Summary assessment of safety
Gotu kola is a safe herb. Other than infrequent gastrointestinal upset or contact dermatitis, almost no adverse effects are seen with this herb.
Technical data
Botany
Centella asiatica, a member of the Apiaceae (Umbelliferae, celery) family, is a tropical, evergreen, perennial creeping herb with reddish stems. The 2 to 3 cm long and 3 to 4 cm wide fleshy leaves are reniform with crenated margins and long, thin petioles with sheating bases. Leaves cluster at each stem node. The plant produces tiny pale pink flowers in small white umbels from June to September (northern hemisphere). Its non-branching rhizomes have many root hairs. Gotu kola produces an intricate network of stolons. Fruit is a disk-shaped, reticulate, diachene. The whole plant is aromatic and tastes spicy and sweet.11
Adulteration
There is no common adulterant, although theoretically Hydrocotyle spp. adulteration could occur. Usually these plants have single leaves at nodes and separate stipules, not sheathing leaf bases. Unintentional adulteration may occur due to confusion over the identity of ‘brahmi’, a common name used for both gotu kola and Bacopa monnieri in India. Trade samples of gotu kola herb often contain unacceptably low levels of triterpenes.
Key constituents
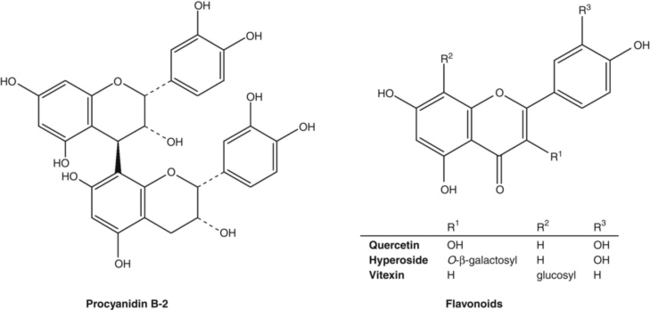
• Triterpene saponins (centellosides), mainly madecassoside and asiaticoside, together with their respective aglycones madecassic and asiatic acids12
• Monoterpenoids and sesquiterpenoids, including myrcene, farnesene, germacrene, caryophyllene and pinene;12 a range of polyacetylenes12
• Flavonoids, including quercetin and kaempferol glucosides.12
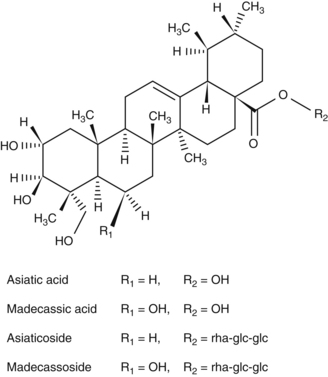
Most work has focused on the three major triterpenoids in gotu kola, which comprise 1% to 8% of the plant.13 One study found that leaves had the highest concentration of triterpenoids compared to roots and petioles.14
Pharmacodynamics
Wound-healing activity
Several in vitro studies have examined the impact of gotu kola or its components on cells involved in wound healing. Early research found that ‘asiaticoside’ (probably TECA) activated the dermal layer and stimulated keratinisation in the epidermis of cultured pig skin.15 Fifty-four genes with known functions for cell proliferation and synthesis of extracellular matrix (ECM) were significantly upregulated when skin fibroblasts were exposed to pure asiaticoside in much later work.16,17 Exposure of human foreskin fibroblasts to TECA and human skin fibroblasts to TTF led to increased collagen synthesis in both cases.18,19 Both type I and type III collagen production was stimulated in human skin fibroblasts by gotu kola saponins.20 The aglycones were also able to stimulate type I collagen synthesis in vitro.21 Ageing of the skin is primarily related to reductions in the level of type I collagen.22 Asiaticoside was found to induce human type I collagen synthesis via activation of the transforming growth factor beta (TGF-beta) receptor I kinase-independent Smad pathway. The Smad signalling cascade is known to perform an important function in human collagen production and this might explain how asiaticoside inhibits hypertrophic scar formation.23
Systemic doses of gotu kola extract or its phytochemical constituents have been shown to improve healing or strengthen connective tissue in various animal models. In an early study TECA (100 mg/kg, oral) shortened the healing time of iterative wounds in rats.24 A crude ethanol extract of gotu kola (1 mL/day) was applied topically or given orally to rats for 24 days after wounding.25 Increased cellular proliferation and collagen synthesis at the wound site was observed, as evidenced by increases in the DNA, protein and collagen content of granulation tissue. Quicker and better maturation and cross-linking of collagen were found and the wounds healed faster. Compared with topical application, the oral use of gotu kola was generally more active for the above parameters.
Topical and oral asiaticoside were significantly more effective at promoting wound healing in guinea pigs than vehicle controls.26 Applications of 0.2% asiaticoside solution increased the proline and collagen content of the healing tissue, increased its tensile strength and enhanced epithelialisation. Diabetic animals also showed improved wound healing after the application of a 0.4% asiaticoside solution. In the chick chorioallantoic membrane assay, 40 μg/disk of asiaticoside increased angiogenesis.
Gotu kola extract (800 mg/kg/day, oral) significantly improved wound strength, connective tissue content and healing rates in rats.27 It also overcame the suppressive action of dexamethasone on wound healing, something that was not observed for asiaticoside alone at oral doses of 2 mg/kg.28 However, oral doses of asiaticoside alone did dose-dependently increase the tensile strength of wounds after 4 and 6 days.28
Osteoarthritis is a degenerative joint disease in which focal cartilage destruction is a primary feature. A triterpene isolate of gotu kola containing 42% asiaticoside and 55% aglycones (0.3 mg/mouse/day, oral for 11 days) inhibited zymosan-induced cartilage degeneration without affecting inflammatory cell filtration and joint swelling.29 This suggests a chondroprotective activity, which was supported by additional in vitro experiments on bovine cartilage explants and chondrocytes.
The impact of oral asiaticoside (10 to 20 mg/day for 10 to 15 days) on parameters of dermal wound healing was studied in 15 patients in an early human pharmacological study.30 The higher dose for longer periods tended to increase leucine aminopeptidase activity and decrease thiol groups in the skin, which were considered to be favourable for accelerated healing.
As already touched on above, gotu kola or its components have also enhanced wound healing when applied topically. Three formulations (ointment, cream and gel) of an aqueous extract of gotu kola increased wound cellular proliferation, collagen synthesis and rates of healing in rats.31 The gel formulation appeared to confer greatest activity. Local application of TECA improved rates of connective tissue regeneration and remodelling in a rat model.32 Glycosaminoglycan synthesis was also increased. Asiaticoside exerted a preferential stimulation of collagen synthesis and was active at low doses only.32 A study in rats found that asiaticoside application (0.2%, topical) twice daily for 7 days led to increased enzymatic (such as catalase and superoxide dismutase) and non-enzymatic (ascorbic acid and vitamin E) antioxidants in the wounds of rats.33 However, these differences were not apparent after 14 days of continuous application, leading the authors to suggest that enhancement of antioxidant levels by asiaticoside early in healing may be an important factor in its wound-healing ability.
Topical application of asiaticoside at 0.5 or 1.0% three times daily for 1 to 3 months remarkably alleviated scar formation in the rabbit ear model.34 TGF-beta1 expression was decreased and inhibitory Smad7 expression was remarkably enhanced (see earlier).
One early study of burns failed to find any benefit of intraperitoneal injection of TECA on wound healing or mortality in mice compared with the control group.35 However, the topical application of a 70% ethanolic extract of gotu kola or isolated asiaticoside (at quite low concentrations) significantly accelerated burn wound repair in mice, possibly via enhanced angiogenesis.36 Oral doses of madecassoside (6, 12 and 24 mg/kg) facilitated burn wound closure in a dose-dependent manner in mice.37 Investigations suggested that several mechanisms may be involved, including antioxidant activity, regulation of collagen synthesis and promotion of angiogenesis.
Oral doses of gotu kola may also improve healing of gastric ulcers. An aqueous extract of gotu kola (0.1 to 0.25 g/kg, oral) and asiaticoside (5 to 10 mg/kg, oral) accelerated healing in rats with acetic-acid-induced gastric ulcers.38 This was associated with increased expression of basic fibroblast growth factor, faster epithelialisation and increased angiogenesis in the healing ulcer crater. The same research group found that the same doses of gotu kola given to rats before ethanol administration significantly inhibited gastric lesion formation and decreased mucosal myeloperoxidase activity (an indicator of neutrophil infiltration).39 These results suggested that the herb strengthened the gastric mucosal barrier and indirectly reduced free radical damage. Later in vivo research by this group suggested that an anti-inflammatory activity might also be involved.27 Increased mucosal defence was supported by another study that used fresh gotu kola juice.40
Antifibrotic effects
Interestingly, given the data cited above clearly showing that gotu kola and its constituents can increase collagen synthesis in wounds, other research suggests it can decrease collagen in situations of fibrosis. Using an in vitro model of overactive hepatic stellate cells, asiatic acid and asiaticoside both inhibited excessive collagen formation.41 Antifibrotic effects were also seen after applying gotu kola extract in vitro to mammalian renal cells.42 One in vitro study found that gotu kola and its saponins inhibited the growth of keratinocytes, suggesting antipsoriatic activity.43 There is also the suggestion from one model that asiaticoside might decrease post-burn hypertrophic scars.44 Rats given TTF by gastric lavage also exhibited significantly less liver fibrosis following administration of dimethylnitrosamine.45 Given these amphoteric effects, gotu kola could perhaps act as a collagen modulator (see also the Clinical trials section regarding scleroderma and keloid formation).
Anti-inflammatory and analgesic activities
Asiatic acid has been shown in vitro to moderately inhibit multiple inflammatory pathways, including inhibiting NF-kappaB and the expressions of cyclo-oxygenase-2 and inducible nitric oxide synthase, and by blocking production of pro-inflammatory cytokines in stimulated macrophages.46 Asiaticoside was less active than asiatic acid in this test system. Madecassic acid exerted similar activity in vitro using stimulated macrophages.47
Centellosides given orally (50 to 200 mg/kg) or subcutaneously (50 to 100 mg/kg) to rats after implantation of glass rods dose-dependently reduced granuloma after 3 weeks.48 All four individual compounds tested in this model were more or less equally effective.
Aqueous extract of gotu kola (4 and 10 mg/kg, ip) was effective at inhibiting PGE2-induced inflammation in rats (including more effectively than mefenamic acid in the case of the higher dose).49 Pain was inhibited in mice by an aqueous extract of gotu kola (assessed by the acetic acid and hot plate tests) in this same study.
Madecassoside (3, 10 and 30 mg/kg/day, oral from days 21 to 42) attenuated the inflammatory response in collagen-induced arthritis in mice.50 A similar study by a different research group confirmed this finding.51 The latter authors suggested that gotu kola might have clinical value in rheumatoid arthritis.
Asiatic acid blocks the binding of angiotensin II to its receptor in vitro.52 This may also have implications in terms of gotu kola reducing or preventing hypertension.
Central and peripheral nervous system activity
Various extracts of gotu kola and pure asiaticoside have demonstrated anxiolytic activity in rats and mice.53,54 In mice, asiaticoside 10 mg/kg (oral) was as effective as diazepam 0.3 mg/kg.54 Anxiolytic activity was not linked to sedation in rats.53 Anxiolytic effects were seen in two older studies in rodents, as well as anticonvulsant activity (in one case as potent as that of diazepam).55,56 In both of these studies, gotu kola extracts also potentiated the sedative effects of barbiturates. Mice given 10 to 20 mg/kg asiaticoside (oral) exhibited similar antidepressant effects to clomipramine at 50 mg/kg.57 Some of these results suggested gotu kola acts as a GABAergic agent, although in other research aqueous gotu kola extracts appeared to acts by decreasing the turnover of catecholamines.58
In vitro, an aqueous extract of gotu kola increased phosphorylation of cAMP-response element binding protein (CREB) in neuroblastoma cells expressing beta-amyloid.59 CREB is a crucial regulator of genes involved in memory formation. Various synthetic derivates of asiaticoside directly protected neurons against the toxic effects of beta-amyloid in vitro.60 This potential activity in Alzheimer-type dementia has been further explored in animal models. An aqueous extract of gotu kola (100, 200 and 300 mg/kg/day, oral for 21 days) prevented streptozotocin-induced cognitive deficit and oxidative stress in rats.61 In mice spontaneously expressing beta-amyloid plaque formation, an undefined extract of gotu kola (2.5 and 5.0 g/kg, oral) decreased beta-amyloid formation after 8 months, partially by modulating oxidative stress and protecting against DNA damage.62
The potential neuroprotective activity of gotu kola has also been explored in experimental models. Asiatic acid exerted a significant neuroprotective effect in cultured cortical cells challenged with glutamate-induced exitotoxicity.63 In vivo, a chloroform-methanolic extract of gotu kola (100 and 200 mg/kg, oral) protected against the free radical generation and excitotoxicity induced by feeding monosodium glutamate to rats.64 Asiatic acid (37, 75 and 165 mg/kg, oral) demonstrated neuroprotective activity in a mouse model of cerebral ischaemia65 and a 50% ethanolic gotu kola extract (300 mg/kg, oral) protected against MPTP-induced neurotoxicity in aged rats (a model of parkinsonism).66
As alluded to above, much of the neuroprotective activity of gotu kola might result from its ability to enhance endogenous antioxidant protective mechanisms. This hypothesis was further examined in several experimental models. An aqueous extract of gotu kola (200 mg and 300 mg/kg, oral) increased brain levels of glutathione and catalase and decreased measures of oxidative damage in rats.67 This was coupled with improved cognition in a series of tests, which the authors suggested was linked to the observed antioxidant effects. Age-related decline in cerebral antioxidant defences and commensurable increases in measures of brain oxidation were considerably countered in aged rats by a gotu kola extract (300 mg/kg/day, oral for 60 days).68 Similar antioxidant findings were observed in the brains of young mice fed a gotu kola extract (0.5% and 1.0% of diet) for 4 weeks.69 Short-term oral intake of an aqueous extract of gotu kola (5 mg/kg/day for 10 days) conferred marked resistance against 3-nitropropionic acid-induced oxidative stress and mitochondrial dysfunction in the brains of young mice.70,71
There is even a suggestion from experimental models that gotu kola might possess neuroregenerative and neurodevelopmental activities. A gotu kola ethanolic extract (300 to 330 mg/kg/day, oral for 18 days) accelerated peripheral nerve regeneration following damage in rats.72 More rapid functional recovery and increased axonal regeneration (larger calibre axons and greater numbers of myelinated axons) were observed compared with controls, indicating that axons grew at a faster rate. The same investigation found that an ethanolic extract and asiatic acid increased neurite outgrowth in vitro, but an aqueous extract was inactive.
Fresh leaf extract of gotu kola (4 and 6 mL/kg/day) fed to rat pups for 4 to 6 weeks increased dendritic length and branching in hippocampal neurons, a region of the brain involved in learning and memory.73,74 Similar findings were also observed by the same research group in adult rats.75 This might explain observations of enhanced learning and memory in young rodents fed gotu kola juice or aqueous extract76,77 and suggests neuroplastic effects from the herb.
Anticonvulsant activity for gotu kola has also been observed in a few studies. An aqueous extract of gotu kola (300 mg/kg, oral) decreased pentylenetetrazole-induced seizures in rats and showed improvement in the associated learning deficit.78 A different research group found this activity was associated with a recovery of brain levels of acetylcholine and acetylcholinesterase. A study in mice found the ethyl acetate extract of gotu kola exerted an anticonvulsant effect that was additive to some antiepileptic drugs.79
Circulatory activity
A review of pharmacological investigations, including human pharmacological studies, discussed the desirable properties of gotu kola (specifically TTF) in the context of the management of CVI, a more severe clinical manifestation of varicose veins.80 These included an action on fibroblasts in the vein wall (improving collagen synthesis and remodelling), enhanced microcirculation with decreased oedema, improved lymphatic drainage, and a possible decrease in endothelial cell damage.
More details of some of these studies in human volunteers follow. An early open study found that 60 mg/day of an unspecified gotu kola extract (probably an isolate) for 30 days increased venous return in patients with CVI.81 In 20 patients with varicose veins, TTF (60 mg/day for 90 days) significantly lowered the elevated serum levels of uronic acids and lysosomal enzymes.82 These results were interpreted as an indirect confirmation of improved connective tissue metabolism in the vein wall.
The vacuum suction chamber produces a wheal on the skin, with the disappearance time (DT) determined by capillary filtration and permeability. After 2 weeks of 180 mg/day of TTF there was a significant decrease in DT in the limbs of patients with either superficial (n=22) or deep (n=12) venous incompetence. This was confirmed by laser Doppler flowmetry and clinical signs and symptoms.83 TTF (90 mg/day for 3 weeks) also reduced the number of circulating endothelial cells in the veins of 15 patients with a history of deep vein thrombosis in an open, controlled study.84 This finding was interpreted as indicating a reduction of endothelial cell injury and an improvement in vascular integrity in these patients with diseased veins.
Some cardioprotective activity has been observed for gotu kola and madecassoside. Pretreatment with a hydroethanolic extract of gotu kola whole plant (100, 500 and 1000 mg/kg/day, oral for 7 days) dose-dependently reduced left ventricular necrosis and measures of oxidative stress in a rat model of myocardial ischaemia-reperfusion injury.85 Madecassoside demonstrated similar protective activity in analogous models in rats86 and rabbits.87 Pretreatment with an aqueous extract of gotu kola (200 mg/kg, oral) protected against the cardiotoxicity of doxorubicin in rats by improving mitochondrial function88 and improving antioxidant responses.89
Antimicrobial and antiparasitic activities
Aqueous but not ethanolic extracts (and pure asiaticoside) of gotu kola were active against herpes simplex virus I and II in vitro.90,91 Two in vitro studies failed to find any activity for gotu kola extracts against Mycobacterium leprae or M. tuberculosis.92,93 One study found that liposomal asiaticoside was significantly more active against M. leprae in vitro.94 A tincture of gotu kola administered orally to dogs (30 mg/kg/day) dramatically reduced blood levels of infection with the microfilarial parasite Dirofilaria immitis without causing any adverse effects.95
Antitumour activity
Gotu kola extracts and triterpenoids have demonstrated activity against a range of cancer cells in vitro, including melanoma, uterine cancer and gastric cancer.96,97 Asiaticoside in vitro induced apoptosis in a human colon cancer cell line.98 A methanolic extract of gotu kola exerted the same effect on MCF-7 human breast cancer cells.99 Asiaticoside has also been shown to enhance the lethality of vincristine against a range of multidrug resistant cancer cell lines.100 Life-span was increased in mice with solid and ascites tumours when they were fed gotu kola crude (1 g/kg) and purified (40 mg/kg) extracts.101 Only simultaneous or prophylactic dosing was protective. Aqueous gotu kola extract (10 and 100 mg/kg, oral) decreased the incidence of colon cancers formed in rats exposed to the carcinogen azoxymethane.102
Other activity
The aqueous extract103 (10, 100 and 300 mg/kg/day, orally for 7 days in mice) or suspension (100 mg/kg/day, orally for 7 days in rats)104 of gotu kola enhanced both cellular and humoral immune responses. In mice, injections of a methanolic extract of gotu kola enhanced phagocytic activity without affecting B lymphocytes or antibody responses.105
Traditionally, gotu kola has been regarded as somewhat of an adaptogen and there is experimental support for this. Cold and restraint stress effects were countered in rats106 and an ethanolic extract (100 mg/kg, oral) exhibited significant antistress activity in rats in a range of experimental models.107,108
Gotu kola may exert antitoxic activity against arsenic. Its aqueous extract (100, 200 or 300 mg/kg, oral) protected against arsenic toxicity in rats, especially in terms of measures of oxidative stress.109 Arsenic concentrations in tissues were not changed.110
Protection against radiation-induced taste aversion was noted in a study in rats (100 mg/kg gotu kola extract, ip).111 Gotu kola extract (100 mg/kg, oral) improved survival against radiation in mice112 and protected against radiation-induced liver damage.113
Pretreatment with asiaticoside (5 to 20 mg/kg/day, oral for 3 days) demonstrated ‘remarkable’ hepatoprotective activity following lipopolysaccharide and galactosamine administration to mice, possibly related to inhibition of TNF (tumour necrosis factor)-alpha and cell kinases.114
Pharmacokinetics
An early study in rats using radioactively labelled compounds found that asiatic and madecassic acids were well absorbed, up to 50% of a single oral dose.115 Once absorbed, the acids were subject to phase II conjugation to form glucuronides and sulphates and largely excreted via the bile. Oral asiaticoside is first converted to asiatic acid by the bowel microflora, leading to the slow and prolonged appearance of asiatic acid in rat plasma.
Oral administration of a gotu kola extract to beagles (containing 540 mg saponins of which asiaticoside was about 72%) revealed the following pharmacokinetic parameters for asiatic acid: half-life 4.29 h, Tmax 2.7 h and Cmax 0.74 µg/mL.116
In a double blind, randomised, crossover, multiple-dose study, six healthy male and six healthy female volunteers each received asiatic acid (6 mg) or asiaticoside (12 mg) twice daily for 9 days, with a single dose on the tenth day.117 Pharmacokinetic parameters (for asiatic acid in plasma) were assessed only after the single dose on the tenth day and were as follows: Cmax 0.98 μg/mL and 0.65 mg/mL, Tmax 4.0 and 5.4 h and pre-dose level 0.39 and 0.50 μg/mL for administration of asiatic acid and asiaticoside, respectively. Despite these differences, the steady state area-under-the-curve values (reflecting total absorption) were similar for the two compounds. (Note that although the mg doses of the two compounds differed, their molar quantities were similar, owing to the higher molecular weight of asiaticoside compared to its aglycone asiatic acid.)
After oral administration of TTF at 30 or 60 mg twice daily for 7 days to 12 healthy men, the time to maximum plasma concentrations of asiatic acid was stable at just over 4 h, while the half-life and area-under-the-curve increased significantly from baseline to the end of study, reaching 6.3 or 10.3 h and 10.5 or 20.8 μg/mL/h for the 30 or 60 mg doses, respectively.118 There were significant intra-individual differences in all parameters studied. Steady state Cmax values for asiatic acid were 1.03 and 1.69 μg/mL, respectively. Asiaticoside was determined to be converted to asiatic acid.
Clinical trials
Vascular diseases
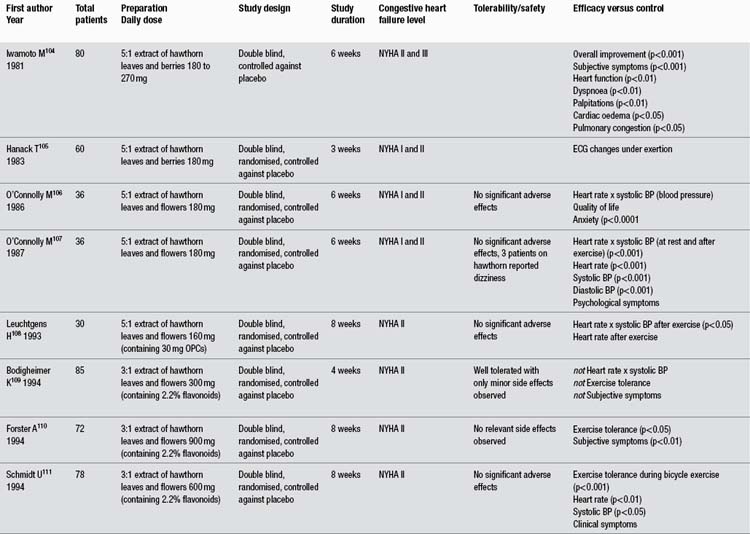
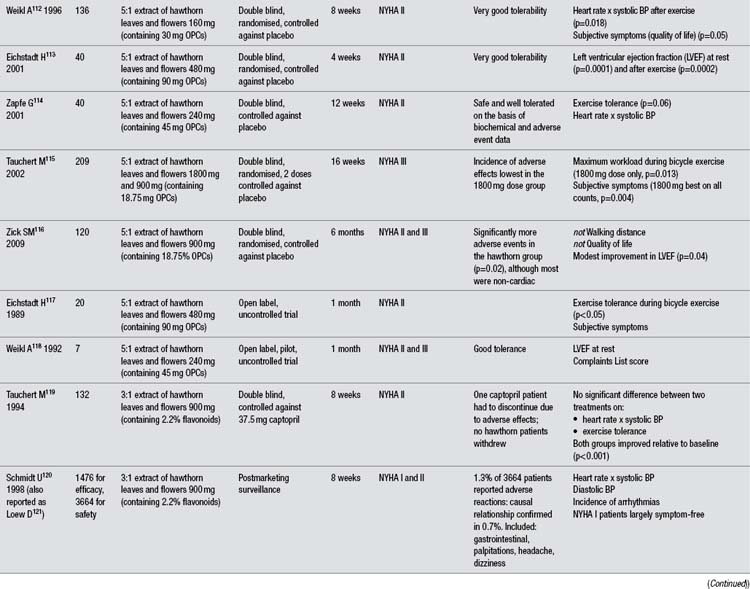
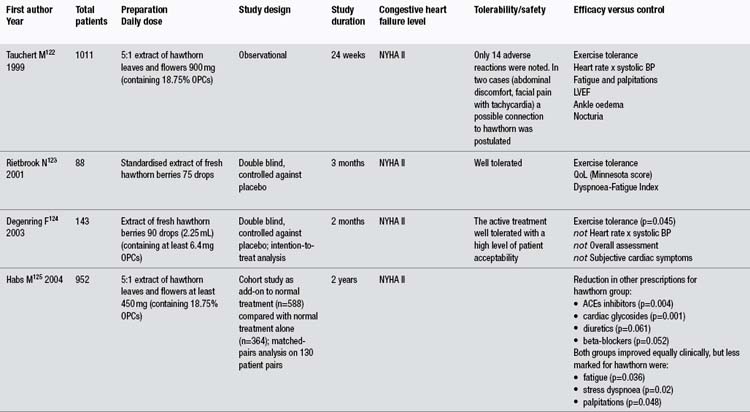
In more than any other patient group, gotu kola has demonstrated a clinical ability to help people with chronic problems with varicose veins in the lower limbs accompanied by oedema, itch, skin atrophy and ulceration, and discomfort or pain (known as chronic venous insufficiency or CVI). The larger clinical trials have been summarised in Table 1. Unfortunately, all key studies to date have been conducted by the same research group and have not been well designed. As noted in the table, apparently one of those studies has been published twice and another essentially three times, with considerable time having elapsed between the first and last publications. Several less rigorous trials have also been reported using TTF for CVI patients, but only those including 40 or more patients were included in this review and Table 1.
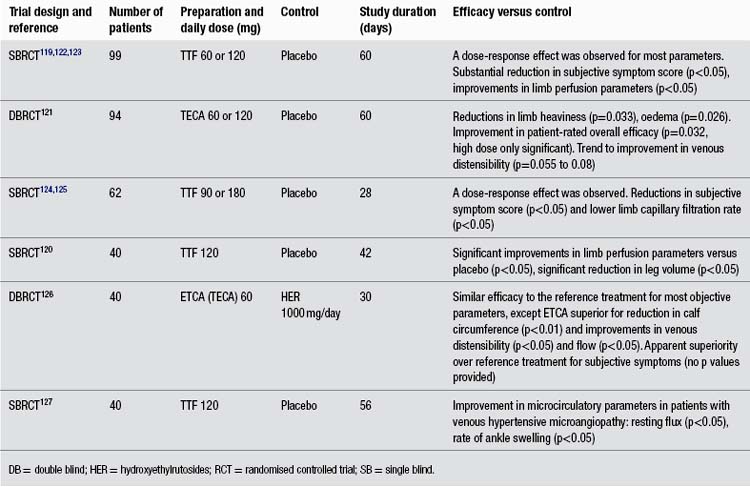
The reasonably methodologically sound randomised clinical trials compared TTF to placebo in 99 and 40 patients (both used 60 mg twice per day, and one study also included a group receiving only 60 mg/day).119,120 However, both were only single blind trials. A more rigorous double blind, randomised trial assessed TECA, 60 or 120 mg/day against placebo.121 This trial found a modest benefit for both doses of the extract over placebo at improving symptomatology. However, this benefit was not as clinically significant as reported in the less rigorous trials.
It is useful to reflect here on why a herb with a well-established reputation for promoting healing has developed into a treatment for CVI. Varicose veins and CVI have long been regarded as disorders of valvular incompetence. However, recent evidence suggests that changes in the vein wall could well precede such incompetence. For example, varicosities are often observed below competent valves and often can occur before valvular incompetence.128 Defects in ECM and collagen composition in the vein wall are thought to be part of this process.129 Endothelial damage from abnormally sustained venous pressure could contribute as well. Given the above, the noted pharmacological properties of gotu kola in terms of effects on fibroblasts, collagen and ECM in the vein wall, together with its potential for reducing endothelial damage and improving microcirculation, would suggest that it is indeed a plausible (and perhaps disease-modifying) treatment for CVI.
Two controlled trials have investigated the activity of oral TTF in patients with microvascular damage due to diabetes. The largest trial involved 100 patients with or without neuropathy and compared TTF (120 mg/day) with placebo over 12 months and also in 40 healthy controls.130 TTF was significantly more effective at improving microcirculatory parameters and oedema (p<0.05). A smaller trial with 50 patients compared 60 mg TTF twice daily with placebo or no treatment for 6 months.131 TTF resulted in significant improvement in parameters linked to microscopic vascular damage, including capillary permeability (p<0.01 to p<0.05).
In a randomised trial, the impact of TTF (180 mg/day) for 2 days before, the day of, and 1 day after, a long airline flight (up to 14 h) against no treatment was compared in patients with mild-to-moderate chronic venous insufficiency.132 Ankle oedema was significantly less in the TTF group (p<0.05), as were microcirculatory parameters such as rate of ankle swelling and venoarteriolar reflex (p<0.05). All parameters deteriorated with the length of flight, a phenomenon that was only partially compensated by the herbal treatment.
An open trial of TTF (60 mg/day) combined with bulk-forming laxatives (as appropriate) was rated as effective in relieving symptoms in over 90% of 210 patients with first or second degree haemorrhoids.133
Two interesting trials suggest that gotu kola may be able to stabilise vulnerable arterial plaque, indicating a possible role for the herb in modifying cardiovascular outcomes. Most atherosclerotic plaques do not trigger life-threatening events. A vulnerable plaque is one that is at high short-term risk of rupture. Plaque rupture is by far the most frequent cause of arterial thrombosis. It is deemed responsible for about 75% of coronary thrombi leading to heart attacks or death and about 90% of thrombosed carotid plaques causing ischaemic stroke. Only plaque with very thin fibrous caps is at risk of rupture and even just a small lesion is life-threatening. These plaques are essentially unstable because of a deficiency of connective tissue in their matrix. Even in the presence of widespread arterial disease, rarely more than a few plaques appear to be at risk of rupture at any given moment.134 One group of researchers observed: ‘It is not clear why some plaques lead to clinical manifestations, whereas many others remain asymptomatic and heal with subsequent fibrosis…’.135 In one sense arterial plaque is a type of wound on the blood vessel wall and vulnerable plaque can be viewed as a wound that is either not healing appropriately or in the early stages of healing (fibrosis).
In two placebo-controlled clinical trials, TTF stabilised low-density carotid136 and femoral artery plaques.137 The dose used in both trials was 180 mg/day for 12 months. These clinical outcomes were assessed by significant and marked increases in the ultrasound echogenicity of plaques compared with placebo (p<0.05). For the patients with carotid lesions, symptomatic cerebral events were observed in 6.5% of patients taking TTF compared with 11% of controls (p<0.05) and positive magnetic resonance images (indicating cerebral ischaemic lesions) were 7% in the herbal group, versus 17% for the control group (p<0.05). Arterial plaque which is echolucent (low echogenicity by ultrasound) has a limited connective tissue component and the plaque is weaker and prone to ulceration, rupture and embolisation. This is not quite the same as vulnerable coronary artery plaque, but shares many similarities.
On the basis of these findings it would be interesting to examine the impact of the long-term intake of gotu kola on health outcomes in patients with coronary artery disease.
Wounds, ulcers, and other skin and epithelial disorders
Early publications comprise around 30 mostly small, uncontrolled, open label clinical trials or case observation studies, dating from the late 1950s. These papers variously refer to the use of ‘asiaticoside’ or a proprietary triterpenoid isolate product, which were probably identical and represent an early version of TECA. The TECA was typically administered by intramuscular (and sometimes intralesional) injection and was often applied topically as well. Although injection was used, the good oral bioavailability of the gotu kola triterpenoids, and the fact that typical oral doses tend to be much higher than the injected doses used, suggest that these early data still have clinical relevance to the oral use of the herb. Conditions with favourable outcomes included bladder ulcers,138 corneal lesions,139 episiotomies140 and other postsurgical lesions,141 leg ulcers,142 skin grafts,143 skin ulcers144 and traumatic injury.145 Not all studies yielded positive outcomes.146,147 A few of the larger, less dated and/or interesting studies are featured below.
Following a positive series of published case reports148 and their experience with a burn patient who demonstrated a substantial reduction in hypertrophic scars, a group of Canadian clinicians conducted a clinical investigation of TECA as an antikeloid agent.149 Their first clinical evaluations involved the injectable form, but later they were able to report on 227 patients with keloids or hypertrophic scars who were treated with the oral form of TECA (60 to 90 mg/day, but up to 150 mg/day in stubborn cases). In an open label trial, 116 of 139 patients were found to respond after 2 to 18 months, either by relief of symptoms or by a gradual disappearance of inflammation. These positive results remained after the cessation of therapy. In most cases the treatment was started on well-established, non-responsive lesions, some of many years’ duration. A small, concurrent double blind study demonstrated a significant benefit over placebo (p<0.05). The authors also studied the preventative action of TECA in 88 patients undergoing surgical scar revision, with treatment started a few weeks prior to surgery and maintained for 3 months after. Improvement was noted in 79% of patients, but there was no comparative control group to account for the benefit of surgery alone.
In a recent clinical trial conducted in Thailand, 200 patients with diabetic foot ulcer were randomised to receive gotu kola extract or placebo.150 General symptoms, wound characteristics, wound size and wound depths were examined at days 7, 14 and 21. Wound contraction was determined by the decrease in the volume of the wound. Twenty cases dropped out from the study and 10 cases were terminated before the end of treatment because termination criteria were met. (Termination criteria included patient refusal, wound infection, delayed primary sutured wound and secondary healing wound.) Results for the 170 patients who completed the trial were analysed.
By the end of each week, wound contraction was significantly better in the group treated with gotu kola compared with those treated with placebo (p≤0.001). The trend to good wound contraction occurred earlier for the herb group than in the placebo group: for example, by day 7, 28.5% of patients receiving gotu kola had good wound contraction, compared with 12.8% in the placebo group; by day 14, the results for good contraction were 38.1% and 18.6% for herbal and placebo treatment, respectively. More granulation tissue formed in the placebo group than the herb group, and the difference between the groups was significant at days 7, 14, and 21 (p<0.001). There were no systemic side effects reported and there was no significant difference in wound infection between the two groups. The dose used was unclear, with the capsules variously described as containing ‘50 mg of extracted asiaticoside’ and ‘50 mg freeze dry lyophilized extract’. The trial author disclosed the dose in a personal communication (March 2011) as 300 mg/day of extract, corresponding to 11.2 g of fresh leaf.
There is some suggestion from case reports in the 1970s that gotu kola might assist liver pathologies. In one study, 30 patients with clinical hepatic cirrhosis (mainly due to bilharzia) and previous splenectomy were given TECA (20 mg/day, im) for 55 days.151 The nodularity and firm consistency of the liver decreased markedly in all the cases and biopsy revealed a substantial reduction in collagen fibres in both the fine and medium portal tracts. Serum alkaline phosphatase and glycoproteins were also reduced. In a second study, ongoing administration of TECA (90 to 150 mg/day, presumably oral) resulted in improvement in 5 of 12 patients with recently diagnosed alcoholic cirrhosis or chronic liver disease.152 Benefits were mainly observed in the patients with alcoholic cirrhosis, where biopsy revealed reduced sclerosis or steatosis.
Gotu kola might have value in obstetrics according to studies from the 1960s. Results for 131 cases of episiotomies treated with TECA (25 mg/day, im on the second, fourth and sixth days after surgery) from an open label trial demonstrated improved healing and reduced symptoms in 106 cases compared with other (unspecified) cases treated by different means.153 Clinical observation of 114 patients with perineal lesions (especially following episiotomy) found injected TECA (with concurrent topical use in some cases) accelerated healing, resulting in reduced hospital stays.140 Accelerated healing of perineal tears was found in another study.154
Some novel uses of gotu kola preparations have also been investigated. Open trials have demonstrated the benefit of topical madecassoside and either intramuscular madecassoside or asiaticoside in patients with previously non-healing wounds or skin ulcers.155,156 In a prospective, open clinical trial, 30 patients with active leprosy were given 500 mg powdered gotu kola (for 12 months), 60 mg asiaticoside (for 6 months) or dapsone three times per day.157 Improvement in leprosy ulcers was similar in all groups. Intramuscular (20 mg/day) and/or topical application of TECA in 90 patients with perforating leg ulcers due to leprosy were compared against placebo, and both treatments were found to be significantly superior at healing the ulcers (p<0.01).158 A combination of dapsone, hydnocarpic acid (from Hydnocarpus wightiana) and isolated asiaticoside (50 mg/day) for 6 to 11 months in 10 patients with active leprosy was just as effective, but less toxic, than standard multidrug treatments.159
The value of gotu kola compounds in the connective tissue disorder scleroderma has been assessed. An open trial involving 13 women with scleroderma and using either 20 or 60 mg/day TECA (im) observed improvement of symptoms in 11.160 TECA 30 mg/day (oral), sometimes with topical TECA ointment, improved local and systemic scleroderma in the majority of 54 patients investigated in another open trial.161 Benefits were also seen in another seven patients with scleroderma (TECA, 30 to 60 mg/day, oral),162 but a review of 15 cases in children found little benefit from current treatments, including asiaticoside (probably TECA).163
Some of the early studies on skin disorders also focused on topical application. Topical application of a cream of gotu kola cleared psoriasis lesions in five and improved the remaining two of seven patients in one case series.164 A cream containing 1% of an extract of gotu kola completely healed previously non-healing, chronic, infected skin ulcers in 17 out of 22 patients after 3 weeks in an open trial.165 One double blind trial in 18 patients failed to find that a 2% TECA powder had any better effect than placebo on healing after skin graft removal.143
In recently published studies there has been a further focus on the topical use of gotu kola compounds, probably thereby underestimating the unique value of the internal use of the herb. In a preliminary open trial, 12 healthy volunteers had a small wound induced on their forearms and then 0.3% madecassoside combined with copper, zinc and manganese was applied daily for 22 days.166 The researchers concluded that inflammation was resolved more quickly, epidermal regrowth was more rapid, and there was less scarring, apparently compared with no treatment. An ointment containing extracts of gotu kola, Viola tricolor and Mahonia aquifolium was not superior to a base cream in a randomised double blind, vehicle-controlled, half-side comparison in 88 patients with mild-to-moderate atopic dermatitis.167
Some of the topical trials are more cosmetic in nature. One early double blind trial involving 80 pregnant women investigated the effects of a topical cream containing gotu kola, vitamin E and collagen-elastin hydrolysate compared with placebo at preventing striae gravidarum (stretch marks).168 The cream was applied once per day. After 12 weeks, the group applying the active cream developed significantly fewer striae than the placebo group.
In a double blind, randomised clinical trial, a gel containing gotu kola extract, Rosmarinus officinalis extract, tetrahydrocurcumin, and dimethylaminoethanol was compared with placebo in 28 women.169 Each woman treated half their face with the gel or placebo for 4 weeks. At the end of this time, skin was significantly softer on the gel-treated skin compared with placebo, as rated both objectively and subjectively. Three women left the trial due to mild skin irritation. A cream with 0.1% asiaticoside applied around the eyes of 27 women for 12 weeks in an open trial was associated with improvement or elimination of wrinkles in 18.170
A 6-month randomised, double blind study was conducted on photo-aged (sun-damaged) skin in 20 female volunteers to assess the impact of a topical treatment containing 5% vitamin C and 0.1% madecassoside.171 There was a significant improvement in the clinical score for deep and superficial wrinkles, suppleness, firmness, roughness and skin hydration. These results were corroborated by objective tests. The reappearance of a normally structured, ‘young’ elastic fibre network was observed.
Nervous system and adaptogenic effects
A single dose of 12 g of crude gotu kola was more effective than placebo at reducing the acoustic startle response in 40 healthy adults in one double blind, placebo-controlled, randomised trial.172 An uncontrolled trial observed that a 70% ethanolic extract of gotu kola (500 mg/day corresponding to about 4.5 g of starting herb) over 2 months significantly improved measures of stress, anxiety, adjustment, depression and attention (all p<0.01 compared with baseline).173 Results after 2 months were substantially better than after 1 month.
An uncontrolled trial used gotu kola to treat 33 volunteers with generalised anxiety disorder.173 They received gotu kola extract (about 9 g/day dried herb equivalent) for 60 days. Participants were initially assessed for mental status using the Brief Psychiatric Rating Scale. Results using self-assessment questionnaires revealed significant improvements (p<0.01) from baseline in anxiety, stress, depression, adjustment and attention at day 30 and day 60.
Early studies focusing on cognitive function yielded conflicting results. A daily 500 mg tablet of gotu kola herb for 12 weeks significantly improved mental function and behaviour in 30 intellectually impaired children in a placebo-controlled trial.174 However, a double blind, placebo-controlled trial using the same tablet daily for 1 year observed no effect on the mental ability of children with normal baseline intelligence.175
A later trial was more positive. Twenty-eight healthy volunteers with a mean age of 65 years were randomly divided into four groups.176 One group received a placebo and the others were given respectively 250, 500 and 750 mg/day of a gotu kola extract for 2 months. The gotu kola extract was standardised for total phenolic content and also contained around 5% asiaticoside and asiatic acid. Assessment using a battery of cognitive performance tests was undertaken 1 h after the first dose (for acute effects) and after 1 and 2 months (for chronic effects).
For the acute effect (single dose) assessment, only the highest dose of gotu kola demonstrated a significant effect above placebo for just two of the nine tests used. These were the reaction time of spatial memory and the percentage accuracy of numeric working memory. Based on what is known about these tests, the authors suggested that these acute effects of gotu kola might partly occur via the modulation of dopamine and norepinephrine in the prefrontal cortex, together with the modulation of acetylcholine and serotonin in the hippocampus.
The repeated administration of gotu kola for a further 2 months also showed the same significant increases in spatial memory reaction time and percentage accuracy of numeric working memory. In addition, significant improvements were seen above placebo for choice reaction time, numeric working memory reaction time, accuracy of word recognition and accuracy of picture recognition. These effects were most consistently demonstrated at the highest dose of gotu kola. There was no significant effect on simple reaction time, the digit vigilance test, the accuracy of spatial memory, word recognition reaction time and picture recognition reaction time. The above effects were interpreted by the authors as gotu kola enhancing working memory. The extract not only improved cognitive performance, but also anxiety: the highest dose increased calmness and alertness after both 1 and 2 months of treatment.
When gotu kola powder (dosage not specified) was administered to 43 healthy but impoverished East Indian adults for 6 months, it elevated erythrocytes and haemoglobin and decreased blood urea nitrogen in a low-quality placebo-controlled trial.177,178 Gotu kola extract at 250, 500 and 750 mg/day significantly improved physical strength and fitness measured by the 30-second chair stand test in 80 healthy older volunteers (average age around 65 years). The two higher doses also improved the physical subscale of a quality of life scale (SF-36). This was a placebo-controlled, randomised, double blind trial and implies an adaptogenic activity for the herb.179
Other conditions
An uncontrolled study examined the effect of 60 mg/day oral TECA and 20 mg three times per week (im) in 64 patients with gastric and duodenal ulcers.180 After 10 weeks, all but two patients had healed completely (and the majority had healed after 6 weeks). In an open randomised trial, patients who had extracts of gotu kola and Punica granatum (pomegranate) applied to periodontal pockets experienced significantly better healing than those who received only standard medical care.181 This confirmed the preliminary results of an earlier study using the same extracts.182
Toxicology and other safety data
Toxicology
Gotu kola has been consumed as a leafy vegetable, particularly in Bangladesh, Thailand, Indonesia (West Java), Sri Lanka and South Africa,183–185 and appears to have no harmful effect when used as a food.186 The leaf and stolon are eaten raw and cooked.187
Acute toxicity testing indicated a low toxicity following oral administration to rats (LD50 >675 mg/kg of gotu kola extract, equivalent to >4 g/kg dried leaf). Chronic administration of 150 mg/kg/day of extract (equivalent to 0.9 g/kg dried leaf) for a period of 30 days did not produce any adverse effects.188 Mice receiving up to 1 g/kg of gotu kola extract (2.5 g/kg dried plant) by mouth did not exhibit adverse effects.55 Aqueous ethanol extract of gotu kola entire plant demonstrated a maximum tolerated dose value of 250 mg/kg after ip injection in mice.189 Subcutaneous injection of 40 to 50 mg/kg of asiaticoside was toxic to mice and rabbits, while 20 to 250 mg/kg resulted in increased bleeding time. An oral dose of 1 g/kg was well tolerated.190
The local toxicity of asiaticoside was investigated by the measurement of skin respiration and histological analysis. (The death of a cell is accompanied by loss of respiratory activity.) Compared with other therapeutic agents, the toxicity of asiaticoside was not excessive and was comparable to that of many common antibiotics. Histological effects on guinea-pig skin indicated that moderate concentrations of asiaticoside produced swollen, abnormally staining cells. Higher concentrations resulted in necrotic cultures, showing signs of ‘thickening’ of the epidermis, even though the cells had mostly disintegrated. This may have been due to the cells becoming rapidly keratinised. Although fairly high concentrations of asiaticoside were required to produce this effect, it occurred in vivo (5 mg, sc) as well as in vitro.35
An ethanol extract of gotu kola exhibited mutagenic activity to strain TA98 (Salmonella/microsome test) only in the presence of S9 mix.191 A water extract of gotu kola was not toxic towards TA98 and TA100 with or without addition of S9 mix at the tested concentration (5 mg/plate). Gotu kola weakly inhibited the mutagenicity of the indirect mutagen IQ (2-amino-3-methylimidazo[4,5-f]quinoline).192 In another experiment, gotu kola water extract (1 mL of a 1:5 decoction) showed mutagenic activity in strain TA 98 with metabolic activation only.193 Gotu kola methanol extract induced abnormal metaphases and increased chromosome aberrations in the Vicia faba root meristem assay.194
Asiaticoside was found to be a weak tumour promoter in the hairless mouse epidermis model and was very weakly carcinogenic to the dermis after topical application.195
Interactions
No clinically relevant interactions have been found. In an in vivo study investigating wound healing with drugs, the anti-inflammatory drugs dexamethasone and phenylbutazone individually combined with asiaticoside caused a reduction in the tensile strength (and hence therapeutic effect) produced by asiaticoside alone.28 In this study the test substances were administered by intramuscular injection (asiaticoside is a saponin and has surfactant activity), so it is not known if the observed results extrapolate to the topical use of asiaticoside or oral use of gotu kola in humans.
In open trials, asiaticoside has been used topically in combination with an antibiotic and corticosteroid196 and was well tolerated.197
Use in pregnancy and lactation
Category B1 – no increase in frequency of malformation or other harmful effects on the fetus from limited use in women. No evidence of increased fetal damage in animal studies.
Gotu kola has been traditionally used in Bengal as a contraceptive agent. A reduction in conception rate was observed in female mice fed gotu kola (juice of whole plant, equivalent to 20 to 80 g fresh whole plant per kg) by gavage. Two sets of animals received the herb for 14 days (7 days before and 7 days during cohabitation) and 21 days (7 days before and 14 days during cohabitation). In the first set, sterile mating occurred for 50% to 60% of animals versus 15% in the controls, and for the second set 50% to 55% versus 20%. An isolated triterpenoid glycoside (40 to 120 mg/kg) and a compound derived from it also demonstrated antifertility activity. In all treatment groups there was no significant decrease in the number of young per litter and birth weight of the young were normal. The authors noted that the isolated glycoside and the compound derived from it caused a consistent reduction of fertility.198 These were very high doses, well above those normally used in clinical practice. Moreover, an antifertility effect does not imply harm during pregnancy.
Antifertility activity was also demonstrated in vivo in an early study for gotu kola (part undefined). Teratological effects have been studied in the rabbit and found to be negative for that animal.149
Side effects
As with all herbs rich in saponins, oral use may cause irritation of the gastric mucous membranes and reflux.
Cases of allergic contact dermatitis have been reported from the use of gotu kola, TTF and asiaticoside, but they are considered to be low-risk treatments.199–206 Both the extract and the triterpene constituents are weak sensitisers,149 although asiaticoside has been classified as a contact allergen.207 Patch tests in many cases confirmed that gotu kola or its constituents were responsible,199,200,203,204 although in some cases other constituents in the preparations were also responsible (such as propylene glycol,202 geraniol, lavender essence and neomycin201).
Traditional sources indicate that gotu kola may produce photosensitisation when used in tropical areas, although whether from use by oral or topical application is not indicated.5,208 Occasional gastrointestinal intolerance has been observed.149 A review of the use of TTF for the treatment of chronic venous insufficiency indicated that it was safe and well tolerated. Most trials used dosage of TTF in the range of 60 to 120 mg/day.80
Three cases of idiosyncratic hepatotoxicity have been reported from Argentina in association with extracts of gotu kola, including at least one that was clearly triggered by rechallenge.209 However, no other such cases are on record and the identity of the herb was not confirmed in these cases. One case of night-eating syndrome was attributed to use of a 70% ethanolic tincture of gotu kola, possibly with other herbs.210
Overdosage
There are no reliable reports of overdose with gotu kola.
An early case (prior to 1896) is recorded concerning a ‘Dr Boiteau who, in treating himself, progressively increased the dose and found that after two months the drug had produced all the effects of a violent, cumulative poison. … the plant, properly prepared and administered, is a powerful stimulant of the circulatory system, its action chiefly affecting the vessels of the skin and mucous membrane’.211 Traditional sources writing in the early to mid-20th century indicate that the plant can be a stupefying narcotic in large doses and in some cases produces headache or vertigo with a tendency to coma.207
Safety in children
Adverse effects are generally not expected. Gotu kola dried herb has been assessed in a clinical trial in India as a mental tonic for mentally disabled children.173 Gotu kola is used in leaf concentrate meals, which are prepared as porridge for preschool children in Sri Lanka to combat nutritional deficiencies.212 (Although the leaf composition varies with location, fresh gotu kola leaves typically contain 2% protein, 7 mg/100 g of vitamin C, 0.09 mg/100 g of vitamin B1 and 5.6 mg/100 g of iron.)213
However, one case of gotu kola-induced hepatotoxicity in a child has been recently reported.214
Regulatory status in selected countries
Gotu kola is official in the Pharmacopoeia of the People’s Republic of China 1997. The usual adult dosage, typically administered in the form of a decoction, is listed as 15 to 30 g, or 30 to 60 g of the fresh herb.
Gotu kola for external use is included on the General Sale List in the UK. It was not included in the Commission E assessment. Gotu kola is official in the British Pharmacopoeia 2011 and the European Pharmacopoeia 2011.
Gotu kola does not have GRAS status in the USA. However, it is freely available as a ‘dietary supplement’ in the USA under DSHEA legislation (Dietary Supplement Health and Education Act of 1994).
Gotu kola is not included in Part 4 of Schedule 4 of the Therapeutic Goods Regulations and is freely available for sale in Australia.
References
1. Nadkarni AK, Nadkarni KM. Indian Materia Medica. Bombay: Popular Prakashan, 1976. pp. 662–666
2. Grieve M, A Modern Herbal, New York, Dover Publications, 1971;vol 1.
3. Sharma PV. Dravyaguna-vijnana. Varanasi: Chaukambha Bharati Academy, 2003. pp. 3–6
4. Chen JK, Chen TT. Chinese Medical Herbology and Pharmacology. CA: Art of Medicine Press, 2004. pp. 147–148
5. British Herbal Medicine Association Scientific Committee. British Herbal Pharmacopoeia. Bournemouth: BHMA, 1983.
6. Felter HW, Lloyd JU. King’s American Dispensatory. ed 18, rev 3, 1905. Portland: reprinted Eclectic Medical Publications; 1983.
7. Farnsworth NR, Bunyapraphatsara N, eds. Thai Medicinal Plants. Bangkok: Medicinal Plant Information Center, 1992.
8. Dharma AP. Indonesian Medicinal Plants. Jakarta: Balai Pustaka, 1987.
9. Cambie RC, Ash J. Fijian Medicinal Plants. Australia: CSIRO, 1994.
10. van Wyk B-E, van Oudtshoorn B, Gericke N. Medicinal Plants of South Africa. Arcadia: Briza Publications, 1997.
11. Applequst W. The Identification of Medicinal Plants: A Handbook of the Morphology of Botanicals in Commerce. St Louis: Missouri Botanical Garden Press, 2006. pp. 45–46
12. Tang W, Eisenbrand G. Chinese Drugs of Plant Origin. Berlin: Springer-Verlag, 1992. pp. 273–275
13. Brinkhaus B, Linder M, Schuppan D, et al. Phytomedicine. 2000;7(5):427–448.
14. Zainol NA, Voo SC, Sarmidi MR, et al. Malaysian J Anal Sci. 2008;12(2):322–327.
15. May A. Eur J Pharmacol. 1968;4(3):331–339.
16. Lu L, Ying K, Wei S, et al. Br J Dermatol. 2004;151(3):571–578.
17. Lu L, Ying K, Wei S, et al. Int J Dermatol. 2004;43(11):801–807.
18. Marquart FX, Bellon G, Gillery P, et al. Connect Tissue Res. 1990;24:107–120.
19. Tenni R, Zanaboni G, De Agostini MP, et al. Ital J Biochem. 1988;37:69–77.
20. Bonte F, Dumas M, Chaudagne C, et al. Ann Pharm Fr. 1995;53(1):38–42.
21. Bonte F, Dumas M, Chaudagne C, et al. Planta Med. 1994;60(2):133–135.
22. Lee J, Jung E, Kim Y, et al. Planta Med. 2006;72(4):324–328.
23. Qi SH, Xie JL, Pan S, et al. Clin Exp Dermatol. 2008;33(2):171–175.
24. Poizot A, Dumez D. C R Acad Sci. 1978;286:789–792.
25. Suguna L, Sivakumar P, Chandrakasan G. Indian J Exp Biol. 1996;34(12):1208–1211.
26. Shukla A, Rasik AM, Jain GK, et al. J Ethnopharmacol. 1999;65:1–11.
27. Shetty BS, Udupa SL, Udupa AL, Somayaji SN. Int J Low Extrem Wounds. 2006;5(3):137–143.
28. Velasco M, Romero E. Curr Ther Res Clin Exp. 1976;19(1):121–125.
29. Hartog A, Smith HF, van der Kraan PM, et al. Exp Biol Med (Maywood). 2009;234(6):617–623.
30. Tincani GP, Riva PC, Baldini E. G Ital Dermatol. 1963;104:429–440.
31. Sunilkumar, Parameshwaraiah S, Shivakumar HG. Indian J Exp Biol. 1998;36(6):569–572.
32. Maquart FX, Chastang F, Simeon A, et al. Eur J Dermatol. 1996;9(4):289–296.
33. Shukla A, Rasik AM, Dhawan BN. Phytother Res. 1999;13(1):50–54.
34. Ju-Lin X, Shao-Hai Q, Tian-Zeng L, et al. J Cutan Pathol. 2009;36(2):234–239.
35. Lawrence JC. Eur J Pharmacol. 1967;1:414–424.
36. Kimura Y, Sumiyoshi M, Samukawa K, et al. Eur J Pharmacol. 2008;584(2–3):415–423.
37. Liu M, Dai Y, Li Y, et al. Planta Med. 2008;74(8):809–815.
38. Cheng CL, Guo JS, Luk J, Koo MWL. Life Sci. 2004;74:2237–2249.
39. Cheng CL, Koo MW. Life Sci. 2000;67(21):2647–2653.
40. Sairam K, Rao CV, Goel RK. Indian J Exp Biol. 2001;39(2):137–142.
41. Dong MS, Jung SH, Kim HJ, et al. Arch Pharm Res. 2004;27:512–517.
42. Wojcikowski K, Wohlmuth H, Johnson DW, et al. Nephrology (Carlton). 2009;14(1):70–79.
43. Qi S, Xie J, Li T. Zhonghua Shao Shang Za Zhi. 2000;16(1):53–56.
44. Zhang T, Rong XZ, Yang RH, et al. Nan Fang Yi Ke Da Xue Xue Bao. 2006;26(1):67–70. [In Chinese]
45. Ming ZJ, Liu SZ, Cao L. Zhongguo Zhong Xi Yi Jie He Za Zhi. 2004;24(8):731–734. [In Chinese]
46. Yun KJ, Kim JY, Kim JB, et al. Int Immunopharmacol. 2008;8:431–441.
47. Won JH, Shin JS, Park HJ, et al. Planta Med. 2010;76(3):251–257.
48. Vogel HG, De Souza N, D’Sa A. Acta Therapeutica. 1990;16:285–298.
49. Somchit MN, Sulaiman MR, Zuraini A, et al. Indian J Pharmacol. 2004;36(6):377–380.
50. Li H, Gong X, Zhang L, et al. Phytomedicine. 2009;16(6–7):538–546.
51. Liu M, Dai Y, Yao X, et al. Int Immunopharmacol. 2008;8(11):1561–1566.
52. Caballero-George C, Vanderheyde PM, Okamoto Y, et al. Phytother Res. 2004;18:729–736.
53. Wijeweera P, Arnason JT, Koszycki D, et al. Phytomedicine. 2006;13:668–676.
54. Chen SW, Wang WJ, Li WJ, et al. Pharmacol Biochem Behav. 2006;85:339–344.
55. Sakina MR, Dandiya PC. Fitoterapia. 1990;61:291–296.
56. Diwan PV, Karwande I, Singh AK. Fitoterapia. 1991;3:253–257.
57. Liang X, Huang YN, Chen SW, et al. Pharmacol Biochem Behav. 2008;89:444–449.
58. Nalini K, Aroor AR, Karanth KS, et al. Fitoterapia. 1992;63(3):232–237.
59. Xu Y, Cao Z, Khan I, et al. J Alzheimers Dis. 2008;13:341–349.
60. Mook-Jung I, Shin JE, Yun SH, et al. J Neurosci Res. 1999;58(3):417–425.
61. Veerendra Kumar MH, Cupta YK. Clin Exp Pharmacol Physiol. 2003;30(5–6):336–342.
62. Dhanasekaran M, Holcomb LA, Hitt AR, et al. Phytother Res. 2009;23(1):14–19.
63. Lee MK, Kim SR, Sung SH, et al. Res Commun Mol Pathol Pharmacol. 2000;108(1–2):75–86.
64. Ramanathan M, Sivakumar S, Anandvijayakumar PR, et al. Indian J Exp Biol. 2007;45(5):425–431.
65. Krishnamurthy RG, Senut MC, Zemke D, et al. J Neurosci Res. 2009;87(11):2541–2550.
66. Haleagrahara N, Ponnusamy K. J Toxicol Sci. 2010;35(1):41–47.
67. Veerendra Kumar MH, Gupta YK. J Ethnopharmacol. 2002;79(2):253–260.
68. Subathra M, Shila S, Devi MA, et al. Exp Gerontol. 2005;40(8–9):707–715.
69. Shinomol GK, Muralidhara. Phytomedicine. 2008;15(11):971–984.
70. Shinomol GK, Muralidhara. Neurotoxicology. 2008;29(6):948–957.
71. Shinomol GK, Ravikumar H, Muralidhara. Phytother Res. 2010;24(6):885–892.
72. Soumyanath A, Zhong YP, Gold SA, et al. J Pharm Pharmacol. 2005;57(9):1221–1229.
73. Mohandas Rao KG, Muddanna Rao S, Gurumadhva Rao S. Evid Based Complement Altern Med. 2006;3(3):349–357.
74. Mohandas Rao KG, Muddanna Rao S, Gurumadhva Rao S. Evid Based Complement Altern Med. 2009;6(2):203–210.
75. Gadahad MR, Rao M, Rao G. J Chin Med Assoc. 2008;71(1):6–13.
76. Rao MKG, Rao MS, Rao GS. Neuroanatomy. 2005;4:18–23.
77. Rao SB, Chetana M, Uma Devi P. Physiol Behav. 2005;86:449–457.
78. Gupta YK, Veerendra Kumar MH, Srivastava AK. Pharmacol Biochem Behav. 2003;74(3):579–585.
79. Vattanajun A, Watanabe H, Tantisira MH, et al. J Med Assoc Thai. 2005;88(suppl 3):S131–S140.
80. Incandela L, Cesarone MR, Cacchio M, et al. Angiology. 2001;52(suppl 2):S9–S13.
81. Cospite M, Ferrara F, Milio G, et al. Giorn Ital Angiol. 1984;4(3):200–205.
82. Arpaia MR, Ferrone R, Amitrano M, et al. Int J Clin Pharmacol Res. 1990;10(4):229–233.
83. Belcaro GV, Grimaldi R, Guidi G. Angiology. 1990;41(7):533–540.
84. Montecchio GP, Samaden A, Carbone S. Haematologica. 1991;76(3):256–259.
85. Pragada RR, Veeravalli KK, Chowdary KP, et al. J Ethnopharmacol. 2004;93(1):105–108.
86. Bian GX, Li GG, Yang Y, et al. Biol Pharm bull. 2008;31(3):458–463.
87. Li GG, Bian GX, Ren JP, et al. Yao Xue Xue Bao. 2007;42(5):475–480.
88. Gnanapragasam A, Yogeeta S, Subhashini R, et al. Mol Cell Biochem. 2007;294(1–2):55–63.
89. Gnanapragasam A, Ebenezar KK, Sathish V, et al. Life Sci. 2004;76(5):585–597.
90. Zheng MS. J Trad Chin Med. 1989;9:113–116.
91. Yoosook C, Bunyapraphatsara N, Boonyakiat Y, et al. Phytomedicine. 2000;6(6):411–419.
92. Herbert D, Paramsivan CN, Prabhakar R, et al. Indian J Leprosy. 1994;66:65–68.
93. Newton SM, Lau C, Gurcha SS, et al. J Ethnopharmacol. 2002;79:57–67.
94. Medda S, Das N, Mahato SB, et al. Indian J Biochem Biophys. 1995;32(3):147–151.
95. Chakraborty T, Sinha Babu SP, Sukul NC. Fitoterapia. 1995;57(2):110–112.
96. Park BC, Bosire KO, Lee ES, et al. Cancer Lett. 2005;218(1):81–90.
97. Yoshida M, Fuchigami M, Nagao T, et al. Biol Pharm Bull. 2005;28(1):173–175.
98. Tang XL, Yang XY, Jung HJ, et al. Biol Pharm Bull. 2009;32(8):1399–1405.
99. Babykutty S, Padikkala J, Sathiadevan PP, et al. Afr J Tradit Complement Altern Med. 2008;6(1):9–16.
100. Huang YH, Zhang SH, Zhen RX, et al. Ai Zheng. 2004;23(12):1599–1604. [In Chinese]
101. Babu TD, Kuttan G, Padikkala J. J Ethnopharmacol. 1995;48(1):53–57.
102. Bunpo P, Kataoka K, Arimochi H, et al. Food Chem Toxicol. 2004;42(12):1987–1997.
103. Punturee K, Wild CP, Kasinrerk W, et al. Asian Pac J Cancer Prev. 2005;6(3):396–400.
104. Patil JS, Nagavi BG, Ramesh M, et al. Ind Drugs. 1998;38:711–714.
105. Jayathirtha MG, Mishra SH. Phytomedicine. 2004;11(4):361–365.
106. Chatterjee TK, Chakraborty A, Pathak M, et al. Indian J Exp Biol. 1992;30(10):889–891.
107. Sarma DNK, Khosa RL. Phytother Res. 1996;10(2):181–183.
108. Sarma DNK, Khosa RL, Chansauria JPN, et al. Phytother Res. 1995;9(8):589–590.
109. Gupta R, Flora SJ. J Appl Toxicol. 2006;26(3):213–222.
110. Flora SJ, Gupta R. Phytother Res. 2007;21(10):980–988.
111. Shobi V, Goel HC. Physiol Behav. 2001;73(1–2):19–23.
112. Sharma J, Sharma R. Phytother Res. 2002;16(8):785–786.
113. Sharma J, Sharma R. Phytother Res. 2005;19(7):605–611.
114. Zhang L, Li HZ, Gong X, et al. Phytomedicine. 2010;17(10):811–819.
115. Chasseaud LF, Fry BJ, Hawkins DR, et al. Arzneimittelforschung. 1971;21(9):1379–1384.
116. Zheng XC, Wang SH. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877(5–6):477–481.
117. Rush WR, Murray GR, Graham DJ. Eur J Drug Metab Pharmacokinet. 1993;18(4):323–326.
118. Grimaldi R, De Ponti F, D’Angelo L, et al. J Ethnopharmacol. 1990;8:235–241.
119. Incandela L, Cesarone MR, Cachio M, et al. Angiology. 2001;52(10 suppl 2):S61–S67.
120. Cesarone MR, Belcaro G, Rulo A, et al. Angiology. 2001;52(suppl 1–2):S45–S48.
121. Pointel JP, Boccalon H, Cloarec M, et al. Angiology. 1987;38(1 pt 1):46–50.
122. Belcaro G, Laurora G, Cesarone MR, et al. Curr Ther Res. 1989;46(6):1015–1026.
123. Cesarone MR, Laurora G, De Sanctis MT, et al. Minerva Cardioangiol. 1994;42(6):299–304.
124. De Sanctis MT, Belcaro G, Incandela L, et al. Angiology. 2001;52(10 suppl 2):S55–S59.
125. Belcaro GV, Rulo A, Grimaldi R. Angiology. 1990;41(1):12–18.
126. Monteverde A, Occhipinti P, Rossi F, et al. Acta Ther. 1987;13:629–636.
127. Cesarone MR, Belcaro G, De Sanctis MT, et al. Angiology. 2001;52(suppl 2):S15–S18.
128. Lim CS, Davies AH. Br J Surg. 2009;96(11):1231–1242.
129. Naoum JJ, Hunter GC. Vascular. 2007;15(5):242–249.
130. Incandela L, Belcaro G, Cesarone MR, et al. Angiology. 2001;52(suppl 2):S27–S31.
131. Cesarone MR, Incandela L, De Sanctis MT, et al. Angiology. 2001;52(suppl 1–2):S49–S54.
132. Cesarone MR, Incandela L, De Sanctis MT, et al. Angiology. 2001;52(suppl 1–2):S33–S37.
133. Guarerio F, Sansonetti G, Donzelli R, et al. Giorn Ital Angiol. 1986;6(1):46–52.
134. Thim T, Hagensen MK, Bentzon JF, et al. J Intern Med. 2008;263(5):506–516.
135. Schoenhagen P, Tuzcu EM, Ellis SG. Circulation. 2002;106(7):760–762.
136. Cesarone MR, Belcaro G, Nicolaides AN, et al. Angiology. 2001;52(suppl 2):S19–S25.
137. Incandela L, Belcaro G, Nicolaides AN, et al. Angiology. 2001;52(suppl 2):S69–S73.
138. Etrebi A, Ibrahim A, Zaki K. J Egypt Med Assoc. 1975;58(5–6):324–327.
139. Peyresblanques J. Bull Soc Ophtalmol Fr. 1959;8:771–781.
140. Baudon-Glanddier B. Gaz Med Fr. 1963;70:2463–2464.
141. Sevin P. Progr Med (Paris). 1962;90:23–24.
142. Huriez CL. Lille Med. 1972;3(suppl. 17):574–579.
143. O’Keeffe P. Br J Plast Surg. 1974;27(2):194–195.
144. Balina LM, Cardama JE, Gatti JC, et al. Dia Med. 1961;33:1693–1696.
145. Stassen P. Rev Med Liege. 1964;19:305–308.
146. Mayall RC, Mayall AC, Bertolotti JG, et al. Rev Bras Med. 1975;32:26–29.
147. Mekkawi MF. Bull Ophthalmol Soc Egypt. 1975;68:77–79.
148. El-Hefnawi H. Dermatologica. 1962;125:387–392.
149. Bosse JP, Papillon J, Frenette G, et al. Ann Plast Surg. 1979;3(1):13–21.
150. Paocharoen V. J Med Assoc Thai. 2010;93(7):S166–S170.
151. El-Zawahry MD, Khalil AM, El-Banna MH. Bull Soc Int Chir. 1975;34(4):296–297.
152. Darnis F, Orcel L, de Saint-Maur PP, et al. Sem Hop. 1979;55(37–38):1749–1750.
153. Castellani L, Gillet JY, Lavernhe G, et al. Bull Fed Soc Gynecol Obstet Lang Fr. 1966;18(2):184–186.
154. Torre MP, Donnadieu JM, Braditch JL. Clinique (Paris). 1963;58:203–206.
155. Kiesewetter H. Wien Med Wochenschr. 1964;114:124–126. [In German]
156. Wolfram ST. Wien Med Wochenschr. 1965;115:439–442. [In German]
157. Chakrabarty T, Deshmukh S. Sci Culture. 1976;11:573.
158. Nebout M. Bull Soc Pathol Exot Filiales. 1974;67:471–478. [In French]
159. Chaudhury S, Hazra S, Podder GC, et al. Indian J Dermatol. 1987;32(3):63–67.
160. Sasaki S, Shinkai H, Akashi Y, et al. Jap J Clin Derm. 1971;25(6):585–593.
161. Guseva NG, Starovoitova MN, Mach ES. Ter Arkh. 1998;70(5):58–61. [In Russian]
162. Szczepanski A, Dabrowska H, Blaszczyk M. Przegl Dermatol. 1974;61(5):701–703.
163. Frati Munari AC, Culebro Nieves G, Velazquez E, et al. Bol Med Hosp Infant Mex. 1979;36(2):201–214.
164. Natarajan S, Paily PP. Indian J Dermatol. 1973;18(4):82–85.
165. Kosalwatna S, Shaipanich C, Bhanganada K. Siriraj Hosp Gaz. 1988;40(6):455–461.
166. Rougier A, Humbert F. J Am Acad Dermatol. 2008;2 suppl 2(58): AB144. Abstract P3109
167. Klövekorn W, Tepe A, Danesch U. Int J Clin Pharmacol Ther. 2007;45(11):583–591.
168. Mallol J, Belda MA, Costa D, et al. Int J Cos Sci. 1991;13:51–57.
169. Sommerfeld B. Phytomedicine. 2007;14:711–715.
170. Lee J, Jung E, Lee H, et al. Int J Cos Sci. 2008;30(3):167–173.
171. Haftek M, Mac-Mary S, Le Bitoux MA, et al. Exp Dermatol. 2008;17(11):946–952.
172. Bradwejn J, Zhou Y, Koszychki D, et al. J Clin Psychopharmacol. 2000;20:680–684.
173. Jana U, Sur TK, Maity LN, et al. Nepal Med Coll J. 2010;12(1):8–11.
174. Appa Rao MVR, Srinivasan K, Koteswara Rao T. J Res Indian Med. 1973;8(4):9–15.
175. Kuppurajan K, Srinivasan K, Janaki K. J Res Indian Med. 1978;13(1):37–41.
176. Wattanathorn J, Mator L, Muchimapura S, et al. J Ethnopharmacol. 2008;116(2):325–332.
177. Appa Rao MVR, Usha SP, Rajagopalan SS, et al. J Res Indian Med. 1967;2:79–85.
178. Appa Rao MVR, Rajapopalan SS, Srinivasan VR, et al. Nagarjun. 1969;12:33–41.
179. Mato L, Wattanathorn J, Muchimapura S, et al. Evid Based Complement Altern Med, 2009. At <http://ecam.oxfordjournals.org/cgi/reprint/nep177v1>. Accessed 6.7.10.
180. Rhee J, Choi KW. Korean J Gastroenterol. 1981;13(1):35–40.
181. Sastravaha G, Gassmann G, Sangtherapitikul P, et al. J Int Acad Periodontol. 2005;7(3):70–79.
182. Sastravaha G, Yotnuengnit P, Booncong P, et al. J Int Acad Periodontol. 2003;5(4):106–115.
183. Bagchi CD, Puri HS. Herba Hung. 1988;27(2–3):137–140.
184. Dharma AP. Indonesian Medicinal Plants. Jakarta: Balai Pustaka, 1987. pp. 24–25
185. van Wyk B-E, Gericke N. People’s Plants: A Guide to Useful Plants of Southern Africa. Arcadia: Briza Publications, 2000. pp. 68, 142
186. Rattanapanone V, Sanpitak N, Phornphibul B. Chiang Mai Med Bull. 1971;10:17–23.
187. Kays SJ, Silva Dias JC. Econ Bot. 1995;49(2):115–152.
188. dede Lucia R, Sertie JAA. Fitoterapia. 1997;68(5):413–416.
189. Dhar ML, Dhar MM, Dhawan BN, et al. Indian J Exp Biol. 1968;6(4):232–247.
190. Boiteau P, Ratsimamanga AR. Therapie. 1956;11:125–149.
191. Ieamworapong C, Kangsadalumpai K, Rojanapo W. Environ Mol Mutagen. 1989;14(suppl 15):93.
192. Yen GC, Chen HY, Peng HH. Food Chem Toxicol. 2001;39:1045–1053.
193. Rivera IG, Martins MT, Sanchez PS, et al. Environ Toxicol Water Qual. 1996;9(2):87–93.
194. Gopalan HNB, Wairimu AN. Environ Mol Mutagen. 1989;14(suppl 15):73.
195. Laerum OD, Iversen OH. Cancer Res. 1972;32(7):1463–1468.
196. Kartnig T. Herb Spice Med Plant. 1988;3:145–173.
197. Hadida E, Sayag J, Bonerandi JJ, et al. Bull Soc Fr Dermatol Syphiligr. 1970;77(4):522–525.
198. Dutta T, Basu UP. Indian J Exp Biol. 1968;6:181–182.
199. Hausen BM. Contact Dermatitis. 1993;29(4):175–179.
200. Danese P, Carnevali C, Bertazzoni MG. Contact Dermatitis. 1994;31(3):201.
201. Santucci B, Picardo M, Cristaudo A. Contact Dermatitis. 1985;13(1):39.
202. Izu R, Aguirre A, Gil N, et al. Contact Dermatitis. 1992;26(3):192–193.
203. Eun HC, Lee AY. Contact Dermatitis. 1985;13(5):310–313.
204. Huriez C, Martin P. G Ital Dermatol Minerva Dermatol. 1969;44(9):463–464.
205. Gonzalo Garijo MA, Revenga Arranz F, Bobadilla Gonzalez P. Allergol Immunopathol. 1996;24(3):132–134.
206. Vena GA, Angelini GA. Contact Dermatitis. 1986;15(2):108–109.
207. Goossens A, Beck MH, Haneke E, et al. Contact Dermatitis. 1999;40:112–113.
208. Chopra RN, Chopra IC, Handa KL, et al. Chopra’s Indigenous Drugs of India, 2nd ed., 1958. Calcutta: reprinted Academic Publishers; 1982. pp. 351–353.
209. Jorge OA, Jorge AD. Rev Esp Enferm Dig. 2005;97(2):115–124.
210. O’Brien B. Ir Med J. 2005;98(10):250–251.
211. Chopra RN, Badhwar RL, Ghosh S, Poisonous Plants of India, New Delhi, Indian Council of Agricultural Research, 1965;vol I. pp. 433–434
212. Cox DN, Rajasuriya S, Soysa PE, et al. Int J Food Sci Nutr. 1993;44:123–132.
213. Peiris KHS, Kays SJ. Hort Tech. 1996;6(1):13–18.
214. Dantuluri S, North-Lewis P, Karthik SV. Dig Liver Dis. 2011;43(6):500.