Inotropes and vasopressors
The pharmacological support of the failing circulation is a fundamental part of critical care. The principal aim of these drugs is to restore inadequate systemic and regional perfusion to physiological levels.
Definitions
Inotropic agents are defined as drugs that act on the heart by increasing the velocity and force of myocardial fibre shortening. The consequent increase in contractility results in increased cardiac output and blood pressure. Characteristics of the ideal inotrope are shown in Box 90.1.
Vasopressors are drugs that have a predominantly vasoconstrictive action on the peripheral vasculature, both arterial and venous. These drugs are used primarily to increase mean arterial pressure.
The distinction between these two groups of drugs is often confusing. Many of the commonly used agents such as the catecholamines have both inotropic and variable effects on the peripheral vasculature that include venoconstriction, arteriolar vasodilatation and constriction.
Vasoregulatory agents may modulate the responsiveness of the peripheral vasculature to vasoactive drugs in pathological states such as sepsis. These agents include vasopressin and corticosteroids.
Given the overlap of pharmacodynamic effects of these drugs, the term ‘vasoactive therapy’ is a more appropriate description.
The failing circulation
Physiology
Traditionally, cardiac output is discussed in terms of factors that govern cardiac function. These include preload, afterload, heart rate and rhythm, and contractility. Although this perspective is helpful in managing patients whose circulatory function is limited by cardiac disease, it is incomplete.
Cardiac output is controlled by venous return to the heart from the peripheral vasculature at an equal rate to that ejected during each cardiac cycle1 (Fig. 90.1).
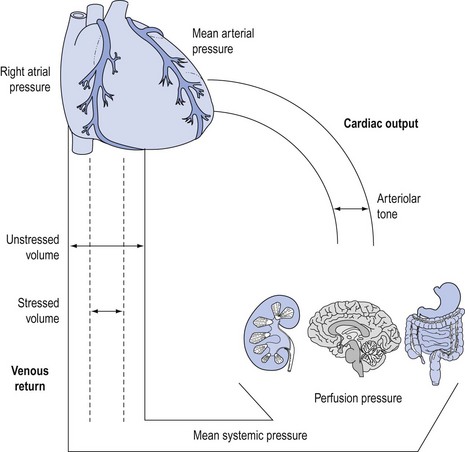
Blood is pumped down a pressure gradient that is determined by the force of myocardial ejection (contractility) and impedance to ventricular ejection (afterload). The resultant mean arterial pressure is the major ‘afferent’ determinant of regional perfusion pressure. Twenty per cent of the blood volume is contained in the arterial (‘conducting’) vessels. There is a marked drop in perfusion pressure and flow across the capillary beds to allow diffusion of substrates and oxygen. The difference between mean arterial pressure and the pressure in end capillaries (‘efferent’ perfusion pressure) determines regional, or organ-specific, perfusion pressure.
Blood enters the venous system and is returned to the heart via a pressure gradient determined by mean systemic pressure and right atrial pressure. The amount of blood returned to the heart determines the degree of ventricular filling prior to systole (preload), which subsequently determines stroke volume and cardiac output.
Under physiological conditions, the venous (‘capacitance’) system contains approximately 70% of the total blood volume, which acts as a physiological reservoir (‘unstressed’ volume). Under conditions where circulatory demands increase, increased sympathetic tone will cause contraction of this reservoir.2 The resultant autotransfusion (‘stressed’ volume) may increase venous return by approximately 30% and subsequently cardiac output.3
Both the arterial and venous systems are integrated under complex neurohormonal influences. These include the adrenergic, renin–angiotensin–aldosterone, vasopressinergic and glucocorticoid systems in addition to local mediators such as nitric oxide, endothelin, endorphins and the eicosanoids.4
Pathophysiology
Circulatory dysfunction or failure may be considered in terms of the major determinants of cardiac output, although there is marked interdependence between these factors.
Heart rate failure
Profound bradycardia will reduce both cardiac output and mean arterial pressure if sympathetic tone is compromised. Inotropes will increase both rate and speed of conduction, in addition to augmenting peripheral venous return, thereby restoring cardiac output and mean arterial pressure.
Tachycardia is associated with decreased left coronary artery perfusion, due to reduction of diastolic time, during which coronary perfusion occurs. This may exacerbate myocardial ischaemia in patients with coronary artery disease, particularly if mean arterial pressure, specifically diastolic blood pressure, is compromised. Therefore drugs that shorten diastolic time or compromise coronary perfusion should be used with caution in susceptible patients.
Preload failure
Loss of intravascular blood volume or extracellular fluid is the most common cause of inadequate ventricular preload. This is corrected with fluids to maintain and restore a euvolaemia. Hypovolaemia must be recognised and treated as soon as possible, preferably before vasoactive therapy is used, although the early use of vasoactive therapy is increasingly being recommended.
There are other determinants of ventricular preload and venous return. Factors such as loss of muscle tone, positive intrathoracic pressure, loss of atrial contraction (atrial fibrillation) and ablation of sympathetic tone will compromise preload by reducing venous return. Under these circumstances, volume replacement alone may be insufficient to maintain adequate preload and vasoactive therapy may be required to increase venous return.
Myocardial failure
Myocardial or ‘pump’ failure may be divided into disorders of systolic ejection (systolic dysfunction) and diastolic filling (diastolic dysfunction).
Systolic dysfunction occurs as a result of reduced effective myocardial contractility. This may be due to primary myocardial factors such as ischaemia, infarction or cardiomyopathy. Myocardial depression of both right and left ventricular function may occur in severe sepsis or following prolonged infusions of catecholamines. Increased impedance to ventricular ejection (e.g. hypertensive states) or structural abnormalities (e.g. aortic stenosis or hypertrophic obstructive cardiomyopathy) may cause systolic dysfunction.
Diastolic dysfunction is characterised by reduced ventricular compliance or increased resistance to ventricular filling during diastole. It may be due to mechanical factors such as structural abnormalities of the ventricle (e.g. restrictive cardiomyopathy) or to impaired diastolic relaxation that occurs with myocardial ischaemia or severe sepsis. This results in elevated end-diastolic pressure and pulmonary venous congestion. Episodic or ‘flash’ pulmonary oedema is a common clinical sign of diastolic dysfunction. Tachycardia that shortens diastolic time may exacerbate diastolic failure. Diastolic dysfunction frequently accompanies systolic failure, in both acute and chronic cardiac failure, particularly in elderly patients.
In the presence of systolic dysfunction, adequate stroke volume may be maintained by an increase in left ventricular end-diastolic volume (the Frank–Starling relationship), provided diastolic function is optimal. However, if the loss of effective myocardial mass is critical, the ventricle will be unable to maintain an adequate stroke volume and cardiac output will fall. In this situation, systolic dysfunction usually requires treatment with inotropic agents in order to augment stroke volume, thereby increasing cardiac output and mean arterial pressure.
Vasoregulatory failure
Disruption or impairment of regulation of the peripheral vasculature may result in circulatory failure. This includes acute sympathetic denervation, such as high quadriplegia, epidural or total spinal anaesthesia (‘spinal’ shock); distributive failure such as anaphylaxis; or pathological ‘vasoplegia’ that occurs in severe sepsis.
These syndromes are characterised by reduced responsiveness of the peripheral circulation to endogenous or exogenous sympathetic stimulation. This results in pooling in the venous circulation due to the inability to provide a ‘stressed’ volume.5
Management of these conditions has traditionally focused on the arterial circulation with attempts to increase systemic vascular resistance. However, the problem is predominantly impaired venous return, compounded to a lesser extent by pathological arteriolar vasodilatation.
Vasoactive agents may have a role in restoring vasoregulatory tone once adequate volume has been established.
Classification
The common ultimate cellular mechanism of action of these agents involves an influence on the release, utilisation or sequestration of intracellular calcium (Fig. 90.2). These agents are divided into two main groups based on whether or not their actions depend upon increases in intracellular cyclic adenosine 3,5-monophosphate (cAMP) and are outlined in Table 90.1.
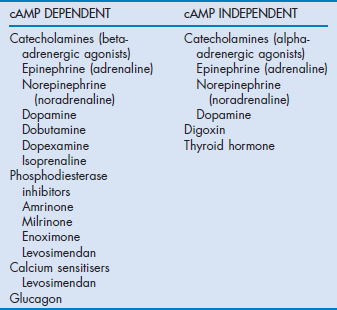
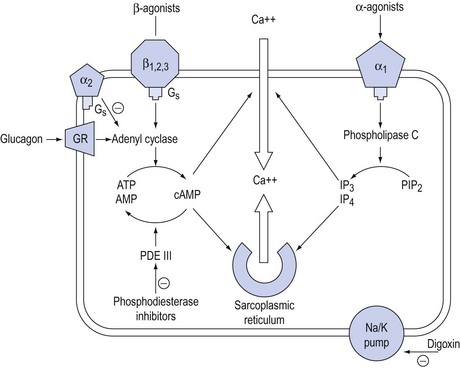
Figure 90.2 Schematic representation of the action of inotropic drugs on intracellular calcium in myocytes. GR = glucagon receptor; Gs = G protein complex; ATP = adenosine triphosphate, AMP = adenosine monophosphate; cAMP = cyclic AMP; PDE III = phosphodiesterase III; IP3 = inositol phosphate 3, PIP2 = phosphoinositol diphosphate.
Catecholamines
Sympathomimetic amines are the most frequently used vasoactive agents in the intensive care unit (ICU) and include the naturally occurring catecholamines dopamine, norepinephrine (noradrenaline) and epinephrine (adrenaline), and the synthetic substances dobutamine, isoprenaline and dopexamine.
Receptor biology
Agonists bind to populations of adrenergic receptors, largely divided into α and β subgroups. Further subgroups of α- (α1A, α1B, α2A, α2B, α2C) and β-receptors (β1, β2, β3) have been identified.4
Signal transduction from agonist–receptor occupation to the effector cell is modulated by conformational changes in G proteins associated with these receptors. Under the additional influence of second messengers such as nitric oxide, endothelin and eicosanoids, these conformational changes promote the release of calcium from intracellular stores and increase membrane calcium permeability. Subsequent phosphorylation of substrate proteins via protein kinases act as third messengers to trigger a cascade of events that lead to specific cardiovascular effects.
β-Receptor occupancy predominantly activates adenyl cyclase to increase the conversion of adenosine triphosphate to cAMP. α-Receptor occupancy acts independently of cAMP by activation of phospholipase C, which increases inositol phosphates (IP3 and IP4) and diacyl glycerol.
This complex agonist–receptor–effector relationship is responsible for homeostatic mechanisms such as physiological responses to stress and autoregulation.
The activity and function of this system is dynamic and may be markedly influenced by pathological states. This may result in qualitative changes in the agonist–receptor–effector organ relationship (desensitisation) where receptors no longer respond to physiological or pharmacological sympathetic stimulation to the same extent. Quantitative changes such as reduced receptor density, receptor sequestration and enzymatic uncoupling (down-regulation) may also result in impaired responses.6
Biosynthesis
The biosynthesis and chemical structures of the naturally occurring catecholamines are shown in Figure 90.3a.
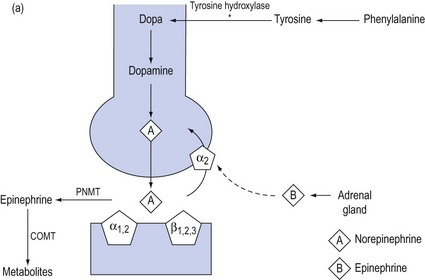
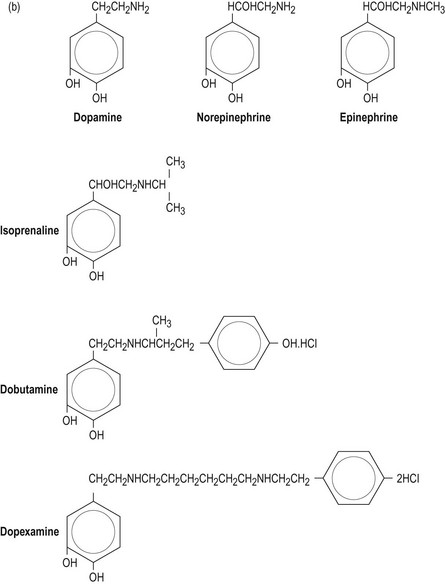
Figure 90.3 (a) Biosynthesis of catecholamines in sympathetic terminals. * = rate-limiting step by tyrosine hydroxylase; PNMT = phenethanolamine-N-methyltransferase, COMT = catechol-o-methyl-transferase. (b) Chemical structure of endogenous and synthetic catecholamines.
Catecholamines consist of an aromatic ring attached to a terminal amine by a carbon chain. The configuration of each drug is important for determining affinity to respective receptors.
Dopamine is hydroxylated to form norepinephrine, which is the predominant peripheral sympathetic chemotransmitter in humans, acting at all adrenergic receptors. The release of norepinephrine from sympathetic terminals is controlled by reuptake mechanisms mediated via α2-receptors and augmented by epinephrine released from the adrenal gland at times of stress. Norepinephrine is converted to form epinephrine, which is subsequently metabolised in liver and lung.
All catecholamines have very short biological half-lives (1–2 minutes) and a steady state plasma concentration is achieved within 5–10 minutes after the start of a constant infusion. This allows rapid titration of drug to a clinical end-point such as mean arterial pressure.
Epinephrine and norepinephrine infusions produce blood concentrations similar to those produced endogenously in shock states, whereas dopamine infusions produce much higher concentrations than those naturally encountered. Dopamine may exert much of its effect by being converted to norepinephrine, thus bypassing the rate-limiting (tyrosine hydroxylase) step in catecholamine synthesis.
The synthetic catecholamines are derivatives of dopamine (Fig. 90.3b). These agents are characterised by increased length of the carbon chain, which confers affinity for β-receptors. Dobutamine is a synthetic derivative of isoprenaline. These agents have relatively little affinity for α-receptors due to the configuration of the terminal amine, which differs from the endogenous catecholamines.
Epinephrine, norepinephrine and isoprenaline all have hydroxyl groups on the β-carbon atom of the side chain, and this is associated with 100-fold greater potency than dopamine or dobutamine.
Systemic effects
The systemic effects of any of these agents will vary greatly between patients and within individuals at different times. Adequacy of response is often unpredictable and depends on the aetiology of circulatory failure and systemic co-morbidities. In some patients, dramatic responses to small doses may occur, whereas in others large doses of inotropes may be required to support the failing circulation.
The classification of sympathomimetic agents into alpha and beta agonists, based on the above structure–function relationships, is only a crude predictor of systemic effects.
Epinephrine, norepinephrine and dopamine are all predominantly beta agonists at low doses, with increasing α-effects becoming evident as the dose is increased.
The synthetic catecholamines are all predominantly beta agonists.
Cardiovascular
The cardiovascular effects of the catecholamines under physiological conditions are shown in Table 90.2.

Norepinephrine, epinephrine and dopamine all tend to increase stroke volume, cardiac output and mean arterial pressure, with little change in heart rate. Dopamine is associated with an increased incidence of dysrhythmias.7 The effects on the peripheral vasculature are similar, with all agents increasing venous return without significant changes in calculated systemic vascular resistance.
Isoprenaline increases cardiac output predominantly by increasing heart rate and by moderate inotropy. This occurs without a significant change in blood pressure due to predominant β2-receptor-induced veno- and vasodilatation.
The profile of dobutamine is similar to isoprenaline, although increases in heart rate are not as pronounced. Both of these agents may decrease mean arterial pressure, particularly in hypovolaemic patients, due to reduced venous return caused by venodilatation. The adverse effects of dobutamine and isoprenaline on heart rate and mean arterial pressure may compromise patients with ischaemic heart disease.
In the failing myocardium, particularly in patients with cardiac failure following cardiopulmonary bypass or septic shock, endogenous stores of norepinephrine are markedly reduced.8 Furthermore, there may be significant desensitisation and down-regulation of cardiac β-receptors. In these situations, α1- and α2-receptors have an important role in maintaining inotropy and peripheral vasoresponsiveness.9 This may be expressed clinically as ‘tolerance’ or tachyphylaxis to catecholamines, particularly with predominantly beta agonists such as dobutamine. This phenomenon may explain the requirement for high doses of catecholamines in refractory shock states. Consequently, the role of beta agonists in patients with severe myocardial failure has been questioned.
Catecholamines have a significant effect on the venous circulation. These drugs primarily restore or maintain ‘stressed volumes’ of the capacitance vessels under pathological conditions, thereby maintaining or increasing cardiac output and mean arterial pressure. This is important in ‘vasoplegic’ states such as septic shock.5
In clinically used doses, intravenously administered catecholamines have minimal direct vasoconstrictive effects on conducting arterial vessels.
The development of peripheral gangrene in refractory septic shock has been attributed to catecholamine-induced vasoconstriction. There is little evidence to support this as the development of tissue gangrene in these situations primarily occurs as a consequence of intravascular thrombosis caused by sepsis-mediated coagulopathy.
Cerebral
Under physiological conditions, catecholamines do not normally cross the blood–brain barrier. Cerebral blood flow is maintained at a constant rate over a range of perfusion pressure by cerebral autoregulation. Under conditions where the integrity of the blood–brain barrier is altered, such as following traumatic brain injury and aneurysmal subarachnoid haemorrhage, exogenous catecholamines may directly enter the cerebral circulation.
The degree by which these agents directly affect the cerebral circulation following head injury is unknown, although there is some evidence suggesting that dopamine has a direct effect causing increased cerebral blood flow and intracranial pressure.10
Renal
The kidney is an efficient autoregulator, maintaining constant glomerular filtration and renal blood flow by neurohumoral mechanisms such as the renin–angiotensin–aldosterone system.11 All catecholamines will increase renal blood flow to a similar extent because of increased cardiac output and mean arterial pressure with a resultant natriuresis. Catecholamine-mediated increases in renal blood flow do not affect glomerular filtration rate.
A direct natriuresis may also result from inhibition of cAMP in the renal tubules. This effect has been attributed primarily to low doses of dopamine (2 µg/kg per min), although this occurs to a similar extent with epinephrine, norepinephrine and dobutamine. The use of low-dose ‘renal’ dopamine has been shown to be ineffective in preventing renal dysfunction in susceptible patients.12
Splanchnic
Splanchnic autoregulation is not as robust as the brain and kidney. Perfusion is more dependent on mean arterial pressure and the duality of the mesenteric and portal systems. Concerns about catecholamine-induced splanchnic vasoconstriction with mesenteric and hepatic ischaemia have not been substantiated.13
Dopamine and dopexamine have been promoted as selective splanchnic vasodilators, but there are no conclusive studies indicating a significant benefit over norepinephrine or epinephrine. Many studies have used gastric intramucosal pH (pHi), a surrogate measurement of splanchnic blood flow, as the primary end-point. However, as pHi remains an unvalidated measurement, the results of many of the comparative trials of the different sympathomimetics are inconclusive. All catecholamines are equally effective in increasing splanchnic perfusion by improving cardiac output and mean arterial pressure.
Metabolic
Catecholamine-mediated β-stimulation may result in hyperglycaemia, hypokalaemia and hypophosphataemia, which may need monitoring and correcting.
Epinephrine is associated with the development of hyperlactataemia and acidaemia, due to activation of pyruvate dehydrogenase. Although pH may fall to levels around 7.2, the acidosis is not associated with impaired tissue perfusion or cellular dysoxia. In most patients who are haemodynamically stable, this is a self-limiting phenomenon and is not associated with adverse outcomes.14,15
Non-catecholamines
Phosphodiesterase inhibitors
Phosphodiesterase inhibitors are compounds that cause non-receptor-mediated competitive inhibition of phosphodiesterase isoenzymes (PDE), resulting in increased levels of cAMP (see Fig. 90.2). Importantly, cAMP also affects diastolic heart function through the regulation of phospholamban, the regulatory subunit of the calcium pump of the sarcoplasmic reticulum. This enhances the rate of calcium resequestration and thereby diastolic relaxation.
For cardiovascular tissue, inhibition of isoenzyme PDE III is responsible for the therapeutic effects. Cardiac effects are characterised by positive inotropy and improved diastolic relaxation. The latter is termed lusitropy and may be beneficial in patients with reduced ventricular compliance or predominant diastolic failure.16
These agents also cause potent vasodilatation with reductions in preload, venous return and afterload as well as a reduction in pulmonary vascular resistance. The term ‘inodilation’ has been used to describe this dual haemodynamic effect.
Tolerance is not a feature. These agents may have a place in the management of patients with β-receptor down-regulation by causing intrinsic inotropic stimulation and by sensitising the myocardium to beta agonists. Other actions include inhibition of platelet aggregation and reduction of post-ischaemic reperfusion injury.
The pharmacokinetics of phosphodiesterase inhibitors are markedly different from catecholamines. Drug half-lives may be prolonged and excretion is predominantly renal. Hypotension may result from vasodilatation and combined use with catecholamines (e.g. norepinephrine or epinephrine) may be necessary to maintain mean arterial pressure.
Phosphodiesterase inhibitors that have been used in clinical practice include the bipyridine derivatives amrinone and milrinone, the imidazolones enoximone and piroximone, and the calcium sensitiser levosimendan. The cardiovascular effects are similar.
Milrinone is the most commonly used drug in clinical practice, with the latter exhibiting more inotropic effects than vasodilatation. Enoximone is more rapidly metabolised, but the metabolite is active and its cardiovascular effects persist for some hours.
Levosimendan is a dose-dependent selective phosphodiesterase inhibitor with a unique action on myocardial calcium metabolism.17 By increasing myofilament calcium sensitivity throughout the cardiac cycle by binding to cardiac troponin C, associated conformational changes induce improved inotropic and lusitropic function. Vasodilatation is also induced through ATP-sensitive potassium channels. Calcium-sensitive actions predominate at low doses, whereas PDE-inhibition effects predominate at higher doses. The half-life of levosimendan is shorter than that of older PDE III inhibitors (approximately 1 hour) and it may be given by infusion.
Historical drugs
Digoxin
Digitalis glycosides have been used for the treatment of heart failure for 200 years and the vagotonic effects used to control the ventricular response in selected supraventricular tachyarrhythmias. Its use in the ICU has been superseded by catecholamines and antiarrhythmics such as amiodarone, although the role of digoxin in chronic heart failure is well established.
The role of digoxin in acute cardiac failure is limited. It has minimal effects as an inotrope and evaluation of its efficacy is difficult. The potential for toxicity in the critically ill patient is increased by hypokalaemia, hypomagnesaemia, hypercalcaemia, hypoxia and acidosis. Toxicity is manifested by dysrhythmias that may assume any form including supraventricular tachyarrhythmias, bradycardia, ventricular ectopy and conduction block at any level.
Glucagon
Glucagon is a naturally occurring polypeptide that directly stimulates adenyl cyclase via specific receptors to increase cAMP concentration in myocardial cells resulting in positive inotropy without producing myocardial excitability. No definitive cardiovascular role for this agent has been established apart from anecdotal reports of its use in severe beta-blocker and tricyclic poisoning.
Thyroid hormone
Thyroid hormone is required for synthesis of contractile proteins and normal myocardial contraction. It is also a regulator of the synthesis of adrenergic receptors. Its use is limited in critically ill patients, but may have a limited, although unproven, role in haemodynamic support of brain-dead organ donors as a catecholamine-sparing agent.
Selective vasopressors
Vascular responsiveness is mediated via adrenergic receptors: α-mechanisms predominantly cause vasoconstriction; β-mechanisms, specifically β2-receptors, mediate vasodilatation (see Table 90.2).
Norepinephrine, epinephrine and dopamine have variable effects on the peripheral vasculature and should not be regarded principally as vasoconstrictors or vasopressors.
There are few selective ‘vasoconstrictors’ that predominantly have a role in vasodilated states such as regional (epidural) anaesthesia or selected patients with acute spinal injury. Their utility in critically ill patients remains limited and they have an unproven role in patients with catecholamine-resistant septic shock and cardiopulmonary resuscitation.
Phenylephrine and metaraminol
These agents are direct-acting alpha-2 agonists that are selective vasoconstrictors, both venous and arterial, with minimal beta activity. They have similar pharmacokinetics to catecholamines and may be given by infusion. In patients with normal sympathetic tone, these drugs may cause reflex bradycardia, particularly following bolus administration.
Vasoregulatory agents
In addition to adrenergic regulation, neurohumoral influences have a ‘permissive’ or regulatory role in maintaining vasomotor tone. These are mediated through the renin–aldosterone–angiotensin axis and local mediators such as vasopressin, corticosteroids, nitric oxide and endothelin.
The response of the whole neurohumoral system may become blunted in conditions such as severe sepsis, where qualitative and quantitative changes may occur. In this context, failure of vasomotor responsiveness may be considered as part of the multiple organ failure.
Vasopressin
Specific vasopressinergic receptors (V1, V2) have been identified in association with sympathetic terminals. Vasopressin is a naturally occurring peptide secreted by the posterior pituitary gland. Reduced serum levels of vasopressin have been demonstrated in septic shock and following cardiopulmonary bypass, suggesting an inflammatory-mediated mechanism. Levels are maintained during cardiogenic shock.
A proportion of patients with severe septic shock requiring high levels of catecholamines to support the circulation may respond to low doses of infused vasopressin (0.04 U/h). However, although vasopressin may improve catecholamine responsiveness in patients with less-severe shock, no benefit in mortality has been demonstrated and there is uncertainty about its use in shock states.18
Steroids
The role of steroid supplementation in circulatory failure has been studied for many years. Although immunosuppressive or anti-inflammatory doses have been shown to be ineffective, particularly in septic shock, replacement of ‘stress response’ doses (approximately 100–200 mg hydrocortisone per day) have been shown to improve catecholamine vasoresponsiveness in selected patients with septic shock.19 However, the role of steroids in shock states remains uncertain, despite two large trials, and they are not routinely recommended.20,21
Clinical uses
Currently, there are no definitive studies comparing the efficacy of one inotrope (or combination of inotropes) over another in terms of improving survival.22
Drug selection
In most instances, individual experience and preference determine selection of inotrope(s).
On a pathobiological basis, exogenous catecholamines are essentially used to augment endogenous mechanisms that may be failing at a number of levels. In this context, vasoactive therapy should be regarded as ‘cardiovascular neuroendocrine augmentation therapy’.
Norepinephrine or epinephrine may be considered as the first-line drug in most causes of circulatory failure. An emerging body of evidence confirms this statement.7,14,15
Dopamine predominantly acts as a precursor of norepinephrine and may be used as an alternative to norepinephrine, although it is associated with an increased incidence of arrhythmias.7
Prediction of the response of an individual to a catecholamine is problematic as marked inter- and intra-individual variability to the response of inotropic agents may occur.
The haemodynamic and metabolic response of an agent must be carefully monitored and evaluated. If there is not a satisfactory response, or if undesirable effects are obtained, the dose or agent should be changed.
Monitoring
Accurate monitoring of the circulation is essential in patients with circulatory failure to assess baseline parameters and the response of vasoactive drugs.
Clinical assessment of the circulation remains the cornerstone of monitoring these patients and includes frequent assessment and recording of pulse rate and rhythm, blood pressure, adequacy of peripheral perfusion, skin turgor, level of consciousness and urine output.
The majority of patients with circulatory failure managed in the ICU require haemodynamic monitoring as clinical signs may be masked or influenced by sedation, ventilation or organ failure. As a minimum, all patients receiving vasoactive drugs in all but trivial doses should have accurate monitoring of mean arterial pressure, ideally with an intra-arterial catheter referenced to the aortic root and an assessment of volume status such as central venous pressure.
In selected patients with primary myocardial failure, an assessment of cardiac output may be useful to quantify baseline function and to assess the response of the heart to drug therapy. Measurement of cardiac output may be done non-invasively using transthoracic or transoesophageal echocardiography or invasively using a pulmonary catheter, ideally using a continuous cardiac output display system, although the use of these catheters has decreased substantially.
Systemic vascular resistance is frequently calculated and used as a surrogate index of afterload. However, the clinical utility of systemic vascular resistance is limited to providing a crude estimate of global vascular tone, as it does not reflect afterload, arteriolar tone or venous return. Consequently, systemic vascular resistance should not be used as a criterion for selection of vasoactive drug or as a titratable end-point.
Restoration of reduced tissue perfusion is a primary aim of vasoactive therapy. This may be assessed clinically by improvements in urine output, serum urea and creatinine, reversal of metabolic acidosis and reduction in serum lactate.
Dosages and drug administration
Vasoactive drugs, such as catecholamines or vasopressors, are administered by continuous infusion through a dedicated central venous catheter using drug delivery systems such as infusion pumps or syringe drivers.
Infusion lines should be free of injection portals and clearly marked with identifying labels.
Concentrations of infusions should be standardised in accordance with individual unit protocols. Suggested infusion concentrations are shown in Table 90.3.
Table 90.3
Infusion concentrations of commonly used vasoactive drugs
| AGENT | INFUSION CONCENTRATION | DOSE |
| Epinephrine | 6 mg/100 mL 5% dextrose | Titrate mL/h (= µg/min) |
| Norepinephrine | 6 mg/100 mL 5% dextrose | Titrate mL/h (= µg/min) |
| Dopamine | 400 mg/100mL 5% dextrose | Titrate mL/h (approx µg/kg/min) |
| Dobutamine | 500 mg/100 mL 5% dextrose | Titrate mL/h (approx µg/kg/min) |
| Isoprenaline | 6 mg/100 mL 5% dextrose | Titrate mL/h (= µg/min) |
| Milrinone | 10 mg/100 mL 5% dextrose | Loading dose: 50 µg/kg over 20 min Infusion: 0.5 µg/kg/min |
| Levosimendan | 25 mg/100 mL 5% dextrose | Loading dose: 24 µg/kg over 10 min Infusion: 0.1 µg/kg/min |
| Phenylephrine | 10 mg/100 mL 5% dextrose | Titrate mL/h ( = 100 µg/h) |
| Metaraminol | 100 mg/100 mL 5% dextrose | Titrate mL/h (= mg/h) |
| Ephedrine | 300 mg/100 mL 5% dextrose | Titrate mL/h (= 3 mg/h) |
| Vasopressin | 20 units/20 mL 5%dextrose | 2.4 mL/h (0.04 u/min) |
| Hydrocortisone | 100 mg/100 mL 5% dextrose | Loading dose: 100 mg Infusion: 0.18 mg/kg/h |
These infusions in mL/hour approximate µg/min. Absolute doses with regard to body weight are not relevant; rather it is the titrated clinical effect. Vasoactive drugs are usually prescribed as a titration against a desired mean arterial pressure.
Cardiopulmonary resuscitation
Epinephrine has been used for circulatory collapse at least since 1907. The International Liaison Committee on Resuscitation guidelines recommends epinephrine as first-line vasoactive drug in cardiopulmonary resuscitation. Doses are 1 mg intravenously every 3 minutes.23
Epinephrine is recommended as first-line therapy for ‘medical pacing’ for severe bradyarrhythmias that do not respond to atropine. Isoprenaline has traditionally been used for this purpose; however, its use has been superseded by epinephrine due to concerns about efficacy and lack of alpha-adrenergic activity.
Cardiogenic shock
Theoretically, catecholamine infusions may confer some advantages in cardiogenic shock, particularly in association with acute myocardial infarction. In patients with systolic heart failure, epinephrine, norepinephrine, dopamine and dobutamine have been shown to cause satisfactory short-term effects. This may allow the myocardium time to recover from post-ischaemic ‘stunning’, particularly after revascularisation. However, no increased long-term survival due to their use has been demonstrated.24
The role of phosphodiesterase inhibitors (e.g. milrinone) and calcium sensitisers (e.g. levosimendan) in acute heart failure has yet to be determined, but they may have a potential role in patients with diastolic heart failure, particularly those with associated high impedance states such as aortic stenosis and pulmonary hypertension. Due to their non-adrenergic mechanism of action, these agents may be useful in patients who are ‘resistant’ to catecholamines.
Separation from cardiopulmonary bypass
Numerous combinations of catecholamines have been used successfully to wean patients from cardiopulmonary bypass. However, there are no definitive studies demonstrating significant benefits of one catecholamine over another. Similarly, the question whether mechanical support devices, such as intra-aortic counterpulsation, offer a significant advantage over inotropes following cardiac surgery remains unanswered.
Epinephrine and norepinephrine have been found to increase cardiac output with little increase in heart rate or afterload and are often regarded as first-line drugs. Dobutamine may be associated with vasodilatation and hypotension.
There is no conclusive evidence that the catecholamines, including norepinephrine, cause vasospasm of arterial conduits in clinically used doses.
Phosphodiesterase inhibitors, such as milrinone and levosimendan, either as sole agents or in conjunction with epinephrine or norepinephrine, have been used with success. These may have a role following mitral valve replacement in patients with pulmonary hypertension or preoperative diastolic failure.25
Cardiopulmonary bypass may be associated with a systemic inflammatory response syndrome characterised by a hyperdynamic, vasodilated ‘low systemic vascular resistance’ state. Norepinephrine is frequently advocated as a ‘vasopressor’ agent in this context to restore mean arterial pressure, which may be reduced as a consequence. Although catecholamines may be required to achieve appropriate target mean arterial pressure and cardiac output, caution should be applied if high doses (e.g. >30 µg/min norepinephrine) are required. This may be associated with tachyphylaxis and potentiation of catecholamine dependency.26
Right ventricular failure
Right ventricular infarction and major pulmonary embolism may be associated with acute right ventricular failure. Right ventricular depression may also occur in severe sepsis. Restoration of preload is critical in these conditions, as the failing right ventricle is particularly susceptible to reductions in preload.27
Inotropes such as norepinephrine and epinephrine are regarded as first-line drugs in these situations in order to maintain adequate mean arterial pressure so that right coronary artery perfusion, which occurs throughout the cardiac cycle, is maintained.
Septic shock
The cardiovascular effects of the sepsis syndrome and septic shock are complex and range from a hyperdynamic, vasodilated state to one of increasing myocardial failure and paralysis of the peripheral vasculature (vasoplegia). The latter represents inability of the venous circulation to respond to endogenous or exogenous catecholamines with resultant venous pooling.
An increasing body of literature now recommends the use of catecholamines as first-line agents in the treatment of septic shock. The Surviving Sepsis Campaign guidelines recommend either norepinephrine or epinephrine be used as the first-line choice vasopressor.28 However, the basis for this recommendation is limited as there is no high-quality evidence to recommend one catecholamine over another.22 Concerns about adverse metabolic and splanchnic side-effects of epinephrine are unsubstantiated, and there is an emerging body of evidence that norepinephrine and epinephrine are equally effective in treating septic shock.14,15 There is evidence that dopamine may be associated with adverse outcomes7 and its use is being questioned in the absence of superiority to norepinephrine or epinephrine.
The efficacy of dobutamine as a sole agent or in combination with norepinephrine in septic shock is questionable and appears to add little to the efficacy of norepinephrine or epinephrine in terms of resolution of shock or mortality.
Doses required to achieve adequate mean arterial pressure in septic shock may vary: norepinephrine or epinephrine infusions (up to 40 µg/min) may be necessary. Patients who develop marked catecholamine dependency, in the absence of other acute remediable causes such as active infection, may respond to low doses of vasopressin or ‘stress response’ doses of hydrocortisone, although there is marked inter-individual variation in response to these drugs.
Anaphylaxis
Epinephrine is the drug of choice for anaphylactic reactions and for life-threatening bronchospasm, as it blocks mediator release and specifically reverses end-organ effects (see Ch. 66).
A dose of 0.1 mg, as 1 mL of 1 : 10 000 solution, may be injected subcutaneously, intramuscularly or intravenously. Repeated doses or infusions of up to 100 µg/min may be required. A strong slowing pulse indicates a pressor effect and provides a useful clinical end-point for the rate of infusion. This alpha-agonist effect is probably also of considerable importance in anaphylaxis, as deaths are frequently due to prolonged refractory hypotension caused by acute biventricular failure. Early intravenous fluid therapy is also important.
Renal protection
‘Renal’ dose dopamine (2 µg/kg per min) had been advocated for many years as a renal protective agent by causing renal vasodilatation. However, this has not been substantiated in controlled clinical trials in susceptible patients or as an adjunctive agent with other inotropes in septic shock.12 Consequently low-dose dopamine is no longer recommended.
Cerebral perfusion pressure
Augmentation of cerebral perfusion pressure is an important strategy in patients with pathological reductions in cerebral blood flow, such as traumatic brain injury and subarachnoid haemorrhage. Norepinephrine and epinephrine have been used to augment cerebral perfusion pressure, although there is no conclusive evidence to recommend one drug over another.10
References
1. Guyton, AC, Lindsay, AW, Kaufmann, BN. Effect of mean circulatory filling pressure and other peripheral circulatory factors on cardiac output. Am J Physiol. 1955; 180:463–468.
2. Hamzauoi, O, Georger, J-F, Monnet, X, et al. Early administration of norepinephrine increases cardiac preload and cardiac output in septic patients with life-threatening hypotension. Crit Care. 2010; 14(4):R142.
3. Henderson, WR, Griesdale, DE, Walley, KR, et al. Clinical review: Guyton – the role of mean circulatory filling pressure and right atrial pressure in controlling cardiac output. Crit Care. 2010; 14:243.
4. Hein, L. Adrenoceptors and signal transduction in neurons. Cell Tissue Res. 2006; 326:541–551.
5. Monnet, X, Jabot, J, Maizel, J, et al. Norepinephrine increases cardiac preload and reduces preload dependency assessed by passive leg raising in septic shock patients. Crit Care Med. 2011; 39:689–694.
6. Lamba, S, Abraham, WT. Alterations in adrenergic receptor signaling in heart failure. Heart Fail Rev. 2000; 5:7–16.
7. De Backer, D, Biston, P, Devriendt, J, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med. 2010; 362:779–789.
8. Levy, RJ, Deutschman, CS. Evaluating myocardial depression in sepsis. Shock. 2004; 22:1–10.
9. Jensen, BC, O'Connell, TD, Simpson, PC. Alpha-1-adrenergic receptors: targets for agonist drugs to treat heart failure. J Mol Cell Cardiol. 2011; 51:518–528.
10. Myburgh, JA. Driving cerebral perfusion pressure with pressors: how, which, when? Crit Care Resusc. 2005; 7:200–205.
11. Bellomo, R, Wan, L, Langenberg, C, et al. Septic acute kidney injury: the glomerular arterioles. Contrib Nephrol. 2011; 174:98–107.
12. Bellomo, R, Chapman, M, Finfer, S, et al. Low-dose dopamine in patients with early renal dysfunction: a placebo-controlled randomised trial. Australian and New Zealand Intensive Care Society (ANZICS) Clinical Trials Group. Lancet. 2000; 356:2139–2143.
13. Duranteau, J, Sitbon, P, Teboul, JL, et al. Effects of epinephrine, norepinephrine, or the combination of norepinephrine and dobutamine on gastric mucosa in septic shock. Crit Care Med. 1999; 27:893–900.
14. Myburgh, JA, Higgins, A, Jovanovska, A, et al. A comparison of epinephrine and norepinephrine in critically ill patients. Intensive Care Med. 2008; 34:2226–2234.
15. Annane, D, Vignon, P, Renault, A, et al. Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomised trial. Lancet. 2007; 370:676–684.
16. Erhardt, L. An emerging role for calcium sensitisation in the treatment of heart failure. Expert Opin Investig Drugs. 2005; 14:659–670.
17. Innes, CA, Wagstaff, AJ. Levosimendan: a review of its use in the management of acute decompensated heart failure. Drugs. 2003; 63:2651–2671.
18. Russell, JA, Walley, KR, Singer, J, et al. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med. 2008; 358:877–887.
19. Annane, D, Bellissant, E, Bollaert, PE, et al. Corticosteroids in the treatment of severe sepsis and septic shock in adults: a systematic review. JAMA. 2009; 301:2362–2375.
20. Annane, D, Sebille, V, Charpentier, C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002; 288:862–871.
21. Sprung, CL, Annane, D, Keh, D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008; 358:111–124.
22. Havel, C, Arrich, J, Losert, H, et al. Vasopressors for hypotensive shock. Cochrane Database Syst Rev. (5):2011.
23. International Liason Committee on Resuscitation. Online. Available http://www. ilcor. org, 2012.
24. Mann, HJ, Nolan, PE, Jr. Update on the management of cardiogenic shock. Curr Opin Crit Care. 2006; 12:431–436.
25. Raja, SG, Rayen, BS. Levosimendan in cardiac surgery: current best available evidence. Ann Thorac Surg. 2006; 81:1536–1546.
26. Mebazaa, A, Pitsis, AA, Rudiger, A, et al. Clinical review: practical recommendations on the management of perioperative heart failure in cardiac surgery. Crit Care. 2010; 14:201.
27. Haji, SA, Movahed, A. Right ventricular infarction-diagnosis and treatment. Clin Cardiol. 2000; 23:473–482.
28. Dellinger, RP, Levy, MM, Rhodes, A, et al. Surviving Sepsis Campaign: international guidelines for the management of severe sepsis and septic shock: 2012. Intensive Care Med. 2013; 39:165–228.