Other Lymphoproliferative and Myeloproliferative Diseases
Benign Lymphoctic Infiltrates
Lymphocytic Infiltrate of Jessner
• A benign skin-limited disorder that overlaps with other entities, including dermal lupus erythematosus, cutaneous lymphoid hyperplasia, and polymorphic light eruption (PMLE).
• Males = females; onset typically during middle age; very rare in children.
• Most commonly appears on the head, neck, and upper back as one or several asymptomatic, erythematous papules, plaques (often annular) > nodules (Fig. 99.1); no secondary or surface changes (such as scale or crust).
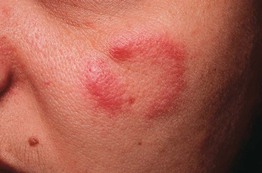
Fig. 99.1 Lymphocytic infiltrate of Jessner. Annular erythematous plaque with central clearing on the face.
• May last several weeks to months and will typically resolve spontaneously over months to years without sequelae.
• DDx: see Fig. 99.2.
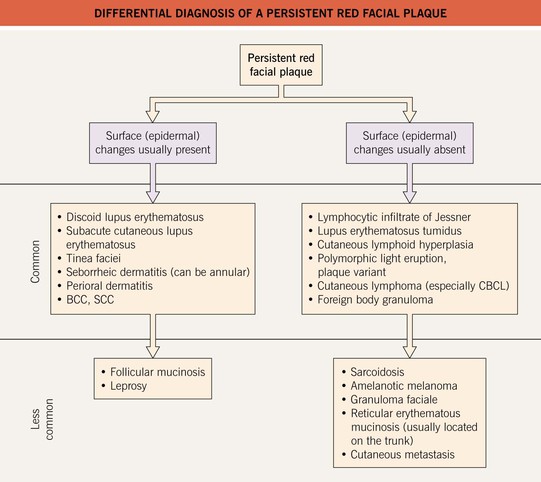
Fig. 99.2 Differential diagnosis of a persistent red facial plaque. BCC, basal cell carcinoma; CBCL, cutaneous B-cell lymphoma; SCC, squamous cell carcinoma.
• Rx: watch-and-wait approach is often acceptable; if Rx desired, consider topical (class I) or intralesional CS; less often hydroxychloroquine is used.
Cutaneous Lymphoid Hyperplasia (CLH); also known as ‘Pseudolymphoma’ or ‘Lymphocytoma Cutis’
• A benign, reactive lymphocytic proliferation that is thought to represent an exaggerated local immunologic reaction to a trigger that is usually unidentified.
• Females > males; adults > children.
• Inciting agents that have been reported include arthropod bites, tattoos, vaccinations, medications (e.g. antihistamines, antidepressants, angiotensin II receptor blockers), and in Europe, Borrelia burgdorferi infection.
• Presents most often on the head, neck, and upper extremities as one or several firm, erythematous to violaceous papules, plaques, or nodules (Fig. 99.3); usually no surface changes.
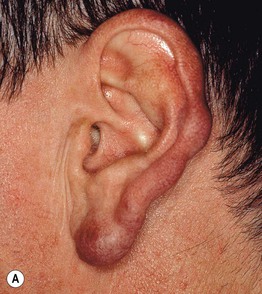
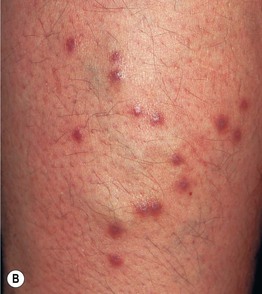
Fig. 99.3 Cutaneous lymphoid hyperplasia (lymphocytoma cutis). A Violet papulonodules on the helix and lobe of the ear (unknown etiology). B Multiple red-brown to violet papules at the sites of Hirudo medicinalis (medicinal leech) application. B, Courtesy, Josef Smolle, MD.
• Often spontaneously resolves without scarring.
• DDx (see Fig. 99.2): lymphocytic infiltrate of Jessner, lymphoma cutis, leukemia cutis, solid organ metastases, and occasionally adnexal tumors.
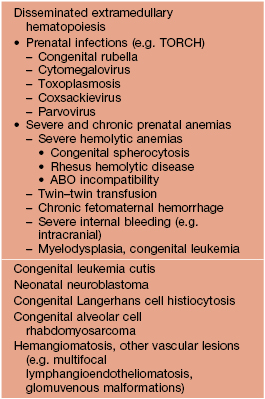
Extramedullary Hematopoiesis
• Most commonly seen in neonates secondary to underlying bone marrow dysfunction; rarely in adults secondary to myelofibrosis, other myeloproliferative disorders or after splenectomy.
• Most often associated with TORCH (toxoplasmosis, other [e.g. parvovirus B19], rubella, cytomegalovirus) infections in neonates.
• Clinically presents with erythematous to violaceous papules and nodules > plaques, ulcers, or nasal polyps (Fig. 99.4).
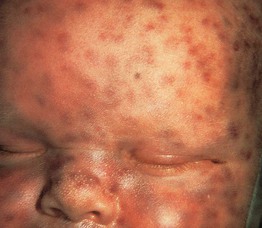
Fig. 99.4 Extramedullary hematopoiesis. ‘Blueberry muffin baby’ secondary to congenital rubella. Courtesy, James Graham, MD.
• When widely disseminated (typically in neonates) it leads to the classic ‘blueberry muffin baby’ presentation (Table 99.1).
• Diagnosis is made by histopathology (dermal infiltrates of immature erythrocytes, leukocytes, and megakaryocytes).
• DDx: see Table 99.1 for neonates, leukemia cutis in adults.
• Rx: treat underlying bone marrow dysfunction; spontaneous resolution typically occurs in cases related to viral infections.
Malignant Hematopoietic Infiltrates
Leukemia Cutis
• Cutaneous lesions associated with both acute and chronic leukemias may be either (1) nonspecific reactive skin lesions (Table 99.2) or (2) specific infiltrates, called ‘leukemia cutis’.
Table 99.2
‘Inflammatory’ disorders associated with acute and chronic leukemias.
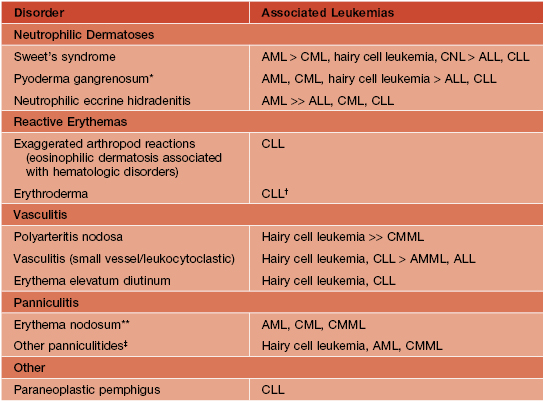
* Especially bullous.
† Many probably represent Sézary syndrome.
** Isolated case reports; may occur concomitantly with Sweet's syndrome.
‡ Sweet's syndrome may have a subcutaneous component.
ALL, acute lymphoblastic leukemia; AML, acute myelogenous leukemia; AMML, acute myelomonocytic leukemia; CLL, chronic lymphocytic leukemia; CML, chronic myelogenous leukemia; CMML, chronic myelomonocytic leukemia; CNL, chronic neutrophilic leukemia.
• Leukemia cutis typically presents as erythematous to violaceous firm papules or nodules (Fig. 99.5); less often purpura, ulcers, and rarely bullae; lesions may be hemorrhagic due to thrombocytopenia.
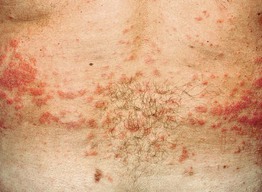
Fig. 99.5 Leukemia cutis. Multiple red-brown papules and plaques in a patient with hairy cell leukemia.
• Favors head, neck, trunk, and sites of scars or trauma.
• Less common presentations of leukemia cutis: chloromas, gingival hyperplasia.
• In most patients with acute leukemia, cutaneous lesions (if present) appear at the time of diagnosis or recurrence.
• Occasionally, leukemia cutis antedates the appearance of leukemia in the peripheral smear (‘aleukemic leukemia cutis’); rarely predates apparent bone marrow involvement by months.
• DDx: lengthy list because of the protean clinical presentations of leukemia cutis, but may include lymphoma cutis, infectious emboli, vasculitis, drug eruptions, Sweet's syndrome and other neutrophilic disorders, extramedullary hematopoiesis.
Hodgkin Disease (HD; Hodgkin Lymphoma)
• Primary or isolated cutaneous HD is very unusual; typically presents as one or several ulcerated nodules and usually follows a more indolent course, with prognosis similar to those patients with early stage HD.
• Secondary cutaneous HD, occurring in patients with advanced systemic disease, constitutes the vast majority of cutaneous HD cases and as expected carries a worse prognosis.
• Secondary cutaneous HD usually presents with the rapid appearance of multiple, disseminated papulonodules and plaques, often on the trunk (Fig. 99.6), along with locations distal to affected lymph nodes.
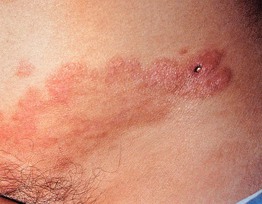
Fig. 99.6 Cutaneous Hodgkin disease (HD). A large pink plaque with central clearing is noted on the lower abdomen.
Angioimmunoblastic T-Cell Lymphoma (AITL)
• Previously referred to as ‘angioimmunoblastic lymphadenopathy’ (AILD).
• Now known to be the second most common peripheral T-cell lymphoma with the recent identification of the follicular helper T (TFH) cell as the cell of origin.
• Unique in that it characteristically presents acutely with widespread, non-bulky lymphadenopathy; fever; and a diffuse, pruritic morbilliform eruption mimicking an acute viral infection or drug eruption.
• Other systemic findings include hepatosplenomegaly, polyclonal hypergammaglobulinemia, immune dysregulation resulting in autoantibody production (e.g. ANA, rheumatoid factor, antiphospholipid antibodies, false (+) Lyme serologies), cytopenias and hypereosinophilia.
• The morbilliform eruption and lymphadenopathy typically wax and wane, making suspicion and diagnosis of this malignancy even more elusive.
• The usual course is aggressive with a poor prognosis (median survival is <3 years); up to 30% may have longer-term survival.
• DDx: reactive lymphadenopathy due to a viral infection; drug eruption, infectious mononucleosis, or other systemic inflammatory diseases.
• Rx: combination chemotherapy; high-dose chemotherapy with autologous stem cell support; allogeneic stem cell transplantation, and newer immunomodulators.
Lymphomatoid Granulomatosis (LYG)
• A rare, EBV-associated, angio-centric/destructive lymphoproliferative disorder that primarily affects the lungs, skin, and nervous system (less often the kidneys and gastrointestinal tract).
• Although previously classified as a reactive process, in most patients it represents a form of large B-cell lymphoma.
• LYG can be divided into grades 1, 2, or 3 depending on the proportion of CD20(+) large lymphoid cells.
• Most often seen in adults; males > females; can occur in the setting of immune dysfunction (e.g. Sjögren's syndrome, rheumatoid arthritis, renal transplantation, HIV infection).
• Frequently presents with cough, dyspnea, chest pain, fever, weight loss, malaise, arthralgia, and myalgia.
• Cutaneous lesions are present in ~25–50% of patients, usually ranging from nodules to ulcerated plaques (Fig. 99.7); rarely an exanthematous eruption develops.
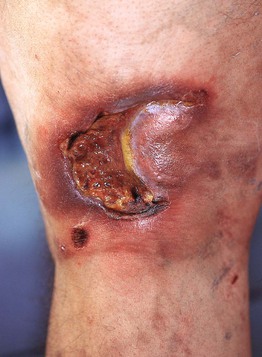
Fig. 99.7 Lymphomatoid granulomatosis. Ulcerated violaceous plaque in the popliteal fossa. Courtesy, Jean L. Bolognia, MD.
• Typically follows an aggressive, fatal course with a 5-year mortality rate of 60–90%.
• DDx: other cutaneous lymphomas, infections (e.g. tuberculosis), pyoderma gangrenosum, medium vessel vasculitis, Wegener's granulomatosis, sarcoidosis.
• Rx: rituximab, multidrug chemotherapy, or perhaps interferon-α in earlier stage disease.
For further information see Ch. 121. From Dermatology, Third Edition.