Histiocytoses
• Group of disorders in which the predominant cell type is a Langerhans cell, a mononuclear cell/macrophage, or a dermal dendrocyte.
• Two major groups: Langerhans cell histiocytoses and non-Langerhans cell histiocytoses.
• Within these two main groups, the disorders overlap and form a clinicopathologic spectrum.
Langerhans Cell Histiocytoses
• Langerhans cells, which represent the major antigen-presenting cells of the epidermis, are CD1a-positive, S-100 protein-positive, langerin (CD207)-positive.
• Langerhans cells have Birbeck granules by electron microscopy.
• Common in children ages 1–3 years, but can occur at any age.
• Traditionally classified into the following categories but they exist along a clinical spectrum.
Letterer–Siwe Disease (Multifocal, Multisystem)
• Multisystem involvement (skin, lung, liver, lymph nodes, bone, bone marrow).
• Classically in those <1–2 year(s) of age.
• Small, 2- to 3-mm, skin-colored to pink papules, which can coalesce; sometimes admixed with pustules or vesicles.
• Favors scalp, flexural neck, axilla, perineum, trunk (Fig. 76.1).
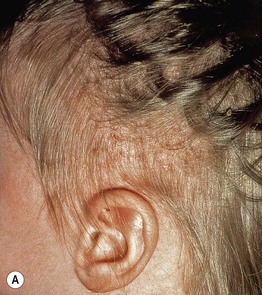
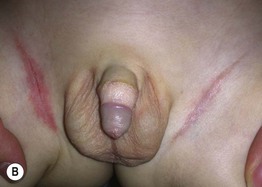
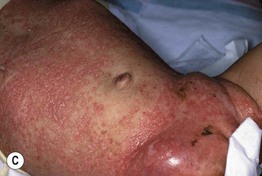
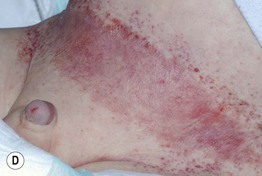
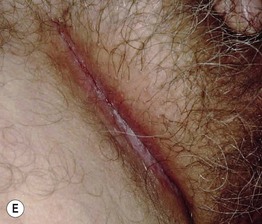
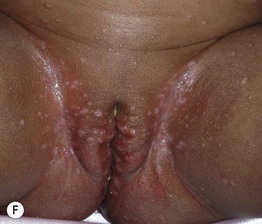
Fig. 76.1 Langerhans cell histiocytosis – clinical spectrum. A Scalp involvement may initially be diagnosed as seborrheic dermatitis; however, there are usually more discrete papules and crusting. B Pink, thin plaques with fissuring along the inguinal crease can also resemble seborrheic dermatitis. C Advanced disease with coalescence of papules into large plaques and prominent inguinal lymphadenopathy. D The presence of petechiae and purpuric papules is a clue to the diagnosis. E In an adult, the clinical presentation of inguinal involvement is similar to that of infants. F In patients with darkly pigmented skin, the papules can be hypopigmented. B, Courtesy, Richard Antaya, MD; D, F, Courtesy, Julie V. Schaffer, MD.
• Secondary changes include scale, crusts, petechiae/purpura.
• DDx includes seborrheic dermatitis, intertrigo, scabies, impetigo, atopic dermatitis, contact dermatitis.
Hand–Schüller–Christian Disease
• Triad of diabetes insipidus (in 30%), exophthalmos, osteolytic bone lesions (especially in cranium).
• Chronic, progressive course.
• Classically in children ages 2–6 years.
• 30% with skin lesions, sometimes resembling those of Letterer–Siwe disease; older lesions often more xanthomatous (i.e. yellow in color due to accumulations of lipid within macrophages) and occasionally lesions are ulcerated.
Eosinophilic Granuloma (Unifocal)
• Localized variant (Fig. 76.2).
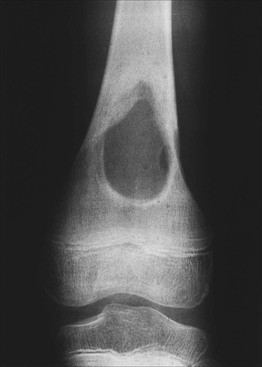
Fig. 76.2 Langerhans cell histiocytosis (eosinophilic granuloma of the bone). Radiography of the femur shows a large well-circumscribed osteolytic lesion. Courtesy, Edward McCarthy, MD.
• Classically in children, ages 7–12 years.
• Skin and mucous membrane involvement rare.
• Often presents as a single, asymptomatic bony lesion (cranium > ribs > vertebrae > pelvis > scapulae > long bones).
Hashimoto–Pritzker Disease (Congenital Self-Healing Reticulohistiocytosis)
• Limited to skin, rapidly self-healing.
• At birth or during first few days of life.
• Variable presentation from a single nodule to widespread red-brown papules/nodules that crust and involute after several weeks (Fig. 76.3).
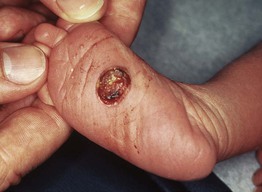
Fig. 76.3 Congenital self-healing reticulohistiocytosis (Hashimoto–Pritzker disease). Solitary eroded nodule on the plantar surface of a neonate. Courtesy, Richard Antaya, MD.
• Longitudinal follow-up is recommended because a small percentage of patients can develop systemic disease.
Non-Langerhans Cell Histiocytoses
Juvenile Xanthogranuloma
• Most common of the non-Langerhans cell histiocytoses.
• Appears primarily in infants and children (75% present during the first year of life).
• Usually a single papule or nodule (Fig. 76.4).
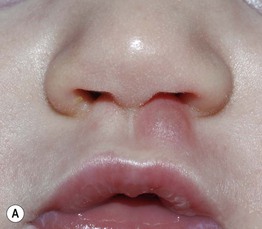
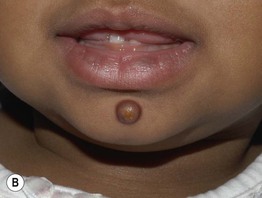
Fig. 76.4 Juvenile xanthogranuloma. A Pink nodule with a few telangiectasias representing an early lesion. B Well-developed yellow-brown papulonodule on the chin, with the color reflecting the accumulation of lipid within histiocytes. Courtesy, Julie V. Schaffer, MD.
• The color is initially pink to red-brown, but over time a yellow hue develops due to accumulation of intracellular lipid.
• Head/neck > trunk > upper extremities > lower extremities.
• Self-limiting; no treatment necessary.
• If multiple lesions, consider ophthalmologic examination for ocular involvement.
• If multiple café-au-lait macules are present, then consider the rare triple association with neurofibromatosis type I and an increased risk of developing juvenile myelomonocytic leukemia.
Benign Cephalic Histiocytosis
• Rare, generally in infants <1 year old.
• 2- to 5-mm red-brown macules or papules on the face and neck, rarely elsewhere (Fig. 76.5).
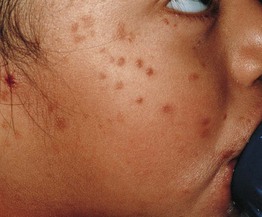
• Self-limiting after months to years; no treatment necessary.
Generalized Eruptive Histiocytoma
• Recurrent crops of tens to hundreds of small red-brown, firm papules (Figs. 76.6 and 76.7).
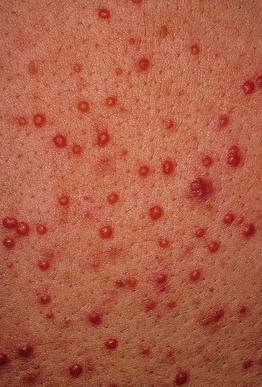
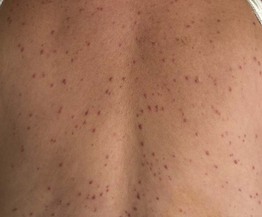
Fig. 76.7 Generalized eruptive histiocytoma. Multiple firm pink to red-brown papules on the trunk. The arms and legs were extensively involved. Courtesy, Ingo Haase, MD, and Iliana Tantcheva-Poor, MD.
Indeterminate Cell Histiocytosis
• Clinically indistinguishable from generalized eruptive histiocytoma.
• Diagnosis made by evaluation of biopsy specimen, with cells expressing CD1a, S100-protein, and CD68 (a marker of macrophages); lack of Birbeck granules ultrastructurally.
Necrobiotic Xanthogranuloma
• Rare, generally in adults >50 years of age.
• Firm, yellowish plaques or papulonodules; sometimes ulcerated.
• Classically periorbital (Fig. 76.8).
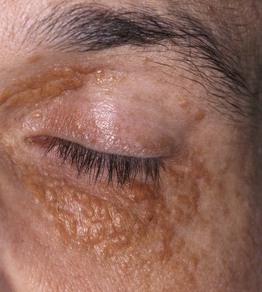
Fig. 76.8 Necrobiotic xanthogranuloma. Yellow-brown papules and plaques in a periorbital distribution in a patient with chronic lymphocytic leukemia. Such lesions may initially be mistaken for xanthelasma. Courtesy, Kalman Watsky, MD.
• IgG monoclonal gammopathy in 80% of patients; hepatosplenomegaly, increased ESR, leukopenia, hypocomplementemia.
• Underlying plasma cell dyscrasia common; also increased risk of lymphoproliferative disorders.
Multicentric Reticulohistiocytosis/Giant Cell Reticulohistiocytoma
– A skin-colored to pink papulonodule often on the head.
• When multiple (Fig. 76.9):
– Pink to red-brown papulonodules that favor the hands (especially periungual) and elbows; occasionally photodistributed.
– 50% with mucous membrane involvement.
– Associated with destructive arthritis that can clinically resemble rheumatoid arthritis; in up to 50%, arthritis is preceded by skin disease.
– Up to one third of affected patients can have an associated malignancy.
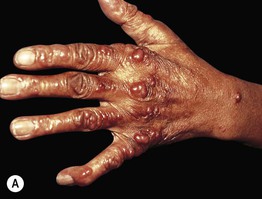
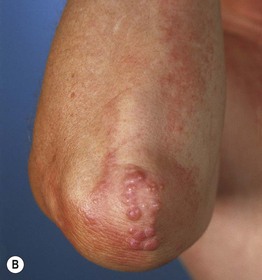
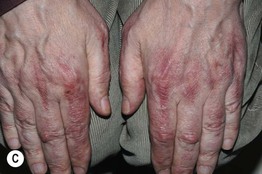
Fig. 76.9 Multicentric reticulohistiocytosis. A Grouped firm pink papules on the dorsal surface of the fingers, hand, and wrist of a 73-year-old African-American woman. B Grouped pink papulonodules on the elbow in a second patient. C A more subtle presentation in a third patient, with small pink papules and thin plaques that favor the skin overlying the small joints of the hands; this is the form that can initially be confused with dermatomyositis. A, Courtesy, Susan D. Laman, MD; B, Courtesy, Jean L. Bolognia, MD; C, Courtesy, Kalman Watsky, MD.
Rosai–Dorfman Disease (Sinus Histiocytosis with Massive Lymphadenopathy)
• Generally in children and young adults.
• Nonspecific red, red-brown, or yellow papulonodule(s) (Fig. 76.10).
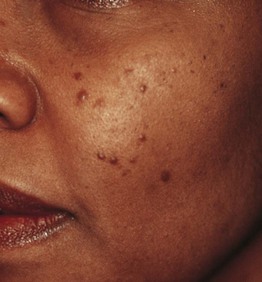
• Favor eyelids and malar area.
• Skin-limited form being increasingly recognized.
• Often self-limited, but there may be a protracted course.
• Systemic disease presents as massive bilateral cervical lymphadenopathy; fever and IgG polyclonal hypergammaglobulinimia may be present.
Xanthoma Disseminatum
• Rare, generally before age 25 years.
• Triad of cutaneous xanthomas, mucous membrane xanthomas (in 40–60%), and diabetes insipidus (in 40%).
• Symmetric eruption of tens to hundreds of yellow, red, or brown papules and plaques.
• Favors face and flexural areas, including major folds (Fig. 76.11).
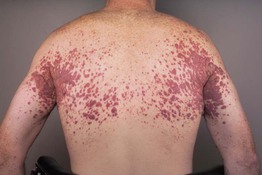
Fig. 76.11 Xanthoma disseminatum. Symmetric involvement of the major flexures is a characteristic finding. Note the yellow discoloration of some of the coalescing papulonodules. Courtesy, David Wetter, MD.
• Rarely sclerosis (Fig. 76.12).
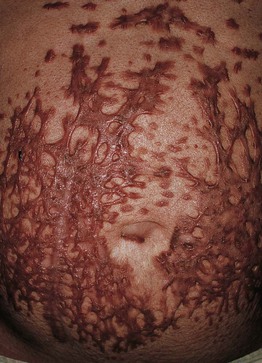
Fig. 76.12 Xanthoma disseminatum. Sclerotic form of xanthoma disseminatum in a patient who developed multiple myeloma.
• Mucous membrane involvement: oral, upper airway, ocular (may threaten vision).
Progressive Nodular Histiocytoma
Although we have made all these distinctions in the non-Langerhans cell histiocytoses, patients may have overlapping clinical and histopathologic features (including immunohistochemical staining). As a result, the evaluation of a patient with a non-Langerhans cell histiocytosis should include a total body skin examination and general physical examination, inclusive of eyes, mucosae, and lymph nodes. In addition, laboratory testing should include serum protein electrophoresis, immunofixation electrophoresis, and evaluation for diabetes insipidus.
For further information see Ch. 91. From Dermatology, Third Edition.