Chelidonium
Synonyms
Greater celandine (Engl), Chelidonii herba (Lat), Schöllkraut, Goldwurz (Ger), chélidoine (Fr), cinerognolle (Ital), svaleurt (Dan), baiqucai (Chin).
What is it?
Chelidonium has a long history of use as a therapeutic plant. It was mentioned by Pliny, to whom we owe the tradition of calling the plant Chelidonium, derived from the Greek chelidon (a swallow). This is apparently because it comes into flower when the swallows arrive and fades when the swallows depart. Pliny reported that its acrid juice was used to remove films from the cornea of the eye and alchemists believed it was beneficial for jaundice because of its intense yellow colour. Although the root also contains the characteristic alkaloids and is utilised to a limited extent medicinally, the aerial parts are more widely used and are the main focus of this monograph.
Effects
Assists liver and gallbladder function, protects against hepatic injury; spasmolytic to the gastrointestinal tract; stimulates bile flow; active topically against fungal infections and warts; decreases benign and malignant tumours (topically and internally).
Traditional view
Chelidonium was employed to treat conditions of the liver such as jaundice, hepatic congestion and biliary dyspepsia. It was also used for bilious and migraine headaches and haemorrhoids.1 The herb was often used in the form of a poultice or ointment for the treatment of cutaneous problems and traumatic inflammation,1 and the fresh milky juice was used topically in the treatment of warts, ringworm and corns.1,2
Summary actions
Choleretic, cholagogue, spasmolytic, mild laxative, anti-inflammatory, antineoplastic, antiviral and vulnerary (topically).
Can be used for
Indications supported by clinical trials
Disorders of the liver and gallbladder; cramp-like pain of the gastrointestinal tract and gall ducts, including irritable bowel syndrome; as an enema for colonic polyposis; as a topical application for warts (mostly uncontrolled studies); functional dyspepsia (alone and in combination).
Traditional therapeutic uses
Gallbladder disease, gallstones; liver disease, jaundice; to aid detoxification via the liver and bowel; hepatic and splenic congestion; migraine, bilious headaches and supraorbital neuralgia; skin conditions including warts, fungal growths and ringworm (especially the fresh juice).1
In China Chelidonium is also used for gastritis, gastric ulcer, enteritis, jaundice and abdominal pain as well as bronchitis and whooping cough.3,4
Preparations
Dried herb as a decoction, liquid extract and tablets or capsules for internal use. Decoction, extract or fresh juice for external use.
Dosage
1 to 2 mL/day of 1:2 liquid extract, 2 to 4 mL/day of 1:5 tincture. Short-term use of higher doses up to the equivalent of 3 to 4 g per day may be necessary (as per the Berlin clinical trial).
The dose used in China is 3 to 9 g per day or even higher. However, these doses are generally administered by decoction and this method may not extract the Chelidonium alkaloids as efficiently as alcohol and water.
Duration of use
High doses should be restricted to short-term use; long-term use of normal doses is not recommended.
Summary assessment of safety
No significant adverse effects have been noted for short-term use, but excessive intake of the decoction may cause nausea and other gastrointestinal symptoms. Long-term use is associated with a low risk of a moderate idiosyncratic hepatotoxic reaction. The herb should not be given to patients with pre-existing liver damage.
Technical data
Botany
Chelidonium majus is a member of the Papaveraceae (poppy) family and is a perennial herb approximately 50 to 90 cm in height with a branched woody taproot. The fragile stems are branched, with scattered hairs and contain an orange latex. The leaves are pinnatisect, with up to seven oblong or ovate leaflets with a bluish green underside. The flowers contain four bright yellow petals and are grouped in small clusters. The fruit capsule is one-celled, up to 5 cm long and contains black seeds with a white appendage.5
Key constituents
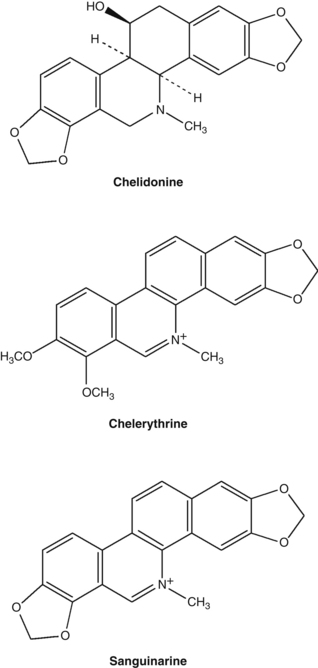
• Isoquinoline alkaloids (0.35 to 1.3%),6 including the major alkaloids chelidonine (>0.07%), chelerythrine, sanguinarine, berberine, coptisine and dl-stylopine
• Other alkaloids: sparteine (which is usually found in the Leguminosae (pea) family)6,7
• Flavonoids, phenolic acids.7
The isoquinoline group of alkaloids contains many structural types including the benzophenanthridines (chelidonine, chelerythrine, sanguinarine) and protoberberines (berberine, coptisine). An analysis of 20 Chelidonium samples from different parts of China found the total alkaloids varied from 0.89% to 1.70%, with coptisine the highest alkaloid present (average content of 0.5%) and berberine the lowest at 0.013%.8
The milky orange sap of Chelidonium contains defensive proteins, including an extracellular peroxidase with nuclease activity.9,10
Pharmacodynamics
Hepatoprotective and choleretic activity
Oral administration of an alcohol extract of dried Chelidonium reduced carbon tetrachloride-induced liver injury in rats.11 Significant reductions in elevated plasma levels of liver enzymes and bilirubin occurred in the treated group. A follow-up study was undertaken to clarify the underlying aspects of tissue recovery. There was an absence of fibrotic changes in the Chelidonium-treated rats (125 mg/kg/day for 3 weeks, oral), which was thought to be related to a reduced degree of cellular necrosis and a reduction in fibroblast-stimulating factors.12
Extracts of dried Chelidonium were tested for choleretic activity using the isolated perfused rat liver. The 70% ethanolic extract of the herb significantly induced choleresis (bile flow). However, it did this without increasing the total output of bile acids (that is, there was an increased flow of more dilute bile). In contrast, the phenolic and alkaloidal fractions of the total extract, tested individually and in combination, did not significantly increase bile flow, although small increases were observed. The authors concluded that the increased bile flow is due to an additive effect from all compounds in the total extract of Chelidonium, not one or two specific active constituents or fractions.13
Antimicrobial activity
Isolated chelerythrine and an alkaloid fraction from the dried roots of Chelidonium containing chelerythrine and sanguinarine were found to be ineffective against Gram-negative bacteria in vitro. However, a significant antimicrobial effect was observed against Gram-positive bacteria such as Staphylococcus aureus and two strains of Streptococcus, and also against the fungus Candida albicans.14 Chelerythrine inhibited the adherence of Streptococcus mutans and was thereby considered to possess significant anticariogenic activity.15 This alkaloid also inhibited the growth of this organism in vitro with an MIC (minimum inhibitory concentration) of 0.78 mg/mL, but chelidonine was inactive.16 Sanguinarine is also well described as an alkaloid with activity against dental plaque.17–19
In an in vitro screening of the ethanolic extracts from 12 Siberian herbs for antimicrobial activity against five common pathogenic organisms, the extract of Chelidonium aerial parts was not active. In contrast, the root extract demonstrated marked activity against Bacillus cereus, Candida albicans and Salmonella enteritidis.20
Extracts of Chelidonium were found to exert antiviral effects in vitro against adenovirus types 12 and 5 and herpes simplex virus type 1 (HSV-1). The most promising antiviral alkaloid was found in greater concentrations in the fresh and aerial plant samples. This alkaloid, which belonged to the benzophenanthridine group, was not identified.21 A crude extract of Chelidonium was found to inhibit HIV-1 from infecting cells in vitro.22 A low sulphated polyglycosaminoglycan appeared to be responsible for this action and protected mice from the negative effects of murine retrovirus infection after iv administration. Such a large molecule is unlikely to have oral activity.
Alkaloids from Chelidonium and sanguinarine inhibited the growth of Trichomonas vaginalis in vitro. Sanguinarine also caused the protozoa to undergo deformation followed by disintegration within 2 h.23
An in vitro study demonstrated that the alkaloids extracted from Chelidonium, chelerythrine, and a mixture of chelerythrine and sanguinarine, exerted an antifungal effect on some Trichophyton strains, Microsporum canis, Epidermophyton floccosum and Aspergillus fumigatus.24 Another in vitro study investigated the effect of Chelidonium extracts on several Candida species and other dermatophytes. Liquid extracts of Chelidonium prepared from dried plant material collected in late July and early September (Europe) were compared. Both extracts showed greater than average antifungal activity against some organisms involved in skin infections. The July extract was active against Candida albicans, but the September extract showed no activity.25
When six species of clinically resistant yeast fungi were exposed to isolated Chelidonium alkaloids, 8-hydroxydihydrosanguinarine and 8-hydroxydihydrochelerythrine demonstrated potent activity, with MIC ranges of 2 to 80 and 4 to100 μg/mL, respectively.26 Other alkaloids also had some degree of antifungal activity. The two above compounds were also quite active against methicillin-resistant Staph. aureus.27
Fusarium species have the capacity to cause opportunistic human infections. Extracts of Chelidonium showed some activity against certain strains of this organism in vitro, with Chelidonium root extracts being more active than shoot extracts.28
Antitumour activity
An ethanolic extract of rhizomes and roots of Chelidonium exhibited cytotoxicity against a carcinoma of the nasopharynx in vitro. One of the cytotoxic principles was found to be the alkaloid coptisine.29 Chelidonium exerted an antimutagenic effect in vitro against several mutagens in the Ames test.30 The extract caused changes in the mitotic index of transplanted ascitic cells, showing marked antimitotic activity.3
Chelidonine and sanguinarine induce apoptosis in leukaemia cells in vitro, but only the former induced cell cycle arrest.31 Sanguinarine and chelerythrine induce DNA damage and cytotoxic effects in normal and cancer cells, whereas chelidonine does not.32 Chelidonine may inhibit tumour cell growth by reducing telomerase activity.33 A methanolic extract of Chelidonium also induced apoptosis in two leukaemia cell lines.34
The milky sap from fresh Chelidonium contains two nuclease enzymes with apoptotic activity against the HeLa tumour cell line, but not CHO cells.35 A lectin isolated from Chelidonium inhibited growth of two tumour cell lines, but not normal mouse fibroblasts.36
The activities of aqueous and alcoholic extracts of Chelidonium, the partially purified methanol extract and chelidonine and protopine were screened using transplanted tumours in mice. The water-soluble, purified methanol extract of dried Chelidonium demonstrated high tumour inhibition with relatively mild cytotoxic side effects. Intraperitoneal administration of 700 mg/kg for 7 days resulted in 55% inhibition of sarcoma 180 and Ehrlich carcinoma. The aqueous extract showed insignificant activity, and chelidonine and protopine (both of which are insoluble in water) showed negligible tumour inhibition and were associated with cytotoxic side effects. The crude methanol extract also showed more pronounced toxic side effects.37
The numbers of stomach tumours in rats treated with oral doses of a Chelidonium extract after initial exposure to a carcinogen were significantly lower, compared with untreated but exposed animals.38 An ethanolic whole plant extract of Chelidonium (0.1 mL/mouse of a 1:20 diluted extract) reduced the incidence of chemically induced liver cancer in mice after 60 and 120 days.39
Ukrain is described as a semi-synthetic eastern European anticancer drug derived from Chelidonium. It purportedly contains one molecule of thiophosphoric acid conjugated (bonded) to three molecules of chelidonine and is administered by intravenous injection.40 However, chemical analysis revealed that some commercial samples of Ukrain were just a mixture of Chelidonium alkaloids, with no trimeric structure evident.41 This was also the case 6 years later in 2006, when another research team found that the Ukrain sample they tested lacked the purported trimeric ‘Ukrain molecule’ and instead resembled an alkaloidal extract of Chelidonium, with chelidonine, sanguinarine and chelerythrine present as major components.42 Their in vitro studies suggested chelidonine was a particularly active inducer of tumour cell apoptosis.
A 2005 review of the anticancer research on Ukrain identified 36 in vitro studies and 46 in vivo experiments. These publications suggest that it has the capacity to exert selective cytotoxic and cytostatic effects on tumour cells, while favourably modifying the immune response.43 Specifically, diminished DNA, RNA and protein synthesis, inhibition of cellular oxygen consumption, inhibition of tubulin polymerisation44 and induction of apoptosis have all been described.45 Ukrain also modifies the host immune response via an increase in T cells and normalisation of the T-helper/T-suppressor lymphocyte ratio. Tumour mass reductions have also been demonstrated in vivo.45 More recent examples of in vivo studies include growth inhibitory effects against B16 melanoma cells46 and Ehrlich’s carcinoma,47 both in mice. It is uncertain how much, if any, of this research would have relevance to the oral use of Chelidonium extracts.
Anti-inflammatory activity
The alkaloids sanguinarine and chelerythrine are potent in vitro inhibitors of 5-lipoxygenase in polymorphonuclear leucocytes and 12-lipoxygenase in mouse epidermis. An extract of Chelidonium also inhibited 5-lipoxygenase. The inhibitory effects against lipoxygenase enzymes appear to be due to a specific enzyme interaction, rather than a non-specific redox mechanism.48
The Chelidonium alkaloids chelerythrine and sanguinarine (5 and 10 mg/kg, oral and sc) demonstrated anti-inflammatory activity in the carrageenan rat paw oedema test.14 A Chelidonium extract (40 and 400 mg/kg/day for 28 days, oral) suppressed the progression of collagen-induced arthritis in mice. Decreased levels of cytokines and activated immune cells were observed, together with increased numbers of regulatory T cells.49
Other effects
Chelidonium extract and isolated components (coptisine and caffeoylmalic acid) weakly antagonised experimentally induced contraction of isolated rat ileal smooth muscle.50 Two ethanolic dry extracts of Chelidonium and their three main alkaloids were studied in different antispasmodic test models on isolated guinea-pig ileum.51 Both extracts induced relaxation in barium chloride, carbachol and electric field stimulated ileum, as did the alkaloids.
Radioreceptor assays suggest that the alkaloids sanguinarine, chelerythrine, stylopine, allcryptopine and particularly protopine interact with the chloride channel of the GABA-A receptor.52 Chelidonium extract inhibited GABA-activated current via G proteins in vitro, suggesting an analgesic mechanism (see below).53 Studies in mice found that allocryptopine and protopine increased the brain concentration of the neurotransmitter GABA and the activity of its synthesising enzyme GAD.54
Chelidonium alkaloids acted as irreversible inhibitors of liver mitochondrial monoamine oxidase in vitro. Chelidonine was the strongest inhibitor.55 The same alkaloids also reversibly inhibited acetylcholinesterase in vitro, with sanguinarine and berberine exhibiting the strongest activity.56 Of the minor alkaloids, 8-hydroxydihydrochelerythrine and 8-hydroxydihydrosanguinarine had potent acetylcholinesterase inhibitory activity.57
Chelerythrine chloride exerted an in vitro antiplatelet effect that was believed to be due to inhibition of thromboxane formation and phosphoinositide breakdown.58
An extract of Chelidonium inhibited human keratinocyte proliferation, with sanguinarine being the most potent constituent. The mechanism of action appears to be inhibition of the inflammatory mediators leukotriene B4 and 12(S)-HETE, both of which have a known role in stimulating epidermal keratinocyte proliferation. Although the alkaloids have demonstrated cytotoxic activity in low concentrations, sanguinarine and chelerythrine did not cause more damage to cell membranes than the antipsoriatic drug anthralin, as observed by the release of lactate dehydrogenase activity (an indicator of plasma membrane damage).59
The antinociceptive action of aminophenazone in mice was potentiated by the Chelidonium alkaloids allocryptopine, chelidonine and sanguinarine.60 Chelidonium extract suppressed glycine-induced responses and elevated those induced by glutamate in isolated rat periaqueductal grey neurons.61 This might activate the descending pain control system leading to an analgesic effect.
A liquid extract of Chelidonium (2.5, 5 and 10 mL/kg, oral) dose-dependently protected against indomethacin-induced gastric ulceration in rats.62 However, it was not as active as other digestive herbs such as licorice and peppermint.
High doses of Ukrain caused slight osteopenic effects in rats, possibly due to inhibition of locomotor activity.63 However, it was anabolic on bone in ovariectomised female rats (doses 7, 14 and 28 mg/kg, ip).
Clinical trials
Spasmolytic and cholagogue effects
In an early uncontrolled trial, a Chelidonium extract exerted good to very good results in two-thirds of patients treated for cholangitis, cholelithiasis and cholecystitis without stones. Forty patients received 3 mL/day (for 43 to 50 days) of a fresh plant tincture standardised to 20 mg alkaloids/100 mL.64 An early clinical trial investigated the effect of a suspension of Silybum marianum, Chelidonium and Curcuma on 28 patients. Compared to a control liquid, the herbal mixture demonstrated a greater increase in bile flow and pancreatic secretion.65
In 60 Berlin practices, 608 patients were treated in an uncontrolled study over a 3-month period with a standardised preparation of dried Chelidonium, which acted as a plant-based spasmolytic. The main presenting symptoms were cramp-like pains in the gastrointestinal tract (43%) or gall ducts (48.2%), but also included dyspeptic symptoms. Each Chelidonium tablet contained 125 mg of a 5:1 to 7:1 hydroethanolic extract with 2.85 mg of total alkaloids, including 0.79 mg of chelidonine. The dose was initially 5 tablets/day and this was reduced to 3 tablets/day in patients who responded to treatment. The average duration of treatment was 22 days and the longest treatment time was 2.5 months. A good or very good therapeutic effect on symptoms with a quick response was observed in 87.4% of cases. In most cases symptom relief occurred within 30 minutes of taking the herbal medication (62.3%). In 46.1% of patients, the average duration of efficacy of each tablet dose was more than 3 h. This study suggests value for Chelidonium in the treatment of cramp-like abdominal pains associated with irritable bowel syndrome and other causes.66
In a retrospective, open label study conducted over a 6-month period, 206 patients with epigastric complaints related to gallstones or gallbladder removal were evaluated.67 Patients received either a Chelidonium capsule (125 mg/day of a hydromethanolic extract containing 0.68 mg of chelidonine) or a liquid (three times 20 drops daily containing 0.15 mg chelidonine). There was a noted improvement in symptoms such as flatulence, diarrhoea, constipation and upper abdominal pain. Pain-free intervals were increased and inflammatory markers decreased.
The efficacy and tolerability of a standardised Chelidonium extract was investigated in a randomised, placebo-controlled, double blind trial involving 60 patients with functional epigastric symptoms.68 These included cramp-like pains, sensation of pressure or fullness, flatulence and nausea. Patients receiving active treatment took 6 tablets/day, each containing 66 to 167 mg of a Chelidonium dry extract (5.3:1 to 7.5:1) delivering 4 mg of total alkaloids calculated as chelidonine. Following 6 weeks of treatment, there was a clear difference in physician-rated response rates: 60% in the Chelidonium group versus 27% in placebo (p=0.0038). The treatment was without significant side effects compared with the placebo.
A double blind, placebo-controlled trial investigated the impact of a Chelidonium and turmeric root combination in 76 patients with upper abdominal pain attributed to functional disorders of the biliary system.69 Patients received either 3 capsules/day (each containing Chelidonium (104 to 131 mg of a 5:1 to 10:1 extract containing 4 mg of alkaloids calculated as chelidonine) and turmeric (45 mg of a 12.5:1 to 25:1 extract)) or a matching placebo for 3 weeks. In the first week there was a significant reduction in pain for the patients receiving the herbal treatment, compared with placebo. No other symptoms changed significantly relative to placebo.
A proprietary formula known as STW 5 contains liquid extracts of Chelidonium, Matricaria recutita (chamomile) flower, Iberis amarus (bitter candywort) herb, Carum carvi (caraway) fruit, Angelica archangelica (garden angelica) root, Glycyrrhiza glabra (licorice) root, Silybum marianum (milk thistle) fruit, Melissa officinalis (lemon balm) leaf and Mentha × piperita (peppermint) leaf and has been extensively researched for indigestion and functional dyspepsia. Two meta-analyses of the clinical studies on this formula have been published.70,71 They found that STW 5, at a dose of 1 mL 3 times per day, significantly reduced symptoms compared with placebo. It also demonstrated similar efficacy to cisapride and metoclopramide.
Warts, polyps
In a small, uncontrolled trial, an infusion of dried Chelidonium was administered as an enema for colonic polyposis. Administration of 10 or more enemas resulted in the complete disappearance of colonic polyps in several cases.72 In a later study, the fresh plant was made into a paste and administered 2 or 3 h after an evacuant enema. In most cases, two or three courses (consisting of 10 to 20 enemas each) were deemed to be necessary. This regime was ineffective for treating malignant regenerated or degenerated polyps. Over a 2-year period treating 149 patients with various forms of polyposis, 59% showed improvement with 27% making a complete recovery.73
An ethanolic extract of Chelidonium was used as a topical application to treat nursing mothers for warts, papillomas, condylomas and nodules in an uncontrolled trial. The extract was applied to the affected area approximately 200 times per day for 2 to 3 weeks or until improvement was observed. Complete resolution of the warts occurred after 15 to 20 days in 135 women.74
Respiratory conditions
Chelidonium was given as a syrup or extract (equivalent to 15 g of herb per day) to patients with chronic bronchitis in an uncontrolled study. The effective response rate was around 80%. It was more effective in the simple type than the asthmatic type.3 Chelidonium syrup or a decoction of the fresh herb was used to treat whooping cough in an uncontrolled study. Dosages were: infants under 6 months, 5 to 8 mL; 6 to 12 months, 8 to 10 mL; 1 to 3 years, 10 to 15 mL; 3 to 6 years, 5 to 20 mL; and above 6 years, 20 to 30 mL. Treatment was for a course of 8 to 10 days. Of 500 cases so treated, 355 were ‘cured’ and 116 improved.3
Chelidonium tincture improved tonsillar function and immunity and reduced recurrence of infection in an open comparative study in children with tonsillitis (article in Russian).75
Anticancer activity
Chelidonium was one of three herbs used to examine the efficacy of traditional Chinese herbs on squamous cell carcinoma of the oesophagus. A 30 mL dose of a decoction of Chelidonium (equivalent to 30 g of crude herb) was given orally to 30 patients twice daily for 2 weeks prior to surgery. Histological examination of the excised tissue demonstrated a greater degree of stromal lymphoid cell infiltration and cancer tissue degeneration in the patients given Chelidonium than in those given the herb plus endoxan or in the control group. The antitumour action of Chelidonium was thought to be due to the activation of an immunological rejection mechanism.76
A systematic review of seven clinical trials on Ukrain given intravenously to patients with colorectal, pancreatic, bladder and breast cancer found benefit in improving survival compared with various control groups.40 Trials were generally of low quality and clear interpretation of results was difficult due to various methodological and reporting flaws. As noted previously, the relevance of Ukrain to the clinical use of Chelidonium is uncertain.
HIV infection
An uncontrolled trial reported on the efficacy of a combination of freeze-dried Chelidonium, Ulmus rubra (slippery elm) bark and Sanguinaria canadensis (bloodroot) in 13 HIV positive patients.77 Each capsule contained 175 mg Chelidonium, 20 mg slippery elm and 5 mg bloodroot. The dose used was 9 capsules per day. The most dramatic response was a general improvement in lymphadenopathy in the patients affected by this. Minor improvements in CD4+ T cell counts and energy levels were also noted.
Toxicology and other safety data
Toxicology
No harmful or toxic effects from therapeutic doses have been established. The LD50 of the decoction in mice by intraperitoneal injection is 9.5 g/kg3 and the LD50 of the alkaloids in mice is 300 mg/kg (subcutaneous).4
In an antitumour experiment, intraperitoneal administration of a methanol extract of Chelidonium (350 mg/kg/day for 7 days) to mice resulted in a 20% mortality rate.37 After 4 weeks of feeding Chelidonium (1.5 and 3 g/kg/day) to rats, no toxic or hepatotoxic effects were observed.78 This suggests the herb is not inherently hepatotoxic and that the observed adverse hepatic reactions are rare, idiosyncratic responses.
Special warnings and precautions
Given the nature of the alkaloid content of this herb and the rare hepatotoxic reactions, long-term use (except topical) is not preferred. Caution should be observed during pregnancy and lactation and in patients with gallstones. Use of Chelidonium should not be combined with heavy alcohol consumption.
Use in pregnancy and lactation
Category C-has caused or is associated with a substantial risk of causing harmful effects on the fetus or neonate without causing malformations.
Intramuscular injection of Ukrain on days 6 to 11 of gestation to hamsters and on days 6 to 15 of gestation to rats (0.1 to 28 mg/kg/day) did not produce teratogenic effects in either species compared with controls. Slight embryotoxic effects (increased post-implantation losses), and in consequence decreased number of average litter size, were noted in hamsters exposed to Ukrain at doses that were otherwise not embryotoxic to rats.79
Chelidonium use is strongly discouraged during breastfeeding.
Side effects
The potential association of Chelidonium with idiosyncratic hepatotoxicity was first reported in 1996. A 69-year-old woman developed symptoms of acute hepatitis after taking tablets containing several herbs including Chelidonium over a period of 6 weeks. Symptoms returned with rechallenge.80 Three additional cases were then reported (1997, 1998).81–83 In one series of observations over 2 years (1997–1999) in an area of approximately 1 million inhabitants in Germany, preparations of Chelidonium apparently induced 10 cases of acute hepatitis. Investigations and tests excluded viral causes, alcohol intake and hereditary causes. Although immunological factors could not be safely excluded, the evidence, including liver biopsy, suggested a treatment-related pathology. Cholestasis was observed in half the cases, but there were no cases of liver failure and the condition improved quickly in all cases when the Chelidonium was stopped. In one case a rechallenge led to a second attack of hepatitis.84 Three cases of acute hepatitis associated with Chelidonium were then reported in the literature in 2002 and May 2003,85,86 and another in 2006.87 Again, patients returned to normal when the herbal treatment was ceased. Another case report of cholestatic hepatitis (with complete recovery) including a review of 16 cases documented in the literature, was published in 2009.88 Assessment of causality for this case suggested a probable relationship with Chelidonium consumption. Generally these hepatotoxic reactions have been observed after using higher-dose German products.
A case of contact dermatitis has been linked to exposure to the plant.89
A case of haemolytic anaemia was reported after the oral ingestion of Chelidonium extract. The patient was treated with corticosteroids, blood transfusions and haemodialysis and recovered after about 12 days.90
When Chelidonium was used in traditional Chinese medicine studies, various degrees of dry mouth, dizziness, gastric discomfort, diarrhoea, abdominal distension, nausea and mild leucopenia were reported in a minority of patients. Symptoms generally disappeared within 3 to 5 days without the discontinuation of treatment.3
Overdosage
Critical consideration of the often-cited fatal case of poisoning in a 4-year-old boy recorded in 1936 suggests that it is by no means certain that Chelidonium was involved. More than 500 g of Chelidonium is said to be required to cause toxic effects in horses and cattle.91
Regulatory status in selected countries
Chelidonium is covered by a positive Commission E Monograph and can be used for cramp-like disorders of the biliary and gastrointestinal tracts.
Chelidonium is not on the UK General Sale List. Under the terms of the British Medicines Act 1968 and the Statutory Instrument SI 2130 (Retail Sale or Supply of Herbal Remedies) Order 1977 (Schedule Part III), the sale of Chelidonium is restricted to herbal practitioners. It may be prescribed at a maximum dosage of 2 g three times per day.
Chelidonium does not have GRAS status. However, it is freely available as a ‘dietary supplement’ in the USA under DSHEA legislation (1994 Dietary Supplement Health and Education Act).
Chelidonium is not included in Part 4 of Schedule 4 of the Therapeutic Goods Act Regulations of Australia and is freely available for sale. However, Chelidonium-containing products must contain the following warning: ‘Greater Celandine may harm the liver in some people. Use only under the supervision of a healthcare practitioner.’
References
1. Felter HW, Lloyd JU. King’s American Dispensatory, Vol 1. 18th ed., rev 3. Portland: 1905. Reprinted by Eclectic Medical Publications; 1983. pp. 491–493.
2. Grieve M, A Modern Herbal, New York, Dover Publications, 1971;Vol 1. pp. 178–179
3. Chang HM, But PP, Pharmacology and Applications of Chinese Materia Medica, Singapore, World Scientific, 1987;vol 1. pp. 390–394
4. Huang KC. The Pharmacology of Chinese Herbs. Boca Raton: CRC Press, 1993. pp. 144–145
5. Launert EL. The Hamlyn Guide to Edible and Medicinal Plants of Britain and Northern Europe. London: Hamlyn, 1981. p. 26
6. Wagner H, Bladt S. Plant Drug Analysis: A Thin Layer Chromatography Atlas, 2nd ed. Berlin: Springer-Verlag, 1996. p. 10
7. Colombo ML, Bosisio E. Pharmacol Res. 1996;33(2):127–134.
8. Gu Y, Qian D, Duan JA, et al. J Sep Sci. 2010;33(8):1004–1009.
9. Nawrot R, Kalinowski A, Gozdzicka-Jozefiak A. Phytochemistry. 2007;68(12):1612–1622.
10. Nawrot R, Lesniewicz K, Pienkowska J, et al. Fitoterapia. 2007;78(7-8):496. 501
11. Mitra S, Gole M, Samajdar K, et al. Int J Pharmacog. 1992;30(2):125–128.
12. Mitra S, Sur RK, Roy A, et al. Phytother Res. 1996;10(4):354–356.
13. Vahlensieck U, Hahn R, Winterhoff H, et al. Planta Med. 1995;61(3):267–271.
14. Lenfeld J, Kroutil M, Marsalek E, et al. Planta Med. 1981;43(10):161–165.
15. Cheng RB, Chen X, Liu SJ, et al. Shanghai Kou Qiang Yi Xue. 2007;16(1):68–72.
16. Cheng RB, Chen X, Liu SJ, et al. Shanghai Kou Qiang Yi Xue. 2006;15(3):318–320.
17. Kuftinec MM, Mueller-Joseph LJ, Kopczyk RA. J Can Dent Assoc. 1990;15(suppl 7):31–33.
18. Laster LL, Lobene RR. J Can Dent Assoc. 1990;56(suppl 7):19–30.
19. Hannah JJ, Johnson JD, Kuftinec MM. Am J Orthod Dentofacial Orthop. 1989;96(3):199–207.
20. Kokoska L, Polesny Z, Rada V, et al. J Ethnopharmacol. 2002;82(1):51–53.
21. Kery A, Horvath J, Nasz I, et al. Acta Pharm Hung. 1987;57(1–2):19–25.
22. Gerencer M, Turecek PL, Kistner O, et al. Antiviral Res. 2006;72(2):153–156.
23. Bodalski T, Pelozarska H, Ujec M. Arch Immunol Terapii Doswiadcjalny. 1958;6(4):705–711.
24. Hejtmánková N, Walterova D, Preininger V. Fitoterapia. 1984;55(5):291–294.
25. Vukusic I, Pepeljnjak S, Kustrak D, et al. Planta Med. 1991;57(suppl 2):A46.
26. Meng F, Zuo G, Hao X, et al. J Ethnopharmacol. 2009;125(3):494–496.
27. Zuo GY, Meng FY, Hao XY, et al. J Pharm Pharm Sci. 2008;11(4):90–94.
28. Matos OC, Baeta J, Silva MJ, et al. J Ethnopharmacol. 1999;66(2):151–158.
29. Kim HK, Farnsworth NR, Blomster RN, et al. J Pharm Sci. 1969;58(3):372–374.
30. Shi GZ. Chung-Hua Yu Fang I Hsueh Tsa Chih. 1992;26(3):165–167.
31. Pilchenkov A, Kaminskyy V, Zavelevich M, et al. Toxicol In Vitro. 2008;22(2):287–295.
32. Kaminskyy V, Lin KW, Filyak Y, et al. Cell Biol Int. 2008;32(2):271–277.
33. Noureini SK, Wink M. World J Gastroenterol. 2009;15(29):3603–3610.
34. Nadova S, Miadokova E, Alfoldiova L, et al. Neuro Endocrinol Lett. 2008;29(5):649–652.
35. Nawrot R, Wolun-Cholewa M, Gozdzicka-Jozefiak A. Folia Histochem Cytobiol. 2008;46(1):79–83.
36. Fik E, Wolun-Cholewa M, Kistowska M, et al. Folia Histochem Cytobiol. 2001;39(2):215–216.
37. Sokoloff B, Saelhof CC, Yoshichi MD, et al. Growth. 1964;28:225–231.
38. Kim DJ, Ahn B, Han BS, et al. Cancer Lett. 1997;112(2):203–208.
39. Biswas SJ, Bhattacharjee N, Khuda- Bukhsh AR. Food Chem Toxicol. 2008;46(5):1474–1487.
40. Ernst E, Schmidt K. BMC Cancer. 2005;5:69.
41. Panzer A, Joubert AM, Eloff JN, et al. Cancer Lett. 2000;160(2):237–241.
42. Habermehl D, Kammerer B, Handrick R, et al. BMC Cancer. 2006;6:14.
43. Uglyanitsa KN, Nefyodov LI, Doroshenko YM, et al. Drugs Exp Clin Res. 2000;26(5–6):341–356.
44. Panzer A, Hamel E, Joubert AM, et al. Cancer Lett. 2000;160(2):149–157.
45. Jagiello-Wojtowicz E, Kleinrok Z, Urbanska EM. Drugs Exp Clin Res. 1998;24(5–6):213–219.
46. Skivka L, Susak Y, Trompak O, et al. J Oncol Pharm Pract, 2010. [Epub ahead of print]
47. Susak YM, Skivka LM, Rudik MP, et al. Exp Oncol. 2010;32(2):107–110.
48. Vavreckova C, Gawlik I, Müller K. Planta Med. 1996;62(5):397–401.
49. Lee YC, Kim SH, Roh SS, et al. J Ethnopharmacol. 2007;112(1):40–48.
50. Boegge SC, Kesper S, Verspohl EJ, et al. Planta Med. 1996;62(2):173–174.
51. Hiller KO, Ghorbani M, Schilcher H. Planta Med. 1998;64(8):758–760.
52. Häberlein H, Tschiersch KP, Boonen G, et al. Planta Med. 1996;62(3):227–231.
53. Kim Y, Shin M, Chung J, et al. Am J Chin Med. 2001;29(2):265–279.
54. Jagiello-Wójtowicz EWA, Feldo M, Kleinrok Z. Polish J Pharmacol Pharm. 1992;44(suppl):144.
55. Iagodina OV, Mikol’skaia EB, Faddeeva MD. Tsitologiia. 2003;45(10):1032–1037.
56. Kuznetsova LP, Sochilina EE, Faddeeva MD, et al. Ukr Biokhim Zh. 2005;77(2):147–153.
57. Cho KM, Yoo ID, Kim WG. Biol Pharm Bull. 2006;29(11):2317–2320.
58. Ko FN, Chen IS, Wu SJ, et al. Biochim Biophys Acta. 1990;1052:360–365.
59. Vavreckova C, Gawlik I, Müller K. Planta Med. 1996;62(6):491–494.
60. Jagiello-Wójtowicz EWA, Feldo M, Chodkowska A, et al. Polish J Pharmacol Pharm. 1992;44(Suppl):143.
61. Shin MC, Jang MH, Chang HK, et al. Clin Chim Acta. 2003;337(1–2):93–101.
62. Khayyal MT, el-Ghazaly MA, Kenawy SA, et al. Arzneimittelforschung. 2001;51(7):545–553.
63. Jablonski M. Drugs Exp Clin Res. 2000;26(5–6):317–320.
64. Neumann-Mangoldt P. Med Welt. 1977;28(4):181–185.
65. Baumann JC, Heintze K, Muth HW. Arzneimittelforschung. 1971;21(1):98–101.
66. Kniebel R, Urlacher W. Zeit Allg Med. 1993;69(25):680–684.
67. Ardjah H. Fortschr Med Suppl. 1991;115:2–8.
68. Ritter R, Schatton WFH, Gessner B, et al. Comp Ther Med. 1993;1:189–193.
69. Niederau C, Gopfert E. Med Klin (Munich). 1999;94(8):425–430.
70. Melzer J, Rösch W, Reichling J, et al. Aliment Pharmacol Ther. 2004;20(11–12):1279–1287.
71. Rösch W, Liebregts T, Gundermann KJ, et al. Phytomedicine. 2006;13(suppl 1):114–121.
72. Aminev AM, Stoliarenko AI. Vop Onkol. 1960;6(8):81–82.
73. Aminev AM. Am J Proctol. 1963;14(1):25–27.
74. Demchenko PF. Vrachebn Delo. 1957;12:1335–1338.
75. Khmel’nitsakaia NM, Vorob’ev KV, Kliachko LL, et al. Vestn Otorinolaringol. 1998;4:39–42.
76. Xian MS, Hayashi K, Lu JP, et al. Acta Med Okayama. 1989;43(6):345–351.
77. D’Adamo P. J Naturopathic Med. 1992;3(1):31–34.
78. Mazzanti G, Di Sotto A, Franchitto A, et al. J Ethnopharmacol. 2009;126(3):518–524.
79. Juszkiewicz T, Minta M, Wlodarczyk B, et al. Drugs Exp Clin Res. 1992;18(suppl):23–29.
80. De Smet PA, Van den Eertwegh AJ, Lesterhuis W, et al. BMJ. 1996;313(7049):92.
81. Greving I, Niedereichholz U, Meister V, et al. Poster No. PO19, Europäischer Pharmakovigilanz Kongress, Berlin: 1997.
82. Greving I, Meister V, Monnerjahn C, et al. Pharmacoepidemiol Drug Saf. 1998;7:S66–S69.
83. Strahl S, Ehret V, Dahm HH, et al. Dtsch Med Wochenschr. 1998;123(47):1410–1414.
84. Benninger J, Schneider HT, Schuppan D, et al. Gastroenterology. 1999;117(5):1234–1237.
85. Crijns AP, de Smet PA, van den Heuvel M, et al. Ned Tijdschr Geneeskd. 2002;146(3):124–128.
86. Stickel F, Poschl G, Seitz HK, et al. Scand J Gastroenterol. 2003;38(5):565–568.
87. Rifai K, Flemming P, Manns MP, et al. Internist (Berl). 2006;47(7):749–751.
88. Moro PA, Cassetti F, Giugliano G, et al. J Ethnopharmacol. 2009;124(2):328–332.
89. Etxenagusia MA, Anda M, Gonzales-Mahave I, et al. Contact Dermatitis. 2000;43(1):47.
90. Pinto Garcia V, Vicente PR, Barez A, et al. Sangre (Barc). 1990;35(5):401–403.
91. Frohne D, Pfander HJ. A Colour Atlas of Poisonous Plants: A Handbook for Pharmacists, Doctors, Toxicologists, and Biologists. London: Wolfe Publishing, 1984. translated from the 2nd German edition by NG Bisset. pp. 160–162