Miscellaneous Bone Conditions
acroosteolysis: resorption of bone that involves the distal regions of the limbs but most commonly the phalanges of the fingers.
algodystrophy syndrome: association of pain and dystrophic changes in bone.
apophysitis: inflammation of an apophysis. Depending on the location, specific types may be referred to as:
Iselin d.: base of the fifth metatarsal
Osgood-Schlatter d.: tibial apophysis at the insertion of the patellar tendon
Sever d.: apophysitis of the heel bone at the insertion of the Achilles tendon
Scheuermann d.: osteochondrosis of the vertebral epiphysis in juveniles
aseptic necrosis: osteonecrosis (bone death) caused by vascular insult, usually at the end of a bone. In adults, the most common symptom producing aseptic necrosis occurs in the femoral head (avascular necrosis). In children, aseptic necrosis is called epiphyseal ischemic necrosis or epiphyseal aseptic necrosis. The precise pathologic condition of these diseases is debatable; therefore the term is used here in reference to epiphyseal osteochondritis or by area:
Assmann d.: head of first metatarsal
Buchman d.: medial cuneiform
Freiberg d.: second metatarsal head
Iselins d.: base of the fifth metatarsal
Kappis d.: talus
Kienböck d.: lunate bone of wrist
Köhler d.: tarsal navicular; sometimes of the patella
Kümmell disease: vertebral body
Lance d.: cuboid
Legg-Calvé-Perthes d.: femoral head; also called Legg-Perthes d., Perthes d., and coxa plana
Panner d.: capitellum of the humerus
Silfverskiöld d.: calcaneus
Thiemann d.: proximal phalanges
Wagner d.: base of the first metatarsal
Aseptic necrosis is also called avascular necrosis. The following are classification systems for aseptic necrosis.
International Classification for Osteonecrosis of the Femoral Head
Stage 0: bone biopsy results consistent with avascular necrosis; normal findings in all other tests.
Stage I: positive scintiscan or magnetic resonance image (MRI); lesions subdivided into medial, central, or lateral depending on location and involvement of femoral head.
I-A: < 15% involvement of femoral head
I-B: 15%–30% involvement of femoral head
I-C: > 30% involvement of femoral head
Stage II: radiographic abnormalities (mottled appearance of femoral head, osteosclerosis, cyst formation, and osteopenia); no signs of collapse of femoral head on radiographs or computerized tomography scan; positive scintiscan and MRI; no changes in acetabulum; lesions subdivided into medial, central, or lateral depending on location of involvement of femoral head.
II-A: < 15% involvement of femoral head
II-B: 15%–30% involvement of femoral head
II-C: > 30% involvement of femoral head
Stage III: crescent sign; lesions subdivided into medial, central, or lateral depending on location of involvement of femoral head.
III-A: < 15% crescent sign or < 2-mm depression of femoral head
III-B: 15%–30% crescent sign or 2- to 4-mm depression of femoral head
III-C: > 30% crescent sign or 4-mm depression of femoral head
Stage IV: Articular surface flattened radiographically and joint space shows narrowing; change in acetabulum with evidence of osteosclerosis, cyst formation, and marginal osteophytes.
Lichtman Classification for Osteonecrosis of the Lunate in the Wrist
Stage I: linear line possibly seen across lunate or no radiographic finding.
Stage II: definite density changes in lunate compared with other carpal bones.
Stage III: lunate collapse with proximal migration of capitate.
Stage IV: Stage III plus degenerative changes in the carpus.
Steinberg Classification for Ostenecrosis of the Hip (Fig. 2-4)
Stage 0: normal radiograph, bone scan, and MRI
Stage I: normal radiograph; abnormal bone scan or MRI
Stage II: sclerosis or cyst formation in femoral head
Stage III: subchondral collapse (crescent sign) without flattening
Stage IV: flattening of femoral head without joint narrowing or acetabular involvement
A: mild (15% of surface and < 2-cm depression)
B: moderate (15%–30% of surface and 2- to 4-mm depression)
C: severe (> 30% of surface or > 4-mm depression)
Table Continued
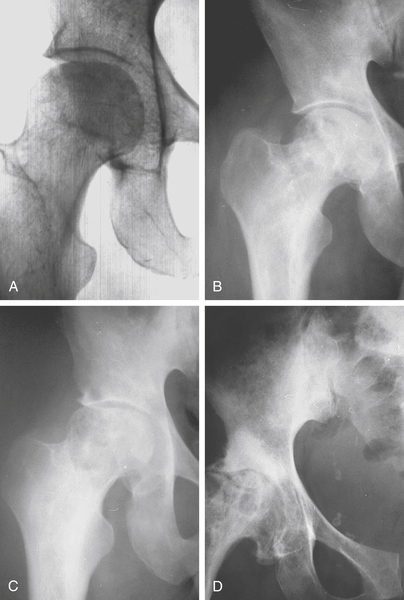
Stage V: flattening of head with joint narrowing or acetabular involvement
A: mild (15% of surface and < 2-cm depression)
B: moderate (15%–30% of surface and 2- to 4-mm depression)
C: severe (> 30% of surface or > 4-mm depression)
Stage VI: advanced degenerative changes
bone infarct: area of bone where blood supply is interrupted.
bone island: small areas of compact but microscopically normal bone that appear as 0.5- to 1-cm areas on radiographs.
bone spur: ossification of ligamentous or muscular attachment to bone. Generally applied to any bony excrescence seen on radiographs, but specifically refers to a portion of ligament or tendon that has turned to bone at the attachment to bone. The most common areas include the heel, patella, humeral epicondyles, and vertebral body margins.
brown tumor: brown-appearing lesion in bone secondary to hyperparathyroidism; also called osteoclastoma.
caisson disease: avascular necrosis of bone (and soft tissue) caused by sudden increase and release in air pressure causing infarct; also called diver’s disease.
cleidocranial dysostosis: autosomal dominant defect of a core binding protein (cfba-1, RUNX2) resulting in failure to form collarbones and portions of the skull.
condensing osteitis of the clavicle: rare and benign disorder of unknown origin affecting the medial clavicle, characteristically in women of late childbearing age. The level of discomfort varies, and radiographs reveal a slight expansion of the medial one third of the clavicle.
congenital pseudarthrosis: inborn propensity for breakdown of integrity of midshaft of tibia with formation of a false joint. There are six types:
Type I: congenital anterior bowing and defect in tibia. There may be other congenital deformities.
Type II: congenital anterior bowing and hourglass deformity of tibia. Fracture occurs usually before 2 years of age.
Type III: bone cyst forms first, then bowing or fracture.
Type IV: sclerotic bone, then fracture occurs.
Type V: dysplastic fibula may develop pseudarthrosis of tibia or fibula.
Type VI: intraosseous neurofibroma or schwannoma may develop, but usually does not result in pseudarthrosis.
cortical fibrous dysplasia: benign anomaly of bone cortex, usually found in children and characterized by a cystic-appearing lesion on radiographs. Microscopically, this lesion is characterized by a fibrous replacement of cortex with some trabecular bone; also called ossifying fibroma of long bone, intracortical fibrous dysplasia.
exostosis: excess bone formation, usually near a joint.
hypertrophic exostosis: sometimes used to describe excess bone formation in osteoarthritis.
multiple hereditary exostoses: autosomal dominant disorder of EXT or EXT2 genes resulting in multiple bony excrescences growing from cortical surfaces. May be pedunculated or sessile. Also called Jaffe d. and Beggel-Hansen d.
fibrodysplasia ossificans progressiva: autosomal dominant disease of BMP 4 and autosomal recessive disorder of BMP 1 receptor type 1A (ACVR1) resulting in connective tissue disease in which soft tissue ossification leads to skeletal malformation and disability; also called myositis ossificans progressiva, hyperplasia fascialis ossificans progressiva, myositis fibrosa generalisata, and fibrositis ossificans progressiva.
fibrogenesis imperfecta ossium: rare inherited disorder in which bone in adults is replaced with collagen-deficient tissue throughout the skeleton, resulting in multiple fractures.
giant cell reparative granuloma: common benign lesion of the jaw characterized by a fibrous background within which there are scattered multinucleated giant cells. A multicentric form is seen in the small bones of the hands and feet.
heterotopic ossification: formation of normal bone at ectopic soft tissue locations. In the acquired form, abnormal bone formation usually follows trauma, burns, or surgery, and will sometimes occur around joints after a closed head injury. Two rare heritable and developmental forms are fibrodysplasia ossificans and progressive osseous heteroplasia.
Brooker Classification for Heterotopic Ossification
Class I: islands of bone within the soft tissues about the hip.
Class II: bone spurs from the pelvis or proximal end of the femur, leaving at least 1 cm between opposing bone surfaces.
Class III: bone spurs from the pelvis or proximal end of the femur, reducing the space between opposing bone surfaces to less than 1 cm.
Class IV: apparent bone ankylosis of the hip.
Hamblen Classification of Heterotopic Ossification
Grade 0: no ossification.
Grade I: formation of new bone involving less than one third of the area of the hip occupied by the femoral head and capsule.
Grade II: formation of new bone involving between one- and two-thirds of the same area of the hip.
Grade III: formation of new bone involving more than two-thirds of that area of the hip.
hypophosphatasia: rare (1 in 100,000) usually autosomal-recessive bone disorder resulting from deficient alkaline phosphatase activity, characterized by defective mineralization of the skeletal and dental structures leading to an appearance of rickets and very low alkaline phosphatase. There are four types based on the severity of alkaline phosphatase abnormality: perinatal hypophosphatasia with neonatal death, infantile hypophosphatasia presenting in the first year of life with severe skeletal fractures, childhood hypophosphatasia with rickets appearance and abnormal teeth, and adult hypophosphatasia with osteoporosis and fractures.
infantile cortical hyperostosis: painful hyperostosis with involvement of long bones and the mandible. This usually occurs in infants 5 months of age and younger, and is associated with irritability, fever, and soft tissue swelling; also called Caffey d.
intraosseous lipoma: rare condition of benign fatty tumor that develops in the medullary canal of a long bone.
intraosseous lipomatosis: very rare disorder of progressive systemic development of leg and foot lipomas in medullary canals, producing bone pain and pathologic fractures.
intraosseous pneumatocyst: rare benign small pocket of air that appears usually in iliac bone. No treatment is required.
longitudinal epiphyseal bracket: ossification anomaly in which an abnormal arcuate or C-shaped secondary ossification center brackets a tubular bone in the hand or foot.
malacoplakia: disease that usually involves the gut and has a probable infectious cause. Histiocytes respond with the formation of Michaelis-Gutmann bodies. Bone lesions are rare and can be destructive.
melorheostosis: form of osteosclerosis or hyperostosis (dense bone); linear longitudinal thickenings of the shaft of long bones, very rare, resulting in a candle wax (dripping) appearance of the bone.
milk-alkali disease: excess calcium in tissue resulting from heavy ingestion of milk and certain antacids.
myelofibrosis: replacement of bone marrow by fibrous tissue.
ochronosis: hereditary error of protein metabolism marked by accumulation of homogentisic acid resulting in degenerative arthritis and a characteristic blackening of cartilage.
ossifying fibroma of the jaw: fibroma of the jaw with histologic appearance of small areas of ossification. This is in distinction to the rare ossifying fibroma of long bones.
pachydysostosis: enlargement of fibular length resulting in bowing of the leg.
periostitis: inflammation of bone covering (periosteum); usually the result of an infection such as syphilis.
progressive diaphyseal dysplasia: neuromuscular dystrophy associated with general wasting; abnormally formed shafts of long bones; also called Engelmann d.
progressive osseous heteroplasia: heritable disease characterized by focal dermal ossification with progression to intramembranous ossification of subcutaneous fat and deeper tissues leading to ankylosis. Hemimelic progressive osseous heteroplasia is a very rare condition in which only one side of the body is involved.
pulmonary osteodystrophy: hypertrophic cortical bone changes occurring near joints in the long bones of patients with chronic lung disease.
pyknodysostosis: condition marked by patchy areas of thickening of the cortex of bone.
regional migratory osteoporosis: most often seen in middle-aged men; characterized by arthralgias of lower limb joints, severe intercurrent osteoporosis, with symptoms lasting 6 to 9 months with recovery.
rickets: failure of deposition of bone salts within the organic matrix of cartilage and bone associated with stunting of growth and bone deformities.
Rothmund-Thomson syndrome (RTS): characterized by growth retardation, thin eyebrows and lashes, juvenile cataracts, sunlight sensitivity, hypogonadism, and teeth abnormalities. This is associated with an increased risk for cancer, such as cutaneous epitheliomas (basal, squamous), gastric adenocarcinoma, fibrosarcoma, and osteosarcoma.
skeletal amyloidosis: deposition of amyloid β-microglobulin material in bone that produces bubbly appearing lesions on radiographs. Condition is associated with plasma cell dyscrasias, primary systemic amyloidosis, focal amyloidosis, and those undergoing hemodialysis for chronic renal insufficiency.
slipped capital femoral epiphysis (SCFE): gradual or sudden movement of the femoral head toward a posterior and medial direction; usually occurs in preteenage children. In the United States, the acronym SCFE is so common that the slang word “skiffy” is used.
solitary fibromatosis of bone: similar to generalized fibromatosis, except bone lesion is isolated to one location. Systemic symptoms do not occur.
subperiosteal giant-cell reparative granuloma: self-limited condition seen in older adults characterized by subperiosteal bony lesions located in cortical bone of the diaphysis with giant cells and reparative cells with bone formation; also called subperiosteal ABC, periostitis ossificans, florid reactive periostitis, and pseudomalignant fibroosseous tumors.
transient osteopenia: decreased bone mass usually following immobilization or injury. Most often, bone mass is recovered. It may be seen as a spontaneous event in the juvenile hip.
uncommitted metaphyseal lesion: benign but radiologically aggressive-appearing lesion seen in the proximal metaphysis of children, microscopically characterized by whorls of fibrous tissue, new bone, giant cells, and vascular components.
unicameral bone cyst: benign bone anomaly in which a fluid-filled cavity is seen in the metaphysis of a long bone of a child. Microscopically, the cavity is lined with fibrous stroma, curlicues of trabecular bone similar to fibrous dysplasia, and cholesterol clefts.
weaver’s bottom: ischial gluteal bursitis often seen in patients with a sedentary occupation.