Defects in Metabolism of Lipids
Disorders of Mitochondrial Fatty Acid β-Oxidation
Charles A. Stanley, Michael J. Bennett
Mitochondrial β-oxidation of fatty acids is an essential energy-producing pathway. It is a particularly important pathway during prolonged periods of starvation, and during periods of reduced caloric intake because of gastrointestinal illness or increased energy expenditure during febrile illness. Under these conditions, the body switches from using predominantly carbohydrate to predominantly fat as its major fuel. Fatty acids are also important fuels for exercising skeletal muscle and are the preferred substrate for the heart. In these tissues, fatty acids are completely oxidized to carbon dioxide and water. The end products of hepatic fatty acid oxidation are the ketone bodies β-hydroxybutyrate and acetoacetate. These cannot be oxidized by the liver but are exported to and serve as important fuels in peripheral tissues, particularly the brain, which can partially substitute ketone bodies for glucose during periods of fasting.
Genetic defects have been identified in nearly all of the known steps in the fatty acid oxidation pathway; all are recessively inherited (Table 86-1).
Table 86-1
Mitochondrial Fatty Acid Oxidation Disorders—Clinical and Biochemical Features
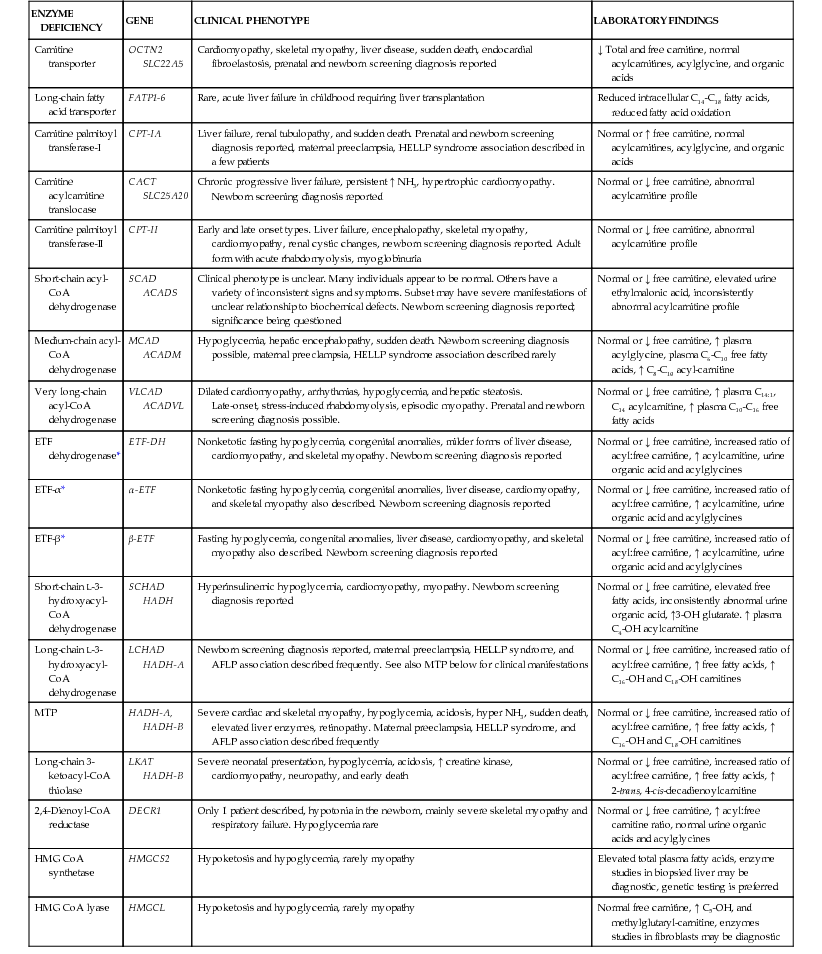
| ENZYME DEFICIENCY | GENE | CLINICAL PHENOTYPE | LABORATORY FINDINGS |
| Carnitine transporter | OCTN2 SLC22A5 | Cardiomyopathy, skeletal myopathy, liver disease, sudden death, endocardial fibroelastosis, prenatal and newborn screening diagnosis reported | ↓ Total and free carnitine, normal acylcarnitines, acylglycine, and organic acids |
| Long-chain fatty acid transporter | FATP1-6 | Rare, acute liver failure in childhood requiring liver transplantation | Reduced intracellular C14-C18 fatty acids, reduced fatty acid oxidation |
| Carnitine palmitoyl transferase-I | CPT-IA | Liver failure, renal tubulopathy, and sudden death. Prenatal and newborn screening diagnosis reported, maternal preeclampsia, HELLP syndrome association described in a few patients | Normal or ↑ free carnitine, normal acylcarnitines, acylglycine, and organic acids |
| Carnitine acylcarnitine translocase | CACT SLC25A20 | Chronic progressive liver failure, persistent ↑ NH3, hypertrophic cardiomyopathy. Newborn screening diagnosis reported | Normal or ↓ free carnitine, abnormal acylcarnitine profile |
| Carnitine palmitoyl transferase-II | CPT-II | Early and late onset types. Liver failure, encephalopathy, skeletal myopathy, cardiomyopathy, renal cystic changes, newborn screening diagnosis reported. Adult form with acute rhabdomyolysis, myoglobinuria | Normal or ↓ free carnitine, abnormal acylcarnitine profile |
| Short-chain acyl-CoA dehydrogenase | SCAD ACADS | Clinical phenotype is unclear. Many individuals appear to be normal. Others have a variety of inconsistent signs and symptoms. Subset may have severe manifestations of unclear relationship to biochemical defects. Newborn screening diagnosis reported; significance being questioned | Normal or ↓ free carnitine, elevated urine ethylmalonic acid, inconsistently abnormal acylcarnitine profile |
| Medium-chain acyl-CoA dehydrogenase | MCAD ACADM | Hypoglycemia, hepatic encephalopathy, sudden death. Newborn screening diagnosis possible, maternal preeclampsia, HELLP syndrome association described rarely | Normal or ↓ free carnitine, ↑ plasma acylglycine, plasma C6-C10 free fatty acids, ↑ C8-C10 acyl-carnitine |
| Very long-chain acyl-CoA dehydrogenase | VLCAD ACADVL | Dilated cardiomyopathy, arrhythmias, hypoglycemia, and hepatic steatosis. Late-onset, stress-induced rhabdomyolysis, episodic myopathy. Prenatal and newborn screening diagnosis possible. | Normal or ↓ free carnitine, ↑ plasma C14:1, C14 acylcarnitine, ↑ plasma C10-C16 free fatty acids |
| ETF dehydrogenase* | ETF-DH | Nonketotic fasting hypoglycemia, congenital anomalies, milder forms of liver disease, cardiomyopathy, and skeletal myopathy. Newborn screening diagnosis reported | Normal or ↓ free carnitine, increased ratio of acyl:free carnitine, ↑ acylcarnitine, urine organic acid and acylglycines |
| ETF-α* | α-ETF | Nonketotic fasting hypoglycemia, congenital anomalies, liver disease, cardiomyopathy, and skeletal myopathy also described. Newborn screening diagnosis reported | Normal or ↓ free carnitine, increased ratio of acyl:free carnitine, ↑ acylcarnitine, urine organic acid and acylglycines |
| ETF-β* | β-ETF | Fasting hypoglycemia, congenital anomalies, liver disease, cardiomyopathy, and skeletal myopathy also described. Newborn screening diagnosis reported | Normal or ↓ free carnitine, increased ratio of acyl:free carnitine, ↑ acylcarnitine, urine organic acid and acylglycines |
| Short-chain L-3-hydroxyacyl-CoA dehydrogenase | SCHAD HADH | Hyperinsulinemic hypoglycemia, cardiomyopathy, myopathy. Newborn screening diagnosis reported | Normal or ↓ free carnitine, elevated free fatty acids, inconsistently abnormal urine organic acid, ↑3-OH glutarate. ↑ plasma C4-OH acylcarnitine |
| Long-chain L-3-hydroxyacyl-CoA dehydrogenase | LCHAD HADH-A | Newborn screening diagnosis reported, maternal preeclampsia, HELLP syndrome, and AFLP association described frequently. See also MTP below for clinical manifestations | Normal or ↓ free carnitine, increased ratio of acyl:free carnitine, ↑ free fatty acids, ↑ C16-OH and C18-OH carnitines |
| MTP | HADH-A, HADH-B | Severe cardiac and skeletal myopathy, hypoglycemia, acidosis, hyper NH3, sudden death, elevated liver enzymes, retinopathy. Maternal preeclampsia, HELLP syndrome, and AFLP association described frequently | Normal or ↓ free carnitine, increased ratio of acyl:free carnitine, ↑ free fatty acids, ↑ C16-OH and C18-OH carnitines |
| Long-chain 3-ketoacyl-CoA thiolase | LKAT HADH-B | Severe neonatal presentation, hypoglycemia, acidosis, ↑ creatine kinase, cardiomyopathy, neuropathy, and early death | Normal or ↓ free carnitine, increased ratio of acyl:free carnitine, ↑ free fatty acids, ↑ 2-trans, 4-cis-decadienoylcarnitine |
| 2,4-Dienoyl-CoA reductase | DECR1 | Only 1 patient described, hypotonia in the newborn, mainly severe skeletal myopathy and respiratory failure. Hypoglycemia rare | Normal or ↓ free carnitine, ↑ acyl:free carnitine ratio, normal urine organic acids and acylglycines |
| HMG CoA synthetase | HMGCS2 | Hypoketosis and hypoglycemia, rarely myopathy | Elevated total plasma fatty acids, enzyme studies in biopsied liver may be diagnostic, genetic testing is preferred |
| HMG CoA lyase | HMGCL | Hypoketosis and hypoglycemia, rarely myopathy | Normal free carnitine, ↑ C5-OH, and methylglutaryl-carnitine, enzymes studies in fibroblasts may be diagnostic |
Clinical manifestations characteristically involve those tissues with a high β-oxidation flux, including liver, skeletal, and cardiac muscle. The most common presentation is an acute episode of life-threatening coma and hypoglycemia induced by a period of fasting because of defective hepatic ketogenesis. Other manifestations may include chronic cardiomyopathy and muscle weakness or exercise-induced acute rhabdomyolysis. The fatty acid oxidation defects can often be asymptomatic during periods when there is no fasting stress. Acutely presenting disease may be misdiagnosed as Reye syndrome or, if fatal, as sudden unexpected infant death. Fatty acid oxidation disorders are easily overlooked because the only specific clue to the diagnosis may be the finding of inappropriately low concentrations of urinary ketones in an infant who has hypoglycemia. Genetic defects in ketone body utilization may also be overlooked because ketosis is an expected finding with fasting hypoglycemia. In some circumstances, clinical manifestations appear to arise from toxic effects of fatty acid metabolites rather than inadequate energy production. These include disorders (long chain 3-hydroxyacyl dehydrogenase [LCHAD], carnitine palmitoyltransferase-1A [CPT-IA], mitochondrial trifunctional protein [MTP; also known as TFP] deficiencies) in which the presence of a homozygous affected fetus increases the risk of a life-threatening illness in the heterozygote mother, resulting in acute fatty liver of pregnancy or preeclampsia with HELLP (hemolysis, elevated liver enzymes, low platelets) syndrome. Malformations of the brain and kidneys have been described in severe electron transfer flavoprotein (ETF), ETF dehydrogenase (ETF-DH), and carnitine palmitoyltransferase-2 (CPT-II) deficiencies that might reflect in utero toxicity of fatty acid metabolites or a developmental role for these enzymes. Progressive retinal degeneration, peripheral neuropathy, and chronic progressive liver disease have been identified in LCHAD and MTP deficiency. Newborn screening programs using tandem mass spectrometry detect characteristic acylcarnitines seen in many of these disorders and permit presymptomatic diagnosis. Screening programs have provided evidence to demonstrate that all the fatty acid oxidation disorders combined are among the most common inborn errors of metabolism.
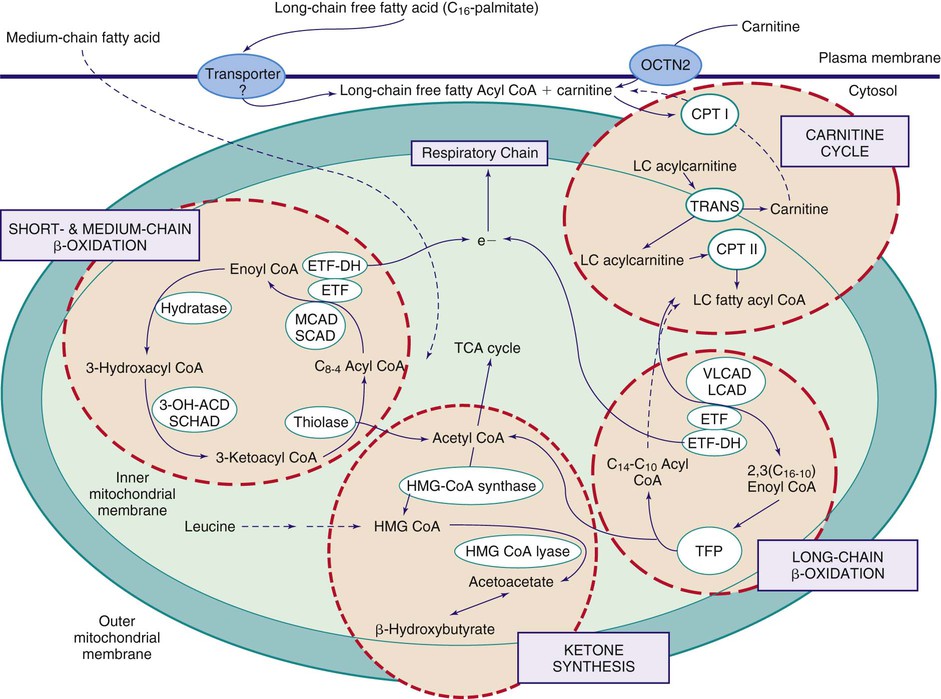
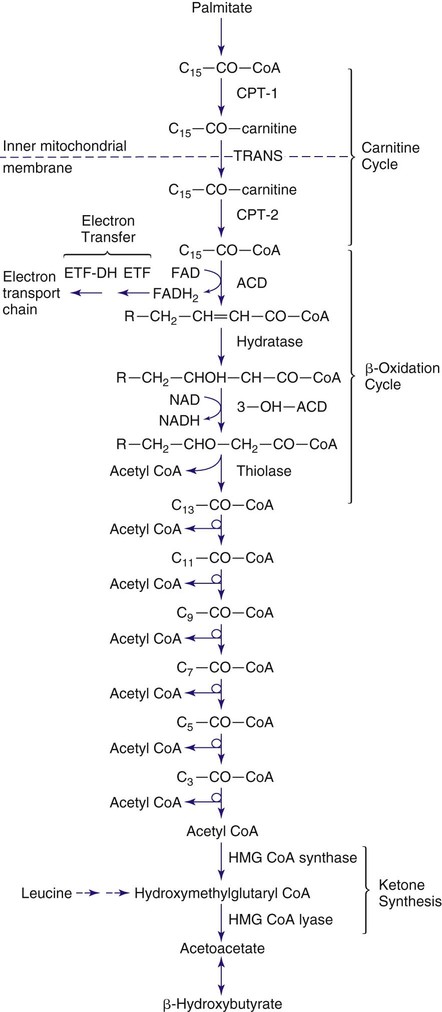
Figures 86-1 and 86-2 outline the steps involved in the oxidation of a typical long-chain fatty acid. In the carnitine cycle, fatty acids are transported across the barrier of the inner mitochondrial membrane as acylcarnitine esters. Within the mitochondria, successive turns of the 4-step β-oxidation cycle convert the coenzyme A (CoA)-activated fatty acid to acetyl-CoA units. Two to 3 different chain-length specific isoenzymes are needed for each of these β-oxidation steps to accommodate the different-sized fatty acyl-CoA species. The electron transfer pathway carries electrons generated in the first β-oxidation step (acyl-CoA dehydrogenase) to the electron transport chain at the level of coenzyme Q for adenosine triphosphate production, while electrons generated from the third step (3-hydroxyacyl-CoA dehydrogenase) enter the electron transport chain at the level of complex 1. Most of the acetyl-CoA generated from hepatic β-oxidation flows through the pathway of ketogenesis to form β-hydroxybutyrate and acetoacetate.
Defects in the β-Oxidation Cycle
Medium-Chain Acyl-Coenzyme A Dehydrogenase Deficiency
Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency is the most common fatty acid oxidation disorder. The disorder shows a strong founder effect; most patients have a northwestern European ancestry, and the majority of patients are homozygous for a single common missense mutation, an A-G transition at complementary DNA position 985 that changes a lysine to glutamic acid at residue 329 (K329E).
Clinical Manifestations
Previously undiagnosed affected patients usually present in the first 3 mo-5 yr of life with episodes of acute illness triggered by prolonged fasting (longer than 12-16 hr). Signs and symptoms include vomiting and lethargy, which rapidly progress to coma or seizures and cardiorespiratory collapse. Sudden unexpected infant death may occur. The liver may be slightly enlarged with fat deposition. Attacks are rare until the infant is beyond the first few months of life, presumably because of more frequent feedings at a younger age. Affected older infants are at higher risk of illness as they begin to fast through the night or are exposed to fasting stress during an intercurrent childhood illness. Presentation in the first days of life with neonatal hypoglycemia has been reported in newborns that were fasted inadvertently. Diagnosis of MCAD has occasionally been documented in previously healthy teenage and adult individuals, indicating that even patients who have been asymptomatic in infancy are still at risk for metabolic decompensation if exposed to sufficient periods of fasting. An unknown number may remain asymptomatic. Prior to routine newborn screening testing, as many as 25% of MCAD deficient cases died or suffered severe brain damage from their first episode. Most patients are now diagnosed in the newborn period by blood spot acylcarnitine screening, allowing the initiation of early treatment and prevention of many of the severe signs and symptoms.
Laboratory Findings
During acute episodes, hypoglycemia is usually present. Plasma and urinary ketone concentrations are inappropriately low (hypoketotic hypoglycemia). Because of the hypoketonemia, there is little or no metabolic acidosis, which is expected to be present in many children with hypoglycemia. Tests of liver function are abnormal, with elevations of liver enzymes (alanine aminotransferase, aspartate aminotransferase), elevated blood ammonia, and prolonged prothrombin and partial thromboplastin times. Liver biopsy at times of acute illness shows microvesicular or macrovesicular steatosis from triglyceride accumulation. During fasting stress or at times of acute illness, urinary organic acid profiles by gas chromatography/mass spectrometry show inappropriately low concentrations of ketones and elevated levels of medium-chain dicarboxylic acids (adipic, suberic, and sebacic acids) that derive from microsomal and peroxisomal omega oxidation of accumulated medium-chain fatty acids. Plasma and tissue concentrations of total carnitine are reduced to 25-50% of normal, and the fraction of total esterified carnitine is increased. This pattern of secondary carnitine deficiency is seen in most fatty acid oxidation defects and reflects competition between increased acylcarnitine levels and free carnitine for transport at the plasma membrane. Significant exceptions to this rule are the plasma membrane carnitine transporter, CPT-IA and β-hydroxy-β-methylglutaryl-CoA (HMG-CoA) synthase deficiencies that do not manifest secondary carnitine deficiency.
Diagnostic metabolite patterns include increased plasma C8:0, C10:0, and C10:1 acylcarnitine species and increased urinary acylglycines including hexanoyl-propionyl, suberyl-propionyl, and 3-phenylpropionyl glycines. Newborn screening programs using tandem mass spectrometry, which almost all babies born in the United States receive, can diagnose presymptomatic MCAD deficiency based on the detection of the abnormal acylcarnitines in filter paper blood spots. In many cases, the diagnosis can be confirmed by finding the common A985G mutation. A second common variant, T199C, has been detected in infants with characteristic acylcarnitines in newborn screening tests. Interestingly, this allele has not been seen to date in symptomatic MCAD patients; it may represent a milder mutation.
Treatment
Acute illnesses should be promptly treated with intravenous fluids containing 10% dextrose to treat or prevent hypoglycemia and to suppress lipolysis as rapidly as possible (see Chapter 92). Chronic therapy consists of avoiding fasting. This usually requires simply adjusting the diet to ensure that overnight fasting periods are limited to <10-12 hr. Restricting dietary fat or treatment with carnitine is controversial. The necessity for active therapeutic intervention for individuals with the T199C variant has not yet been established.
Prognosis
Up to 25% of unrecognized patients may die during their first attack of illness. There is frequently a history of a previous sibling death that is presumed to be from an unrecognized MCAD deficiency. Some patients may suffer permanent brain injury during an attack of profound hypoglycemia. The prognosis for survivors without brain damage is excellent because progressive cognitive impairment or cardiomyopathy does not occur in MCAD deficiency. Muscle pain and reduced exercise tolerance may become evident with increasing age. Fasting tolerance improves with age and the risk of illness decreases. Because as many as 35% of affected patients have never had an episode, testing of siblings of affected patients is important to detect asymptomatic family members.
Very-Long-Chain Acyl-Coenzyme A Dehydrogenase Deficiency
Very-long-chain acyl-CoA dehydrogenase (VLCAD) deficiency is the second most commonly diagnosed disorder of fatty acid oxidation. It was originally termed long-chain acyl-CoA dehydrogenase deficiency before the existence of the inner mitochondrial membrane-bound VLCAD was known. All patients previously diagnosed as having long-chain acyl-CoA dehydrogenase deficiency have VLCAD gene defects. Patients with VLCAD deficiency have no ability to oxidize physiologic long-chain fatty acids and are usually more severely affected than those with MCAD deficiency who have a milder oxidative defect. VLCAD deficiency presents earlier in infancy and has more chronic problems with muscle weakness or episodes of muscle pain and rhabdomyolysis. Cardiomyopathy may be present during acute attacks provoked by fasting. The left ventricle may be hypertrophic or dilated and show poor contractility on echocardiography. Sudden unexpected death has occurred in several patients, but most who survived the initial episode showed improvement, including normalization of cardiac function. Other physical and routine laboratory features are similar to those of MCAD deficiency, including secondary carnitine deficiency. The urinary organic acid profile shows a nonketotic dicarboxylic aciduria with increased levels of C6-12 dicarboxylic acids. Diagnosis may be suggested by an abnormal acylcarnitine profile with plasma or blood spot C14:0, 14:1, 14:2 acylcarnitine species; however, the specific diagnosis requires mutational analysis of the VLCAD gene. Treatment is based primarily on avoidance of fasts for longer than 10-12 hr. Continuous intragastric feeding is useful in some patients.
Short-Chain Acyl-Coenzyme A Dehydrogenase Deficiency
A small number of patients with 2 clear null mutations in the short-chain acyl-CoA dehydrogenase (SCAD) gene have been described with variable phenotype. Most individuals classified as being SCAD deficient have polymorphic DNA changes in the SCAD gene; for example, 2 common polymorphisms are G185S and R147W, which are homozygously present in 7% of the population. Some investigators argue that these may be susceptibility changes, which require a second, as yet unknown, genetic mutation to express a clinical phenotype; while others believe that SCAD deficiency is a harmless biochemical condition. This autosomal recessive disorder presents with neonatal hypoglycemia and may have normal levels of ketone bodies. The diagnosis is indicated by elevated levels of butyrylcarnitine (C4-carnitine) on newborn blood spots or plasma and increased excretion of urinary ethylmalonic acid and butyrylglycine. These metabolic abnormalities are most pronounced in patients with null mutations and variably present in patients who are homozygous for the common polymorphisms.
The necessity for treatment in SCAD deficiency has not yet been established. It has been proposed that long-term evaluation of asymptomatic individuals is necessary to determine whether this is or is not a real disease. Although most individuals with SCAD deficiency remain asymptomatic throughout life, it has been proposed that there is a subset of individuals with SCAD deficiency with severe manifestations, including dysmorphic facial features, feeding difficulties/failure to thrive, metabolic acidosis, ketotic hypoglycemia, lethargy, developmental delay, seizures, hypotonia, dystonia, and myopathy.
Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase/Mitochondrial Trifunctional Protein Deficiency
The LCHAD enzyme is part of a MTP, which also contains 2 other steps in β-oxidation: long-chain enoyl CoA hydratase and long-chain β-ketothiolase. It is a heterooctameric protein composed of 4 α and 4 β chains that derive from distinct contiguous genes with a common promoter region. In some patients, only the LCHAD activity of the MTP is affected (LCHAD deficiency), whereas others have deficiencies of all 3 activities (MTP deficiency).
Clinical manifestations include attacks of acute hypoketotic hypoglycemia similar to MCAD deficiency; patients often show evidence of more severe disease, including cardiomyopathy, muscle cramps and weakness, and abnormal liver function (cholestasis). Toxic effects of fatty acid metabolites may produce pigmented retinopathy leading to blindness, progressive liver failure, peripheral neuropathy, and rhabdomyolysis. Life-threatening obstetric complications, acute fatty liver of pregnancy, and HELLP syndrome are observed in heterozygous mothers carrying homozygotic fetuses affected with LCHAD/MTP deficiency. Sudden unexpected infant death may occur. The diagnosis is indicated by elevated levels of blood spot or plasma 3-hydroxy acylcarnitines of chain lengths C16-C18. Urinary organic acid profile in patients may show increases in levels of 3-hydroxydicarboxylic acids of chain lengths C6-C14. Secondary carnitine deficiency is common. A common mutation in the α subunit, E474Q, is seen in more than 60% of LCHAD-deficient patients. This mutation in the fetus is especially associated with the obstetric complications, but other mutations in either subunit may also be linked to maternal illness.
Treatment is similar to that for MCAD or VLCAD deficiency; that is, avoiding fasting stress. Some investigators have suggested that dietary supplements with medium-chain triglyceride oil to bypass the defect in long-chain fatty acid oxidation and docosahexaenoic acid (for protection against the retinal changes) may be useful. Liver transplantation has been attempted in cases with severe liver failure, but does not ameliorate the metabolic abnormalities or prevent the myopathic or retinal complications.
Short-Chain 3-Hydroxyacyl-Coenzyme A Dehydrogenase Deficiency
Only 12 patients with proven mutations of short-chain 3-hydroxyacyl-CoA dehydrogenase (SCHAD) have been reported, although a few additional unpublished cases are known to the authors. Most cases with recessive mutations of the SCHAD gene have presented with episodes of hypoketotic hypoglycemia that was caused by hyperinsulinism. In contrast to patients with other forms of fatty acid oxidation disorders, these cases required specific therapy with diazoxide for hyperinsulinism to avoid recurrent hypoglycemia. A single case with compound heterozygous mutations presented with fulminant hepatic failure at age 10 mo. The SCHAD protein has a nonenzymatic function (moonlighting) in which it directly interacts with glutamate dehydrogenase (GDH) to inhibit its activity. In the absence of an SCHAD protein, this inhibition is removed leading to upregulation of GDH enzyme activity, a recognized cause of hyperinsulinism usually caused by activating mutations of the GDH gene. This severe deficiency of SCHAD protein often presents predominantly as protein sensitive hypoglycemia rather than as fasting hypoglycemia. It appears that if a SCHAD protein is present the inhibition of GDH is maintained even when there is no SCHAD enzyme activity; these patients may present with a more traditional fatty acid oxidation defect. Specific metabolic markers for SCHAD deficiency include elevated plasma C4-hydroxy acylcarnitine and urine 3-hydroxyglutaric acid.
Treatment of SCHAD deficient patients with hyperinsulinism is with diazoxide. There is insufficient experience with the non-hyperinsulinemic form of SCHAD deficiency at present to recommend treatment modalities, but prevention of fasting seems advisable, which is similar to other fatty acid oxidation disorders.
Defects in the Carnitine Cycle
Plasma Membrane Carnitine Transport Defect (Primary Carnitine Deficiency)
Primary carnitine deficiency is the only genetic defect in which carnitine deficiency is the cause, rather than the consequence, of impaired fatty acid oxidation. The most common presentation is progressive cardiomyopathy with or without skeletal muscle weakness beginning at 1-4 yr of age. A smaller number of patients may present with fasting hypoketotic hypoglycemia in the 1st yr of life before the cardiomyopathy becomes symptomatic. The underlying defect involves the plasma membrane sodium gradient-dependent carnitine transporter that is present in heart, muscle, and kidney. This transporter is responsible both for maintaining intracellular carnitine concentrations 20-50–fold higher than plasma concentrations and for renal conservation of carnitine.
Diagnosis of the carnitine transporter defect is aided by the fact that patients have extremely reduced carnitine levels in plasma and muscle (1-2% of normal). Heterozygote parents have plasma carnitine levels approximately 50% of normal. Fasting ketogenesis may be normal because liver carnitine transport is normal, but it may become impaired if dietary carnitine intake is interrupted. The fasting urinary organic acid profile may show a hypoketotic dicarboxylic aciduria pattern if hepatic fatty acid oxidation is impaired, but it is otherwise unremarkable. The defect in carnitine transport can be demonstrated clinically by the severe reduction in renal carnitine threshold or by in vitro assay of carnitine uptake using cultured fibroblasts or lymphoblasts. Mutations in the organic cation/carnitine transporter (OCTN2) underlie this disorder. Treatment with pharmacologic doses of oral carnitine (100-200 mg/kg/day) is highly effective in correcting the cardiomyopathy and muscle weakness, as well as any impairment in fasting ketogenesis. Muscle total carnitine concentrations remain <5% of normal on treatment.
Carnitine Palmitoyltransferase-IA Deficiency
Several dozen infants and children have been described with a deficiency of the liver and kidney carnitine palmitoyltransferase-I (CPT-I) isozyme (CPT-IA). Clinical manifestations include fasting hypoketotic hypoglycemia, occasionally with markedly abnormal liver function tests and, rarely, with renal tubular acidosis. The heart and skeletal muscle are not involved because the muscle isozyme is unaffected. Fasting urinary organic acid profile sometimes shows a hypoketotic C6-C12 dicarboxylic aciduria but may be normal. Plasma acylcarnitine analysis demonstrates mostly free carnitine with very little acylated carnitine. This observation has been used to establish CPT-IA diagnosis on newborn screening by tandem mass spectrometry. CPT-IA deficiency is the only fatty acid oxidation disorder in which plasma total carnitine levels are elevated often to 150-200% of normal. This may be explained by the fact that the inhibitory effects of long-chain acylcarnitines on the renal tubular carnitine transporter are absent in CPT-IA deficiency. The enzyme defect can be demonstrated in cultured fibroblasts or lymphoblasts. CPT-IA deficiency in the fetus has been associated with acute fatty liver of pregnancy in the mother in a single case report. A common variant in the CPT-IA gene has been identified in individuals of Inuit background in the United States and First Nations tribes in Canada and Greenland. The variant is detected by a positive newborn acylcarnitine screen; enzyme activity is reduced by 80% and regulation by malonyl-CoA is lost. It has not been established if this is a pathologic DNA variant or an adaptation to ancient Inuit and First Nations high-fat diets. This variant is associated with an increased risk for sudden infant death syndrome. Treatment for the severe form of CPT-IA deficiency is similar to that for MCAD deficiency with avoidance of situations where fasting ketogenesis is necessary.
Carnitine:Acylcarnitine Translocase Deficiency
This defect of the inner mitochondrial membrane carrier protein for fatty acylcarnitines blocks the entry of long-chain fatty acids into the mitochondria for oxidation. The clinical phenotype of this disorder is characterized by a severe and generalized impairment of fatty acid oxidation. Most newborn patients present with attacks of fasting-induced hypoglycemia, hyperammonemia, and cardiorespiratory collapse. All symptomatic newborns have had evidence of cardiomyopathy and muscle weakness. Several patients with a partial translocase deficiency and milder disease without cardiac involvement have also been identified. No distinctive urinary or plasma organic acids are noted, although increased levels of plasma long-chain acylcarnitines of chain lengths C16-C18 are reported. Diagnosis can be confirmed using genetic analysis. Functional carnitine:acylcarnitine translocase activity can be measured in cultured fibroblasts or lymphoblasts. Treatment is similar to that of other long-chain fatty acid oxidation disorders.
Carnitine Palmitoyltransferase-II Deficiency
Three forms of CPT-II deficiency have been described. The severe neonatal lethal presentation of this disorder is associated with a profound enzyme deficiency, and early death has been reported in several newborns with dysplastic kidneys, cerebral malformations, and mild facial anomalies. A milder, second defect, is associated with an adult presentation of episodic rhabdomyolysis. The first episode usually does not occur until late childhood or early adulthood. Attacks may be precipitated by prolonged exercise. There is aching muscle pain and myoglobinuria that may be severe enough to cause renal failure. Serum levels of creatine kinase are elevated to 5,000-100,000 units/L. Fasting hypoglycemia has not been described, but fasting may contribute to attacks of myoglobinuria. Muscle biopsy shows increased deposition of neutral fat. The myopathic presentation of CPT-II deficiency is associated with a common mutation S113L. This mutation produces a heat-labile protein that is unstable to increased muscle temperature during exercise resulting in the myopathic presentation. The third intermediate form of CPT-II deficiency presents in infancy/early childhood with fasting-induced hepatic failure, cardiomyopathy, and skeletal myopathy with hypoketotic hypoglycemia, but does not have the severe developmental changes seen in the neonatal lethal presentation. This pattern is similar to that seen in VLCAD deficiency and management is identical.
Diagnosis of all forms of CPT-II deficiency can be made by a combination of molecular analysis and demonstrating deficient enzyme activity in muscle or other tissues and in cultured fibroblasts.
Defects in the Electron Transfer Pathway
Electron Transfer Flavoprotein and Electron Transfer Flavoprotein Dehydrogenase Deficiencies (Glutaric Acidemia Type 2, Multiple Acyl-Coenzyme A Dehydrogenation Defects)
ETF and ETF-DH function to transfer electrons into the mitochondrial electron transport chain from dehydrogenation reactions catalyzed by VLCAD, MCAD, and SCAD, as well as by glutaryl-CoA dehydrogenase and 4 enzymes involved in branched-chain amino acid oxidation. Deficiencies of ETF or ETF-DH produce illness that combines the features of impaired fatty acid oxidation and impaired oxidation of several amino acids. Complete deficiencies of either protein are associated with severe illness in the newborn period, characterized by acidosis, hypoketotic hypoglycemia, coma, hypotonia, cardiomyopathy, and an unusual odor of sweaty feet caused by isovaleryl-CoA dehydrogenase inhibition. Some affected neonates have had facial dysmorphism and polycystic kidneys similar to that seen in severe CPT-II deficiency, which suggests that toxic effects of accumulated metabolites may occur in utero.
Diagnosis can be made from the urinary organic acid profile, which shows abnormalities corresponding to blocks in oxidation of fatty acids (ethylmalonate and C6-C10 dicarboxylic acids), lysine (glutarate), and branched-chain amino acids (isovaleryl-, isobutyryl-, and α-methylbutyryl-glycine) and by molecular testing. Most severely affected infants do not survive the neonatal period.
Partial deficiencies of ETF and ETF-DH produce a disorder that may mimic MCAD deficiency or other milder fatty acid oxidation defects. These patients have attacks of fasting hypoketotic coma. The urinary organic acid profile reveals primarily elevations of dicarboxylic acids and ethylmalonate, derived from short-chain fatty acid intermediates. Secondary carnitine deficiency is present. Some patients with mild forms of ETF/ETF-DH deficiency benefit from treatment with high doses of riboflavin, which is a cofactor for the pathway of electron transfer.
Defects in Ketone Synthesis Pathway
β-Hydroxy-β-Methylglutaryl-Coenzyme A Synthase Deficiency
See Chapter 85.6.
HMG-CoA synthase is the rate-limiting step in the conversion of acetyl-CoA derived from fatty acid β-oxidation in the liver to ketones. Several patients with this defect have recently been identified. The presentation is one of fasting hypoketotic hypoglycemia without evidence of impaired cardiac or skeletal muscle function. Urinary organic acid profile showed only a hypoketotic dicarboxylic aciduria. Plasma and tissue carnitine levels are normal, in contrast to all the other disorders of fatty acid oxidation. A separate synthase enzyme, present in cytosol for cholesterol biosynthesis, is not affected. The HMG-CoA synthase defect is expressed only in the liver and cannot be demonstrated in cultured fibroblasts. The gene has been cloned, and mutations in the affected patients have been characterized. Avoiding fasting is usually a successful treatment.
β-Hydroxy-β-Methylglutaryl-Coenzyme A Lyase Deficiency
See Chapter 85.6.
Defects in Ketone Body Utilization
The ketone bodies, β-hydroxybutyrate and acetoacetate, are the end products of hepatic fatty acid oxidation and are important metabolic fuels for the brain during fasting. Two defects in utilization of ketones in brain and other peripheral tissues present as episodes of hyperketotic coma, with or without hypoglycemia.
Succinyl-Coenzyme A:3-Ketoacid-Coenzyme A Transferase Deficiency
See Chapter 85.6.
Several patients with succinyl-CoA:3-ketoacid-CoA transferase (SCOT) deficiency have been reported. The characteristic presentation is an infant with recurrent episodes of severe ketoacidosis induced by fasting. Plasma acylcarnitine and urine organic acid abnormalities do not distinguish SCOT deficiency from other causes of ketoacidosis. Treatment of episodes requires infusion of glucose and large amounts of bicarbonate until metabolically stable. Patients usually exhibit inappropriate hyperketonemia even between episodes of illness. SCOT is responsible for activating acetoacetate in peripheral tissues using succinyl CoA as a donor to form acetoacetyl-CoA. Deficient enzyme activity can be demonstrated in brain, muscle, and fibroblasts from affected patients. The gene has been cloned, and numerous mutations have been characterized.
β-Ketothiolase Deficiency
See Chapter 85.6.
Disorders of Very Long Chain Fatty Acids
Gerald V. Raymond
Peroxisomal Disorders
The peroxisomal diseases are genetically determined disorders caused either by the failure to form or maintain the peroxisome or by a defect in the function of a single protein that is normally located in this organelle. These disorders cause serious disability in childhood and occur more frequently and present a wider range of phenotype than has been recognized in the past.
Etiology
Peroxisomal disorders are subdivided into 2 major categories (Table 86-2).
Table 86-2
Classification of Peroxisomal Disorders
A: DISORDERS OF PEROXISOME IMPORT
A1: Zellweger syndrome
A2: Neonatal adrenoleukodystrophy
A3: Infantile Refsum disease
A4: Rhizomelic chondrodysplasia punctata
B: DEFECTS OF SINGLE PEROXISOMAL ENZYME
B1: X-linked adrenoleukodystrophy
B2: Acyl-CoA oxidase deficiency
B3: Bifunctional enzyme deficiency
B4: Peroxisomal thiolase deficiency
B5: Classic Refsum disease
B6: 2-Methylacyl-CoA racemase deficiency
B7: DHAP acyltransferase deficiency
B8: Alkyl-DHAP synthase deficiency
B9: Mevalonic aciduria
B10: Glutaric aciduria type III
B11: Hyperoxaluria type I
B12: Acatalasemia
CoA, coenzyme A; DHAP, dihydroxyacetone phosphate.
In category A, the peroxisomal biogenesis disorders (PBDs), the basic defect is the failure to import 1 or more proteins into the organelle. In category B, defects affect a single peroxisomal protein. The peroxisome is present in all cells except mature erythrocytes and is a subcellular organelle surrounded by a single membrane; more than 50 peroxisomal enzymes are identified. Some enzymes are involved in the production and decomposition of hydrogen peroxide; others are concerned with lipid and amino acid metabolism. Most peroxisomal enzymes are first synthesized in their mature form on free polyribosomes and enter the cytoplasm. Proteins that are destined for the peroxisome contain specific peroxisome targeting sequences (PTSs). Most peroxisomal matrix proteins contain PTS1, a 3-amino acid sequence at the carboxyl terminus. PTS2 is an aminoterminal sequence that is critical for the import of enzymes involved in plasmalogen and branched-chain fatty acid metabolism. Import of proteins involves a complex series of reactions that involves at least 23 distinct proteins. These proteins, referred to as peroxins, are encoded by PEX genes.
Epidemiology
Except for X-linked adrenoleukodystrophy (ALD), all the peroxisomal disorders in Table 86-2 are autosomal recessive traits. ALD is the most common peroxisomal disorder, with an estimated incidence of 1 in 17,000 live births. The combined incidence of the other peroxisomal disorders is estimated to be 1 in 50,000 live births.
Pathology
Absence or reduction in the number of peroxisomes is pathognomonic for disorders of peroxisome biogenesis. In most disorders, there are membranous sacs that contain peroxisomal integral membrane proteins, which lack the normal complement of matrix proteins; these are peroxisome “ghosts.” Pathologic changes are observed in most organs and include profound and characteristic defects in neuronal migration; micronodular cirrhosis of the liver; renal cysts; chondrodysplasia punctata; sensorineural hearing loss; retinopathy; congenital heart disease; and dysmorphic features.
Pathogenesis
It is likely that all pathologic changes are secondary to the peroxisome defect. Multiple peroxisomal enzymes fail to function in the PBDs (Table 86-3). The enzymes that are diminished or absent are synthesized but are degraded abnormally fast because they may be unprotected outside of the peroxisome. It is not clear how defective peroxisome functions lead to the widespread pathologic manifestations.
Table 86-3
Abnormal Laboratory Findings Common to Disorders of Peroxisome Biogenesis
Peroxisomes absent to reduced in number
Catalase in cytosol
Deficient synthesis and reduced tissue levels of plasmalogens
Defective oxidation and abnormal accumulation of very-long-chain fatty acids
Deficient oxidation and age-dependent accumulation of phytanic acid
Defects in certain steps of bile acid formation and accumulation of bile acid intermediates
Defects in oxidation and accumulation of L-pipecolic acid
Increased urinary excretion of dicarboxylic acids
Mutations in 12 different PEX genes have been identified in PBDs. The pattern and severity of pathologic features vary with the nature of the import defects and the degree to which import is impaired. These gene defects lead to disorders that were named before their relationship to the peroxisome was recognized, namely, Zellweger syndrome, neonatal ALD, infantile Refsum disease, and rhizomelic chondrodysplasia punctata (RCDP). The first 3 disorders are considered to form a clinical continuum, with Zellweger syndrome the most severe, infantile Refsum disease the least severe, and neonatal ALD intermediate. They can be caused by mutations in any of the 11 genes involved in peroxisome assembly. The specific gene defects cannot be distinguished on the basis of clinical features. The clinical severity varies with the degree to which protein import is impaired. Mutations that abolish import completely are often associated with the Zellweger syndrome phenotype, whereas a missense mutation, in which some degree of import function is retained, leads to the somewhat milder phenotypes. A defect in PEX7, which involves the import of proteins that utilize PTS2, is associated with RCDP. PEX7 defects that leave import partially intact are associated with milder phenotypes, some of which resemble classic Refsum disease.
The genetic disorders that involve single peroxisomal enzymes usually have clinical manifestations that are more restricted and relate to the single biochemical defect. The primary adrenal insufficiency of ALD is caused by accumulation of very-long-chain fatty acids (VLCFAs) in the adrenal cortex, and the peripheral neuropathy in Refsum disease is caused by the accumulation of phytanic acid in Schwann cells and myelin.
Peroxisomal Biogenesis Disorders with Milder or Atypical Phenotypes
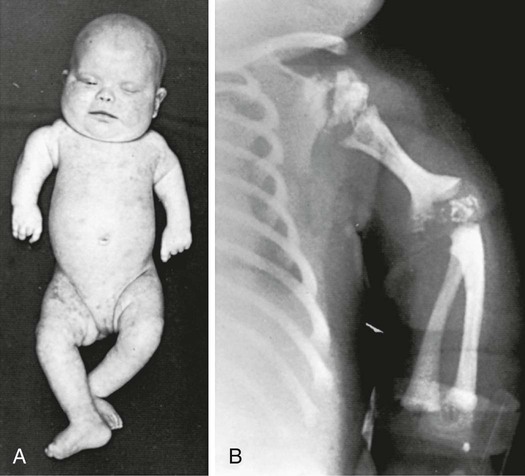
Newborn infants with Zellweger syndrome show striking and consistent recognizable abnormalities. Of central diagnostic importance are the typical facial appearance (high forehead, unslanting palpebral fissures, hypoplastic supraorbital ridges, and epicanthal folds; Fig. 86-3), severe weakness and hypotonia, neonatal seizures, and eye abnormalities. Because of the hypotonia and craniofacial appearance, Down syndrome may be suspected. Infants with Zellweger syndrome rarely live more than a few months. More than 90% show postnatal growth failure. Table 86-4 lists the main clinical abnormalities.
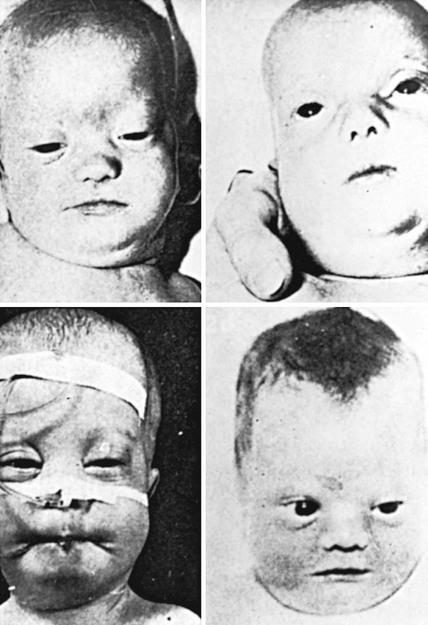
Table 86-4
Main Clinical Abnormalities in Zellweger Syndrome
| ABNORMAL FEATURE | Cases in Which the Feature Was Present | |
| NO. | % | |
| High forehead | 58 | 97 |
| Flat occiput | 13 | 81 |
| Large fontanelle(s), wide sutures | 55 | 96 |
| Shallow orbital ridges | 33 | 100 |
| Low/broad nasal bridge | 23 | 100 |
| Epicanthus | 33 | 92 |
| High arched palate | 35 | 95 |
| External ear deformity | 39 | 97 |
| Micrognathia | 18 | 100 |
| Redundant skin fold of neck | 13 | 100 |
| Brushfield spots | 5 | 83 |
| Cataract/cloudy cornea | 30 | 86 |
| Glaucoma | 7 | 58 |
| Abnormal retinal pigmentation | 6 | 40 |
| Optic disc pallor | 17 | 74 |
| Severe hypotonia | 94 | 99 |
| Abnormal Moro response | 26 | 100 |
| Hyporeflexia or areflexia | 56 | 98 |
| Poor sucking | 74 | 96 |
| Gavage feeding | 26 | 100 |
| Epileptic seizures | 56 | 92 |
| Psychomotor retardation | 45 | 100 |
| Impaired hearing | 9 | 40 |
| Nystagmus | 30 | 81 |
Patients with neonatal ALD show fewer, less-prominent craniofacial features. Neonatal seizures occur frequently. Some degree of psychomotor developmental delay is present; function remains in the severely or profoundly retarded range, and development may regress after 3-5 yr of age, probably from a progressive leukodystrophy. Hepatomegaly, impaired liver function, pigmentary degeneration of the retina, and severely impaired hearing are invariably present. Adrenocortical function is usually impaired and may require adrenal hormone replacement. Chondrodysplasia punctata and renal cysts are absent.
Patients with infantile Refsum disease have survived to adulthood. They are able to walk, although gait may be ataxic and broad based. Cognitive function is in the severely impaired range. All have sensorineural hearing loss and pigmentary degeneration of the retina. They have moderately dysmorphic features that may include epicanthal folds, a flat bridge of the nose, and low-set ears. Early hypotonia and hepatomegaly with impaired function are common. Levels of plasma cholesterol and high- and low-density lipoprotein are often moderately reduced. Chondrodysplasia punctata and renal cortical cysts are absent. Postmortem study in infantile Refsum disease reveals micronodular liver cirrhosis and small hypoplastic adrenals. The brain shows no malformations, except for severe hypoplasia of the cerebellar granule layer and ectopic locations of the Purkinje cells in the molecular layer. The mode of inheritance is autosomal recessive.
Some patients with PBDs have milder and atypical phenotypes. They may present with peripheral neuropathy or with retinopathy, impaired vision, or cataracts in childhood, adolescence, or adulthood and have been diagnosed to have Charcot-Marie-Tooth disease or Usher syndrome. Some patients have survived to the 5th decade. Defects in PEX7, which most commonly lead to the RCDP phenotype, may also lead to a milder phenotype with clinical manifestations similar to those of classical Refsum disease (phytanoyl-CoA hydroxylase deficiency).
Rhizomelic Chondrodysplasia Punctata
RCDP is characterized by the presence of stippled foci of calcification within the hyaline cartilage and is associated with dwarfing, cataracts (72%), and multiple malformations caused by contractures. Vertebral bodies have a coronal cleft filled by cartilage that is a result of an embryonic arrest. Disproportionate short stature affects the proximal parts of the extremities (Fig. 86-4A). Radiologic abnormalities consist of shortening of the proximal limb bones, metaphyseal cupping, and disturbed ossification (Fig. 86-4B). Height, weight, and head circumference are less than the 3rd percentile, and these children have a severe intellectual disability. Skin changes such as those observed in ichthyosiform erythroderma are present in approximately 25% of patients.
Isolated Defects of Peroxisomal Fatty Acid Oxidation
The disorders labeled B1 through B3 (see Table 86-2) each involve 1 of 3 enzymes involved in peroxisomal fatty acid oxidation. Their clinical manifestations resemble those of the Zellweger spectrum disorder continuum; they can be distinguished from disorders of peroxisome biogenesis only by laboratory tests. Defects of bifunctional enzyme are common and are found in approximately 15% of patients with the Zellweger spectrum disorder. Patients with isolated acyl-CoA oxidase deficiency have a somewhat milder phenotype that resembles and come to attention because of the development of a childhood leukodystrophy.
Isolated Defects of Plasmalogen Synthesis
Plasmalogens are lipids in which the first carbon of glycerol is linked to an alcohol rather than a fatty acid. They are synthesized through a complex series of reactions, the first 2 steps of which are catalyzed by the peroxisomal enzymes dihydroxyacetone phosphate alkyl transferase and synthase. Deficiency of either of these enzymes (B4 and B5 in Table 86-2) leads to a phenotype that is clinically indistinguishable from the peroxisomal import disorder RCDP. This latter disorder is caused by a defect in PEX7, the receptor for PTS2. It shares the severe deficiency of plasmalogens with disorders B4 and B5 but, in addition, has defects of phytanic oxidation. The fact that disorders B4 and B5 are associated with the full phenotype of RCDP suggests that a deficiency of plasmalogens is sufficient to produce it.
Classic Refsum Disease
The defective enzyme (phytanoyl-CoA oxidase) is localized to the peroxisome. The manifestation of classic adult Refsum disease includes impaired vision from retinitis pigmentosa, ichthyosis, peripheral neuropathy, ataxia, and, occasionally, cardiac arrhythmias. In contrast to infantile Refsum disease, cognitive function is normal and there are no congenital malformations. Classic Refsum disease often does not manifest until young adulthood, but visual disturbances such as night blindness, ichthyosis, and peripheral neuropathy may already be present in childhood and adolescence. Early diagnosis is important because institution of a phytanic acid-restricted diet can reverse the peripheral neuropathy and prevent the progression of the visual and central nervous system manifestations. The classic Refsum disease phenotype may also be caused by defects in PEX7.
2-Methylacyl-Coenzyme A Racemase Deficiency
This disorder is caused by an enzyme defect that leads to the accumulation of the branched-chain fatty acids (phytanic and pristanic acid) and bile acids. Patients present with adult-type peripheral neuropathy and may also have pigmentary degeneration of the retina.
Laboratory Findings
Laboratory tests for peroxisomal disorders can be viewed at 3 levels of complexity.
Level 1: Does the Patient Have a Peroxisomal Disorder?
This can be resolved by noninvasive tests that are generally available (see Table 86-4). Measurement of plasma VLCFA is the most commonly used assay. Whereas plasma VLCFA levels are elevated in many patients with peroxisomal disorders, this is not always the case. The most important exception is RCDP, in which VLCFA levels are normal, but plasma phytanic acid levels are increased and red blood cell plasmalogen levels are reduced. In some other peroxisomal disorders, the biochemical abnormalities are still more restricted. Therefore, a panel of tests is recommended and includes plasma levels of VLCFA and phytanic, pristanic, and pipecolic acids and red blood cell levels of plasmalogens. Tandem mass spectrometry techniques also permit convenient quantitation of bile acids in plasma and urine. This panel of tests can be performed on 2 mL samples of venous blood and permits detection of most peroxisomal disorders. Furthermore, normal results make the presence of the typical peroxisomal disorder unlikely.
Level 2: What Is the Precise Nature of the Peroxisomal Disorder?
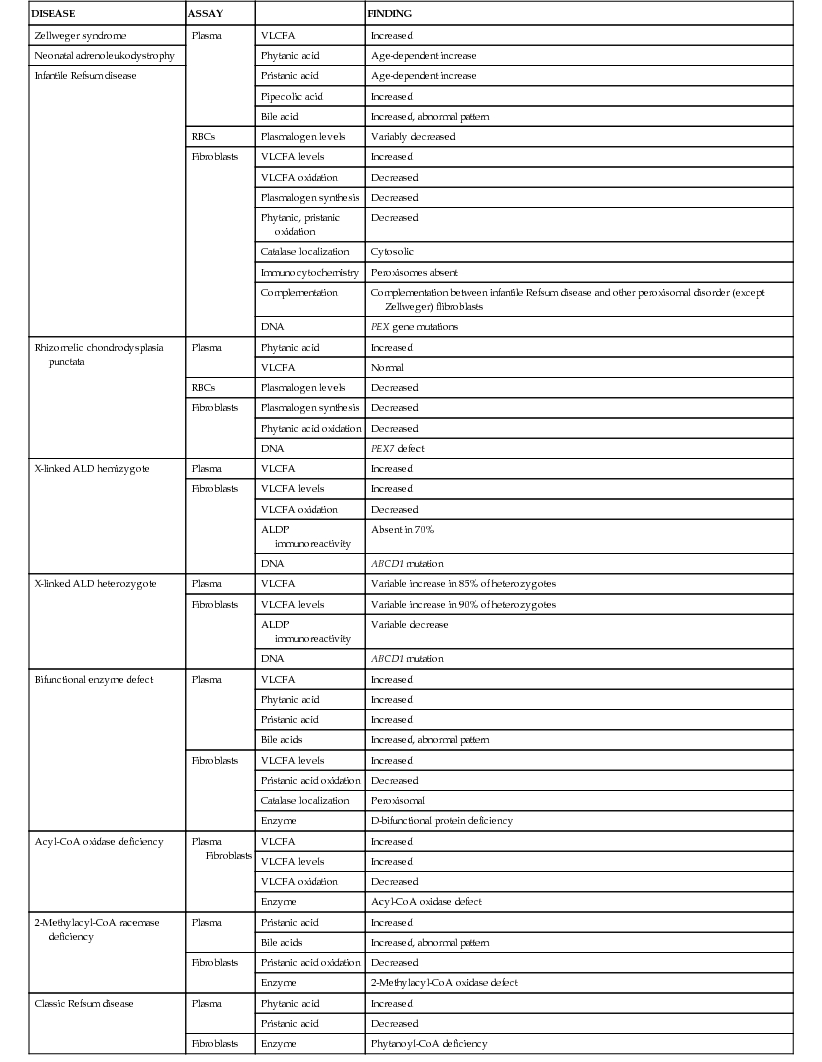
Table 86-5 lists the main biochemical abnormalities in the various peroxisomal disorders. When combined with the clinical presentation, the panel of level 1 tests (see above) is often sufficient to identify the precise nature of the defect. Marked reduction of erythrocyte plasmalogen levels combined with elevated plasma phytanic acid permits precise diagnosis in a patient with the clinical features of RCDP. Classic Refsum disease can be diagnosed by demonstration of increased plasma phytanic acid combined with normal or reduced levels of pristanic acid levels, while in D-bifunctional enzyme deficiency and 2-methylacyl-CoA racemase deficiency, the levels of pristanic and phytanic acid are both increased. Precise identification of some peroxisomal disorders may require more extensive studies in cultured skin fibroblasts. This may be required for the differentiation of PBDs from defects in bifunctional enzyme. In PBDs, the patient's peroxisomes are absent and catalase is in the soluble fraction, whereas in bifunctional enzyme defect, peroxisomes are present and catalase is in the particulate fraction. Fibroblast studies are required to identify the nature of the molecular defect in PBDs. Whether such specialized studies are clinically warranted depends on individual circumstances. Precise definition of the defect in a proband may improve the precision of prenatal diagnosis in at-risk pregnancies, and it is required for carrier detection. It is also of value in setting prognosis.
Table 86-5
Peroxisomal Disorders That Involve Fatty Acid Oxidation: Diagnostic Assays
| DISEASE | ASSAY | FINDING | |
| Zellweger syndrome | Plasma | VLCFA | Increased |
| Neonatal adrenoleukodystrophy | Phytanic acid | Age-dependent increase | |
| Infantile Refsum disease | Pristanic acid | Age-dependent increase | |
| Pipecolic acid | Increased | ||
| Bile acid | Increased, abnormal pattern | ||
| RBCs | Plasmalogen levels | Variably decreased | |
| Fibroblasts | VLCFA levels | Increased | |
| VLCFA oxidation | Decreased | ||
| Plasmalogen synthesis | Decreased | ||
| Phytanic, pristanic oxidation | Decreased | ||
| Catalase localization | Cytosolic | ||
| Immunocytochemistry | Peroxisomes absent | ||
| Complementation | Complementation between infantile Refsum disease and other peroxisomal disorder (except Zellweger) flibroblasts | ||
| DNA | PEX gene mutations | ||
| Rhizomelic chondrodysplasia punctata | Plasma | Phytanic acid | Increased |
| VLCFA | Normal | ||
| RBCs | Plasmalogen levels | Decreased | |
| Fibroblasts | Plasmalogen synthesis | Decreased | |
| Phytanic acid oxidation | Decreased | ||
| DNA | PEX7 defect | ||
| X-linked ALD hemizygote | Plasma | VLCFA | Increased |
| Fibroblasts | VLCFA levels | Increased | |
| VLCFA oxidation | Decreased | ||
| ALDP immunoreactivity | Absent in 70% | ||
| DNA | ABCD1 mutation | ||
| X-linked ALD heterozygote | Plasma | VLCFA | Variable increase in 85% of heterozygotes |
| Fibroblasts | VLCFA levels | Variable increase in 90% of heterozygotes | |
| ALDP immunoreactivity | Variable decrease | ||
| DNA | ABCD1 mutation | ||
| Bifunctional enzyme defect | Plasma | VLCFA | Increased |
| Phytanic acid | Increased | ||
| Pristanic acid | Increased | ||
| Bile acids | Increased, abnormal pattern | ||
| Fibroblasts | VLCFA levels | Increased | |
| Pristanic acid oxidation | Decreased | ||
| Catalase localization | Peroxisomal | ||
| Enzyme | D-bifunctional protein deficiency | ||
| Acyl-CoA oxidase deficiency | Plasma Fibroblasts | VLCFA | Increased |
| VLCFA levels | Increased | ||
| VLCFA oxidation | Decreased | ||
| Enzyme | Acyl-CoA oxidase defect | ||
| 2-Methylacyl-CoA racemase deficiency | Plasma | Pristanic acid | Increased |
| Bile acids | Increased, abnormal pattern | ||
| Fibroblasts | Pristanic acid oxidation | Decreased | |
| Enzyme | 2-Methylacyl-CoA oxidase defect | ||
| Classic Refsum disease | Plasma | Phytanic acid | Increased |
| Pristanic acid | Decreased | ||
| Fibroblasts | Enzyme | Phytanoyl-CoA deficiency |
Level 3: What Is the Molecular Defect?
Definition of the molecular defect in the proband, which is now offered in several laboratories, is essential for carrier detection and speeds prenatal diagnosis. Characterization of the mutation may be of prognostic value in patients with PEX1 defects. This defect is present in approximately 60% of PBD patients, and about half of the PEX1 defects have the G843D allele, which is associated with a significantly milder phenotype than is found in other mutations.
Diagnosis
There are several noninvasive laboratory tests that permit precise and early diagnosis of peroxisomal disorders (see Table 86-4). The challenge in PBDs is to differentiate them from the large variety of other conditions that can cause hypotonia, seizures, failure to thrive, or dysmorphic features. Experienced clinicians can readily recognize classic Zellweger syndrome by its clinical manifestations. However, more mildly affected PBD patients often do not show the full clinical spectrum of disease and may be identifiable only by laboratory assays. Clinical features that serve as indications for these diagnostic assays include severe intellectual disability; weakness and hypotonia; dysmorphic features; neonatal seizures; retinopathy, glaucoma, or cataracts; hearing deficits; enlarged liver and impaired liver function; and chondrodysplasia punctata. The presence of 1 or more of these abnormalities increases the likelihood of this diagnosis. Atypical milder forms presenting as peripheral neuropathy have also been described.
Some patients with the isolated defects of peroxisomal fatty acid oxidation (group B) resemble those with group A disorders and can be detected by the demonstration of abnormally high levels of VLCFAs.
Patients with RCDP must be distinguished from patients with other causes of chondrodysplasia punctata. In addition to warfarin embryopathy and Zellweger syndrome, these disorders include the milder autosomal dominant form of chondrodysplasia punctata (Conradi-Hünermann syndrome), which is characterized by longer survival, absence of severe limb shortening, and usually intact intellect; an X-linked dominant form; and an X-linked recessive form associated with a deletion of the terminal portion of the short arm of the X chromosome. RCDP is suspected clinically because of the shortness of limbs, psychomotor retardation, and ichthyosis. The most decisive laboratory test is the demonstration of abnormally low plasmalogen levels in red blood cells and an impaired capacity to synthesize plasmalogens in cultured skin fibroblasts. These biochemical defects are not present in other types of chondrodysplasia punctata. Chondrodysplasia punctata may also be associated with a defect of 3β-hydroxysteroid-Δ8,Δ7-isomerase, an enzyme involved in biosynthesis of cholesterol.
Complications
Patients with Zellweger cerebrohepatorenal syndrome have multiple disabilities involving muscle tone, swallowing, cardiac abnormalities, liver disease, and seizures. These conditions are treated symptomatically, but the prognosis is poor, and most patients succumb in the first year of life. Patients with RCDP may develop quadriparesis owing to compression at the base of the brain.
Treatment
The most effective therapy is the dietary treatment of classic Refsum disease with a phytanic acid-restricted diet.
For patients with the somewhat milder variants of the peroxisome import disorders, success has been achieved with multidisciplinary early intervention, including physical and occupational therapy, hearing aids or cochlear implants, alternative communication, nutrition, and support for the parents. Although most patients continue to function in the severely delayed range, some make significant gains in self-help skills, and several are in stable condition in their teens or even early 20s.
Attempts to mitigate some of the secondary biochemical abnormalities include the oral administration of docosahexaenoic acid. The levels of this substance are greatly reduced in patients with disorders of peroxisome biogenesis and this therapy normalizes the plasma levels of this substance. Although there were anecdotal reports of clinical improvement, a randomized placebo-controlled study failed to find benefit.
Genetic Counseling
All the peroxisomal disorders, except hyperoxaluria type 1, can be diagnosed prenatally in the 1st or 2nd trimester. The tests are similar to those described for postnatal diagnosis (see Table 86-5) and use chorionic villus sampling or amniocytes. Because of the 25% recurrence risk, couples with an affected child must be advised about the availability of prenatal diagnosis. Heterozygotes can be identified in ALD and in those disorders in which the molecular defect has been identified.
Adrenoleukodystrophy (X-Linked)
ALD is a genetically determined disorder associated with the accumulation of saturated VLCFAs and a progressive dysfunction of the adrenal cortex and central and peripheral nervous system white matter.
Etiology
The key biochemical abnormality is the tissue accumulation of saturated VLCFAs, with a carbon chain length of 24 or more. Excess hexacosanoic acid (C26:0) is the most striking and characteristic feature. This accumulation of fatty acids is caused by genetically deficient peroxisomal degradation of fatty acid. The gene that is defective (ABCD1) codes for a peroxisomal membrane protein (ALDP, the ALD protein). Most families have a mutation that is “private” (unique to that kindred) and these are updated on the website, http://x-ald.nl. The mechanism by which the ALDP defect leads to VLCFA accumulation appears to be a disruption of transport of saturated fatty acids into the peroxisome with resultant continued elongation of progressively longer fatty acids.
Epidemiology
The minimum incidence of ALD in males is 1 in 21,000, and the combined incidence of ALD males and heterozygous females in the general population is estimated to be 1 in 17,000. All races are affected. The various phenotypes often occur in members of the same kindred.
Pathology
Characteristic lamellar cytoplasmic inclusions can be demonstrated with the electron microscope in adrenocortical cells, testicular Leydig cells, and nervous system macrophages. These inclusions probably consist of cholesterol esterified with VLCFA. They are most prominent in cells of the zona fasciculata of the adrenal cortex, which at first are distended with lipid and later atrophy.
The nervous system can display 2 types of lesions. In the severe childhood cerebral form and in the rapidly progressive adult forms, demyelination is associated with an inflammatory response manifested by the accumulation of perivascular lymphocytes that is most intense in the parietooccipital region. In the slowly progressive adult form, adrenomyeloneuropathy, the main finding is a distal axonopathy that affects the long tracts in the spinal cord. The inflammatory response is mild or absent.
Pathogenesis
The adrenal dysfunction is probably a direct consequence of the accumulation of VLCFAs. The cells in the zona fasciculata are distended with abnormal lipids. Cholesterol esterified with VLCFA is relatively resistant to adrenocorticotropic hormone (ACTH)-stimulated cholesterol ester hydrolases, and this limits the capacity to convert cholesterol to active steroids. In addition, C26:0 excess increases the viscosity of the plasma membrane and this may interfere with receptor and other cellular functions.
There is no correlation between the neurologic phenotype and the nature of the mutation or the severity of the biochemical defect as assessed by plasma levels of VLCFAs or between the degree of adrenal involvement and nervous system involvement. The severity of the illness and rate of progression correlate with the intensity of the inflammatory response. The inflammatory response may be cytokine mediated and may involve an autoimmune response triggered in an unknown way by the excess of VLCFAs. Mitochondrial damage and oxidative stress also contribute. Approximately half of the patients do not experience the inflammatory response; this difference is not understood.
Clinical Manifestations
There are 5 relatively distinct phenotypes, 3 of which are present in childhood with symptoms and signs. In all the phenotypes, development is usually normal in the first 3-4 yr of life.
In the childhood cerebral form of ALD, symptoms are first noted most commonly between the ages of 4 and 8 yr. The most common initial manifestations are hyperactivity, which is often mistaken for an attention deficit disorder, and worsening school performance in a child who had previously been a good student. Auditory discrimination is often impaired, although tone perception is preserved. This may be evidenced by difficulty in using the telephone and greatly impaired performance on intelligence tests in items that are presented verbally. Spatial orientation is often impaired. Other initial symptoms are disturbances of vision, ataxia, poor handwriting, seizures, and strabismus. Visual disturbances are often caused by involvement of the cerebral cortex, which leads to variable and seemingly inconsistent visual capacity. Seizures occur in nearly all patients and may represent the first manifestation of the disease. Some patients present with increased intracranial pressure or with unilateral mass lesions. Impaired cortisol response to ACTH stimulation is present in 85% of patients, and mild hyperpigmentation is noted. In most patients with this phenotype, adrenal dysfunction is recognized only after the condition is diagnosed because of the cerebral symptoms. Cerebral childhood ALD tends to progress rapidly with increasing spasticity and paralysis, visual and hearing loss, and loss of ability to speak or swallow. The mean interval between the first neurologic symptom and an apparently vegetative state is 1.9 yr. Patients may continue in this apparently vegetative state for 10 yr or more.
Adolescent ALD designates patients who experience neurologic symptoms between the ages of 10 and 21 yr. The manifestations resemble those of childhood cerebral ALD except that progression is slower. Approximately 10% of patients present acutely with status epilepticus, adrenal crisis, acute encephalopathy, or coma.
Adrenomyeloneuropathy first manifests in late adolescence or adulthood as a progressive paraparesis caused by long tract degeneration in the spinal cord. Approximately half of the patients also have involvement of the cerebral white matter.
The “Addison only” phenotype is an important condition. Of male patients with Addison disease, 25% may have the biochemical defect of ALD. Many of these patients have intact neurologic systems, whereas others have subtle neurologic signs. Many acquire adrenomyeloneuropathy in adulthood.
The term “asymptomatic ALD” is applied to persons who have the biochemical defect of ALD but are free of neurologic or endocrinal disturbances. Nearly all persons with the gene defect eventually become neurologically symptomatic.
Approximately 50% of female heterozygotes acquire a syndrome that resembles adrenomyeloneuropathy but is milder and of later onset. Adrenal insufficiency and cerebral disease are rare.
Cases of typical ALD have occurred in relatives of those with adrenomyeloneuropathy. One of the most difficult problems in the management of X-linked ALD is the common observation that affected individuals in the same family may have quite different clinical courses. For example, in 1 family, 1 affected boy had severe classic ALD culminating in death by age 10 yr; another affected male (a brother) had late-onset adrenomyeloneuropathy, and a third had no symptoms at all.
Laboratory and Radiographic Findings
The most specific and important laboratory finding is the demonstration of abnormally high levels of VLCFA in plasma, red blood cells, or cultured skin fibroblasts. The test should be performed in a laboratory that has experience with this specialized procedure. Positive results are obtained in all male patients with ALD and in approximately 85% of female carriers of ALD. Mutation analysis is the most reliable method for the identification of carriers.
Neuroimaging
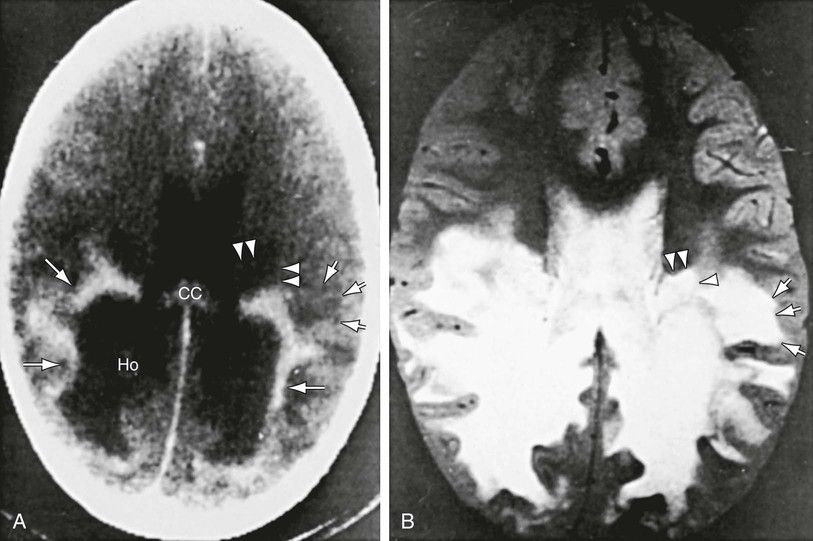
Patients with childhood cerebral or adolescent ALD show cerebral white matter lesions that are characteristic with respect to location and attenuation patterns on MRI. In 80% of patients, the lesions are symmetric and involve the periventricular white matter in the posterior parietal and occipital lobes. Approximately 50% show a garland of accumulated contrast material adjacent and anterior to the posterior hypodense lesions (Fig. 86-5A). This zone corresponds to the zones of intense perivascular lymphocytic infiltration where the blood–brain barrier breaks down. In 12% of patients, the initial lesions are frontal. Unilateral lesions that produce a mass effect suggestive of a brain tumor may occur. MRI provides a clearer delineation of normal and abnormal white matter than does CT (Fig. 86-5B).
Impaired Adrenal Function
More than 85% of patients with the childhood form of ALD have elevated levels of ACTH in plasma and a subnormal rise of cortisol levels in plasma following intravenous injection of 250 µg of ACTH (Cortrosyn).
Diagnosis and Differential Diagnosis
The earliest manifestations of childhood cerebral ALD are difficult to distinguish from the more common attention-deficit disorders or learning disabilities. Rapid progression, signs of dementia, or difficulty in auditory discrimination suggest ALD. Even in early stages, CT or MRI may show strikingly abnormal changes. Other leukodystrophies or multiple sclerosis may mimic these radiographic findings, although early ALD has more of a predilection for the posterior brain than its mimics. Definitive diagnosis depends on demonstration of VLCFA excess, which occurs only in ALD and the other peroxisomal disorders.
Cerebral forms of ALD may present as increased intracranial pressure and unilateral mass lesions. These have been misdiagnosed as gliomas, even after brain biopsy, and several patients have received radiotherapy before the correct diagnosis was made. Measurement of VLCFA in plasma or brain biopsy specimens is the most reliable differentiating test.
Adolescent or adult cerebral ALD can be confused with psychiatric disorders, dementing disorders, or epilepsy. The first clue to the diagnosis of ALD may be the demonstration of white matter lesions by neuroimaging; assays of VLCFA are confirmatory.
ALD cannot be distinguished clinically from other forms of Addison disease; it is recommended that assays of VLCFA levels be performed in all male patients with Addison disease. ALD patients do not usually have antibodies to adrenal tissue in their plasma.
Complications
An avoidable complication is the occurrence of adrenal insufficiency. The most difficult neurologic problems are those related to bed rest, contracture, coma, and swallowing disturbances. Other complications involve behavioral disturbances and injuries associated with defects of spatial orientation, impaired vision and hearing, and seizures.
Treatment
Corticosteroid replacement for adrenal insufficiency or adrenocortical hypofunction is effective. It may be lifesaving and increase general strength and well-being, but it does not alter the course of the neurologic disability.
Bone Marrow Transplantation
Bone marrow transplantation (BMT) benefits patients who show early evidence of the inflammatory demyelination that is characteristic of the rapidly progressive neurologic disability in boys and adolescents with the cerebral ALD phenotype. BMT is a high-risk procedure, and patients must be selected with great care. The mechanism of the beneficial effect is incompletely understood. Bone marrow-derived cells do express ALDP, the protein that is deficient in ALD; approximately 50% of brain microglial cells are bone marrow derived. The favorable effect may be caused by modification of the brain inflammatory response. Five to 10 yr follow-up of boys and adolescents who had early cerebral involvement has shown stabilization. On the other hand, BMT has not shown favorable effects in patients who already had severe brain involvement and may accelerate disease progression under these circumstances. The nonverbal IQ has been found to be of predictive value, and transplant is not recommended in patients with performance IQ significantly below 80. Unfortunately, in more than half the patients who are diagnosed because of neurologic symptoms, the illness is so advanced at the time of diagnosis that they are not candidates for transplant.
Consideration of BMT is most relevant in neurologically asymptomatic or mildly involved patients. Screening at-risk relatives of symptomatic patients identifies these patients most frequently. Screening by measurement of plasma VLCFA levels in patients with Addison disease may also identify candidates for BMT. Because of its risk (10-20% mortality) and the fact that up to 50% of untreated patients with ALD do not develop inflammatory brain demyelination, transplant is not recommended in patients who are free of demonstrable brain involvement. The MRI is also of key importance for the crucial decision of whether transplant should be performed. MRI abnormalities precede clinically evident neurologic or neuropsychologic abnormalities. The brain MRI should be monitored at 6 mo intervals in neurologically asymptomatic boys and adolescents between the ages of 3 and 15 yr. If the MRI is normal, BMT is not indicated. If brain MRI abnormalities develop, the patient should be evaluated at 3 mo intervals to determine if the abnormality is progressive, in combination with careful neurologic and neuropsychologic evaluation; and if early progressive involvement is confirmed, transplant should be considered. Magnetic resonance spectroscopy improves the capacity to determine whether the brain involvement is progressive. It is not known whether BMT has a favorable effect on the noninflammatory spinal cord involvement in adults with the adrenomyeloneuropathy phenotype.
Lorenzo's Oil Therapy
The administration of Lorenzo's oil to asymptomatic boys in an open study reduced the risk of developing the childhood cerebral phenotype by a factor of 2 or more. Lorenzo's oil (4 : 1 mixture of glyceryl trioleate and glyceryl trierucate) combined with a dietary regimen is under investigation for neurologically asymptomatic boys who have a normal brain MRI and are younger than 8 yr old. It has been determined that it must be supervised carefully. Adrenal function and brain MRI must be monitored. Patients who develop progressive MRI abnormalities are evaluated for hematopoietic stem cell transplant when changes are still in an early phase. Lorenzo's oil has not been shown to alter disease progression in patients who already have cerebral involvement.
Supportive Therapy
The progressive behavioral and neurologic disturbances associated with the childhood form of ALD are extremely difficult for the family. ALD patients require the establishment of a comprehensive management program and partnership among the family, physician, visiting nursing staff, school authorities, and counselors. In addition, parent support groups (e.g., United Leukodystrophy Foundation) are often helpful. Communication with school authorities is important because under the provisions of Public Law 94-142, children with ALD qualify for special services as “other health impaired” or “multihandicapped.” Depending on the rate of progression of the disease, special needs might range from relatively low-level resource services within a regular school program to home- and hospital-based teaching programs for children who are not mobile.
Management challenges vary with the stage of the illness. The early stages are characterized by subtle changes in affect, behavior, and attention span. Counseling and communication with school authorities are of prime importance. Changes in the sleep–wake cycle can be benefited by the judicious use at night of medications for sleep.
As the leukodystrophy progresses, the modulation of muscle tone and support of bulbar muscular function are major concerns. Baclofen in gradually increasing doses (5 mg twice a day to 25 mg 4 times a day) is an effective pharmacologic agent for the treatment of acute episodic painful muscle spasms. Other agents may also be used, with care being taken to monitor the occurrence of side effects and drug interactions. As the leukodystrophy progresses, bulbar muscular control is lost. Although initially this can be managed by changing the diet to soft and pureed foods, most patients eventually require a gastrostomy tube. At least 30% of patients have focal or generalized seizures that usually readily respond to standard anticonvulsant medications.
Genetic Counseling and Prevention
Genetic counseling and primary and secondary prevention of ALD are of crucial importance. Extended family screening should be offered to all at-risk relatives of symptomatic patients; one program led to the identification of more than 250 asymptomatic affected males and 1,200 women heterozygous for ALD. The plasma assay permits reliable identification of affected males in whom plasma VLCFA levels are increased already on the day of birth. Identification of asymptomatic males permits institution of steroid replacement therapy when appropriate and prevents the occurrence of adrenal crisis, which may be fatal. Monitoring of brain MRI also permits identification of patients who are candidates for BMT at a stage when this procedure has the greatest chance of success. Plasma VLCFA assay is recommended in all male patients with Addison disease. ALD has been shown to be the cause of adrenal insufficiency in more than 25% of boys with Addison disease of unknown cause. Identification of women heterozygous for ALD is more difficult than that of affected males. Plasma VLCFA levels are normal in 15-20% of heterozygous women, and failure to note this has led to serious errors in genetic counseling. DNA analysis permits accurate identification of carriers, provided that the mutation has been defined in a family member, and is the procedure recommended for the identification of heterozygous women.
Prenatal diagnosis of affected male fetuses can be achieved by measurement of VLCFA levels in cultured amniocytes or chorionic villus cells and by mutation analysis. Whenever a new patient with ALD is identified, a detailed pedigree should be constructed and efforts should be made to identify all at-risk female carriers and affected males. These investigations should be accompanied by careful and sympathetic attention to social, emotional, and ethical issues during counseling.
Disorders of Lipoprotein Metabolism and Transport
William A. Neal, Collin C. John
Epidemiology of Blood Lipids and Cardiovascular Disease
The Seven Countries Study of geographic, social class, and ethnic differences in coronary heart disease (CHD) around the world found strong associations between average intake of saturated fats, plasma cholesterol, and mortality from CHD. Of all common chronic diseases, none is so clearly influenced by both environmental and genetic factors as CHD. This multifactorial disorder is strongly associated with increasing age and male gender, though it is increasingly apparent that heart disease is underrecognized in women. Tobacco use confers a 2-fold higher lifetime risk. Sedentary activity and high intake of saturated fats leading to adiposity increase risk through differences in the plasma levels of lipoproteins that are atherogenic. Family history is a reflection of the combined influence of lifestyle and genetic predisposition to early heart disease. Risk of premature heart disease associated with positive family history is 1.7 times higher than in families with no such history.
The pathogenesis of atherosclerosis begins during childhood. The Johns Hopkins Precursors Study demonstrated that white male medical students with blood cholesterol levels in the lowest quartile showed only a 10% incidence of CHD 3 decades later, whereas those in the highest quartile had a 40% incidence. The Pathobiological Determinants of Atherosclerosis in Youth Study demonstrated a significant relationship between the weight of the abdominal fat pad and the extent of atherosclerosis found at autopsy on subjects 15-34 yr of age. The Bogalusa Heart Study of more than 3,000 black and white children and adolescents has provided the most comprehensive longitudinal data relating the presence and severity of CHD risk factors with semiquantifiable severity of atherosclerosis. Coronary atherosclerosis was present in 8.5% of military autopsies performed following combat or unintentional injuries.
The “fetal origins hypothesis” is based on the observation that infants born with low birthweight have a higher incidence of heart disease as adults. Epidemiologic studies support the idea that prenatal and early postnatal conditions may affect adult health status. Children who are large for gestational age at birth and exposed to an intrauterine environment of either diabetes or maternal obesity are at increased risk of eventually developing the “metabolic syndrome” (insulin resistance, type II diabetes, obesity, CHD). Breastfeeding preterm infants confers a long-term cardioprotective benefit 13-16 yr later. Those adolescents who were breastfed as infants had lower C-reactive protein concentrations and a 14% lower low-density lipoprotein (LDL):high-density lipoprotein (HDL) ratio compared to those fed infant formulas. The impact of early nutrition and other lifestyle variables on gene expression, known as epigenetics, is an important mechanism by which adult metabolism and body composition may be determined.
In addition, secondary causes of hyperlipidemia may be the result of drugs (cyclosporine, steroids, isotretinoin, protease inhibitors, alcohol, thiazide diuretics, β-blocking agents, valproate) or various diseases (nephrotic syndrome, hypothyroidism, Cushing syndrome, anorexia nervosa, obstructive jaundice).
Blood Lipids and Atherogenesis
Numerous epidemiologic studies demonstrate the association of hypercholesterolemia, referring to elevated total blood cholesterol, with atherosclerotic disease. The ability to measure subcomponents within classes of lipid particles, as well as markers of inflammation, have further elucidated the process of atherogenesis and plaque rupture leading to acute coronary syndromes. Atherosclerosis affects primarily the coronary arteries but may also involve the aorta, arteries of the lower extremities, and carotid arteries.
The early stage of development of atherosclerosis is thought to begin with vascular endothelial dysfunction and intima-media thickness, which has been shown to occur in preadolescent children with risk factors such as obesity or familial hypercholesterolemia. The complex process of penetration of the intimal lining of the vessel may be a consequence of a variety of insults, including the presence of highly toxic oxidized LDL particles. Lymphocytes and monocytes penetrate the damaged endothelial lining, where they become macrophages laden with LDL lipids and then become foam cells. Such accumulation is counterbalanced by HDL particles capable of removing lipid deposits from the vessel wall. Fundamental to plaque formation is an inflammatory process (elevated C-reactive protein) involving macrophages and the arterial wall. The deposition of lipid within the subendothelial lining of the arterial wall appears macroscopically as fatty streaks, which may to some degree be reversible. A later stage of plaque development involves disruption of arterial smooth muscle cells stimulated by the release of tissue cytokines and growth factors. The atheroma is composed of a core of fatty substance separated from the lumen by collagen and smooth muscle (Fig. 86-6). Growth of the atherosclerotic plaque may result in ischemia of the tissue supplied by the artery. Chronic inflammation within the atheroma, results in plaque instability and subsequent rupture. Platelet adherence leads to clot formation at the site of rupture, resulting in myocardial infarction or a cerebrovascular event.
Plasma Lipoprotein Metabolism and Transport
Abnormalities of lipoprotein metabolism are associated with diabetes mellitus and premature atherosclerosis. Lipoproteins are soluble complexes of lipids and proteins that effect transport of fat absorbed from the diet, or synthesis by the liver and adipose tissues, for utilization and storage. Dietary fat is transported from the small intestine as chylomicrons. Lipids synthesized by the liver as very-low-density lipoproteins (VLDLs) are catabolized to intermediate-density lipoproteins (IDLs) and LDLs. HDLs are fundamentally involved in VLDL and chylomicron metabolism and cholesterol transport. Nonesterified free fatty acids are metabolically active lipids derived from lipolysis of triglycerides stored in adipose tissue bound to albumin for circulation in the plasma (Fig. 86-7).
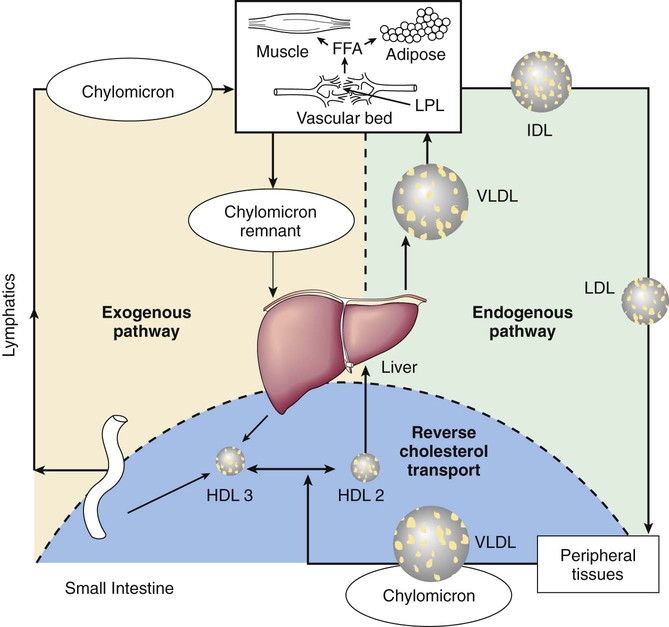
Lipoproteins consist of a central core of triglycerides and cholesteryl esters surrounded by phospholipids, cholesterol, and proteins (Fig. 86-8). The density of the several classes of lipoproteins is inversely proportional to the ratio of lipid to protein (Fig. 86-9). Lipoproteins consist of a central core of triglycerides and cholesteryl esters surrounded by phospholipids, cholesterol, and proteins.
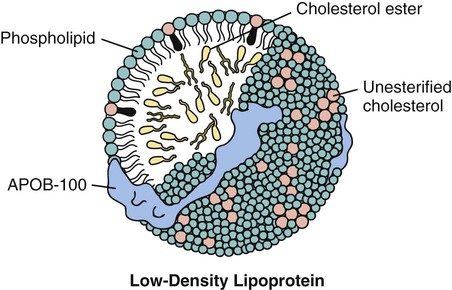
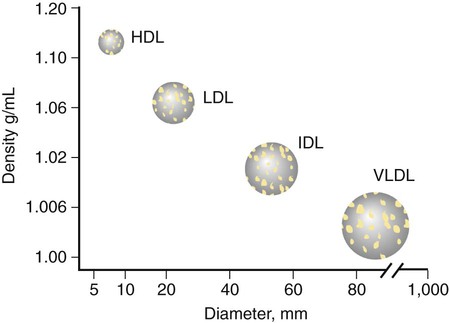
Constituent proteins are known as apolipoproteins (Table 86-6). They are responsible for a variety of metabolic functions in addition to their structural role, including cofactors or inhibitors of enzymatic pathways, and mediators of lipoprotein binding to cell surface receptors. ApoA is the major apolipoprotein (Apo) of HDL. ApoB is present in LDL, VLDL, IDL, and chylomicrons. ApoB-100 is derived from the liver, whereas apoB-48 comes from the small intestine. ApoC-I, C-II, and C-III are small peptides important in triglyceride metabolism. Loss of function and disruptive mutations of the APOC3 gene are associated with low levels of triglycerides and a reduced risk of ischemic CHD. Likewise, apoE, which is present in VLDL, HDL, chylomicrons, and chylomicron remnants, plays an important role in the clearance of triglycerides.
Table 86-6
Characteristics of the Major Lipoproteins
| LIPOPROTEIN | SOURCE | SIZE (nm) | DENSITY (g/mL) | Composition % | ||
| PROTEIN | LIPID | APOLIPOPROTEINS | ||||
| Chylomicrons | Intestine | 80-1,200 | <0.95 | 1-2 | 98-99 | C-I, C-II, C-III, E, A-I, A-II, A-IV, B-48 |
| Chylomicron remnants | Chylomicrons | 40-150 | <1.0006 | 6-8 | 92-94 | B-48, E |
| VLDL | Liver, intestine | 30-80 | 0.95-1.006 | 7-10 | 90-93 | B-100, C-I, C-II, C-III |
| IDL | VLDL | 25-35 | 1.006-1.019 | 11 | 89 | B-100, E |
| LDL | VLDL | 18-25 | 1.019-1.063 | 21 | 79 | B-100 |
| HDL | Liver, intestine VLDL, Chylomicrons | 5-20 | 1.125-1.210 | 32-57 | 43-68 | A-I, A-II, A-IV C-I, C-II, C-III D, E |
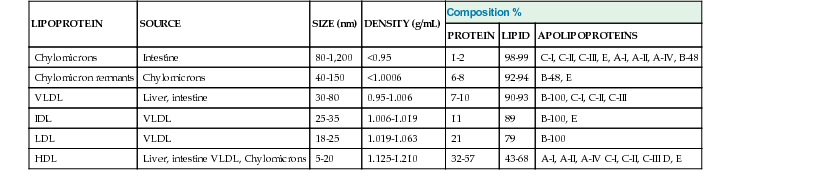
Transport of Exogenous (Dietary) Lipids
All dietary fat with the exception of medium-chain triglycerides is efficiently carried into the circulation by way of lymphatic drainage from the intestinal mucosa. Triglyceride and cholesteryl esters combine with apoA and apoB-48 in the intestinal mucosa to form chylomicrons, which are carried into the peripheral circulation via the lymphatic system. HDL particles contribute apoC-II to the chylomicrons, required for the activation of lipoprotein lipase (LPL) within the capillary endothelium of adipose, heart, and skeletal muscle tissue. Free fatty acids are oxidized, esterified for storage as triglycerides, or released into the circulation bound to albumin for transport to the liver. After hydrolysis of the triglyceride core from the chylomicron, apoC particles are recirculated back to HDL. The subsequent contribution of apoE from HDL to the remnant chylomicron facilitates binding of the particle to hepatic LDL receptors (LDL-R). Within the hepatocyte, the chylomicron remnant may be incorporated into membranes, resecreted as lipoprotein back into the circulation, or secreted as bile acids. Normally, all dietary fat is disposed of within 8 hr after the last meal, an exception being individuals with a disorder of chylomicron metabolism. Postprandial hyperlipidemia is a risk factor for atherosclerosis. Abnormal transport of chylomicrons and their remnants may result in their absorption into the blood vessel wall as foam cells, caused by the ingestion of cholesteryl esters by macrophages, the earliest stage in the development of fatty streaks.
Transport of Endogenous Lipids from the Liver
The formation and secretion of VLDL from the liver and its catabolism to IDL and LDL particles describe the endogenous lipoprotein pathway. Fatty acids used in the hepatic formation of VLDL are derived primarily by uptake from the circulation. VLDL appears to be transported from the liver as rapidly as it is synthesized, and it consists of triglycerides, cholesteryl esters, phospholipids, and apoB-100. Nascent particles of VLDL secreted into the circulation combine with apoC and apoE. The size of the VLDL particle is determined by the amount of triglyceride present, progressively shrinking in size as triglyceride is hydrolyzed by the action of LPL, yielding free fatty acids for utilization or storage in muscle and adipose tissue. Hydrolysis of approximately 80% of the triglyceride present in VLDL particles produces IDL particles containing an equal amount of cholesterol and triglyceride. The remaining remnant IDL is converted to LDL for delivery to peripheral tissues or to the liver. ApoE is attached to the remnant IDL particle to allow binding to the cell and subsequent incorporation into the lysosome. Individuals with deficiency of either apoE2 or hepatic triglyceride lipase accumulate IDL in the plasma.
LDL particles account for approximately 70% of the plasma cholesterol in normal individuals. LDL receptors are present on the surfaces of nearly all cells. Most LDL is taken up by the liver, and the rest is transported to peripheral tissues such as the adrenal glands and gonads for steroid synthesis. Dyslipidemia is greatly influenced by LDL-R activity. The efficiency with which VLDL is converted into LDL is also important in lipid homeostasis.