of autoreactive T-cell responses to bullous pemphigoid antigen 2 in patients and healthy controls. J Clin Invest. 1998; 102(12):2082–2089. [Dec 15].
Bunn, C.C., Denton, C.P., Shi-Wen, X. Anti-RNA polymerases and other autoantibody specificities in systemic sclerosis. Br J Rheumatol. 1998; 37(1):15–20. [Jan].
Burgdorf, W.H., Dick, G.F., Soderberg, M.D., Goltz, R.W. Focal dermal hypoplasia in a father and daughter. J Am Acad Dermatol. 1981; 4(3):273–277. [Mar].
Burge, S.M., Wilkinson, J.D. Darier-White disease: a review of the clinical features in 163 patients. J Am Acad Dermatol. 1992; 27(1):40–50. [Jul].
Burge, S.M. Hailey-Hailey disease: the clinical features, response to treatment and prognosis. Br J Dermatol. 1992; 126(3):275–282. [Mar].
Burke, J.P., Duggirala, R., Hale, D.E., et al. Genetic basis of acanthosis nigricans in Mexican Americans and its association with phenotypes related to type 2 diabetes. Hum Genet. 2000; 106(5):467–472. [May].
Burkhart, C.G., Burkhart, C.N. Tazarotene gel for Darier’s disease. J Am Acad Dermatol. 1998; 38(6 Pt 1):1001–1002. [Jun].
Burkhart, N.W., Burkes, E.J., Burker, E.J. Meeting the educational needs of patients with oral lichen planus. Gen Dent. 1997; 45(2):126–132. [quiz 143-44, Mar-Apr].
Burlakow, P., Medak, H., McGraw, E.A., Tiecke, R. The cytology of vesicular conditions affecting the oral mucosa Part 2: keratosis follicularis. Acta Cytol, Baltimore. 1969; 13:407.
Burns, J.C., Joffe, L., Sargent, R.A., Glode, M.P. Anterior uveitis associated with Kawasaki syndrome. Pediatr Infect Dis. 1985; 4(3):258–261. [May-Jun].
Burns, R.A., Reed, W.B., Swatek, F.E., Omieczynski, D.T. Familial benign chronic pemphigus. Arch Dermatol. 1967; 96:254.
Butterworth, T., Stream, L.P. Clinical Genodermatology. Williams and Wilkins, Baltimore. 1962.
Bye, A.M., Goodfellow, A., Atherton, D.J. Transient zinc deficiency in a full-term breast-fed infant of normal birth weight. Pediatr Dermatol. 1985; 2(4):308–311. [Jul].
Byers, P.H. An exception to the rule. New Engl J Med. 2001; 345(16):1203–1205. [Oct 18].
Byers, P.H., Barsh, G.S., Holbrook, K.A. Molecular pathology in inherited disorders of collagen metabolism. Hum Pathol. 1982; 13:89.
Callen, J.P., Fowler, J.F., Kulick, K.B. Serologic and clinical features of patients with discoid lupus erythematosus: relationship of antibodies to single-stranded deoxyribonucleic acid and of other antinuclear antibody subsets to clinical manifestations. J Am Acad Dermatol. 1985; 13(5 Pt 1):748–755. [Nov].
Callen, J.P., Spencer, L.V., Burruss, J.B., Holtman, J. Azathioprine: an effective, corticosteroid-sparing therapy for patients with recalcitrant cutaneous lupus erythematosus or with recalcitrant cutaneous leukocytoclastic vasculitis. Arch Dermatol. 1991; 127(4):515–522. [Apr].
Callen, J.P. Chronic cutaneous lupus erythematosus: clinical, laboratory, therapeutic, and prognostic examination of 62 patients. Arch Dermatol. 1982; 118(6):412–416. [Jun].
Callen, J.P. Management of antimalarial-refractory cutaneous lupus erythematosus. Lupus. 1997; 6(2):203–208.
Callen, J.P. Systemic lupus erythematosus in patients with chronic cutaneous (discoid) lupus erythematosus: clinical and laboratory findings in seventeen patients. J Am Acad Dermatol. 1985; 12(2 Pt 1):278–288. [Feb].
Callen, J.P. Treatment of cutaneous lesions in patients with lupus erythematosus. Dermatol Clin. 1994; 12(1):201–206. [Jan].
Cameli, N., Picardo, M., Pisani, A., et al. Characterization of the nail matrix basement membrane zone: an immunohistochemical study of normal nails and of the nails in Herlitz junctional epidemolysis bullosa. Br J Dermatol. 1996; 134(1):182–184. [Jan].
Camisa, C., Helm, T.N. Paraneoplastic pemphigus is a distinct neoplasia-induced autoimmune disease. Arch Dermatol. 1993; 129(7):883–886. [Jul].
Cannell, H. Dyskeratosis congenita. Br J Oral Surg. 1971; 9:8.
Cannon, A.B. White nevus of the mucosa (naevus spongiosus albus mucosae. Arch Dermatol, Syph. 1935; 31:365.
Carney, R.G., Carney, R.G., Jr. Incontinentia pigmenti. Arch Dermatol. 1970; 102:157.
Carney, R.G., Jr. Incontinentia pigmenti (review of world literature). Arch Dermatol. 1976; 112:535.
Carr, R.D., Heisel, E.B., Stevenson, T.D. CRST syndrome: a benign variant of scleroderma. Arch Dermatol. 1965; 92:519.
Cassidy JT, Petty RE. Textbook of Pediatric Rheumatology. WB Saunders, Philadelphia
Cawley, E.P., Kerr, D.A. Lichen planus. Oral Surg. 1952; 5:1069.
Celli, J., Duijf, P., Hamel, B.C., et al. Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell. 1999; 99(2):143–153. [Oct 15].
Champion, R.H., et al. Rook/Wilkinson/Ebling Textbook of Dermatology; 3, 1998. [2668].
Chan, E.S., Thornhill, M., Zakrzewska, J. Interventions for treating oral lichen planus. Cochrane Database Syst Rev. (2):2000. [CD001168].
Chan, L.S., Dorman, M.A., Agha, A., et al. Pemphigoid vegetans represents a bullous pemphigoid variant: patient’s IgG autoantibodies identify the major bullous pemphigoid antigen. J Am Acad Dermatol. 1993; 28(2 Pt 2):331–335. [Feb].
Chan, L.S., Hammerberg, C., Cooper, K.D. Significantly increased occurrence of HLA- DQB1*0301 allele in patients with ocular cicatricial pemphigoid. J Invest Dermatol. 1997; 108(2):129–132. [Feb].
Chan, L.S., Vanderlugt, C.J., Hashimoto, T., et al. Epitope spreading: lessons from autoimmune skin diseases. J Invest Dermatol. 1998; 110(2):103–109. [Feb].
Chan, L.S., Woodley, D.T. Pemphigoid: Bullous and cicatricial. Current Therapy in Allergy, Immunology, and Rheumatology, 93-96. 1996.
Chan, L.S., Yancey, K.B., Hammerberg, C., et al. Immune-mediated subepithelial blistering diseases of mucous membranes. Pure ocular cicatricial pemphigoid is a unique clinical and immunopathological entity distinct from bullous pemphigoid and other subsets identified by antigenic specificity of auto. Arch Dermatol. 1993; 129(4):448–455. [Apr].
Chatkupt, S., Gozo, A.O., Wolansky, L.J., Sun, S. Characteristic MR findings in a neonate with incontinentia pigmenti. Am J Roentgenol. 1993; 160(2):372–374. [Feb].
Chipps, J.E. Erythema multiforme exudativum. Oral Surg. 1951; 4:345.
Christensen, E., Holmstrup, P., Wiberg-Jorgensen, F., Neumann-Jensen, B., et al. Arterial blood pressure in patients with oral lichen planus. J Oral Path. 1977; 6:139.
Chuang, T.Y., Ilstrup, D.M., Perry, H.O., Kurland, L.T. Pityriasis rosea in Rochester, Minnesota, 1969 to 1978. J Am Acad Dermatol. 1982; 7(1):80–89. [Jul].
Ciccarelli, A.O., Rothaus, K.O., Carter, D.M., Lin, A.N. Plastic and reconstructive surgery in epidermolysis bullosa: clinical experience with 110 procedures in 25 patients. Ann Plast Surg. 1995; 35(3):254–261. [Sep].
Clarke, A., Sarfarazi, M., Thomas, N.S., et al. X-linked hypohidrotic ectodermal dysplasia: DNA probe linkage analysis and gene localization. Hum Genet. 1987; 75(4):378–380. [Apr].
Clements, P.J., Furst, D.E.Systemic Sclerosis. Baltimore: Williams and Wilkins, 1996.
Clouston, H.R. A hereditary ectodermal dystrophy. Can Med Assoc J. 1929; 21:18–31.
Cockayne, E.A.Inherited Abnormalities of the Skin and Appendages. London: Oxford University Press, 1933.
Cohenour, W., Gamble, J.W. Acanthosis nigricans: review of literature and report of case. J Oral Surg. 1971; 29:48.
Connolly, M.K. Scleroderma. Dermatol Ther. 2001; 14:81–94.
Connors, T.J., Czarnecki, D.B., Haskett, M.I. Acquired zinc deficiency in a breast-fed premature infant. Arch Dermatol. 1983; 119(4):319–321. [Apr].
Cook, T.J. Hereditary ectodermal dysplasia of anhidrotic type. Am J Orthod Orthod Oral Surg. 1939; 25:1008.
Cooke, B.E.D. The diagnosis of bullous lesions affecting the oral mucosa. Br Dent J. 1960; 109:83. [131].
Cooley, J.E., Briggaman, R.A., Cronce, D.J., et al. Hailey-Hailey disease keratinocytes: normal assembly of cell-cell junctions in vitro. J Invest Dermatol. 1996; 107(6):877–881. [Dec].
Cote, B., Wechsler, J., Bastuji-Garin, S., et al. Clinicopathologic correlation in erythema multiforme and Stevens-Johnson syndrome. Arch Dermatol. 1995; 131(11):1268–1272. [Nov].
Coursin, D.B. Stevens-Johnson syndrome: nonspecific parasensitivity reaction? J Am Med Assoc. 1960; 198:113.
Cousins, R.J., Smith, K.T. Zinc-binding properties of bovine and human milk in vitro: influence of changes in zinc content. Am J Clin Nutr. 1980; 33(5):1083–1087. [May].
Crawford, E.G., Jr., Burkes, E.J., Jr., Briggaman, R.A. Hereditary epidermolysis bullosa: oral manifestations and dental therapy. Oral Surg. 1976; 42:490.
Cullar, M.L., Espinoza, L.R. Psoriatic arthritis: Current developments. J Fla Med Assoc. 1995; 82(5):338–342.
Cunningham, S., Conway, E.E., Jr. Systemic lupus erythematosus presenting as an intracranial bleed. Ann Emerg Med. 1991; 20(7):810–812. [Jul].
Curth, H.O. Classification of acanthosis nigricans. Int J Dermatol. 1976; 15:592.
Curtis, A.C., Slaughter, J.C. The clinical diagnosis of dermatological lesions of the face and oral cavity. Am J Orthod Oral Surg. 1947; 33:218.
D’Alise, M.D., Timmons, C.F., Swift, D.M. Focal dermal hypoplasia (Goltz syndrome) with vertebral solid aneurysmal bone cyst variant: a case report. Pediatr Neurosurg. 1996; 24(3):151–154.
Dajani, A.S., Taubert, K.A., Takahashi, M., et al. Guidelines for long-term management of patients with Kawasaki disease: report from the committee on rheumatic fever, endocarditis, and Kawasaki disease, council on cardiovascular disease in the young, American Heart Association. Circulation. 1994; 89(2):916–922. [Feb].
Danbolt, N. Acrodermatitis enteropathica. Acta Derm Venereol (Stochk). 1956; 36:275.
Danforth, R.A., Green, T.L. Oral warty dyskeratoma. Oral Surg. 1980; 49:523.
Daniels, T.E., Quadra-White, C. Direct immunofluorescence in oral mucosal disease: a diagnostic analysis of 130 cases. Oral Surg. 1981; 51:38.
Darling, T.N., Bauer, J.W., Hintner, H., Yancey, K.B. Generalized atrophic benign epidermolysis bullosa. Adv Dermatol. 1997; 13:87–119. [discussion 120].
Darling, A.I., Crabb, H.S.M. Lichen planus. Oral Surg. 1954; 7:1276.
Darmstadt, G.L., Yokel, B.K., Horn, T.D. Treatment of acanthosis nigricans with tretinoin. Arch Dermatol. 1991; 127(8):1139–1140. [Aug].
Davis, R.K., Baer, P.N., Archard, H.O., Palmer, J.H. Tuberous sclerosis with oral manifestations Report of two cases. Oral Surg. 1964; 17:395.
Dawber, R.P.R., Baran, R., de Berker, D. Disorders of nails. In Champion R.H., et al, eds.: Rook/Wilkinson/ebling: Textbook of Dermatology, 6th, Blackwell: Oxford, 1998. [2834].
De Dobbeleer, G., De Graef, C., M’Poudi, E., et al. Reproduction of the characteristic morphologic changes of familial benign chronic pemphigus in cultures of lesional keratinocytes onto dead deepidermized dermis. J Am Acad Dermatol. 1989; 21(5 Pt 1):961–965. [Nov].
De Luca, A., Terrone, C., Tirri, E. Vesical telangiectasias as a cause of macroscopic hematuria in systemic sclerosis. Clin Exp Rheumatol. 2001; 19(1):93–94. [Jan-Feb].
Derks, B., Gericke, G.S., Louw, M. Focal dermal hypoplasia (Goltz syndrome): case reports. S Afr Med J. 1978; 54(1):27–29. [Jul 1].
Dicken, C.H., Bauer, E.A., Hazen, P.G., et al. Isotretinoin treatment of Darier’s disease. J Am Acad Dermatol. 1982; 6(4 Pt 2 Suppl):721–726. [Apr].
Director, W. Pemphigus vulgaris: a clinicopathologic study. Arch Dermatol Syph. 1952; 65:155.
Do Prado, R.F., Marocchio, L.S., Felipini, R.C. Oral lichen planus versus oral lichenoid reaction: difficulties in the diagnosis. Indian J Dent Res. 2009; 20(3):361–364.
Dokal, I. Dyskeratosis congenita in all its forms. Br J Haematol. 2000; 110(4):768–779. [Sep].
Dokal, I. Dyskeratosis congenita: recent advances and future directions. J Pediatr Hematol Oncol. 1999; 21(5):344–350. [Sep-Oct].
Domloge-Hultsch, N., Anhalt, G.J., Gammon, W.R., et al. Antiepiligrin cicatricial pemphigoid. a subepithelial bullous disorder. Arch Dermatol. 1994; 130(12):1521–1529. [Dec].
Domonkos, A.N., Arnold, H.L., Jr., Odom, R.B. Andrews’ Diseases of the Skin, 7th. Philadelphia: WB Saunders, 1982.
Don, P.C., Carney, P.S., Lynch, W.S., et al. Carbon dioxide laserabrasion: a new approach to management of familial benign chronic pemphigus (Hailey-Hailey disease. J Dermatol Surg Oncol. 1987; 13(11):1187–1194. [Nov].
Drago, F., Ranieri, E., Malaguti, F. Human herpesvirus 7 in patients with pityriasis rosea. Electron microscopy investigations and polymerase chain reaction in mononuclear cells, plasma and skin. Dermatology. 1997; 195(4):374–378.
Drury, Re, Prieto, A. Epidermolysis bullosa dystrophica. Oral Surg. 1964; 18:544.
Duong, D.J., Spigel, G.T., Moxley, R.T., 3rd., Gaspari, A.A. American experience with low- dose thalidomide therapy for severe cutaneous lupus erythematosus. Arch Dermatol. 1999; 135(9):1079–1087. [Sep].
Eisen, D. The clincal features, malignant potential, and systemic associations of oral lichen planus: a study of 723 patients. J Am Acad Dermatol. 2002; 46(2):207–214.
Eisen, D. The evaluation of cutaneous, genital, scalp, nail, esophageal, and ocular involvement in patients with oral lichen planus. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999; 88(4):431–436. [Oct].
Eisen, D. The therapy of oral lichen planus. Crit Rev Oral Biol Med. 1993; 4(2):141–158.
Eisenberg, E. Oral lichen planus: a benign lesion. J Oral Maxillofac Surg. 2000; 58(11):1278–1285. [Nov].
Elder D., Elenitsas R., Jaworsky C., Johnson B., eds. Lever’s Histopathology of the Skin. 8th. Lippincott-Raven, Philadelphia, 1998:356.
Elder, J.T., Henseler, T., Christophers, E., et al. Of genes and antigens: the inheritance of psoriasis. J Invest Dermatol. 1994; 103(5 Suppl):150S–153S. [Nov].
Elkins, L., Gruber, I.E. Senile elastosis. Oral Surg. 1951; 4:1007.
El-Labban, N.G., Kramer, I.R.H. Civatte bodies and the actively dividing epithelial cells in oral lichen planus. Br J Dermatol. 1974; 90:13.
Evans, G.W., Johnson, P.E. Characterization and quantitation of a zinc-binding ligand in human milk. Pediatr Res. 1980; 14(7):876–880. [Jul].
Everett, E.D. Mucocutaneous lymph node syndrome (Kawasaki disease) in adults. J Am Med Assoc. 1979; 242:542.
Fagot-Campagna, A., Pettitt, D.J., Engelgau, M.M., et al. Type 2 diabetes among North American children and adolescents: an epidemiologic review and a public health perspective. J Pediatr. 2000; 136(5):664–672. [May].
Falabella, A.F., Valencia, I.C., Eaglstein, W.H., Schachner, L.A. Tissue-engineered skin (Apligraf) in the healing of patients with epidermolysis bullosa wounds. Arch Dermatol. 2000; 136(10):1225–1230. [Oct].
Farber, E.M., Nall, L. Psoriasis. a review of recent advances in treatment. Drugs. 1984; 28(4):324–346. [Oct].
Farber, E.M. Therapeutic perspectives in psoriasis. Int J Dermatol. 1995; 34(7):456–460. [Jul].
Feinberg, A., Menter, M.A. Focal dermal hypoplasia (Goltz syndrome) in a male. a case report. S Afr Med J. 1976; 50(14):554–555. [Mar 27].
Feinstein, A., Friedman, J., Schewach-Millet, M. Pachyonychia congenita. J Am Acad Dermatol. 1988; 19(4):705–711. [Oct].
Felding, I.B., Bjorklund, L.J. Rapp-Hodgkin ectodermal dysplasia. Pediatr Dermatol. 1990; 7(2):126–131. [Jun].
Fine, J.D., Bauer, E.A., Briggaman, R.A., et al. Revised clinical and laboratory criteria for subtypes of inherited epidermolysis bullosa: a consensus report by the Subcommittee on diagnosis and classification of the national epidermolysis bullosa registry. J Am Acad Dermatol. 1991; 24(1):119–135. [Jan].
Fine, J.D., Bauer, E.A., McGuire, J., Moshell, A.Epidermolysis Bullosa: Clinical, Epidemiologic, and Laboratory Advances and the Findings of the National Epidermolysis Bullosa Registry. John’s Hopkins University Press, 1999.
Fine, J.D., Eady, R.A., Bauer, E.A., et al. Revised classification system for inherited epidermolysis bullosa: report of the second international consensus meeting on diagnosis and classification of epidermolysis bullosa. J Am Acad Dermatol. 2000; 42(6):1051–1066. [Jun].
Fine, J.D., McGrath, J., Eady, R.A. Inherited epidermolysis bullosa comes into the new millenium: a revised classification system based on current knowledge of pathogenetic mechanisms and the clinical, laboratory, and epidemiologic findings of large, well-defined patient cohorts. J Am Acad Dermatol. 2000; 43(1 Pt 1):135–137. [Jul].
Fischman, S.L., Barnett, M.L., Nisengard, R.J. Histopathologic, ultrastructural, and immunologic findings in an oral psoriatic lesion. Oral Surg. 1977; 44:253.
Fisher, B.K., Margesson, L.J. Hailey-Hailey disease (familial benign chronic pemphigus). In: Genital Skin Disorders: Diagnosis and Treatment. Mosby-Year Book; 1998. [68, 117].
Fjellner, B. Focal dermal hypoplasia in a 46, XY male. Int J Dermatol. 1979; 18(10):812–815. [Dec].
Flint, I.D., Spencer, D.M., Wilkin, J.K. Eczema herpeticum in association with familial benign chronic pemphigus. J Am Acad Dermatol. 1993; 28(2 Pt 1):257–259. [Feb].
Fosko, S.W., Stenn, K.S., Bolognia, J.L. Ectodermal dysplasias associated with clefting: significance of scalp dermatitis. J Am Acad Dermatol. 1992; 27(2 Pt 1):249–256. [Aug].
Foster, M.E., Nally, F.F. Benign mucous membrane pemphigoid (cicatricial mucosal pemphigoid): a reconsideration. Oral Surg. 1977; 44:697.
Foster, S.C., Album, M.M. Incontinentia pigmenti: Bloch-Sulzburger, Bloch-Seimens disease. Oral Surg. 1970; 29:837.
Francis, J.S., Sybert, V.P. Incontinentia pigmenti. Semin Cutan Med Surg. 1997; 16(1):54–60. [Mar].
Franzot, J., Kansky, A., Kavcic, S. Pachyonychia congenita (Jadassohn-Lewandowsky syndrome). a review of 14 cases in Slovenia. Dermatologica. 1981; 162(6):462–472.
Freedman, P.D., Lumerman, H., Kerpel, S.M. Oral focal acantholytic dyskeratosis. Oral Surg. 1981; 52:66.
Freire-Maia, N. Ectodermal dysplasias. Hum Hered. 1971; 21(4):309–312.
Frithiof, L., Banoczy, J. White sponge nevus (leukoedema exfoliativum mucosae oris): ultrastructural observations. Oral Surg. 1976; 41:607.
Fritsh, P.O., Elias, P.M. Erythema multiforme and toxic epidermal necrolysis. In: Fitzpatrick T.B., et al, eds. Fitzpatrick’s Dermatology in General Medicine. New York: McGraw-Hill; 1993:585–600.
Fritsh, P.O., Rui-Maldonado, R. Erythema multiforme. In: Freedberg I.M., et al, eds. Fitzpatrick’s Dermatology in General Medicine. New York: McGraw-Hill; 1999:636–644.
Fullmer, H.M., Witte, W.E. Periodontal membrane affected by scleroderma. Arch Pathol. 1962; 73:184.
Galimberti, R.L., Kowalczuk, A.M., Bianchi, O., et al. Chronic benign familial pemphigus. Int J Dermatol. 1988; 27(7):495–500. [Sep].
Gallego, H., Crutchfield, C.E., 3rd., Lewis, E.J., Gallego, H.J. Report of an association between discoid lupus erythematosus and smoking. Cutis. 1999; 63(4):231–234. [Apr].
Gardner, N.G., Hudson, C.D. The disturbances in odontogenesis in epidermolysis bullosa hereditaria letalis. Oral Surg. 1975; 40:483.
Getzler, N.A., Flint, A. Keratosis follicalaris: a study of one family. Arch Dermatol. 93(545), 1966.
Ghiggeri, G.M., Caridi, G., Altieri, P., et al. Are the nail-patella syndrome and the autosomal Goltz-like syndrome the phenotypic expressions of different alleles at the COL5A1 locus? Hum Genet. 1993; 91(2):175–177. [Mar].
Giallorenzi, A.F., Goldstein, B.H. Acute (toxic) epidermal necrolysis. Oral Surg. 1975; 40:611.
Giansanti, J.S., Long, S.M., Rankin, J.L. The “tooth and nail” type of autosomal dominant ectodermal dysplasia. Oral Surg. 1974; 37:576.
Gilliam, J.N., Sontheimer, R.D. Distinctive cutaneous subsets in the spectrum of lupus erythematosus. J Am Acad Dermatol. 1981; 4(4):471–475. [Apr].
Glover, M.T., Atherton, D.J. Transient zinc deficiency in two full-term breast-fed siblings associated with low maternal breast milk zinc concentration. Pediatr Dermatol. 1988; 5(1):10–13. [Feb].
Goldberg, M.F. Macular vasculopathy and its evolution in incontinentia pigmenti. Trans Am Ophthalmol Soc. 1998; 96:55–65. [discussion 65-72].
Goldman, H.M., Bloom, J. Oral manifestations of psoriasis: case reports. Oral Surg. 1951; 4:48.
Goldman, H.M., Bloom, J., Cogen, D.W. Bullous lichen ruber planus. Oral Surg. 1959; 12:1468.
Goltz, R.W. Focal dermal hypoplasia syndrome: an update. Arch Dermatol. 1992; 128(8):1108–1111. [Aug].
Goltz, R.W. Focal dermal hypoplasia. Arch Dermatol. 1962; 86:708–717.
Gorlin, R.J., Anderson, J.A. The characteristic dentition of incontinentia pigmenti. J Pediatr. 1960; 57:78.
Gorlin, R.J., Chaudhry, A.P. The oral manifestation of keratosis follicularis. Oral Surg. 1959; 12:1468.
Gorlin, R.J., Pindborg, J.J.Syndromes of the Head and Neck. New York: McGraw-Hill, 1964.
Gorlin, R.J., Meskin, L.H., Peterson, W.C., Jr., Goltz, R.W. Focal dermal hypoplasia syndrome. Acta Dermatol. 1963; 43:421.
Gorlin, R.J. Epidermolysis bullosa. Oral Surg. 1971; 32:760.
Graham, J.H., Johnson, W.C., Helwig, E.B.Dermal Pathology. Hagerstown: Harper and Row, 1972.
Graham-Brown, R.A., Cochrane, G.W., Swinhoe, J.R., et al. Vaginal stenosis due to bullous erythema multiforme (Stevens-Johnson syndrome): case report. Br J Obstet Gynaecol. 1981; 88(11):1156–1157. [Nov].
Grahame, R. Hypermobility—not a circus act. Int J Clin Pract. 2000; 54(5):314–315. [Jun].
Graves, K., Kestenbaum, T., Kalivas, J. Hereditary acrodermatitis enteropathica in an adult. Arch Dermatol. 1980; 116(5):562–564. [May].
Greaves, M.W., Weinstein, G.D. Treatment of psoriasis. New Engl J Med. 1995; 332(9):581–588. [Mar 2].
Greenbaum, S.S. Oral lesions in pityriasis rosea. Arch Dermatol Syph. 1941; 44:55.
Greenwood, R., Tring, F.C. Treatment of malignant acanthosis nigricans with cyproheptadine. Br J Dermatol. 1982; 106(6):697–698. [Jun].
Grider, A., Mouat, M.F. The acrodermatitis enteropathica mutation affects protein expression in human fibroblasts: analysis by two-dimensional gel electrophoresis. J Nutr. 1998; 128(8):1311–1314. [Aug].
Griffin, C.J., Jolly, M., Smythe, J.D. The fine structure of epithelial cells in normal and pathological buccal mucosa II: colloid body formation. Aust Dent J. 1980; 25:12.
Griffith, M., Kaufman, H.S., Silverman, S., Jr. Studies on oral lichen planus I: serum immunoglobulins and complement. J Dent Res. 1974; 53:623.
Griffiths WAD, Judge MR, Leigh IM. Disorders of Keratinization. Ibidem, 1564-66
Grinspan, D., Diaz, J., Villapol, L.O., Schneiderman, J., et al. Lichen ruber planus de la muqueuse buccale Su association con diabete. Bull Soc Fr Dermatol, Syphiligr. 73(898), 1966.
Grinspan, D., Villapol, L.O., Diaz, J., Bellver, B., et al. Liquen rojo plano erosive de la mucosa bucal Su asociacion con diabetes. Actes Finales del V Congreso Ibero Latino Americano de Dermatologia. 1963; 1243.
Grupper, C., Avril, J. Lichen erosif buccal diabete et hypertension (Syndrome de Grinspan. Bull Soc Fr Dermatol Syphiligr. 1965; 72:721.
Guequierre, J.P., Wright, C.S. Pityriasis rosea with lesions on mucous membranes. Arch Dermatol Syph. 1941; 43:1000.
Guilhou, J.J., Clot, J., Meynadier, J., Lapinski, H. Immunological aspects of psoriasis I: immunoglobulins and anti-IgG factors. Br J Dermatol. 1976; 94:501.
Gupta, A.K., Lynde, C.W., Lauzon, G.J., et al. Cutaneous adverse effects associated with terbinafine therapy: 10 case reports and a review of the literature. Br J Dermatol. 1998; 138(3):529–532. [Mar].
Hailey, H., Hailey, H. Familial benign chronic pemphigus. Arch Dermatol. 1939; 39:679–684.
Hansen, E.R., Hjorting-Hansen, E. Det Kroniske Slimhindepemfigoid med saerligt henblik paorale manifestationer. Tandlaegebladet. 1963; 67:49.
Happle, R., Frosch, P.J. Manifestation of the lines of Blaschko in women heterozygous for X- linked hypohidrotic ectodermal dysplasia. Clin Genet. 1985; 27(5):468–471. [May].
Happle, R. Incontinentia pigmenti versus hypomelanosis of Ito: the whys and wherefores of a confusing issue. Am J Med Genet. 1998; 79(1):64–65. [Aug 27].
Hargraves, M.M., Richmond, H., Morton, R. Presentation of two bone marrow elements: the ‘tart’ cell and the ‘LE’ cell. Proc Staff Meet, Mayo Clin. 1948; 23:25.
Hargraves, M.M. Discovery of the LE cell and its morphology. Mayo Clin Proc. 1969; 44:579.
Harrist, T.J., Murphy, G.F., Mihm, M.C., Jr. Oral warty dyskeratoma. Arch Dermatol. 1964; 116:239.
Haustein, U.F., Anderegg, U. Pathophysiology of scleroderma: an update. J Eur Acad Dermatol Venereol. 1998; 11(1):1–8. [Jul].
Hay, R.J., Wells, R.S. The syndrome of ankyloblepharon, ectodermal defects and cleft lip and palate: an autosomal dominant condition. Br J Dermatol. 1976; 94(3):277–289. [Mar].
Hebra F. On diseases of the skin, including the exanthemata. New Sydenham Society, London, 1866-80
Heiss, N.S., Knight, S.W., Vulliamy, T.J., et al. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet. 1998; 19(1):32–38. [May].
Helou, J., Allbritton, J., Anhalt, G.J. Accuracy of indirect immunofluorescence testing in the diagnosis of paraneoplastic pemphigus. J Am Acad Dermatol. 1995; 32(3):441–447. [Mar].
Hernandez-Perez, E. Familial benign chronic pemphigus. Cutis. 1987; 39(1):75–77. [Jan].
Hertzberg, M.S., Schifter, M., Sullivan, J., Stapleton, K. Paraneoplastic pemphigus in two patients with B-cell non-Hodgkin’s lymphoma: significant responses to cyclophosphamide and prednisolone. Am J Hematol. 2000; 63(2):105–106. [Feb].
Hitchin, A.D., Hall, D.C. Incontinentia pigmenti (Bloch-Sulzberger syndrome) and its dental manifestations. Br Dent J. 1964; 116:239.
Hoff, M. Dental manifestations in Ehlers-Danlos syndrome. Oral Surg. 1977; 44:864.
Holden, J.D., Akers, W.A. Goltz’s syndrome: focal dermal hypoplasia: a combined mesoectodermal dysplasia. Am J Dis Child. 1967; 114:292.
Holmstrom, G., Thoren, K. Ocular manifestations of incontinentia pigmenti. Acta Ophthalmol Scand. 2000; 78(3):348–353. [Jun].
Horn, T.D., Anhalt, G.J. Histologic features of paraneoplastic pemphigus. Arch Dermatol. 1992; 128(8):1091–1095. [Aug].
Howell, J.B. Nevus angiolipomatosus vs focal dermal hypoplasia. Arch Dermatol. 92(238), 1965.
Hud, J.A., Jr., Cohen, J.B., Wagner, J.M., Cruz, P.D., Jr. Prevalence and significance of acanthosis nigricans in an adult obese population. Arch Dermatol. 1992; 128(7):941–944. [Jul].
Huff, J.C., Weston, W.L., Tonnesen, M.G. Erythema multiforme: a critical review of characteristics, diagnostic criteria, and causes. J Am Acad Dermatol. 1983; 8(6):763–775. [Jun].
Huff, J.C., Weston, W.L. Recurrent erythema multiforme. Medicine, Baltimore. 1989; 68(3):133–140. [May].
Huff, J.C. Acyclovir for recurrent erythema multiforme caused by herpes simplex. J Am Acad Dermatol. 1988; 18(1 Pt 2):197–199. [Jan].
Huff, J.C. Erythema multiforme and latent herpes simplex infection. Semin Dermatol. 1992; 11(3):207–210. [Sep].
Hunt, M.J., Salisbury, E.L., Painter, D.M., Lee, S. Vesiculobullous Hailey-Hailey disease: successful treatment with oral retinoids. Australas J Dermatol. 1996; 37(4):196–198. [Nov].
Hurt, W.C. Observation on pemphigus vegetans. Oral Surg. 1965; 20:481.
Ikeda, S., Suga, Y., Ogawa, H. Successful management of Hailey-Hailey disease with potent topical steroid ointment. J Dermatol Sci. 1993; 5(3):205–211. Jun
Ismail, S.B., Kumar, S.K., Zain, R.B. Oral lichen planus and lichenoid reactions: etiopathogenesis, diagnosis, management and malignant transformation. J Oral Sci. 2007; 49(2):89–106.
Jablonska, S., Blaszczyk, M. Scleroderma overlap syndromes. Adv Exp Med Biol. 1999; 455:85–92.
Jacobs, J.C. Systemic lupus erythematosus in childhood. Pediatrics. 1963; 32:257.
Jacobsen, N.J., Lyons, I., Hoogendoorn, B., et al. ATP2A2 mutations in Darier’s disease and their relationship to neuropsychiatric phenotypes. Hum Mol Genet. 1999; 8(9):1631–1636. [Sep].
Jadassohn, J., Lewandowsky, F., Pachyonychia congenita. Keratosis disseminata circumscripta (follicularis). Tylomata. Leukokeratosis linguae. Jacob’s Ikonographia Dermatologica; 1. Urban und Schwarzenberg, Berlin, 1906. [29-30].
Jadinski, J.J., Shklar, G. Lichen planus of the gingiva. J Periodontol. 1976; 47:724.
Jansen, T., Paepe, A.D., Nuytinck, L., Altmeyer, P. Acrogeric phenotype in Ehlers-Danlos syndrome type IV: attributed to a missense mutation in the COL3A1 gene. Br J Dermatol. 2001; 144(5):1086–1087. [May].
Jouet, M., Stewart, H., Landy, S., et al. Linkage analysis in 16 families with incontinentia pigmenti. Eur J Hum Genet. 1997; 5(3):168–170. [May-Jun].
Kampgen, E., Burg, G., Wank, R. Association of herpes simplex virus-induced erythema multiforme with the human leukocyte antigen DQw3. Arch Dermatol. 1988; 124(9):1372–1375. [Sep].
Kansky, A., Dolenc-Volje, M., Bowden, P.E., et al. Mycological examination in pachyonychia congenita. Acta Dermatoven APA. 1998; 7:135–138.
Kasai, T., Kato, Z., Matsui, E., et al. Cerebral infarction in incontinentia pigmenti: the first report of a case evaluated by single photon emission computed tomography. Acta Paediatr. 1997; 86(6):665–667. [Jun].
Kasmann-Kellner, B., Jurin-Bunte, B., Ruprecht, K.W. Incontinentia pigmenti (Bloch- Sulzberger-syndrome): case report and differential diagnosisto related dermato-ocular syndromes. Ophthalmologica. 1999; 213(1):63–99.
Kato, H., Ichinose, E., Yoshioka, F., et al. Fate of coronary aneurysms in Kawasaki disease: serial coronary angiography and long-term follow-up study. Am J Cardiol. 1982; 49(7):1758–1766. [May].
Katz, S.I. Dermatitis herpetiformis: the skin and the gut. Ann Int Med. 1980; 93:857.
Kawasaki, T. [Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children]. Arerugi. 1967; 16(3):178–222. [Mar].
Kere, J., Srivastava, A.K., Montonen, O., et al. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet. 1996; 13(4):409–416. [Aug].
Kerob, D., Assier-Bonnet, H., Esnault-Gelly, P., et al. Recurrent erythema multiforme unresponsive to acyclovir prophylaxis and responsive to valacyclovir continuous therapy. Arch Dermatol. 1998; 134(7):876–877. [Jul].
Khafaga, Y.M., Jamshed, A., Allam, A.A., et al. Stevens-Johnson syndrome in patients on phenytoin and cranial radiotherapy. Acta Oncol. 1999; 38(1):111–116.
Kibar, Z., Der Kaloustian, V.M., Brais, B., et al. The gene responsible for Clouston hidrotic ectodermal dysplasia maps to the pericentromeric region of chromosome 13q. Hum Mol Genet. 1996; 5(4):543–547. [Apr].
Kihiczak, N.I., Leevy, C.B., Krysicki, M.M., et al. Cutaneous signs of selected systemic diseases. J Med. 1999; 30(1-2):3–12.
Klaus, S.N., Winkelmann, R.K. The clinical spectrum of urticaria pigmentosa. Mayo Clin Proc. 1965; 40:923.
Knight, S., Vulliamy, T., Copplestone, A., et al. Dyskeratosis Congenita (DC) registry: identification of new features of DC. Br J Haematol. 1998; 103(4):990–996. [Dec].
Knight, S.W., Heiss, N.S., Vulliamy, T.J., et al. Unexplained aplastic anaemia, immunodeficiency, and cerebellar hypoplasia (Hoyeraal-Hreidarsson syndrome) due to mutations in the dyskeratosis congenita gene, DKC1. Br J Haematol. 1999; 107(2):335–339. [Nov].
Koch, P., Bahmer, F.A. Oral lesions and symptoms related to metals used in dental restorations: a clinical, allergological, and histologic study. J Am Acad Dermatol. 1999; 41(3 Pt 1):422–430. [Sep].
Koszewski, B.J., Hubbard, T.F. Congenital anemia in hereditary ectodermal dysplasia. Arch Dermatol. 1956; 74:159.
Kovesi, G., Banoczy, J. Follow-up studies in oral lichen planus. Int J Oral Surg. 2(13), 1973.
Krutchkoff, D.J., Cutler, L., Laskowski, S. Oral lichen planus: the evidence regarding potential malignant transformation. J Oral Path. 1978; 7:1.
Krutchkoff, D.J., Eisenberg, E. Lichenoid dysplasia: a distinct histopathologic entity. Oral Surg Oral Med Oral Pathol. 1985; 60(3):308–315.
Kumer, L., Loos, H.O. Congenital pachyonychia (Riehl type. Wien Klin Wochenschr. 1935; 48:174–178.
Kurkcuoglu, N., Alli, N. Cimetidine prevents recurrent erythema multiforme major resulting from herpes simplex virus infection. J Am Acad Dermatol. 1989; 21(4 Pt 1):814–815. [Oct].
Kushnick, T., Paya, K., Mamunes, P. Chondroectodermal dysplasia. Am J Dis Child. 103(77), 1962.
Lafontaine, D.L.J., Bousquet-Antonelli, C., Henry, Y., et al. The box H + ACA snoRNAs carry Cbf5p, the putative rRNA pseudouridine synthase. Genes Dev. 1998; 12(4):527–537. [Feb 15].
Lamartine, J., Munhoz Essenfelder, G., Kibar, Z., et al. Mutations in GJB6 cause hidrotic ectodermal dysplasia. Nat Genet. 2000; 26(2):142–144. [Oct].
Laskaris, G., Angelopoulos, A. Cicatricial pemphigiod: direct and indirect immunofluorescent studies. Oral Surg. 1981; 51:48.
Laskaris, G., Nicolis, G. Immunofluorescent studies. Oral Surg. 1980; 50:340.
Laskaris, G., Sklavounou, A., Bovopoulou, O. Juvenile pemphigus vulgaris. Oral Surg. 1981; 51:415.
Lebbe, C., Agbalika, F. Pityriasis rosea and human herpesvirus 7, a true association? Dermatology. 1998; 196(2):275.
Lebwohl, M.G. The evolution of vitamin D analogs for the treatment of psoriasis. Arch Dermatol. 1995; 131(11):1323–1324. [Nov].
Lee, A.G., Goldberg, M.F., Gillard, J.H., et al. Intracranial assessment of incontinentia pigmenti using magnetic resonance imaging, angiography, and spectroscopic imaging. Arch Pediatr Adolesc Med. 1995; 149(5):573–580. [May].
Lee, L.A., David, K.M. Cutaneous lupus erythematosus. Curr Probl Dermatol. 1989; 1:165–200.
Leenutaphong, V., Jiamton, S. UVB phototherapy for pityriasis rosea: a bilateral comparison study. J Am Acad Dermatol. 1995; 33(6):996–999. [Dec].
Leenutaphong, V., Sivayathorn, A., Suthipinittharm, P., Sunthonpalin, P. Stevens- Johnson syndrome and toxic epidermal necrolysis in Thailand. Int J Dermatol. 1993; 32(6):428–431. [Jun].
Lehmann, P., Holzle, E., Kind, P., et al. Experimental reproduction of skin lesions in lupus erythematosus by UVA and UVB radiation. J Am Acad Dermatol. 1990; 22(2 Pt 1):181–187. [Feb].
Leigh, I.M., Mowbray, J.F., Levene, G.M., Sutherland, S. Recurrent and continuous erythema multiforme: a clinical and immunological study. Clin Exp Dermatol. 1985; 10(1):58–67. [Jan].
Leung, D.Y., Meissner, H.C., Fulton, D.R., et al. Toxic shock syndrome toxin-secreting Staphylococcus aureus in Kawasaki syndrome. Lancet. 1993; 342(8884):1385–1388. [Dec, 4].
Leung, D.Y., Schlievert, P.M., Meissner, H.C. The immunopathogenesis and management of Kawasaki syndrome. Arthritis Rheum. 1998; 41(9):1538–1547. [Sep].
Lever, W.F., Schaumberg-Lever, G. Histopathology of the Skin, 5. Philadelphia: JB Lippincott, 1975.
Lever, W.F. Oral lesions in pemphigus. Am J Orthod Oral Surg. 1942; 28:569.
Levin, H.L. Psoriasis of the hard palate. Oral Surg. 1954; 7:280.
Lewis, H.M. Therapeutic progress. II: treatment of psoriasis. J Clin Pharm Ther. 1994; 19(4):223–232. [Aug].
Lewis, I.C., Stevens, E.M., Farquhar, J.W. Epidermolysis bullosa in the newborn. Arch Dis Child. 1955; 30:277.
Linch, D.C., Acton, C.H.C. Ehlers-Danlos syndrome presenting with juvenile destructive periodontitis. Br Dent J. 1979; 147:95.
Lodi, G., Porter, S.R., Scully, C. Hepatitis C virus infection: review and implications for the dentist. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998; 86(1):8–22. [Jul].
Looby, J.P., Burket, L.W. Scleroderma of the face with involvement of the alveolar process. Am J Orthod Oral Surg. 1942; 28:493.
Lowe, L.B., Jr. Hereditary epidermolysis bullosa. Arch Dermatol. 1967; 95:587.
Lozada, F., Silverman, S., Jr. Erythema multiforme. Oral Surg. 1978; 46:628.
Lumley, M.A., Jordan, M., Rubenstein, R., et al. Psychosocial functioning in the Ehlers- Danlos syndrome. Am J Med Genet. 1994; 53(2):149–152. [Nov 1].
Lyell, A. Toxic epidermal necrolysis, an eruption resembling scalding of the skin. Br J Dermatol. 1956; 68:355.
MacCauley, F.J. Incontinentia pigmenti (Bloch-Sulzberger syndrome): a case report. Br Dent J. 1968; 125:169.
MacDermot, K.D., Hulten, M. Female with hypohidrotic ectodermal dysplasia and de novo (X;9) translocation: clinical documentation of the AnLy cell line case. Hum Genet. 1990; 84(6):577–579. [May].
Macey-Dare, L.V., Goodman, J.R. Incontinentia pigmenti: seven cases with dental manifestations. Int J Paediatr Dent. 1999; 9(4):293–297. Dec
Mangano, S., Barbagallo, A. Incontinentia pigmenti: clinical and neuroradiologic features. Brain Dev. 1993; 15(5):362–366. [Sep-Oct].
Mao, J.R., Bristow, J. The Ehlers-Danlos syndrome: on beyond collagens. J Clin Invest. 2001; 107(9):1063–1069. [May].
Marcus-Farber, B.S., Bergman, R., Ben Porath, E., et al. Serum antibodies to parvovirus B19 in patients with pityriasis rosea. Dermatology. 1997; 194(4):371.
Marmary, Y., Glaiss, R., Pisanty, S. Scleroderma: oral manifestations. Oral Surg. 1981; 52:32.
Martens, P.B., Moder, K.G., Ahmed, I. Lupus panniculitis: clinical perspectives from a case series. J Rheumatol. 1999; 26(1):68–72. [Jan].
Maser, E.D. Oral manifestations of pachyonychia congenita: report of a case. Oral Surg. 1977; 43:373.
Mason WH, Takahashi M, Schneider T. Recurrence of Kawasaki disease in a large urban cohort in the United States: Proceedings of the Fourth International Symposium on Kawasaki Disease
Matsuda, I., Hattori, S., Nagata, N., et al. HLA antigens in mucocutaneous lymph node syndrome. Am J Dis Child. 1977; 131(12):1417–1418. [Dec].
McCartan, B., McCreary, C. What is the rationale for treating oral lichen planus? Oral Dis. 1999; 5(3):181–182. [Jul].
McCarthy, P.L., Shklar, G. Benign mucous-membrane pemphigus. New Engl J Med. 1958; 258:726.
McCarthy, P.L., Shklar, G. Diseases of the Oral Mucosa, 2nd. Philadelphia: Lea and Febiger, 1980.
McClatchey, K.D., Silverman, S., Jr., Hansen, L.S. Studies on oral lichen planus III: Clinical and histologic correlations in 213 patients. Oral Surg. 1975; 39:122.
McDaniel, W.H. Epidermolysis bullosa. Arch Dis Child. 1954; 29:334.
McGinnis, J.P., Jr., Turner, J.E. Ultrastructure of the white sponge nevus. Oral Surg. 1975; 40:644.
McGrath, J.A. Dyskeratosis congenita: new clinical and molecular insights into ribosome function. Lancet. 1999; 353(9160):1204–1205. [Apr 10].
McKusick, V. Mendelian Inheritance in Man, 11th. Baltimore: J Hopkins University Press, 1994.
McKusick, V.A., Egeland, J.A., Eldridge, R., Krusen, D.E. Dwarfism in the Amish I: the Ellis-van Creveld syndrome. Bull Hopkins Hosp. 1964; 115:306.
McKusick, V.A. Hereditary Disorders of Connective Tissue, 4th. St Louis: CV Mosby, 1972.
McLean, W.H., Rugg, E.L., Lunny, D.P., et al. Keratin 16 and keratin 17 mutations cause pachyonychia congenita. Nat Genet. 1995; 9(3):273–278. [Mar].
McMichael, A.J., Morhenn, V., Payne, R., Sasazuki, T., et al. HLA C and D antigens associated with psoriasis. Br J Dermatol. 1978; 98:287.
Medak, H., Burlakow, P., McGrew, E.A., Tiecke, R. Mucosa: pemphigus vulgaris. Acta Cytol (Baltimore. 1970; 14:11.
Melish, M.E., Hicks, R.V. Kawasaki syndrome: clinical features, Pathophysiology, etiology and therapy. J Rheumatol Suppl. 1990; 24:2–10. [Sep].
Melish, M.E., Hicks, R.M., Larson, E.J. Mucocutaneous lymph node syndrome in the United States. Am J Dis Child. 1976; 130:599.
Miljkovic, J., Bercic, M., Belic, M. Pityriasis rosea with unusual papulovesicular presentation. Acta Derm Venerol. 1996; 5:61–63.
Miller, R.L., Bernstein, M.L., Arm, R.N. Darier’s disease of the oral mucosa: clinical case report with ultrastructural evaluation. J Oral Path. 1982; 11:79.
Mirowski, G.W., Caldemeyer, K.S. Incontinentia pigmenti. J Am Acad Dermatol. 2000; 43(3):517–518. [Sep].
Mitchell, J.R., Wood, E., Collins, K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999; 402(6761):551–555. [Dec 2].
Mitchell, R.D., Smith, N.H.H. Cicatricial pemphigoid: a review of eleven cases. Aust Dent J. 1979; 4:260.
Miteva, L., Nikolova, A. Incontinentia pigmenti: a case associated with cardiovascular anomalies. Pediatr Dermatol. 2001; 18(1):54–56. [Jan-Feb].
Moldenhaouer, E., Ernst, K. Das Jadassohn-Lewandowsky syndrome. Hautartzt. 1968; 19:441–447.
Monreal, A.W., Ferguson, B.M., Headon, D.J., et al. Mutations in the human homologue of mouse dl cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia. Nat Genet. 1999; 22(4):366–369. [Aug].
Monreal, A.W., Zonana, J., Ferguson, B. Identification of a new splice form of the EDA1 gene permits detection of nearly all X-linked hypohidrotic ectodermal dysplasia mutations. Am J Hum Genet. 1998; 63(2):380–389. [[published erratum appears in Am J Hum Genet 1998 Oct; 63(4):1253-55], Aug].
Morales, A., Livingood, C.S., Hu, F. Familial benign chronic pemphigus. Arch Dermatol. 1966; 93:324.
Morgan, J.D. Incontinentia pigmenti (Bloch-Sulzberger syndrome): a report of four additional cases. Am J Dis Child. 1971; 122:294.
Mork, N.J., Rajka, G., Halse, J. Treatment of acanthosis nigricans with etretinate (Tigason) in a patient with Lawrence-Seip syndrome (generalized lipodystrophy. Acta Derm Venereol. 1986; 66(2):173–174.
Mukhtar, Q., Cleverley, G., Voorhees, R.E., McGrath, J.W. Prevalence of acanthosis nigricans and its association with hyperinsulinemia in New Mexico adolescents. J Adolesc Health. 2001; 28(5):372–376. [May].
Muller, C. On the causes of congenital onychogryphosis. Munchen Med Wochenschr. 1904; 49:2180–2182.
Munro, C.S., Carter, S., Bryce, S., et al. A gene for pachyonychia congenita is closely linked to the keratin gene cluster on 17q12-q21. J Med Genet. 1994; 31(9):675–678. [Sep].
Mutasim, D.F., Pelc, N.J., Anhalt, G.J. Paraneoplastic pemphigus. Dermatol Clin. 1993; 11(3):473–481. [Jul].
Nagase, T., Takanashi, M., Takada, H., Ohmori, K. Extensive vesiculobullous eruption following limited ruby laser treatment for incontinentia pigmenti: a case report. Australas J Dermatol. 1997; 38(3):155–157. [Aug].
Naldi, L. Psoriasis. Dermatol Clin. 1995; 13(3):635–647. [Jul].
Newburger, J.W., Takahashi, M., Burns, J.C., et al. The treatment of Kawasaki syndrome with intravenous gamma globulin. New Engl J Med. 1986; 315(6):341–347. [Aug 7].
Nguyen, T.T., Keil, M.F., Russell, D.L., et al. Relation of acanthosis nigricans to hyperinsulinemia and insulin sensitivity in overweight African American and white children. J Pediatr. 2001; 138(4):474–480. [Apr].
Nicholls, A.C., Valler, D., Wallis, S., Pope, F.M. Homozygosity for a splice site mutation of the COL1A2 gene yields a non- functional pro(alpha)2(I) chain and an EDS/OI clinical phenotype. J Med Genet. 2001; 38(2):132–136. [Feb].
Nisengard, R.J., Jablonska, S., Beutner, E.H., Shu, S., et al. Diagnostic importance of immunofluoresence in oral bullous diseases and lupus erythematosus. Oral Surg. 1975; 40:365.
Paller, A.S., Moore, J.A., Scher, R. Pachyonychia congenita tarda. A late-onset form of pachyonychia congenita. Arch Dermatol. 1991; 127(5):701–703. [May].
Parrish, J.E., Scheuerle, A.E., Lewis, R.A., et al. Selection against mutant alleles in blood leukocytes is a consistent feature in Incontinentia Pigmenti type 2. Hum Mol Genet. 1996; 5(11):1777–1783. [Nov].
Parsons, J.M. Pityriasis rosea update: 1986. J Am Acad Dermatol. 1986; 15(2 Pt 1):159–167. [Aug].
Patibanda, R. Warty dyskeratoma of oral mucosa. Oral Surg. 1981; 52:422.
Pellegrino, R.J., Shah, A.J. Vascular occlusion associated with incontinentia pigmenti. Pediatr Neurol. 1994; 10(1):73–74. [Feb].
Perry, H.O., Brunsting, L.A. Pemphigus foliaceus: further observations. Arch Dermatol. 1965; 91:10.
Person, J.R., Rogers III, R.S. Bullous and cicatricial pemphigoid Clinical, histopathologic, and immunopathologic correlations. Mayo Clin Proc. 1977; 52:54.
Pierard, G.E., Van Neste, D., Letot, B. Hidrotic ectodermal dysplasia. Dermatologica. 1979; 158(3):168–174.
Pietra, B.A., De Inocencio, J., Giannini, E.H., Hirsch, R. TCR V beta family repertoire and T cell activation markers in Kawasaki disease. J Immunol. 1994; 153(4):1881–1888. [Aug 15].
Pindborg, J.J., Gorlin, R.J. Oral changes in acanthosis nigricans (juvenile type). Acta Derm Venereol (Stockh). 1962; 42:63.
Pines, A., Ehrenfeld, M., Fisman, E.Z., et al. Mitral valve prolapse in psoriatic arthritis. Arch Intern Med. 1986; 146(7):1371–1373. [Jul].
Pisanti, S., Ship, I.I. Oral psoriasis. Oral Surg. 1970; 30:351.
Pisanti, S., Sharav, Y., Kaufman, E., Posner, L.N. Pemphigus vulgaris: incidence in Jews of different ethnic groups, according to age, sex, and initial lesion. Oral Surg. 38(382), 1973.
Porter, S.R., Kirby, A., Olsen, I., Barrett, W. Immunologic aspects of dermal and oral lichen planus: a review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997; 83(3):358–366. [Mar].
Prindiville, D.E., Stern, D. Oral manifestations of Darier’s disease. J Oral Surg. 1976; 34:1001.
Proby, C., Fujii, Y., Owaribe, K., et al. Human autoantibodies against HD1/plectin in paraneoplastic pemphigus. J Invest Dermatol. 1999; 112(2):153–156. [Feb].
Puente, J.J. Hereditare familiare Pachyonychie. Zbl Haut Geschl Kr. 1955; 50:309.
Pullon, P.A. Ultrastructure of oral lichen planus. Oral Surg. 1969; 28:365.
Quinn, J.H. Acute pemphigus vulgaris: dental significance. Oral Sure. 1948; 1:751.
Ramanathan, K., Omar-Ahmad, U.D., Kutty, M.K., Ching, L.K., et al. Poroderatosis Mibelli: report of a case. Br Dent J. 1969; 126:31.
Raychaudhuri, S.P., Rein, G., Farber, E.M. Neuropathogenesis and neuropharmacology of psoriasis. Int J Dermatol. 1995; 34(10):685–693. Oct
Reed, R.J., Leone, P. Porokeratosis: a mutant clonal keratosis of the epidermis I histogenesis. Arch Dermatol. 101, 1970. [340].
Reed, W.B., Lopez, D.A., Landing, B. Clinical spectrum of anhidrotic ectodermal dysplasia. Arch Dermatol. 1970; 102:134.
Rees, T.D., Wood-Smith, D., Converse, J.M. The Ehlers-Danlos syndrome: with a report of three cases. Plast Reconstr Surg. 1963; 32:39.
Reynold, J.M., Gold, M.B., Scriver, C.R. The characterization of hereditary abnormalities of keratin: Clouston’s ectodermal dysplasia. Birth Defects Orig Artic Ser. 1971; 7(8):91–95. [Jun].
Rice, J.S., Hurt, W.C., Rovin, S. Pemphigus vegetans Report of an unusual case. Oral Surg. 1963; 16:1383.
Richter, B.J., McNutt, N.S. The spectrum of epidermolysis bullosa acquisita. Arch Dermatol. 1979; 115:1325.
Robinson, H.M., Jr., McCrumb, F.R., Jr. Comparative analysis of the mucocutaneous- ocular syndromes. Arch Dermatol, Syph. 1950; 61:539.
Robison, J.W., Odom, R.B. Bullous pemphigoid in children. Arch Dermatol. 1978; 114:899.
Roenigk, H.H., Jr., Ryan, J.G., Bergfeld, W.F. Epidermolysis bullosa acquisita Report of three cases and review of all published cases. Arch Dermatol. 1971; 103:1.
Rogers, M. The ‘bar code phenomenon’: a microscopic artifact seen in patients with hypohidrotic ectodermal dysplasia [letter]. Pediatr Dermatol. 2000; 17(4):329–330. [Jul-Aug].
Rook, A., Wilkinson, D.S., Eblin, F.J.G. Textbook of Dermatology, 3rd. Oxford: Blackwell Scientific Publications, 1979.
Rosenthal, I.H. Generalized scleroderma (Hidebound disease) its relation to the oral cavity, with case history and dental restoration. Oral Surg. 1948; 1:1019.
Rowley, A.H., Shulman, S.T. Kawasaki syndrome. Pediatr Clin North Am. 1999; 46(2):313–329. [Apr].
Russell, D.L., Finn, S.B. Incontinentia pigmenti (Bloch-Sulzberger syndrome): a case report with emphasis on dental manifestations. J Dent Child. 1967; 34:494.
Russotto, S.B., Ship, I.I. Oral manifestations of dermatitis herpetiformis. Oral Surg. 1971; 31:42.
Sabir, S., James, W.D., Schuchter, L.M. Cutaneous manifestations of cancer. Curr Opin Oncol. 1999; 11(2):139–144. [Mar].
Sadeghi, E.M., Witkop, C.J. Ultrastructural study of hereditary benign intraepithelial dyskeratosis. Oral Surg. 1977; 44:567.
Sakuntabhai, A., Ruiz-Perez, V., Carter, S., et al. Mutations in ATP2A2, encoding a Ca2 + pump, cause Darier disease. Nat Genet. 1999; 21(3):271–277. [Mar].
Salmon, T.N., Robertson, G.R., Jr., Tracy, N.H., Jr., Hiatt, W.R. Oral psoriasis. Oral Surg. 1974; 38:48.
Schiodt, M., Andersen, L., Shear, M., Smith, L.J. Leukoplakia-like lesions developing in patients with oral discoid lupus erythematosus. Acta Odontol Scand. 1981; 39:209.
Schiodt, M., Dabelsteen, E., Ullman, S., Halberg, P. Deposits of immunoglobulins and complement in oral lupus erythematosus. Scand J Dent Res. 1974; 82:603.
Schiodt, M., Halberg, P., Hentzer, B. A clinical study of 32 patients with oral discoid lupus erythematosus. Int J Oral Surg. 1978; 7:85.
Schiodt, M., Holmstrup, P., Dabelsteen, E., Ullman, S. Deposits of immunoglobulins, lichen planus, and leukoplakia. Oral Surg. 1981; 51:603.
Schnyder, U.W., Klunker, W., Erbliche Verhornungsstorungen der Haut Jadassohn JHandbuch der Haut und Geschlechtkrankheiten Erganzungswerk VII. Berlin: Springer, 1966. [905-08].
Scully, C., Beyli, M., Ferreiro, M.C., et al. Update on oral lichen planus: etiopathogenesis and management. Crit Rev Oral Biol Med. 1998; 9(1):86–122.
Sedano, H.O., Sauk, J.J., Jr., Gorlin, R.J. Oral Manifestations of Inherited Disorders Boston, Butterworth. 1977;
Shelly, W.B. Herpes simplex virus as a cause of erythema multiforme. J Am Med Assoc. 1967; 201:153.
Sherertz, E.F. Improved acanthosis nigricans with lipodystrophic diabetes during dietary fish oil supplementation. Arch Dermatol. 1988; 124(7):1094–1096. [Jul].
Shimizu, H., Masunaga, T., Ishiko, A., et al. Autoantibodies from patients with cicatricial pemphigoid target different sites in epidermal basement membrane. J Invest Dermatol. 1995; 104(3):370–373. [Mar].
Shklar, G., Cataldo, E. Histopathology and cytology of oral lesions of pemphigus. Arch, Dermatol. 1970; 101:635.
Shklar, G., McCarthy, P.L. Oral manifestations of benign mucous membrane pemphigus (mucous membrane pemphigoid. Oral Surg. 1959; 12:950.
Shklar, G., Frim, S., Flynn, E. Gingival lesions of pemphigus. J Periodontol. 1978; 49:428.
Shklar, G., Meyer, I., Zacarian, S.A. Oral lesions in bullous pemphigoid. Arch Dermatol. 99(663), 1969.
Shklar, G. Erosive and bullous oral lesions of lichen planus: histologic studies. Arch Dermatol. 1968; 97:411.
Silverman, S., Jr. Oral lichen planus: a potentially premalignant lesion. J Oral Maxillofac Surg. 2000; 58(11):1286–1288. [Nov].
Silverman, S., Jr., Griffith, M. Studies on oral lichen planus II: follow-up on 200 patients, clinical characteristics, and associated malignancy. Oral Surg. 1974; 37:705.
Smith, D.B. Scleroderma: its oral manifestations. Oral Surg. 1958; 11:865.
Solomon, L.M., Keuer, E.J. The ectodermal dysplasias: problems of classification and some newer syndromes. Arch Dermatol. 1980; 116(11):1295–1299. [Nov].
Sorrow, J.M., Jr., Hitch, J.M. Dyskeratosis congenita. Arch Dermatol. 1963; 88:340.
Spouge, J.D., Trott, J.R., Chesko, G. Darier-White’s disease: a cause of white lesions of the mucosa Report of four cases. Oral Surg. 1966; 21:441.
Stafne, E.C., Austin, L.T. A characteristic dental finding in acrosclerosis and diffuse scleroderma. Am J Orthod Oral Surg. 1944; 30:25.
Stern, L., Jr. The diagnosis of pemphigus by its oral signs. Oral Surg. 1949; 2:1443.
Stillman, M.A., Bart, R.S., Lopf, A.W. Squamous cell carcinoma occurring in oral lichen planus. Cutis. 1973; 11:486.
Stuart, C.A., Smith, M.M., Gilkison, C.R., et al. Acanthosis Nigricans among Native Americans: an indicator of high diabetes risk. Am J Public Health. 1994; 84(11):1839–1842. [Nov].
Su, W.P., Chun, S.I., Hammond, D.E., Gordon, H. Pachyonychia congenita: a clinical study of 12 cases and review of the literature. Pediatr Dermatol. 1990; 7(1):33–38. [Mar].
Sugarman, M.M. Lupus erythematosus. Oral Surg. 1953; 6:836.
Sugerman, P.B., Satterwhite, K., Bigby, M. Autocytotoxic T-cell clones in lichen planus. Br J Dermatol. 2000; 142(3):449–456. [Mar].
Sugerman, P.B., Savage, N.W., Zhou, X., et al. Oral lichen planus. Clin Dermatol. 2000; 18(5):533–539. [Sep-Oct].
Suzuki, K., Hu, D., Bustos, T., et al. Mutations of PVRL1, encoding a cell-cell adhesion molecule/herpesvirus receptor, in cleft lip/palate-ectodermal dysplasia. Nat Genet. 2000; 25(4):427–430. [Aug].
Tai'eb, A. X-linked hypohidrotic ectodermal dysplasia in the new born: a diagnostic challenge. Eur J Pediatr Dermatol. 1998; 8:201–204.
Tanaka, N., Sekimoto, K., Naoe, S. Kawasaki disease: relationship with infantile periarteritis nodosa. Arch Pathol Lab Med. 1976; 100:81.
Tanay, A., Mehregan, A.H. Warty dyskeratoma. Dermatologica. 1969; 138:155.
Task Force on Psoriasis. Guidelines of care for psoriasis. Committee on Guidelines of Care. Task Force on Psoriasis. J Am Acad Dermatol. 1993; 28(4):632–637. [Apr].
Taylor, M.H., Peterson, D.S. Kawasaki’s disease. J Am Dent Assoc. 1982; 104:44.
Terezhalmy, G.T. Mucocutaneous lymph node syndrome. Oral Surg. 1979; 47:26.
Theodore, F.H. Ocular-oral syndromes. Oral Surg. 1952; 5:259.
Thurnam, J. Two cases in which the skin, hair and teeth were very imperfectly developed. Proc RM Chir Soc. 1848; 31:71–82.
Tomich, C.E., Burkes, E.J. Warty dyskeratoma. Oral Surg. 1971; 31:798.
Toombs, E.L., Peck, G.L. Electrosurgical treatment of etretinate-resistant Darier’s disease. J Dermatol Surg Oncol. 1989; 15(12):1277–1280. [Dec].
Touraine, A.Eheredite en medicine. Paris: Masson, 1955. [448-49].
Toussant, S., Kareino, H. Pityriasis rosea. In: Elder D., Elenitsas R., Jaworsky C., et al, eds. Lever’s Histopathology of the Skin. 8th. Philadelphia: Lippincott-Raven; 1997:164–165.
Troiano, G., Valente, G., Isoppo, M. Pitiriasi rosea con lesioni eritema polimorfo-simili. Chron Dermatol. 1997; 7:417–421.
Tsubota, K., Satake, Y., Kaido, M., et al. Treatment of severe ocular-surface disorders with corneal epithelial stem-cell transplantation. New Engl J Med. 1999; 340(22):1697–1703. [Jun 3].
Tuffanelli, D.L., Kay, D., Fukuyama, K. Dermalepidermal junction in lupus erythematosus. Arch Dermatol. 1969; 99:652.
Valdimarsson, H., Sigmundsdottir, H., Jonsdottir, I. Is psoriasis induced by streptococcal superantigens and maintained by M-protein-specific T-cells that cross-react with keratin? Clin Exp Immunol. 1997; 107(Suppl 1):21–24. [Jan].
van der Meij, E.H., Mast, H., van der Waal, I. The possible premalignant character of oral lichen planus and oral lichenoid lesions: a prospective five-year follow-up study of 192 patients. Oral Oncol. 2007; 43(8):742–748.
Videni, N., Kansky, A., Basta-Juzbai, A. Pachyonychia congenita (Jadassohn- Lewandowsky syndrome): a review of 25 cases in Croatia. Acta Derm. 1991; 18:173–180.
von Bulow, F.A., Hjorting-Hansen, E., Ulmansky, M. An electronmicroscopic study of oral mucosal lesions in erythema multiforme exudativum. Acta Pathol Microbiol Scand [A]. 1966; 66:145.
Voorhees, J.J., Stawiski, M., Duell, E.A. Increased cyclic GMP and decreased cyclic AMP levels in the hyperplastic, abnormally differentiated epidermis of psoriasis. Life Sci. 1973; 13:639.
Wade, G.W. Scleroderma Dent Progr. 1963; 3:236.
Wald, C., Diner, H. Dyskeratosis congenita with associated periodontal disease. Oral Surg. 1976; 37:736.
Walker, D.M. Immunological processes involving the oral mucosa in lichen planus. Proc Roy Soc Med, 69 7. 1976.
Walls, W.L., Altman, D.H., Winslow, O.P. Chondroectodermal dysplasia (Ellis-Van Creveld syndrome. Am Med Aassoc J Dis Child. 1959; 98:242.
Weathers, D.R., Driscoll, R.M. Darier’s disease of the oral mucosa. Oral Surg. 1974; 37:711.
Weathers, D.R., Baker, G., Archard, H.O., Burkes, E.J., Jr. Psoriasiform lesions of the oral mucosa with emphasis on ‘ectopic geographic tongue’. Oral Surg. 1974; 37:872.
Weathers, D.R., Olansky, S., Sharpe, L.O. Darier’s disease with mucous membrane involvement: a case report. Arch Dermatol. 1969; 100:50.
Weech, A.A. Hereditary ectodermal dysplasia (congenital ectodermal defect. Am J Dis Child. 1929; 37:766–790.
Weinstein, G.D. Tazarotene: a novel retinoid for the topical treatment of psoriasis. Pharmacy and Therapeutics. Aug, 377-81. 1997.
Weisman, R.A., Calcaterra, T.C. Head and neck manifestations of scleroderma. Ann Otol. 1978; 87:332.
Weiss, E., Schmidberger, H., Jany, R., et al. Palliative radiotherapy of mucocutaneous lesions in malignant acanthosis nigricans. Acta Oncol. 1995; 34(2):265–267.
Weiss, R.S., Swift, S. The significance of a positive LE: phenomenon. Arch Dermatol Syph. 1955; 72:103.
Wheeland, R.G., Gilmore, W.A. The surgical treatment of hypertrophic Darier’s disease. J Dermatol Surg Oncol. 1985; 11(4):420–423. [Apr].
Whinston, G.J. Oral lesions of erythema multiform. Oral Surg. 1952; 5:1207.
White, D.K., Leis, H.J., Miller, A.S. Intraoral psoriasis associated with widespread dermal psoriasis. Oral Surg. 1976; 41:174.
White, S.C., Frey, N.W., Blaschke, D.D., Ross, M.D., et al. Oral radiographic changes in patients with progressive systemic sclerosis (scleroderma. J Am Dent Assoc. 1977; 94:1178.
Whitten, J.B. The electron microscopic examination of congenital keratoses of the oral mucous membranes I: white sponge nevus. Oral Surg. 1970; 29:69.
Wiesenfeld, D., Martin, A., Scully, C., Thomson, J. Oral manifestations in linear IgA disease. Br Dent J. 1982; 153:398.
Wilsch, L., Haneke, E., Schaidt, G. Letters:structural hair abnormalities in hidrotic ectodermal dysplasia (HED. Arch Dermatol Res. 1977; 259(1):101–103. [Jul. 21].
Wilson, A.G. Three cases of hereditary hyperkeratosis of the nail bed. Br J Dermatol. 1905; 17:13–14.
Winkelmann, R.K. Classification and pathogenesis of scleroderma. Mayo Clin Proc. 1971; 46:83.
Winter, G.B., Geddes, M. Oral manifestations of chondroectodermal dysplasia (Ellis- van Creveld syndrome): report of a case. Br Dent J. 1967; 122:103.
Witkop, C.J., Jr., Shenkle, C.H., Graham, J.B., Murray, M.R., et al. Hereditary benign intraepithelial dyskeratosis II: oral manifestations and hereditary transmission. Arch Pathol. 1960; 70:696.
WitkopJr, C.J.Genetics and Dental Health. New York: McGraw-Hill, 1962.
Witkop, C.J., Jr., Gorlin, R.J. Four hereditary mucosal syndromes. Arch Dermatol. 1961; 84:762.
Witkop, C.J., Jr. Genetics and dentistry. Eugenics Q. 1958; 5:15.
Wooten, J.W., Tarsitano, J.J., LaVere, A.M. Oral psoriasiform lesions: a possible prosthodontic complication. J Prosthet Dent. 1970; 24:145.
Wright, R.K., Mandy, S.H., Halprin, K.M., Hsia, S.L. Defects and deficiency of adenyl cyclase in psoriatic skin. Arch Dermatol. 1973; 107:47.
Yaghmai, R., Kimyai-Asadi, A., Rostamiani, K., et al. Overlap of dyskeratosis congenita with the Hoyeraal-Hreidarsson syndrome. J Pediatr. 2000; 136(3):390–393. [Mar].
Yanagawa, H., Nakamura, Y., Yashiro, M., et al. Results of the nationwide epidemiologic survey of Kawasaki disease in 1995 and 1996 in Japan. Pediatrics. 1998; 102(6):E65. [Dec].
Yanagawa, H., Yashiro, M., Nakamura, Y., et al. Epidemiologic pictures of Kawasaki disease in Japan: from the nationwide incidence survey in 1991 and 1992. Pediatrics. 1995; 95(4):475–479. [Apr].
Yeh, J.S., Munn, S.E., Plunkett, T.A., et al. Coexistence of acanthosis nigricans and the sign of Leser-Trelat in a patient with gastric adenocarcinoma: a case report and literature review. J Am Acad Dermatol. 2000; 42(2 Pt 2):357–362. [Feb].
Young, L.L., Lenox, J.A. Pachyonychia congenita: a long-term evaluation of associated oral and dermal lesions. Oral Surg. 1973; 36:663.
Zegarelli, D.J., Zegarelli, E.V. Intraoral pemphigus vulgaris. Oral Surg. 1977; 44:384.
Zegarelli, E.V., Everett, F.G., Kutscher, A.H., Gorman, J., et al. Familial white folded dysplasia of the mucous membranes. Arch Dermatol. 1959; 80:59.
Zonana, J. Hypohidrotic (anhidrotic) ectodermal dysplasia: molecular genetic research and its clinical applications. Semin Dermatol. 1993; 12(3):241–246. [Sep].
*Source: Lupus Foundation of America, Inc. http://www.lupus.org/