Acute Hypoxemic Respiratory Failure and ARDS
Warren L. Lee MD, PhD, Arthur S. Slutsky MD
Hypoxemic Respiratory Failure
Hypoxemic respiratory failure has classically been defined as an arterial Po2 of less than 60 mm Hg. It is distinguished from hypercapnic respiratory failure (Pco2 > 45 mm Hg), although the two conditions often coexist. There are a number of important qualifications to this definition that must be made. The threshold of 60 mm Hg is somewhat arbitrary and reflects the shape of the oxyhemoglobin dissociation curve, marking the arterial Po2 below which the saturation of hemoglobin with oxygen falls precipitously in most people. An important caveat is that it is important whether hypoxemic respiratory failure is acute (over hours to days) or chronic (over weeks to months) as it has implications not just for diagnosis and treatment, but also for the physiologic adaptations to hypoxemia that develop over time. For example, an individual living at high altitude may have an arterial Po2 less than 50 mm Hg because of a low inspired partial pressure of oxygen. Such an individual could well be asymptomatic because of acclimatization to that environment and would not be considered to have hypoxemic respiratory failure even if she had an arterial Po2 of 45 mm Hg. It is also important to realize that this definition of hypoxemic respiratory failure (arterial Po2 < 60 mm Hg) encompasses a broad spectrum of severity of illness. For example, both a patient on a general medical ward with community-acquired pneumonia and a mechanically ventilated patient on 100% oxygen in the critical care unit can fulfill the definition.
This definition of hypoxemic respiratory failure (based on the arterial Po2) does not fully address the importance of oxygenation at the level of the tissues. Oxygen delivery to the tissues is the product of cardiac output and oxygen content. Oxygen content is critically dependent on the hemoglobin concentration and its saturation with oxygen; although the oxygen saturation depends on arterial Po2 (as described by the oxyhemoglobin dissociation curve), the direct contribution of dissolved oxygen to blood oxygen content is very low under most conditions. In other words, in the anemic patient or in patients with very low cardiac output, tissue hypoxia may exist despite a seemingly adequate arterial Po2. Finally, as indicated earlier, both hypoxemic and hypercarbic respiratory failure may coexist. A patient may initially present with isolated hypoxemia yet, upon tiring, may develop hypercarbia. Analogously, hypoventilation can cause both hypercarbia and hypoxemia.
This chapter focuses on acute hypoxemic respiratory failure. In particular, much of our discussion pertains to the etiology and management of hypoxemia at the “severe” end of the spectrum. This is not to say that mild hypoxemia is not important; certainly the condition of a patient on the medical ward with pneumonia and mild hypoxemia can deteriorate and the patient would require intubation and mechanical ventilation.
Classification of Hypoxemia
The traditional approach to the causes of arterial hypoxemia classifies them into five pathophysiologic mechanisms: decreased inspired Po2, hypoventilation, impaired diffusion, ventilation-perfusion ( ) mismatch, and right-to-left shunt. Whereas hypoxemia from
) mismatch, and right-to-left shunt. Whereas hypoxemia from  mismatch is responsive to supplemental oxygen, hypoxemia from a right-to-left shunt is not. This physiologic approach is probably most useful in understanding how a particular disease causes hypoxemia, but is not usually very illuminating when trying to make a specific diagnosis, other than in situations in which a patient is hypoxemic due to profound hypercapnia. In a hospital setting, a decreased inspired Po2 can usually be excluded, because most patients will be placed on supplemental oxygen. Hypoventilation can rapidly be excluded if the patient is not hypercapnic. Impaired diffusion by itself is not an important cause of acute hypoxemia, in that oxygen transfer across the alveolar-capillary membrane to red blood cells is usually perfusion-limited, not diffusion-limited. What this means is that there is usually ample time for diffusion of oxygen, even in the presence of intrinsic lung disease. In sum, most patients in the intensive care unit with acute hypoxemic respiratory failure have some combination of
mismatch is responsive to supplemental oxygen, hypoxemia from a right-to-left shunt is not. This physiologic approach is probably most useful in understanding how a particular disease causes hypoxemia, but is not usually very illuminating when trying to make a specific diagnosis, other than in situations in which a patient is hypoxemic due to profound hypercapnia. In a hospital setting, a decreased inspired Po2 can usually be excluded, because most patients will be placed on supplemental oxygen. Hypoventilation can rapidly be excluded if the patient is not hypercapnic. Impaired diffusion by itself is not an important cause of acute hypoxemia, in that oxygen transfer across the alveolar-capillary membrane to red blood cells is usually perfusion-limited, not diffusion-limited. What this means is that there is usually ample time for diffusion of oxygen, even in the presence of intrinsic lung disease. In sum, most patients in the intensive care unit with acute hypoxemic respiratory failure have some combination of  mismatch and right-to-left shunt.
mismatch and right-to-left shunt.
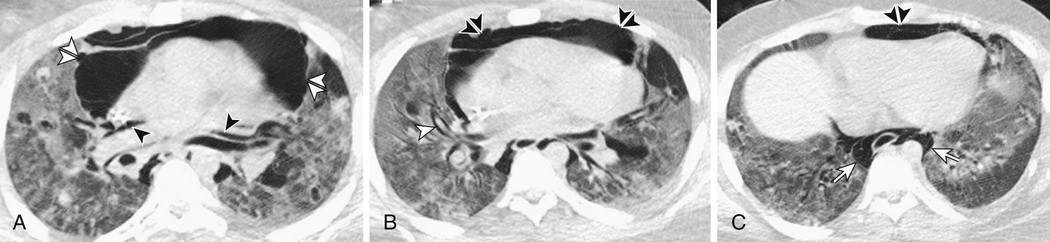
Another classification of acute hypoxemic respiratory failure is structural-anatomic (Fig. 100-1). Causes of acute arterial hypoxemia can be classified based on whether the primary pathology is located in the air spaces, interstitium, heart and pulmonary vasculature, airways, or pleural space. Such an approach would quickly lead one to consider such causes as pulmonary edema or pneumonia, hypersensitivity pneumonitis, pulmonary embolism, bronchospasm, and pneumothorax. Although disorders involving other structures such as the central nervous system and respiratory muscles can lead to hypoxemia, these causes would be expected to have associated hypercarbia.
Clinical Features and Diagnostic Approach
The clinical features of acute hypoxemic respiratory failure vary based on the underlying cause. Assuming an intact drive to breathe and that the patient has not fatigued, the hypoxemic patient is usually tachypneic and tachycardic. Cyanosis of the lips or tongue (so-called central cyanosis) would indicate that the concentration of reduced (deoxygenated) hemoglobin is greater than 5 g/100 mL.
Given the extensive differential diagnosis of acute hypoxemic respiratory failure and the often urgent need for therapy, the clinician must be practical yet thoughtful. A basic history should be obtained to identify risk factors for cardiac dysfunction, pulmonary infection or aspiration, venous thromboembolism, or obstructive lung diseases. In the setting of trauma to the chest, pneumothorax, hemothorax, and pulmonary contusion must be considered. Further questions to identify less common causes of acute hypoxemic respiratory failure can be posed as appropriate. A focused physical examination of the cardiac and respiratory systems often can establish the presence or absence of congestive heart failure or a focal area of consolidation or effusion. Similarly, the diagnosis of a pneumothorax is more satisfying (and prompt) when made on physical examination rather than later, after chest imaging.
Implementation of therapy is simultaneous with the diagnostic workup. As always, this starts with the “ABCs” of airway, breathing, and circulation. Once the ABCs are assured, the patient should be given supplemental oxygen (if coexistent hypercapnia is present, care must be taken in the dose of supplemental oxygen) and intravenous access should be obtained. Continuous cardiac monitoring and pulse oximetry should be available.
The initial investigations are dictated by the findings on history and physical examination. However, all patients should have a chest radiograph, an electrocardiogram, and routine blood work, including a complete blood count with differential and serum chemistry. An arterial blood gas should be obtained and the alveolar-arterial Po2 gradient should be calculated; a normal alveolar-arterial Po2 gradient in the setting of arterial hypoxemia suggests hypoventilation as the sole cause of the hypoxemia. The blood gas is also useful for diagnosing other acid-base disturbances and hemoglobinopathies such as carbon monoxide poisoning. The need for further investigations, including bronchoscopy, computed tomographic angiography of the chest, and echocardiography, depends on the results of the initial assessment. A completely normal chest radiograph in the setting of hypoxemic respiratory failure narrows the differential diagnosis substantially. In this uncommon circumstance, the clinician should consider pulmonary embolism and right-to-left shunts (i.e., intracardiac or pulmonary arteriovenous malformations) as possibilities. A more common scenario is that the chest radiograph of a patient with pneumonia can appear surprisingly normal (or may show only a “small” opacity) due to concomitant intravascular volume depletion. Once intravascular volume has been restored, the extent of the opacity can be appreciated.1,2
Causes of Acute Respiratory Failure
In a large multicenter international prospective cohort study of patients requiring mechanical ventilation, the most common reported causes of acute respiratory failure were postoperative respiratory failure, pneumonia, congestive heart failure, sepsis, and trauma.3 In a small prospective cohort study that included 41 patients with hypoxemic respiratory failure, chronic obstructive pulmonary disease and pneumonia were the most common causes.4 Other data from small randomized controlled trials of noninvasive ventilation identified congestive heart failure, pneumonia, trauma, acute respiratory distress syndrome, and mucous plugging as the most common causes of respiratory failure.5,6 However, in these studies, patients with certain diseases were excluded, including those with chronic obstructive pulmonary disease5,6 and asthma,6 limiting the ability to generalize from these findings. Indeed, in the one randomized study that specifically enrolled patients with acute hypoxemic respiratory failure, only patients with bilateral lung opacities on chest radiography were included.6
For a detailed discussion of many of the specific causes of acute hypoxemic respiratory failure (e.g., pneumonia), refer to the appropriate chapters in this book. The remainder of this chapter focuses on a particular subtype of acute hypoxemic respiratory failure, known as acute respiratory distress syndrome (ARDS).
Acute Respiratory Distress Syndrome
Diagnosis and Epidemiology
Diagnosis
ARDS is characterized by noncardiogenic pulmonary edema, lung inflammation, hypoxemia, and decreased lung compliance. Unlike some disorders (e.g., coronary artery disease), ARDS, as its name suggests, is a syndrome, reflecting a constellation of clinical and physiologic observations thought to represent a common pathology. In coronary artery disease, narrowing of the coronary vasculature and disruption of an unstable atherosclerotic plaque are known to underpin the symptoms of angina and unstable angina, respectively. A diagnostic “gold standard,” namely coronary angiography, exists. In contrast, the pathogenesis of ARDS remains elusive and there is no gold standard diagnostic test. The heterogeneity of the clinical conditions associated with ARDS (discussed later) would be consistent with the possibility that ARDS is in fact a collection of different diseases that have not yet been separately identified. These problems necessarily permeate any discussion or research into ARDS, and this chapter is no exception.
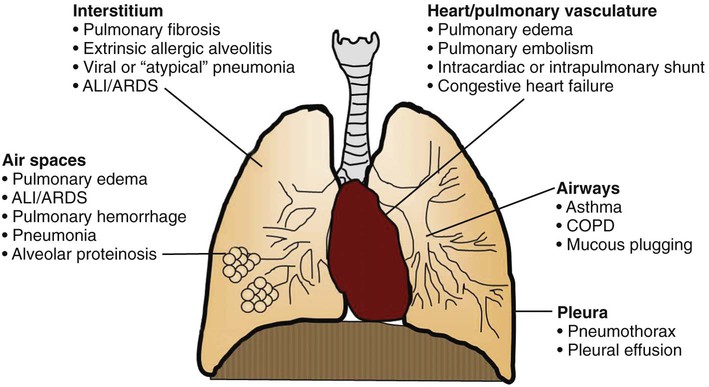
The first description of ARDS appeared in a remarkable case series reported in 1967.7 Ashbaugh and colleagues described 12 patients ranging in age from 11 to 48 years who presented with respiratory distress, hypoxemic respiratory failure, and patchy bilateral opacities on chest radiographs (Fig. 100-2). Most of the cases were preceded by severe trauma or viral infection and the onset of symptoms was relatively rapid, with most patients developing respiratory distress within 48 to72 hours of the beginning of their illness. Many patients required positive pressure ventilation and exhibited low respiratory system compliance, and some experienced improvement in oxygenation with the application of positive end-expiratory pressure (PEEP). This syndrome was initially termed the adult respiratory distress syndrome to distinguish it from the respiratory distress syndrome seen in infants. Subsequently, recognizing that the syndrome can also develop in children, it was renamed the acute respiratory distress syndrome.
The cases described by Ashbaugh and colleagues generated interest and research into ARDS. Unfortunately, the lack of specific diagnostic criteria and an understanding of the pathogenesis of the disorder made it difficult to undertake research and to compare studies. In 1988, a formalized and expanded definition of ARDS was proposed that consisted of three parts: (1) determining whether the illness was acute or chronic, (2) determining whether there were any associated risk factors or medical conditions (e.g., sepsis),8 and (3) assigning points based on the severity of pulmonary dysfunction (the Lung Injury Score), as measured by the degree of hypoxemia, the level of PEEP required, the respiratory system compliance, and the degree of radiographic abnormality. An average score was calculated and a final value greater than 2.5 was used to diagnose ARDS. One advantage of this system was its description of associated medical conditions, which might be pertinent to the etiology of ARDS. Because ARDS from different etiologies might reflect different pathogenesis and perhaps confer a different response to therapy, it was thought that knowing the cause of ARDS might be important in the conduct and comparison of studies. The definition, however, made no attempt to exclude cardiogenic pulmonary edema; it was assumed that clinicians would do so automatically.
In 1994, a consensus conference of American and European investigators published their definition of ARDS, which was widely adopted.9 Aiming for simplicity, ARDS was defined as a syndrome of acute onset, with bilateral opacities on chest radiography consistent with pulmonary edema, pulmonary artery occlusion pressure of 18 mm Hg or less (or absence of clinical evidence of left atrial hypertension), and hypoxemia as measured by the ratio of the arterial partial pressure of oxygen (arterial Po2) to the fraction of oxygen inspired (Fio2). Recognizing that there was a spectrum of severity of the disease, the consensus panel recommended that arterial Po2/Fio2 ratio ≤ 300 would define an entity termed acute lung injury (ALI). ARDS was the most severe form of ALI and was diagnosed when arterial Po2/Fio2 ≤ 200. The simplicity of the definition led to its general acceptance by clinicians and its incorporation into clinical research. At the same time, such a straightforward definition could not take into account the heterogeneity of the disease or the ambiguity of clinical practice.
For example, the requirement for bilateral opacities on chest radiograph is open to interpretation. One study presented a random series of chest radiographs from intubated hypoxemic patients (arterial Po2/Fio2 < 300) to an international group of expert clinicians, the majority of whom conduct clinical research on ARDS. The clinicians were asked to examine each radiograph and to decide whether it fulfilled the American-European Consensus Conference (AECC) definition of ARDS (bilateral opacities consistent with pulmonary edema). This select group of physicians demonstrated only moderate agreement (kappa statistic 0.55) in their classification of the radiographs; when the percentage of radiographs deemed to be consistent with ARDS by each physician was examined, the percentages were evenly distributed from a low of 36% to a high of 71%.10 Another larger study on the issue also demonstrated only moderate agreement when two physicians were asked to rate radiographs on the presence or absence of diffuse bilateral opacities consistent with ARDS. However, with prior training and discussion, the agreement between an intensivist and a radiologist in the rating of the films was excellent.11
The AECC definition has also been criticized for not taking the level of PEEP into consideration. Clinicians have long appreciated that applying PEEP can improve oxygenation, an observation that was noted in the first description of ARDS. This implies that the arterial Po2/Fio2 will change with changes in the level of PEEP; indeed, a patient who meets the AECC criteria for ARDS may no longer meet them once PEEP has been increased.12 It has also been shown that patients meeting the AECC definition for ARDS can be stratified based on their response to standard ventilator settings 24 hours later; many of the patients at that point have a lesser degree of hypoxemia and a significantly lower mortality rate.13 Thus, the AECC definition, while simple to use, encompasses a heterogeneous group of patients.
In 2012, the so-called Berlin definition of ARDS was developed to address some of these limitations (Table 100-1).14,15 The degree of hypoxemia was stratified into mild, moderate, and severe based on the arterial Po2/Fio2 ratio and a requirement for a PEEP level of 5 cm H2O or greater was included (whether applied by endotracheal tube or, in the setting of mild disease, by noninvasive ventilation). The term ALI was eliminated (but is used as needed in this chapter to help readers interpret older literature). Given that the use of pulmonary artery catheters has fallen off dramatically due to a demonstrated lack of benefit,16,17 the need for a pulmonary arterial wedge pressure measurement was eliminated and instead it was recommended that objective testing (e.g., echocardiography) be performed to exclude cardiogenic edema if no risk factor for ARDS could be identified. “Acute” was explicitly defined as ARDS developing within 1 week of a known risk factor. Although considerable effort went into its design, when compared with the AECC criteria, the Berlin definition is only slightly better at predicting mortality attributable to ARDS.14
Table 100-1
Berlin Definition of ARDS
| Criterion | Definition |
| Timing | Within 1 week of a known precipitant, or new/worsening respiratory symptoms |
| Chest imaging (chest radiograph or CT scan) | Bilateral opacities—not fully explained by effusions, lobar/lung collapse, or nodules |
| Origin of edema | Respiratory failure not fully explained by cardiac failure or fluid overload If no risk factor for ARDS is present, need objective assessment (e.g., echocardiogram) to exclude hydrostatic edema |
| Oxygenation | |
| Mild | 200 mm Hg < arterial Po2/Fio2 ≤ 300 mm Hg with PEEP or CPAP ≥ 5 cm H2O |
| Moderate | 100 mm Hg < arterial Po2/Fio2 ≤ 200 mm Hg with PEEP ≥ 5 cm H2O |
| Severe | Arterial Po2/Fio2 ≤ 100 mm Hg with PEEP ≥ 5 cm H2O |
Two additional points should be made about the diagnosis of ARDS. First, while physicians properly think of ARDS as being distinct from cardiogenic pulmonary edema, it is important to acknowledge that many patients with ARDS will have left atrial hypertension during the course of their illness.18 Second, despite the increasing use of B-type natriuretic peptides as diagnostic tools for acute congestive heart failure, their ability to distinguish ARDS from cardiogenic pulmonary edema is unclear.19,20
Incidence
Given the multitude of definitions and the difficulty in making a diagnosis of ARDS, it is not surprising that determining the incidence of the disease has been challenging. The reported incidence of ARDS has ranged from 75 per 100,000 population21 to as low as 1.5 per 100,00022 in Europe; the European numbers appear fairly constant over time.23 Many older studies did not use the AECC definition and did not include ALI; the Berlin definition is too recent to have been widely studied. The incidence of ALI has been estimated to be 78.9 cases per 100,000 person-years.24 One study calculated an incidence of ALI of 22 cases per 100,000 using data from a large prospective trial of ARDS and from the American Hospital Association.25 One of the methodologic strengths of the study was its inclusion of cases of ALI over a 3-year period, thereby decreasing the impact of seasonal variability. The heterogeneity in the literature of the estimates of incidence may reflect differences in the methodology of the various studies, but may also reflect true variation. Regional differences in genetic or environmental factors and specific disease-associations such as cardiopulmonary bypass or lung transplant may account for some of the regional variability in the incidence of ALI.26
Risk Factors
Sepsis, aspiration of gastric contents, and multiple transfusions (>15 units/24 hours in one study) are associated with the highest risk for development of ARDS.27 In particular, ARDS develops in almost 40% of patients with sepsis. The presence of more than one presumed risk factor for ARDS may increase the incidence of ARDS, although the sample sizes of these subsets have been quite small.27 ARDS has been seen to develop in more than one third of patients receiving massive blood transfusion and in one fourth of patients with multiple trauma (one or more pulmonary contusions, multiple fractures, multiple transfusions).28
One of the predisposing factors for the development of ARDS is a history of chronic alcoholism (relative risk, 2.0; 95% confidence interval, 1.3–2.9).29 This relationship has been shown to remain significant after adjustment for gender, at-risk diagnosis, and severity of illness. Among those in whom ARDS developed, patients with a history of alcoholism had a higher mortality rate than those without such history (odds ratio, 6.3; 95% confidence interval, 2.2–20.4).29 The mechanisms of this association are unknown.
The same researchers examined whether patients with diabetes might be protected against development of ARDS, the rationale being that hyperglycemia is known to impair neutrophil function, and neutrophils are thought to be central to the pathogenesis of ARDS (see Pathogenesis). More than 100 patients admitted to the intensive care unit with septic shock were followed prospectively for the development of ARDS. Over a 2-year period, diabetes was associated with a significantly decreased risk of ARDS (relative risk, 0.53; 95% confidence interval, 0.28–0.98), which persisted after adjustment for the source of sepsis and other potential confounders. Interestingly, there was no difference in mortality between patients with ARDS who did and did not have diabetes.30
A systematic overview of studies examining potential risk factors for ARDS concluded that the strongest evidence of a causal relationship was for sepsis, trauma, multiple transfusions, aspiration of gastric contents, pulmonary contusion, pneumonia, and smoke inhalation.31 More recently, active or passive exposure to cigarette smoke has been associated with the development of ARDS after trauma.32
The heterogeneity of risk factors for ARDS is remarkable. At the AECC, investigators divided the known risk factors into those thought to cause direct lung injury (e.g., pneumonia) and those in which the mechanism of lung injury was thought to be indirect (e.g., pancreatitis) (Table 100-2). While conceptually attractive, this classification scheme may not reflect underlying differences in the mechanisms, severity, or outcome of lung injury.33 This issue is discussed in more detail in the section on outcomes.
Table 100-2
Conditions Associated with ARDS, Categorized by Possible Mechanisms of Injury
| Direct injury | Indirect injury |
| Pneumonia | Sepsis |
| Aspiration | Major trauma |
| Pulmonary contusion | Multiple blood transfusions |
| Toxic inhalation | Pancreatitis |
| Near-drowning | Cardiopulmonary bypass |
| Reperfusion injury (e.g., after lung transplantation) | Drug overdose |
| Adverse effect of medication |
Etiology and Pathogenesis
Overview of Pathophysiology
In the past, ARDS was also called noncardiogenic pulmonary edema, a descriptive term that nonetheless reflected what was known of the pathogenesis of the disorder. Unlike congestive heart failure, in which elevated left-sided cardiac pressures cause hydrostatic pulmonary edema, in ARDS, the edema fluid that fills the alveoli is exudative in origin. In other words, the alveolar-capillary barrier exhibits increased permeability, allowing for the leakage of protein-rich fluid into the air spaces. Alveolar filling leads to decreased respiratory system compliance as well as right-to-left shunting and profound hypoxemia. Although arterial Pco2 is generally within the normal range, dead space ventilation is significantly increased, as demonstrated by elevated minute ventilation. Pulmonary hypertension is also commonly observed in ARDS, and a number of mechanisms have been proposed including hypoxic vasoconstriction, intravascular fibrin deposition in the pulmonary capillaries, and compression of blood vessels by the positive pressure ventilation used to treat the disorder. This section reviews the pathology of ARDS and discusses current theories regarding its pathogenesis. Much of the research into ARDS has focused on determining the basis for the increase in alveolar-capillary permeability.
Pathology
The pathologic features of ARDS have classically been described using three overlapping and sequential stages.34 In the first or exudative phase of lung injury, the pathologic findings have been termed diffuse alveolar damage. There are hyaline membranes lining the alveolar walls and protein-rich edema fluid in the alveolar spaces, as well as epithelial disruption and infiltration of the interstitium and air spaces with neutrophils. Areas of hemorrhage and macrophages can also be found in the alveoli. This phase, which is thought to last for approximately 5 to 7 days, is followed in some patients by the so-called proliferative phase. At this point, hyaline membranes are reorganized and fibrosis begins to be observed. Obliteration of pulmonary capillaries and deposition of interstitial and alveolar collagen may be observed, along with a decrease in the number of neutrophils and the extent of pulmonary edema. This proliferative stage has traditionally been described as being followed by a fibrotic phase, essentially emphasizing the appearance of pulmonary fibrosis in a subset of patients with persistent (i.e., >2 weeks) ARDS. More recently, it has been realized that areas of fibrosis may actually develop sooner than usually appreciated: elevated levels of N-terminal procollagen peptide III, thought to represent collagen synthesis, can be detected in bronchoalveolar lavage fluid of patients with ARDS as early as 24 hours into the course of the illness. In addition, the bronchoalveolar lavage fluid from these patients has been shown to stimulate cultured fibroblasts to proliferate.35 These and other observations have led some investigators to hypothesize that fibroproliferation may be initiated simultaneously with (rather than after) inflammatory lung injury.36
Alveolar-Capillary Membrane
If ARDS is a disorder of increased alveolar-capillary permeability, it stands to reason that the pulmonary microvascular endothelium or the alveolar epithelium (or both) must be involved in its pathophysiology. Damage to the alveolar epithelium is thought to be a key event.37 Once ARDS has begun, the importance of the alveolar epithelium is also clear: Type II pneumocytes must differentiate into type I cells to help cover the denuded epithelial surface, while both type I and II cells express the Na+,K+-ATPase, which is thought to be important in clearance of edema fluid (discussed later). Injury to type II pneumocytes impairs the production and metabolism of surfactant. In addition to damage to the alveolar epithelium, loss of lung endothelial barrier integrity is both necessary and sufficient for the development of ARDS.38,39 How the alveolar-capillary membrane is damaged is not known for certain, although mediators of epithelial and endothelial apoptosis40 and neutrophils are suspected of playing a role (see later).
Surfactant.
Surfactant is a complex mixture of phospholipids and surfactant proteins that reduces alveolar surface tension. Many researchers have described alterations to the surfactant obtained from patients with ARDS. There are decreased amounts of surfactant-related protein and decreased dipalmitoylphosphatidylcholine and phosphatidylglycerol in the surfactant of patients with ARDS. The proportion of large (active) to small (inactive) aggregates of surfactant is diminished, due to both decreased production of surfactant and increased conversion of the large to small forms. In addition, plasma proteins that have leaked through the alveolar-capillary barrier may interfere with surfactant function.41 For example, damage to surfactant protein A has been demonstrated in patients with ARDS.42 The impairment in surfactant function observed in ARDS would theoretically predispose alveolar units to collapse. In addition, surfactant proteins have been found to have antimicrobial properties,43,44 although whether the loss of these properties is relevant to the pathogenesis of ARDS is not known. Indeed, despite the litany of abnormalities in surfactant that have been described,45 the extent to which these alterations contribute to the pathogenesis of ARDS remains controversial: unlike the neonatal respiratory distress syndrome, in which a deficiency of surfactant underlies the pathophysiology of the disease, it is possible that the abnormalities of surfactant in ARDS are the result rather than the cause of altered physiology. Indeed, four large randomized controlled trials of exogenous surfactant supplementation in ARDS failed to demonstrate any improvement in mortality or requirement for mechanical ventilation41 (see Treatment). Only one trial, using calfactant (a natural surfactant, distinct from the synthetic surfactants used in previous trials) in pediatric patients with ARDS, has shown any benefit46; whether this is due to the unique surfactant used in this study or to differences between the patients enrolled in this trial and the negative trials is unknown.
Neutrophils and Other Inflammatory Mediators
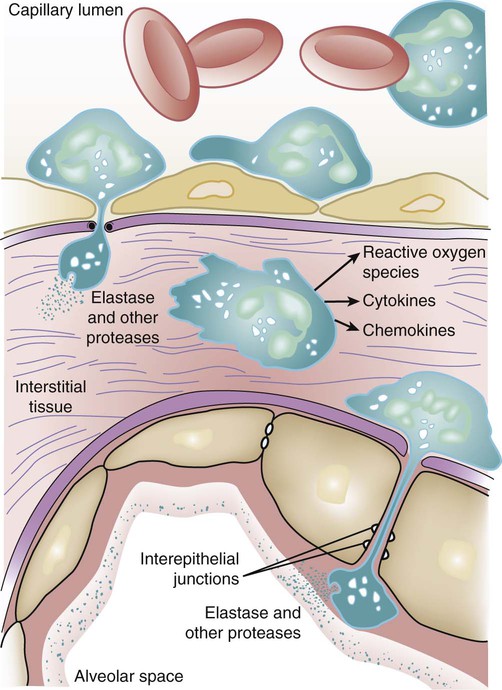
Histologically, one of the hallmarks of ARDS is the accumulation of neutrophils in the microvasculature of the lung.47 As key players in innate immunity, neutrophils can generate an impressive array of cytotoxic compounds. These include reactive oxygen species, cationic peptides (e.g., defensins), eicosanoids, and proteolytic enzymes such as leukocyte elastase. In addition, once activated, neutrophils release growth factors and cytokines (e.g., tumor necrosis factor [TNF]-α and interleukin [IL]-1β) that may enhance the inflammatory response. Given this destructive potential, it has long been theorized that neutrophils may be central to the pathogenesis of ARDS48 (Fig. 100-3). There is an abundance of clinical and preclinical data to support this hypothesis. For example, in humans with ARDS following sepsis, bronchoalveolar lavage neutrophilia is associated with a poor prognosis.49 Neutrophil-depleted mice exposed to hyperoxia develop less lung injury than normal mice.50 Similarly, in a hamster model of ARDS induced by endotoxin inhalation, the administration of a specific inhibitor of neutrophil elastase (even hours after administration of endotoxin) prevented the development of lung injury.51
If neutrophils are involved in the pathogenesis of ARDS, some defect in their regulation must be invoked. For example, in an uncomplicated bacterial pneumonia, the pulmonary inflammatory response is limited by counter-regulatory processes that prevent tissue damage. Whether this regulation is ineffective or insufficient in ARDS is a fascinating and unanswered question.52
One of the earliest manifestations of ARDS, even before hypoxemia, is a transient leukopenia due to sequestration of neutrophils in the lung microvasculature.53 The average pulmonary capillary is smaller than the average neutrophil, and neutrophils therefore have to deform to pass through the microvasculature. Activated neutrophils “stiffen” and cannot negotiate narrow capillary segments.54 Inhibitors of actin polymerization can abrogate this stiffening, implicating a change in the actin cytoskeleton in neutrophil sequestration in the lung.55 Neutrophil sequestration also involves interactions between molecules on the surface of the neutrophil and the lung capillary endothelium. For example, in rabbits, blocking the adhesion molecule L-selectin on the surface of the neutrophil prevented neutrophil sequestration in alveolar capillaries induced by exposure to endotoxin.56
After the initial sequestration, neutrophils must translocate (via diapedesis) across the alveolar-capillary barrier to the alveolar space. The determinants of this seemingly simple movement remain incompletely understood. Integrins on the surface of the neutrophil are thought to mediate emigration of neutrophils from the pulmonary circulation in response to some, but not all, inflammatory stimuli.57 Proteases secreted by the alveolar epithelium, in concert with regional cytokine production and glycosaminoglycans on the epithelial surface, may act together to form a chemotactic gradient for neutrophils to follow.58
Many studies are focusing on the mechanisms of neutrophil activation whereas others are focusing on the interaction between platelets and neutrophils and how mutual activation may contribute to ARDS or sepsis.59-62 Other studies have looked at various components of intracellular signal transduction pathways, such as kinases (enzymes that phosphorylate substrates) and transcription factors. For example, one such kinase is the p38 mitogen-activated protein kinase, which is activated when cells are stimulated with lipopolysaccharide (LPS).63 Activation of this kinase stimulates TNF-α production and macrophage inflammatory protein-2 release (a chemotactic factor for macrophages).64 Interestingly, inhibition of p38 mitogen-activated protein kinase in mice, even hours after the mice are exposed to aerosolized LPS, attenuates neutrophil chemotaxis and migration from the lung microvasculature into alveoli.65 Another kinase often discussed in the context of inflammation is phosphatidylinositol 3-kinase. This enzyme phosphorylates phosphatidylinositol, a lipid-derived second messenger whose phosphorylated forms have been implicated in a myriad of intracellular signaling events. Phosphatidylinositol 3-kinase γ is preferentially activated in neutrophils exposed to IL-8 or fMLP (a bacterial-derived peptide).66 Despite exposure to intraperitoneal endotoxin, phosphatidylinositol 3-kinase γ knockout mice displayed decreased neutrophil accumulation, cytokine production, and lung injury compared to control mice. The neutrophils in the lung of the knockout mice also demonstrated diminished activation of NF-κB, an important transcription factor known to mediate the up-regulation of numerous cytokines and proinflammatory mediators.67
There are many potential mechanisms by which activated neutrophils might mediate ALI. In addition to secreting cytokines and growth factors that might stimulate the local and systemic inflammatory response, neutrophils release defensins68 and generate reactive oxygen species that can mediate tissue damage.69 In animal studies, inhibition of the assembly of NADPH oxidase (the major source of reactive oxygen species) by apocynin has been shown to attenuate sepsis-induced lung injury.70 The nitric oxide synthase (NOS) pathway has also been implicated in mediating lung injury; knockout mice lacking the gene for inducible NOS developed less severe lung injury than wild-type animals upon injection with LPS.71
In addition, neutrophils contain proteolytic enzymes that may be involved in the pathogenesis of ALI. In particular, both neutrophil elastase and metalloproteinases have been extensively studied.52 Because of its ability to degrade multiple substrates including growth factors and cytokines, neutrophil elastase may be involved in regulation of the inflammatory response. It has also been shown to degrade epithelial and endothelial cadherins, proteins that are major components of adherens junctions, which hold cells together. It is possible that elastase-mediated destruction of cadherin could predispose to alveolar flooding. Despite its potential to cause unwanted tissue damage, neutrophil elastase (NE) is likely crucial to host defense. In NE-deficient mice, the administration of intraperitoneal Klebsiella pneumoniae causes 100% mortality within 48 hours, whereas mortality in normal mice was only 50%.72 However, NE-deficient mice are paradoxically resistant to normally lethal doses of LPS. Furthermore, mice that lack both NE and another neutrophil protease (cathepsin G) are protected against alveolar damage induced by endotoxic shock.73 These apparently contradictory results suggest that NE, while important in a regulated inflammatory response, may nonetheless participate in inflammatory injury under certain circumstances. In numerous studies of multiple different experimental models in multiple species of animals, NE has been shown to play an important role in the pathogenesis of ALI. Whether it is as important in the development of ARDS in humans remains unanswered. A clinical trial of a leukocyte elastase inhibitor in ARDS was halted because preliminary analysis suggested lack of efficacy.74
Metalloproteinases may also be important mediators of leukocyte-mediated lung injury. Elevated concentrations of the matrix metalloproteinases gelatinase A and B have been found in the epithelial lining fluid of patients with ARDS.75 Mice lacking the genes for gelatinase B or stromelysin 1 had less severe injury in an animal model of ALI.76 As mentioned earlier, matrilysin (matrix metalloproteinase 7) has been shown to regulate the formation of a chemotactic gradient and the transmigration of neutrophils across the alveolar epithelium in a mouse model of ALI.58 Finally, administration of an inhibitor of elastase and metalloproteinases (chemically modified tetracycline-3) attenuated ALI after cardiopulmonary bypass in pigs.77
Despite the emphasis on neutrophils as potentially causative for lung injury, it is important to note that ARDS has certainly been described in patients with profound neutropenia.78,79 Indeed, it is possible that the neutrophil infiltration observed in some cases of ARDS is adaptive (i.e., a physiologic response to a primary injury) rather than destructive. At present, however, it is generally believed that neutrophils are a causative factor in the development of most cases of ARDS. Comparatively little is known about the mechanisms of neutrophil-independent ARDS and this must be a goal of future investigations.
Inflammation and Coagulation
Because one of the most common precipitants for ARDS is sepsis, it is hoped that therapies for sepsis may either prevent or improve the outcome of ARDS. Our current concept of sepsis is that there is an initial inflammatory state, with up-regulation of inflammatory cytokines such as TNF-α and IL1β and recruitment and activation of inflammatory cells such as neutrophils. This early stage of inflammation is followed by a relatively depressed immunity, in which the patient is vulnerable to nosocomial infections.80 The theory that dysregulation of the inflammatory response is responsible for sepsis has led to the search for agents that would selectively block components of the inflammatory pathway. It has also been appreciated that there are important connections between the molecular cascades that regulate both inflammation and coagulation; for example, TNF-α causes an increase in thrombin and fibrin formation, while fibrin fragments themselves are known to be chemotactic for neutrophils.81 TNF-α increases tissue factor expression on endothelium and inhibits fibrinolysis, both of which favor fibrin formation. Activated protein C (APC), an endogenous anticoagulant, has direct and indirect anti-inflammatory effects, which include reducing levels of IL-6 and attenuating neutrophil activation in sepsis.82,83 Because sepsis is the most common cause of ARDS and because abnormalities of coagulation are very common in sepsis, investigators have wondered whether altered coagulation is involved in the genesis of ARDS. How this might happen is largely speculative, but intraalveolar, interstitial, and intravascular fibrin deposition have all been observed in patients with ARDS. Indeed, fibrin is a major component of hyaline membranes. Intraalveolar fibrin has been postulated to serve as a nidus for fibroblast proliferation and potentially, as ALI resolves, as a stimulus for pulmonary fibrosis. Fibrin might contribute as well to ongoing lung injury through its chemotactic properties, and intravascular fibrin deposition, as in microthrombi, might contribute to the elevated pulmonary vascular pressures observed in ARDS.
Interest in the role of coagulation in the pathogenesis of ARDS was buttressed in part by observations in animal studies that anticoagulation can attenuate the severity of sepsis-induced lung injury,84 although studies in humans have been disappointing. Indeed, a small randomized controlled trial of APC in patients with ALI was stopped prematurely due to lack of efficacy,85 and APC was pulled from the market after a large multicenter trial showed it provided no mortality benefit in patients with septic shock.86
Other molecular determinants of inflammation continue to be elucidated. Some data have implicated transforming growth factor-β in an animal model of ALI.87 In one study, transforming growth factor-β was postulated to cause increased alveolar epithelial cell permeability by depleting intracellular glutathione levels.88 Other investigators have attempted to improve the endogenous counter-inflammatory response; in one study, the gene for heat shock protein 70 was ligated to an adenoviral promoter, and the resultant recombinant construct was administered into the lungs of rats in which ARDS had been induced by cecal ligation and perforation. Administration of the heat shock protein 70 construct increased heat shock protein 70 expression specifically in the lung; remarkably, this increased expression was associated with a significant reduction in pulmonary edema and inflammation and even mortality.89 Indeed, mice deficient in heat shock protein 70 showed increased mortality and lung injury after cecal ligation and perforation.90 The mechanisms underlying this effect are not clear, but may involve suppression of the proinflammatory transcription factor NF-κB or decreased lung parenchymal apoptosis.91
Na+ and Water.
Na+ channels at the apical surface of alveolar epithelial cells mediate intracellular Na+ uptake from the alveoli. This creates an osmotic gradient that causes alveolar fluid to follow, thereby allowing for the clearance of pulmonary edema. Na+,K+-ATPases in the basolateral membrane of the epithelial cell exchange intracellular Na+ for extracellular K+, maintaining the intracellular sodium concentration low enough for the apical Na+ resorption to continue. This process allows for the net removal of Na+ and fluid from the alveolar space, and can be accelerated (at least experimentally) by catecholamines.
Dysfunction of either the apical Na+ channels or the basolateral Na+,K+-ATPases (or both) have been postulated to be involved in the pathogenesis of ARDS. For example, it is known that hypoxia decreases epithelial Na+ reabsorption by impairing the expression of subunits of the epithelial Na+ channel.92 Hypoxia also decreases the activity of both the apical Na+ channel93 and the Na+,K+-ATPase.94 In an animal model of hemorrhagic shock-induced lung injury, the normal up-regulation of alveolar fluid reabsorption in response to catecholamines was absent. This defect was attributed to an increase in nitric oxide (NO) in the lung, possibly due to an effect on β-adrenergic receptor signaling.95 In a study of patients with ARDS, alveolar fluid clearance was calculated from the change in alveolar fluid protein concentrations. When patients were stratified by their rate of fluid clearance, those with the highest rates had the shortest duration of mechanical ventilation and the lowest mortality.96 In a model of ventilator-induced lung injury (VILI) in rats, overexpression of the Na+,K+-ATPase improved the clearance of lung liquid.97 However, attempts to increase alveolar fluid clearance in a clinical setting (e.g., by β-adrenergic agonists) have so far been disappointing98; further details are provided later under Novel Therapies (see also Chapter 9).
Angiopoietins.
Angiopoietins are peptides involved in embryonic vascular development. Of the four angiopoietins that have been identified, angiopoietin 1 and angiopoietin 2 (ANGPT1 and ANGPT2, respectively) are the best described. ANGPT1 is expressed by numerous cell types while ANGPT2 is mostly limited to endothelial cells. Both act on the TIE2 receptor tyrosine kinase that is found predominantly on endothelial cells and hematopoietic stem cells.99 A number of observations have generated interest in the potential role of these proteins in the pathophysiology of ARDS. First, elevated levels of ANGPT2 have been described in patients with sepsis and ARDS100 and its administration to mice causes pulmonary vascular leak.101,102 The effect of ANGPT2 in disrupting endothelial cell integrity could be reversed in vitro by ANGPT1. Second, overexpression of ANGPT1 was protective against both endotoxin-induced septic shock and ALI in mice103; a similar benefit was observed using a synthetic peptide agonist of the TIE2 receptor.38 While the cellular mechanism of these effects remains uncertain and is the subject of intense investigation,104,105 the data to date emphasize the important role for endothelial cells in the development of and recovery from ARDS.
Ventilator-Induced Lung Injury
Although mechanical ventilation for ARDS may be lifesaving, there is abundant preclinical and clinical evidence that it can also be harmful.106,106a While it was quickly recognized that excessive airway pressure could lead to barotrauma (Fig. 100-4), including pneumothorax, pneumomediastinum and subcutaneous emphysema, attention has since turned to subtler but more common manifestations of lung injury related to mechanical ventilation. Mechanical ventilation can induce pulmonary edema by causing increases in both epithelial and endothelial permeability.106 Indeed 40 years ago Webb and Tierney demonstrated that ventilation of rats with high peak airway pressures could lead to severe pulmonary edema,107 and more than 20 years ago, investigators noted that mechanical ventilation could produce a form of increased permeability pulmonary edema remarkably similar to ARDS.108 Now, the accumulated evidence suggests that certain mechanical ventilation strategies may aggravate if not induce ARDS in some patients.109
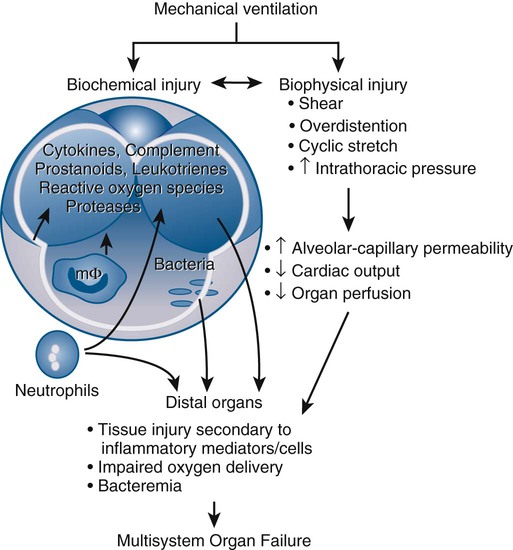
A major mechanism causing VILI is overdistention of lung units, rather than the absolute airway pressure per se. Normal rats ventilated with high airway pressure due to a high tidal volume developed increased permeability, whereas rats ventilated with smaller tidal volumes, but with the same end-inspiratory pressure (obtained by strapping the chest walls of the rats) did not develop increased permeability.106 Rats were also ventilated with low airway pressure using negative inspiratory pressure (applied at the chest wall) and high tidal volumes. Their results demonstrated that the rats ventilated with high tidal volumes had significantly more edema than others. In particular, the negative (low) pressure and high tidal volume group had the worst edema. These important observations, confirmed in other species,110 led to the appreciation that large tidal volumes, rather than high airway pressures per se, are an important determinant of ventilator-induced pulmonary edema. The term volutrauma was created to recognize this fact.
The repetitive opening and closing of terminal lung units associated with mechanical ventilation is also considered to be detrimental. The mechanism of this injury, which has been termed atelectrauma,111 is thought to be the high shear stresses generated at the interface of collapsed and aerated tissue when a collapsed airway is reopened.112 Theoretically, PEEP should be helpful in minimizing this injury by keeping the lung recruited and promoting greater lung homogeneity; any such advantage of PEEP would have to be balanced against the potential harm caused by a potentially greater overdistention of lung units due to the higher PEEP levels (see Video 100-1 ).
).
It is important to point out that patients with ARDS may be especially vulnerable to VILI because of the heterogeneous nature of the pulmonary parenchymal injury. On computed tomography scans of the lungs, normal-appearing lung and densely consolidated injured lung are both seen; as a consequence, there are marked regional differences in lung compliance113 (Fig. 100-5). A tidal volume designed to inflate an entire lung would preferentially inflate the normal-appearing areas, potentially leading to overdistention and volutrauma. Patients with ARDS may similarly be more vulnerable to atelectrauma. Although some evidence suggests that normal lungs can tolerate at least short periods of cyclic opening and closing of airways from mechanical ventilation,114 injured lungs, such as in ARDS, would be exposed to much higher shear stresses and would not be expected to fare as well.115
Over the past 15 years, there has been a growing appreciation that VILI is not only a mechanical injury, but also reflects an underlying complex cellular and molecular response. The term biotrauma has been coined to emphasize this change in thinking.116 In our understanding of VILI, one of the most significant advances made is that mechanical ventilation per se can induce both a local and systemic inflammatory response. In animal studies of ex vivo lungs, it has been shown that, compared to a control ventilation strategy with moderate tidal volumes and PEEP, ventilation either with high tidal volumes or with zero PEEP causes elevations in lung lavage levels of inflammatory cytokines. Lungs ventilated both with high tidal volumes and with zero PEEP had a synergistic elevation in cytokine levels.117 This inflammatory response to an injurious ventilation strategy has also been shown to extend beyond the lung. In an acid aspiration rat model of lung injury, a ventilatory strategy using high tidal volumes and zero PEEP was associated with an increase in blood levels of various cytokines; this was not observed in groups ventilated with a small tidal volume or in the group ventilated with the large tidal volume but higher PEEP levels.118
Most importantly, similar observations have been made in humans with ARDS. In one study, patients were randomized to “lung-protective” ventilation, in which the tidal volume was set to avoid overdistention and PEEP was set above the lower inflection point of the pressure volume curve.119 The control group received a strategy that would have been considered conventional ventilation at the time. While alveolar-lavage and plasma cytokine levels declined in the lung-protective group, they rose significantly in the control group. Post hoc analyses revealed a significantly higher number of ventilator-free days in the lung-protective group than in controls and significant correlations between the development of multisystem organ failure and plasma cytokines. The impact of ventilator strategy on the development of biotrauma can be remarkably quick. Within 1 hour after switching patients from a protective ventilatory strategy, Stuber and colleagues found an increase in a number of proinflammatory cytokines in the lungs and plasma of patients with ARDS.120
Mortality and Complications
As discussed in the section on outcomes, patients with ARDS often die of the systemic inflammatory response syndrome and multiorgan dysfunction. Hence, the observation that mechanical ventilation can influence pulmonary and systemic cytokine levels is intriguing. Taken together, these findings suggest that mechanical ventilation has the potential not only to injure the lungs, but also to lead to a loss of compartmentalization of the pulmonary inflammatory response. Systemic dissemination of this response could be associated with the development of systemic inflammatory response syndrome and potentially with multisystem organ failure (Fig. 100-6).121 In one study, injurious mechanical ventilation in rats not only caused elevated plasma and lung cytokine levels, but also caused increased renal epithelial cell apoptosis and associated renal dysfunction.122 Furthermore, pretreatment with either IL-10 (an anti-inflammatory cytokine) or IL-22 (a member of the IL-10 family that has immunoregulatory and tissue protective properties) reduced lung injury and reduced mortality in animal models of VILI.123
Finally, patients with ARDS often require very high inspired fractions of oxygen. The toxic effects of hyperoxia on the lung have been well described,124 and the histologic appearance mirrors that of human ARDS. Oxygen toxicity is thought to be mediated by the formation of both reactive oxygen and nitrogen species, which can damage tissues by a multitude of mechanisms,69 and hyperoxia appears to worsen VILI.125 Antioxidants have been considered as a potential therapeutic strategy for preventing and treating ARDS, although clinical trials have so far been disappointing (see Therapy).
Genetic Determinants
To date, relatively little is known about which genes might affect the development or prognosis of ARDS. Genome-wide association studies have suggested a number of candidate genes,126-129 including those coding for angiopoietin 2 and the angiotensin-converting enzyme,130 that might affect the incidence and/or outcome of ARDS. Subsequent studies have reported conflicting results.131,132 The heterogeneity of patients with ARDS is likely to make it difficult to identify clinically important genetic associations except in clearly defined subsets of patients.
Mortality and Complications
Mortality
The mortality rate for ARDS has decreased over the past 10 to 15 years, with a number of studies describing a decline in the rate from more than 60% to less than 40% since 1993.133-135 Mortality rates in adults and children with ARDS appear to be similar.136 The reasons for the improvement in survival over time are not known, although some have attributed it to better supportive care in the intensive care unit and the use of lung-protective strategies.
Predictors of Poor Prognosis
Despite the prominence of hypoxemia among the clinical manifestations of ARDS, early trials did not find that the severity of hypoxemia early in the course of the illness was a good predictor of subsequent mortality.137 Pulmonary injury scoring systems, such as the Lung Injury Score and the ARDS score, have been shown to predict a prolonged (>2 weeks) requirement for intubation and ventilation,138 while scoring systems that measure the overall severity of illness, such as the Simplified Acute Physiology Score, correlate better with survival.139 The classic teaching is that patients with ARDS do not usually die of refractory hypoxemia, which may seem paradoxical given that hypoxemia is frequently the focus of resuscitative efforts. In fact, most patients with fatal ARDS die of sepsis and multiorgan failure.140,141 The explanation for this apparent paradox is unknown, although it has been hypothesized that injurious mechanical ventilation during the course of ARDS may be involved.121 As discussed earlier, ventilation using excessive tidal volumes has been shown to cause elevations in pulmonary and systemic cytokine levels, and has been linked in an animal model to apoptosis of renal cells and renal dysfunction.122
The mortality rate from ARDS varies depending on the precipitant. The highest risk of death has consistently been reported in sepsis, while ARDS in the setting of major trauma has a much better prognosis.142 In addition, it is known that chronic liver disease,137 older age,143 chronic alcoholism,29 and nonpulmonary organ dysfunction137 are associated with higher mortality from ARDS. Other predictors of death from ARDS have included a history of organ transplantation and infection with human immunodeficiency virus,144 while one study described a higher mortality rate in men and also in African-Americans compared to non–African-Americans.145
As mentioned earlier, risk factors for ARDS have been classified as being pulmonary or nonpulmonary in origin, thereby leading to an injury to the lung by direct or indirect means. It remains unclear whether this distinction has prognostic importance. In one prospective cohort study of ARDS patients, investigators found a trend toward higher mortality in patients with a pulmonary precipitant, although the difference was not statistically significant.146 In contrast, the ARDS Network investigators retrospectively analyzed the data from their large randomized study of low tidal volume ventilation versus traditional tidal volume ventilation. While confirming that the mortality rate for ARDS was highest in patients with sepsis and lowest in patients with trauma, there was no difference in mortality, days off the mechanical ventilator, or the development of organ failure between patients with pulmonary or nonpulmonary risk factors. In addition, there was no statistically significant evidence that the low tidal volume strategy was less efficacious in any subgroup.142 A meta-analysis came to a similar conclusion.33 The consensus group that developed the Berlin definition of ARDS decided not to include pulmonary versus nonpulmonary ARDS as distinct categories.
Because hypoxemia is not a reliable predictor of mortality from ARDS, investigators have searched for other lung-specific markers of prognosis. In one prospective study of 179 patients with ARDS, a multiple logistic regression was performed to identify which clinical and physiologic variables predicted mortality.147 The analysis found that the dead space fraction (as calculated by the Bohr equation) was elevated in ARDS and was an independent predictor of mortality. For every increase of 0.05 in the dead space fraction, the odds of death from ARDS increased by 45%. The mechanism of this association is not known: it is possible that the pulmonary vascular injury in ARDS that accounts for the increase in dead space may be important to overall outcome.
The development of pulmonary fibrosis is also thought to connote a worse prognosis. Elevated procollagen III levels, thought to be indicative of collagen synthesis, have been found in the pulmonary edema fluid of patients with ARDS and have been shown to correlate with increased mortality.148,149 In another study, 22 of 25 consecutive patients with ARDS underwent transbronchial biopsies of the most abnormally appearing areas on the chest radiographs. In those whose biopsies showed any fibrosis, the mortality rate was significantly higher than in patients whose biopsies showed no fibrosis.150 Of note, there is evidence in animal models that the stress induced by mechanical ventilation may lead to lung fibrosis via epithelial-mesenchymal transition.151
Finally, there is extensive interest in the identification of biomarkers to predict outcome from ARDS. For instance, associations have been reported between elevated levels of cytokines such as IL-6 and IL-8152 and growth factors such as angiopoietin 2100 and a poor prognosis from ARDS. However, even a combination of biomarkers when used together with clinical predictors is only slightly more predictive for mortality than the clinical predictors alone.153
Complications
ARDS is complicated by ventilator-associated pneumonia in about 30% to 65% of cases (see also Chapter 34). In this setting, ventilator-associated pneumonia usually develops more than 5 to 7 days after the onset of mechanical ventilation and is often preceded by colonization of the lower respiratory tract by potential pathogens.154 The likely organisms include nonfermenting gram-negative rods, methicillin-resistant Staphylococcus aureus, and Enterobacteriaceae.155 Although the development of VAP prolongs the duration of mechanical ventilation in ARDS, it does not appear to increase mortality.154,156,157 Making a definitive diagnosis of VAP in patients with ARDS can be challenging, because patients with ARDS already have radiographic abnormalities, and not uncommonly have leukocytosis and fever. If diagnostic techniques such as bronchoalveolar lavage or protected specimen brushes are used, the yield is higher when the lung is sampled bilaterally and when the patient is off antibiotics.157,158
Another feared complication of ARDS is barotrauma (pneumothorax, pneumomediastinum, subcutaneous emphysema, eFig. 100-1) due to the effect of positive pressure ventilation in heterogeneous lungs with diminished compliance. Because most patients with ARDS will be supine (rather than erect), diagnosing a pneumothorax requires vigilance; the radiographic appearance of a pneumothorax is different and can be subtler in the supine patient (e.g., air in the costophrenic angle, the “deep sulcus” sign). Data from a number of prospective studies suggest that the incidence of barotrauma in ARDS currently is about 10% or less.109,159,160
Therapy
Supportive Care
One of the first goals of therapy in ARDS is to treat the underlying cause. In particular, patients with sepsis may respond to aggressive source control, including antibiotics and, when appropriate, surgical débridement and drainage. In patients with ARDS and sepsis of unknown origin, both the lung and the abdomen should be considered and excluded as foci of infection.140,141 Additional goals are the prevention of complications and the provision of supportive care (e.g., nutrition, ventilation) to allow the body time to heal. Typically, such treatment should include prophylaxis against gastrointestinal stress ulceration and deep venous thrombosis.
Hemodynamic Management
The optimal approach to hemodynamic management in ARDS has become less controversial with the publication of studies comparing different strategies. Before these studies, it had been unclear whether clinicians should attempt to diurese to decrease pulmonary edema at the expense of potentially causing hypovolemia and shock or to liberalize fluids to maintain perfusion.
This issue was addressed by a large multicenter randomized controlled trial from the ARDS Network, made up of investigators at multiple American hospitals. The trial randomized 1000 patients with ARDS to one of two highly protocolized fluid strategies: through the administration of fluids, diuretics, or vasoactive agents, the two protocols targeted either a higher or lower intravascular pressure (as measured by a pulmonary artery or central venous catheter) for 7 days. Patients randomized to the conservative fluid strategy (targeting a lower intravascular pressure) had significant improvements in oxygenation and more ventilator-free and intensive care unit–free days than the patients in the liberal strategy. Importantly, patients in the conservative arm did not have a higher incidence of dialysis or shock than those in the liberal arm; in addition, mortality rates in the two arms were similar. Thus, the results of this study suggest that conservative administration of fluids is safe and beneficial to patients with ARDS.
At first, this recommendation may seem at odds with the principle of early goal-directed therapy, whereby patients with sepsis are aggressively resuscitated with fluid in accordance with the randomized controlled trial reported by Rivers and coworkers.161 However, the apparently disparate results of these clinical trials are not difficult to reconcile. First, it is important to note that patients in the ARDSNet study were enrolled an average of 24 hours after meeting the criteria for ALI, much later than the 6-hour window in the study by Rivers. In addition, the study protocol of the ARDSNet trial was strictly designed to avoid aggravating or inciting shock or pulmonary edema.
One last caveat is that the ARDSNet study used a very complex algorithm for fluid administration that may not be widely adopted. A simplified protocol has since been proposed but has not been validated.162 In the meantime, we recommend that clinicians manage fluids conservatively in patients who are not in shock, nonetheless taking care to avoid overdiuresis and hypovolemia.
Clinicians had historically inserted a pulmonary artery catheter into patients with pulmonary edema to help establish the diagnosis and to guide therapy. The consensus from numerous clinical trials is that, for most patients, the information from a pulmonary artery catheter does not improve outcome.16 For this reason, we do not recommend the routine use of these catheters in patients with ARDS.
Which fluid to use in patients with ARDS remains an open question. The use of starch-based colloids has fallen out of favor in patients with severe sepsis, because of a randomized trial that found a higher incidence of renal failure in patients with severe sepsis receiving 10% pentastarch compared to patients receiving Ringer lactate.163 Whether pentastarch has the same detrimental effect in patients with ARDS without severe sepsis is unknown. On a theoretical basis, the use of albumin is appealing, because it would increase intravascular oncotic pressure and predispose to less pulmonary edema. A small placebo-controlled study in ARDS demonstrated that a regimen of albumin and furosemide infusions over 5 days caused a substantial and statistically significant improvement in oxygenation accompanied by a decrease in heart rate. However, most of the patients had ARDS as a result of trauma, with less than 5% having sepsis. There were no differences in important clinical outcomes (e.g., mortality), although the study was not powered to address these issues.164 A follow-up study established that the beneficial effect on oxygenation was due to the albumin, not the furosemide.165 Although a large clinical trial of patients admitted to the intensive care unit has concluded that albumin was as safe as crystalloid,166 the study did not specifically enroll patients with ARDS and only looked at short-term outcomes. It is also important to remember that as a blood product, the administration of albumin is associated with a very small but finite risk of transmissible diseases. Thus, pending further trials, the role of albumin in the management of patients with ARDS is unclear.
Nutrition
It has been hypothesized that manipulations in diet could enhance the immune system and improve the outcome of inflammatory diseases such as sepsis and ARDS. Such strategies have involved supplementing enteral feeds with one or more of arginine, glutamine, omega-3 fatty acids, and antioxidants.
One small randomized study examined the effect of a modified enteral feed containing eicosapentaenoic acid, gamma-linolenic acid, and various antioxidants compared to a control enteral feed in patients with ARDS.167 The authors found that the modified feed improved oxygenation, reduced the number of neutrophils in alveolar lavage fluid, decreased length of stay, and decreased the requirement for mechanical ventilation. In a more recent study, modified enteral feeds (containing eicosapentaenoic acid, gamma-linolenic acid, and various antioxidants) also improved oxygenation, although clinically important outcomes were unchanged.168 Many other studies of modified enteral feeds (often called immunonutrition) have been conducted in less well-defined populations of critically ill patients, with conflicting results. A meta-analysis on the topic highlighted the heterogeneity of the studies and suggested that the effect of immunonutrition varied depending on the group of patients being studied.169 At this point, the role of immunonutrition in the management of ARDS remains unclear.
A related issue in this area has been how much enteral nutrition should be given. In a recent randomized controlled trial, the rate of enteral feeding (e.g., at a regular rate versus a low, so-called trophic rate that delivered one third as many calories) did not affect the outcome of ALI.170
Pharmacotherapy
Attempts to develop pharmacologic therapies for ARDS have been frustrating and largely unsuccessful, with no pharmacotherapies that unequivocally reduce mortality from ARDS, despite a multitude of randomized controlled trials of dozens of potential agents.171 Despite the heterogeneity of the agents that have been evaluated, three generalizations can be made:
1. Despite showing effectiveness in vitro or in animal studies, most potential therapies have failed to reduce mortality or other important clinical outcomes in human clinical trials.
2. A number of agents improve oxygenation but do not affect mortality from ARDS.
3. Post hoc analyses of subsets of patients from a number of studies of various agents suggest benefit, but prospective data are lacking.
The following section reviews the biologic rationale for various potential therapies for ARDS, with an emphasis on evidence from clinical trials when available.
Corticosteroids.
Because of the presumed inflammatory pathophysiology underlying ARDS, a number of trials of high-dose corticosteroids have been performed. In some, the goal was the prevention of ARDS in patients at risk (e.g., with septic shock), while in others, steroids were given in established ARDS. The usual regimen was methylprednisolone 30 mg/kg every 6 hours for 1 to 2 days. None of the trials using this treatment regimen showed any benefit from the use of steroids,172,173 and one showed a higher incidence of infection in patients who received steroids.174 More recently, the use of steroids has been contemplated later in the course of ARDS, during the fibroproliferative phase. Persistently elevated plasma cytokine levels have been shown to correlate with worsened survival from ARDS, prompting some to theorize that late ARDS (>7 days after onset) is characterized by persistent inflammation that might be responsive to treatment with steroids. A small study randomized 24 patients with late ARDS to 2 mg/kg of methylprednisolone (followed by a 32-day taper) or placebo. Patients in the steroid group had lower mortality, improved oxygenation, decreased organ dysfunction, and earlier extubation, but also had a higher (but not statistically significant) rate of infection.175 However, these data are difficult to interpret because of the small sample size and the number of patients who crossed over to the alternate therapy.
A randomized placebo-controlled trial of 180 patients with ARDS of at least 7 days' duration was subsequently performed by the ARDS Network.176 Patients were randomized to either placebo or a single dose of 2 mg/kg of methylprednisolone followed by 0.5 mg/kg every 6 hours for 14 days, then every 12 hours for 7 days, then tapering. Although the patients randomized to steroids showed improvements in the number of ventilator-free and shock-free days (during the first 28 days) as well as in oxygenation, there was no difference in 60-day mortality. A subgroup analysis revealed that patients who received steroids more than 14 days after the onset of ARDS had a significantly increased 60- and 180-day mortality rate. The issue of whether to use steroids in late ARDS remains controversial,177 but it seems reasonable to consider steroids in patients who are not improving between about days 7 and 14 in that benefit has been suggested in subgroups and harm has not been proven.
Vasodilators.
Prostaglandin E1 is a vasodilator that has been studied as a potential therapy for ARDS, based largely on its putative anti-inflammatory properties. In vitro and preclinical studies in animals suggested that prostaglandin E1 given parenterally, especially when administered in a liposome, had the potential to decrease neutrophil activation. Despite promising early data,178 a large randomized double-blind multicenter trial of liposomal prostaglandin E1 found no improvement in survival or in ventilator dependence even though the drug improved oxygenation.179
Prostacyclin is another vasodilator which, when administered by nebulizer, acts selectively on the pulmonary vasculature. Because the aerosolized solution tends to go to the better ventilated areas of the lung, vasodilatation of the branches of the pulmonary artery that supply these areas lead to improved  matching and improved oxygenation. Although prostacyclin has been used as rescue therapy for refractory hypoxemia and is well tolerated,180 there are no large randomized studies and no placebo-controlled studies of its use in ARDS.181,182
matching and improved oxygenation. Although prostacyclin has been used as rescue therapy for refractory hypoxemia and is well tolerated,180 there are no large randomized studies and no placebo-controlled studies of its use in ARDS.181,182
NO is a highly reactive gas formed endogenously by NOS from the amino acid arginine. It stimulates cellular guanylate cyclase, leading to increased cyclic guanosine monophosphate levels. It acts as a potent vasodilator, and when given by inhalation, NO causes vasodilatation of the pulmonary circulation. NO is rapidly inactivated in the bloodstream by combining with hemoglobin to form methemoglobin, which is usually rapidly metabolized and does not accumulate to levels that are thought to be toxic (i.e., methemoglobin < 5%). Because of this rapid inactivation, NO is a selective vasodilator that does not affect the systemic circulation. Like aerosolized prostacyclin, NO causes the most vasodilatation in the areas of the lung that are best ventilated, thereby improving  matching.183 In addition, NO has both antiinflammatory and proinflammatory properties, although the contribution of these properties to its effects in a clinical setting is unclear.184 NO can also react with oxygen and water to form toxic metabolites, such as NO2 and nitrous and nitric acid, although at concentrations of NO of less than 40 ppm, this problem is not usually clinically significant. A soda lime absorber can be placed in the inspiratory limb of the NO circuit in order to remove any NO2 before the inspired gas reaches the patient.
matching.183 In addition, NO has both antiinflammatory and proinflammatory properties, although the contribution of these properties to its effects in a clinical setting is unclear.184 NO can also react with oxygen and water to form toxic metabolites, such as NO2 and nitrous and nitric acid, although at concentrations of NO of less than 40 ppm, this problem is not usually clinically significant. A soda lime absorber can be placed in the inspiratory limb of the NO circuit in order to remove any NO2 before the inspired gas reaches the patient.
In the largest randomized, double-blind, placebo-controlled study of NO in ARDS to date, more than 170 patients were randomized to different doses of NO (from 1.25 to 80 ppm) or placebo. Although approximately 60% of patients had a significant improvement in oxygenation within 4 hours of NO administration, there was no difference in survival or in liberation from mechanical ventilation between patients receiving NO and those in the placebo group.185 In addition, the initial improvement in oxygenation from NO was not sustained over the course of study. There were few adverse effects of NO, and for patients who were administered less than 40 ppm, methemoglobin and NO2 levels were the same as in the placebo group. The results of this study confirmed the findings of other smaller unblinded trials of NO in ARDS186,187 and were repeated in a systematic review.188 In another large randomized but unblinded study of NO in ARDS, a higher proportion of patients in the NO group required renal replacement therapy than in the control group.189 A meta-analysis of trials using NO for ARDS concluded that, while NO caused initial improvement in oxygenation, it had no impact on survival and was associated with an increased risk of renal dysfunction.190 We suggest that NO should not be used routinely in the treatment of patients with ARDS, other than perhaps as a “rescue” therapy for extreme life-threatening hypoxemia. In fact, in a recent individual patient meta-analysis, inhaled NO did not improve mortality in patients with ARDS regardless of severity as assessed by arterial Po2/Fio2 ratios as low as 70.191
Surfactant.
As discussed in the section on pathogenesis, a number of abnormalities of surfactant have been described in ARDS. These include an increase in relatively inactive forms, inactivation of surfactant by proteins that have leaked into the alveolar space, damage to type II epithelial cells (which produce surfactant), and destruction of surfactant constituents by the inflammatory process.45 These changes, along with the efficacy of surfactant supplementation in the neonatal respiratory distress syndrome, led to the hypothesis that surfactant supplementation might be beneficial in ARDS. Data from animal studies and small case series were promising.192 A small randomized controlled trial administered bovine surfactant through an endotracheal catheter in patients with ARDS and showed in one subgroup of patients an improvement in oxygenation and a trend to decreased mortality.193
However, enthusiasm for surfactant was greatly dampened by the results of a large multicenter randomized, blinded, placebo-controlled trial. In this study, investigators administered an aerosolized synthetic (protein-free) surfactant or saline placebo continuously for up to 5 days to patients with new onset (<48 hours) of sepsis-induced ARDS. There was no physiologic or clinical benefit from the surfactant. Despite its methodologic rigor, the study has been criticized because less than 5% of the administered dose of surfactant was thought to have reached the distal lung. In addition, the lack of surfactant proteins in the synthetic surfactant may have diminished its ability to reduce surface tension.194 Because of these issues, the role of surfactant supplementation continues to be studied.
In a phase I/II trial, 40 patients with new-onset ARDS were randomized to a surfactant protein C-based preparation (given up to four times over 24 hours) or to no drug. The surfactant was administered through a catheter placed in the endotracheal tube. The drug had no effect on oxygenation or on ventilator-free days, which were the primary outcomes of the study. The authors reported a significant decrease in IL-6 levels in bronchoalveolar lavage fluid from patients who received the surfactant preparation, although it is unclear whether this was a prespecified end point.195
Finally, two phase III trials of the recombinant surfactant protein C-based preparation have been completed. The studies demonstrated that the surfactant improved oxygenation, but had no impact on mortality or ventilator-free days. Only one trial, using calfactant (a natural surfactant, distinct from previous trials) in pediatric patients with ARDS, has shown any mortality benefit46; whether this is due to the unique surfactant used in this study or to differences between the patients enrolled in this trial and the negative trials (e.g., adult vs. pediatric) is unknown. Whether surfactant will prove to be useful in well-defined subgroups of patients with ARDS remains an open question.41
Antioxidants and Anti-inflammatory Agents (Other Than Steroids).
Oxidative stress has long been postulated to be involved in the pathogenesis of ARDS.69 In fact, lung injury due to hyperoxia is a commonly used model to study ARDS in animals. Reactive oxygen species form as a by-product of activation of neutrophils and macrophages; in addition, the requirement by many patients with ARDS for a high inspired fraction of oxygen may predispose to oxidative stress. Decreased levels of glutathione, a major endogenous scavenger of reactive oxygen species, have been observed in the alveolar fluid of patients with ARDS. Small clinical trials with N-acetylcysteine and procysteine were promising.196 Unfortunately, despite this optimism, larger clinical trials of antioxidants in ARDS have been disappointing. A multi-center trial of the antioxidant procysteine in ARDS showed no beneficial effect of the drug.47
Various agents with putative anti-inflammatory effects have been tested in ARDS. These include ketoconazole, lisofylline, and (in patients with sepsis at risk for ARDS) the nonsteroidal anti-inflammatory drug ibuprofen. In separate well-conducted, randomized, blinded, controlled trials, none of these agents demonstrated benefit in ARDS.197-199 A prospective randomized controlled trial evaluated the effect of recombinant platelet activating factor acetylhydrolase at preventing ARDS in patients with severe sepsis. This trial of more than 100 patients showed no decrease in the development of ARDS in patients who received the drug; however, the mortality rate was lower in patients receiving an intermediate dose of the study drug.200 Other trials of so-called anti-inflammatory therapies in sepsis have been disappointing.80,171
Novel Therapies.
As discussed earlier in the section on Pathogenesis, alveolar fluid clearance can be augmented by catecholamines. A small trial randomized 40 patients with ARDS to intravenous salbutamol or placebo for 7 days.201 Patients receiving salbutamol had a decrease in extravascular lung water but a higher incidence of arrhythmias. A follow-up study was performed in almost 50 intensive care units in the United Kingdom and enrolled more than 300 patients with ARDS. However, the trial was halted earlier than planned because of an increase in mortality in the group receiving intravenous salbutamol.98 A similar-sized trial of aerosolized albuterol (given every 4 hours for up to 10 days) for ARDS found no benefit.202 Thus, routine use of β2-agonists cannot be recommended in ARDS.
Stem cell therapy for ARDS is being explored.203 Beneficial effects have been noted in animals with endotracheal and intravenous delivery of stem cells204 and may be augmented by using stem cells transfected with ANGPT1.205 A schematic of novel potential therapeutic approaches is shown in Figure 100-7.
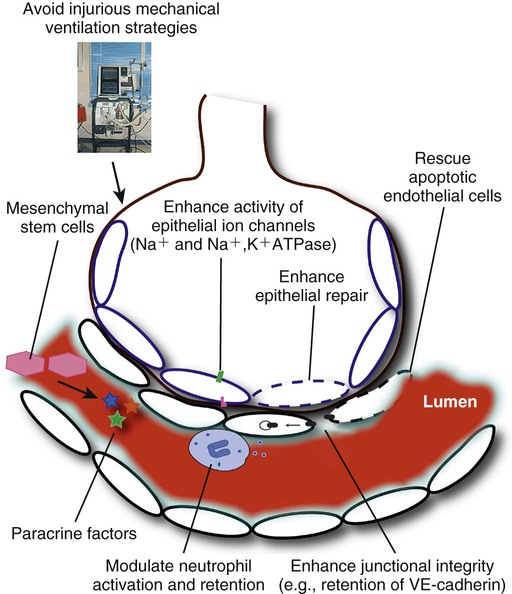
Discrepancies Between Studies in Animals and Humans.
As discussed earlier, virtually all large-scale clinical trials of pharmacotherapy for ARDS have been negative, despite promising and often exciting animal studies. There are a number of reasons why results from these animal studies have not been replicated in human clinical trials. First, in animal studies, the agent being investigated is often administered at the same time or shortly after the lung is injured (e.g., cecal ligation and perforation followed by administration of the drug within a few hours). In contrast, the onset of lung injury in humans is often much more difficult to define, and potential therapies are given many hours after the diagnosis is made. Thus, agents that might have been effective at preventing or attenuating ARDS may have been given too late. Second, most “proof of principle” animal studies are of relatively short duration and do not mimic the complicated clinical course of human ARDS. Third, animals in most studies represent essentially a homogeneous group; in contrast, human patients have multiple comorbidities and have multiple cointerventions. Fourth, the underlying precipitant for ARDS, even in carefully selected subgroups of patients, differ in severity and duration, unlike in animals wherein a uniform injury is applied. This heterogeneity makes it challenging to identify agents that may be of only modest clinical benefit. Finally, as discussed, the current definition of ARDS is problematic, and studies may include many patients with markedly different pathophysiology/biology.
Mechanical Ventilation
Mechanical ventilation is lifesaving and is the standard therapy for ARDS. Ventilatory management of ARDS has undergone dramatic changes over the last 20 years, in large part due to increased use of computed tomography to image the lungs and to advance understanding of VILI.
Pressure and Volume Limitation
Mechanical ventilation of patients under anesthesia for surgery traditionally involves large tidal volumes of 10 to 15 mL/kg, with the dual goals of achieving normal arterial oxygenation and arterial pH. A similar approach to ventilating patients with ARDS was followed in the past. This emphasis on achieving normal physiologic parameters in patients with ARDS was understandable at the time: in addition to being characterized by profound hypoxemia and diminished pulmonary compliance, ARDS was thought to involve the lungs diffusely and homogeneously based on plain radiography.7 A large tidal volume therefore appeared to be the only way to both ventilate patients and maintain oxygenation. Subsequently, studies using computed tomography206 demonstrated that the lungs of many patients with ARDS are actually heterogeneous: instead of the diffuse involvement suggested by plain radiographs, computed tomography scans often show patchy opacities interspersed with more normal-appearing areas of lung (see Fig. 100-5). The heterogeneous distribution of the injury in ARDS implies that the tidal volume administered to a patient will preferentially inflate the more compliant (or normal) areas of lung. These regions of the lung, exposed to tidal volumes meant for an entire lung, are therefore at risk for overdistention and ventilator-associated lung injury. As discussed earlier, mechanical ventilation with excessive tidal volumes can cause pulmonary edema due to increased alveolar-capillary permeability, with remarkably similar histology to ARDS.
Despite the clear physiologic rationale and abundant experimental data to support pressure and volume limitation when ventilating patients with ARDS, it was not until the late 1990s that data from randomized clinical trials in humans began to accrue. Between 1998 and 2000, five randomized controlled trials of ventilation strategies in ARDS were published.109,160,207-209 In all of the trials, patients were randomized either to a strategy involving some degree of tidal volume and pressure limitation, or to a “conventional” ventilation strategy with higher tidal volume and pressure limits.
Of the five studies, the largest was conducted by the ARDS Network, composed of investigators from multiple American hospitals and supported by the National Heart, Lung, and Blood Institute of the United States.109 This study, which was more than seven times larger than any of the other four, randomized 861 patients to lower tidal volumes or traditional tidal volumes. In the traditional group, plateau pressure was also kept below 50 cm H2O. In the small volume group, tidal volume was set at 6 mL/kg of predicted body weight and reduced if necessary to maintain plateau pressure between 25 and 30 cm H2O. Respiratory acidosis was treated aggressively, with the ventilator rate being set at six to 35 breaths per minute to achieve a pH of 7.3 to 7.45. Bicarbonate infusions were allowed for acidosis that persisted despite a ventilator rate of 35 breaths per minute. Tidal volume was increased for refractory acidemia, if pH was less than 7.15. Only preset combinations of PEEP and Fio2 were allowed in the two groups, for a target oxygen saturation of 88% to 95% (Table 100-3). Patient-ventilator dyssynchrony was reduced by sedating the patient, when necessary.
Table 100-3
Ventilation Protocol Used in the ARDS Network Study109
| Parameter | Protocol |
| Mode of ventilation | Volume assist control |
| Tidal volume | ≤6 mL/kg predicted body weight* |
| Plateau pressure | ≤30 cm H2O |
| Frequency | 6–35 breaths/min, titrated for pH 7.3–7.45 |
| I : E ratio | 1 : 1 to 1 : 3 |
| Oxygenation goal | Arterial Po2 55–80 mm Hg, or Spo2 88%–95% |
| Fio2/PEEP (cm H2O) combinations allowed | 0.3/5, 0.4/5, 0.4/8, 0.5/8, 0.5/10, 0.6/10, 0.7/10, 0.7/12, 0.7/14, 0.8/14, 0.9/14, 0.9/16, 0.9/18, 1/18–24 |
| Weaning | By pressure support, required when Fio2/PEEP ≤ 0.4/8 |
The trial, which originally set out to accrue 1000 patients, was stopped early because an interim analysis showed benefit in the small tidal volume group. The mortality rate was 39.8% in the traditional group, compared with 31% in the small tidal volume group (P=0.007). Breathing without assistance at day 28 was also significantly more frequent in the small volume group, and the number of ventilator-free days was higher. The number of days without organ failure was also higher in the small volume arm.
A key factor in comparing trials is to appreciate that the ARDS Network study used predicted body weight, while other studies used ideal body weight or measured body weight to set the tidal volume. The rationale underlying the use of predicted body weight is that it correlates much better with lung size than measured body weight, and the goal in individualizing the tidal volume for any patient is to match the tidal volume to the size of the lung. The distinction is important, since data from the ARDS Network study demonstrate that measured body weight was approximately 20% higher than predicted body weight. In other words, the tidal volumes used in other studies may have been higher than appreciated. Predicted body weight in kilograms can be calculated for male patients as 50 + 0.91(centimeters of height-152.4), and for female patients as 45.5 + 0.91(centimeters of height-152.4).109
The ARDS Network study adopted an aggressive approach to hypercarbia and acidemia, including use of higher respiratory rates, bicarbonate infusions, and loosening of ventilatory restrictions. As a result, hypercarbia and subsequent low pH in the treatment arm was less marked than in some other studies. This too may have contributed to the lower mortality in the lower tidal volume arm.
Some investigators have proposed that mechanical ventilation of a given patient be set based on the static inspiratory pressure-volume curve of the patient's respiratory system.209 In ARDS, such curves often have a sigmoidal shape, with a lower inflection point at low lung volumes, and an upper inflection point at high volumes. Initially, it was thought that the lower inflection point represented the pressure at which collapsed lung units reexpand, accounting for the abrupt change in compliance. The upper inflection point was thought to represent the pressure at which the alveoli become overdistended. On this basis, it was proposed that PEEP should be set higher than the pressure of the lower inflection point, while plateau pressure should be kept lower than the upper inflection point. Although conceptually appealing, this interpretation of the static inspiratory pressure-volume curve is likely erroneous. Recruitment of the lung is known to continue even above the lower inflection point.210 Conversely, during tidal ventilation, alveoli may be continually recruited so that the ventilatory cycle takes place in a region closer to the deflation limb rather on the inflation limb of the pressure-volume curve.211 Thus, limiting tidal volume or plateau pressure based on the inspiratory pressure-volume curve is not used widely.212
Neuromuscular Blockade
Mechanical ventilation of patients with ARDS is often complicated by patient-ventilator dyssynchrony, which can worsen oxygenation and ventilation. To overcome this, clinicians have traditionally sedated patients and even paralyzed them using neuromuscular blocking agents. Many clinicians have since become reluctant to use neuromuscular blocking agents because of concerns of inducing myopathy and prolonging the duration of mechanical ventilation. However, a multicenter randomized controlled trial of more than 300 patients with early (<48 hours) and severe ARDS compared a 48-hour infusion of cisatracurium to placebo.213 Both groups received lung-protective ventilation based on the ARDSNet trial, discussed earlier. The authors reported a significant reduction in 90-day mortality (the primary end point) as well as a number of secondary end points, including barotrauma and organ failure. There was no increase in intensive care unit–acquired weakness. The mechanism of benefit from neuromuscular blocking agents is unknown, but may involve decreased ventilator-induced lung injury.214 These provocative data, while promising, await replication before being widely adopted.
The Role of PEEP and Recruitment Maneuvers
As mentioned earlier, the first description of ARDS commented on the apparent utility of PEEP in improving oxygenation. From a theoretical standpoint, PEEP may be beneficial in avoiding damage from cyclic opening and closing of terminal lung units (atelectrauma, see Pathogenesis), and in allowing for a reduction in tidal volume (and hence volutrauma). In addition, PEEP, by improving oxygenation, may allow for lowering of the Fio2, thereby decreasing the risk for oxygen toxicity. On the other hand, PEEP that is too high can itself cause excessive end-inspiratory lung volume and volutrauma. Many clinicians are also familiar with the potential effect of PEEP to depress cardiac output and blood pressure. The optimal level of PEEP to use in patients with ARDS has therefore been a source of controversy.215-217
Along the same lines, the low tidal volumes and pressures advocated for lung-protective ventilation can lead to progressive de-recruitment of the lung, which can worsen hypoxemia and potentially aggravate atelectrauma. To counteract de-recruitment, so-called recruitment maneuvers have been proposed. These maneuvers involve an increase in airway pressure for a certain duration, although the pressure to be applied and its duration have not been standardized. One example of a recruitment maneuver would be 40 cm H2O of continuous positive airway pressure for 40 seconds. Similar to the problems with setting the level of PEEP, however, it is difficult to predict which patients might benefit from recruitment maneuvers and which might be harmed by overdistention. Overdistention of well-perfused lung units could result in diversion of blood to poorly perfused alveoli, with consequent worsening of right-to-left shunt and hypoxemia.218
A number of randomized controlled trials have addressed the issue of PEEP and recruitment maneuvers in the management of ARDS.
The ARDS Network investigators randomized 549 patients with ARDS to lower or higher PEEP levels, according to preset combinations of PEEP and Fio2. The patients in the lower PEEP arm received the PEEP/Fio2 levels used in the original ARDSNet trial of lung-protective ventilation (see earlier). The precise level of PEEP used in the higher arm varied with the Fio2, but was on average 5 cm H2O higher than the control arm (mean of 13 to 15 cm H2O over the first 7 days). Recruitment maneuvers were initially used in the higher PEEP group but were discontinued after the first 80 patients because they were not very effective.219 The study was stopped early due to perceived futility and the mortality rate in both arms was about 25%, significantly lower than had been reported in other trials of ARDS.220
A second large trial randomized almost 1000 patients with ALI to what was described as an “open lung” strategy versus the ventilation protocol used in the original ARDSNet trial.221 The “open lung” strategy consisted of pressure control ventilation with plateau pressure (Pplat) less than 40, recruitment maneuvers, and high PEEP, together with the low tidal volumes (6 mL/kg) used in the control group. On average, on day 1 of the study, PEEP in the open lung arm was 16 cm H2O, compared with 10 cm H2O in the control group. This trial also found no difference in the mortality rates between the two groups (about 40%), although the open lung group had improvements in secondary end points related to hypoxemia.
Finally, a large French trial randomized 767 patients with ALI to PEEP of 5 to 9 cm H2O or PEEP maximized as long as Pplat was less than 28 to30.222 On day 1 of the study, the mean applied PEEP level in the control arm was about 7 cm H2O, or marginally lower than that of patients in the original ARDSNet trial of lung-protective ventilation, while the higher PEEP arm had an average applied PEEP of 15 cm H2O. The study demonstrated no difference in mortality, but the higher PEEP arm had improved compliance, oxygenation, and a decreased duration of mechanical ventilation and organ failure.222
Taken together, the data from these trials suggest that higher PEEP levels than used in the original ARDSNet trial are safe and may improve oxygenation, but there are insufficient data to argue that higher PEEP improves survival. Why these clinical studies are generally negative despite very strong animal data demonstrating the benefits of higher PEEP levels is somewhat of a puzzle. One possibility is the yin-yang of PEEP. To the extent that PEEP recruits lung units, it may be beneficial; to the extent that it leads to greater overdistention of lung units, it may be harmful. In all studies to date, PEEP has been applied to all patients whether or not their lungs are recruitable. As such, one reasonable approach to be tested in future studies of PEEP is to randomize only patients to a high/low PEEP strategy if they have lungs that are potentially recruitable.217,223 Indeed, a meta-analysis that included these trials found that while higher PEEP was associated with a lower mortality in patients with arterial Po2/Fio2 < 200 mm Hg (i.e., ARDS), there was less benefit and a trend to harm in patients with a higher arterial Po2/Fio2 and hence less severe lung injury.224
The data also suggest that the role of recruitment maneuvers in the management of ARDS is only supportive, and the data currently available do not demonstrate improvement in clinically important outcomes. The major putative benefit of recruitment maneuvers is to decrease ventilator-induced lung injury. Many studies report a variable and transient improvement in oxygenation after some form of recruitment,219 which can be better maintained with a higher level of PEEP after recruitment. It is also possible that the same degree of recruitment could be achieved by simply raising the level of PEEP.225,226 Although recruitment maneuvers are generally safe, the patient must be closely monitored for any adverse effects on hemodynamics or oxygenation.
Permissive Hypercapnia and Tracheal Gas Insufflation
The use of lower tidal volumes (to avoid VILI) often results in respiratory acidemia, an effect that has been termed permissive hypercapnia. The theoretical detrimental effects of hypercapnia include myocardial depression, increased pulmonary vascular resistance, and decreased renal blood flow. Hypercapnia may have an injurious effect on alveolar epithelial cells.227 Perhaps the most clinically important adverse effect of hypercapnia is elevated intracranial pressure from increased cerebral blood flow. However, data also suggest that hypercapnia has protective effects, including attenuation of free radical–mediated lung injury and pulmonary inflammation.228,229
Given the uncertainty about permissive hypercapnia, it is worth remembering that the only large randomized controlled trial to show a reduction in mortality in ARDS treated respiratory acidosis fairly aggressively (discussed earlier).109 In the absence of other data, following the protocol used in that study seems prudent.
One approach that has been used to decrease high levels of arterial Pco2 is tracheal gas insufflation, a technique whereby a gas flow is introduced via a small catheter placed in the endotracheal tube, with its tip near the carina. Tracheal gas insufflation has been proposed as an adjunct to permissive hypercapnia, because the insufflated gas improves CO2 clearance from the anatomic dead space and ventilator tubing. However, the gas flow has the potential to increase alveolar volume and pressure, and may increase PEEP.230 Although there are many case reports describing the use of tracheal gas insufflation,231 there have been no randomized studies in ARDS. Because of technical and monitoring issues, we do not recommend its routine use of tracheal gas insufflation in ARDS.
Mechanical Ventilation of Patients in the Prone Position (Proning)
Placing patients with ARDS in the prone position (proning) was described almost 30 years ago as a means of improving oxygenation. The mechanisms by which the prone position improves oxygenation are multiple, but probably the most important factor is the effect that proning has on chest wall and lung compliance.232 In the supine position, the most dorsal and caudal regions of lung (along the spine and diaphragm) are the worst affected in many patients with ARDS. Some of this is due to gravity, but the weight of the heart and of the abdominal organs on the lungs also contributes. When a patient is placed in the prone position, the anterior chest wall is fixed (by the bed) and becomes less compliant, thus increasing the proportion of ventilation directed to the dorsal lung. In addition, the volume of lung that is being compressed by the heart due to gravity is substantially decreased. The net result is more homogenous ventilation of the lung and presumably improved  matching. Data from a study in dogs suggest that ventilation in the prone position can attenuate the severity of VILI.233
matching. Data from a study in dogs suggest that ventilation in the prone position can attenuate the severity of VILI.233
A number of multicenter randomized controlled trials have now examined the efficacy of proning in patients with ARDS. One study enrolled more than 300 patients with ALI and randomized them to conventional treatment (supine) or treatment in the prone position for 6 or more hours for 10 days. The study found that oxygenation improved in approximately 70% of proning procedures and that most of the improvement was evident within 1 hour of proning. However, although oxygenation improved significantly in prone patients, there was no difference in mortality between the groups. Retrospective analyses of the quartile of patients with the poorest oxygenation or the highest acuity of illness (or the highest tidal volume) showed a lower mortality rate at 10 days in the patients who were prone, but this difference did not persist beyond discharge from the intensive care unit.234 The study has been criticized for the relative short duration of the intervention, as well as the fact that most patients spent the majority of the day supine.235
A subsequent study randomized more than 700 patients with acute hypoxemic respiratory failure (which included but was not limited to patients with ARDS) to ventilation in the prone or supine position; patients were prone an average of 50 hours after intubation for a median of 8 hours per day, for a median of 4 days.236 Prone patients exhibited improved oxygenation but no improvement in mortality or duration of ventilation. These patients also suffered from a higher incidence of complications, including pressure sores, selective intubation, and endotracheal tube obstruction.
A pediatric study (median age 2 years) of proning in ARDS was similarly negative,237 and two Spanish trials of proning for adults with ARDS were inconclusive but suggestive of benefit.238,239
In 2013, a multicenter randomized trial of prolonged (>16 hours) proning was reported in more than 400 patients with early (<36 hours) and severe ARDS.240 ARDS was defined according to the AECC criteria, with severe ARDS denoting arterial Po2/Fio2 < 150 mm Hg with Fio2 ≥ 0.6, PEEP ≥ 5 cm H2O and tidal volume of about 6 mL/kg predicted body weight. Patients were proned within the first hour after randomization and kept prone for at least 16 consecutive hours; proning was performed every day up to day 28. Remarkably, the 28-day survival rate was 16% in the prone group and 33% in the supine group.
These results are very impressive and are likely to increase the adoption of proning by intensivists. While proning in that trial was not associated with any complications, it is important to note that it was conducted in centers with significant expertise in prone-position ventilation, which requires some attention to detail, and that it did not involve use of a rotating bed. Lines and tubes are vulnerable to dislodgement during the process, and sufficient staff should be on hand to assist with the move. Personnel should be ready and capable of immediate reintubation in case the endotracheal tube is displaced. Prone patients are more susceptible to development of pressure sores, and exquisite care must be taken to ensure that no stray objects (e.g., syringes, electrocardiogram leads) are left under the patient, because these leave impressions and even scars on the body. An unstable spinal injury is an absolute contraindication to proning. In addition, if cardiopulmonary resuscitation is required, the patient must be returned to the supine position emergently. Finally, in the most recent trial, there were a number of exclusion criteria, including elevated intracranial pressure, massive hemoptysis, recent tracheal or facial surgery, and low mean arterial pressure (<65 mm Hg).240
Volume-Control Versus Pressure-Control Ventilation
The volume and pressure-limited protocol used in the ARDS Network study employed volume-assist control as the mode of ventilation. The Lung Open Ventilation study (described earlier) demonstrated that pressure-control ventilation could be used with equivalent outcomes.221 Other studies that have examined this issue have also found little difference in physiologic parameters or outcome between the two modes of ventilation.241,242
High-Frequency Jet Ventilation and High-Frequency Oscillation
In high-frequency jet ventilation, a small gauge catheter is used to introduce gas under high pressure into the endotracheal tube. The high velocity of the gas entrains additional oxygen and humidified air from side ports in the system. This form of mechanical ventilation achieves a tidal volume of 2 to 5 mL/kg and involves frequencies of 100 to 200 breaths per minute.243 Exhalation is passive, requiring recoil of the lungs and chest wall. There is little evidence that high-frequency jet ventilation is superior to conventional mechanical ventilation for ARDS. An early randomized trial of over 300 oncology patients with ARDS compared conventional ventilation (using volume-cycled ventilation) with high-frequency jet ventilation. Because the end points of the study differed between the two groups of patients, it is difficult to interpret the data. Nonetheless, the authors found no significant difference in any clinically important outcome.244
In high-frequency oscillatory ventilation (HFO), lung recruitment is maintained using a constant mean airway pressure generated by an inspiratory bias flow and limitation of gas outflow from the circuit. Ventilation is achieved through rapid (e.g., 5 Hz) regular oscillations of a piston or diaphragm. The push and pull action of the piston causes oscillations in pressure in the endotracheal tube and proximal airways, creating peak and trough pressures around the set mean airway pressure. The tidal volumes achieved through HFO are small, on the order of 1 to 5 mL/kg. In theory, HFO seems ideally suited for avoiding VILI. Atelectrauma is minimized because of the relatively high mean airway pressure, which along with small tidal volumes limits derecruitment of the lung, and volutrauma is minimized because the small tidal volumes limit end-inspiratory stretch.245 With HFO, loss of pressure from the circuit can lead to de-recruitment, thus recruitment maneuvers (e.g., application of continuous positive airway pressure for approximately 30 to 40 seconds) should be applied after each patient disconnect from the ventilator (e.g., open suctioning).
In an early randomized trial of HFO in adults, 148 patients with ARDS were randomized to conventional ventilation using pressure-control mode or to HFO.246 The specific ventilatory parameters for both the conventional group and the HFO group were adjusted to achieve adequate oxygenation at a minimum Fio2 (i.e., 88% at Fio2 ≤ 0.60), as well as an arterial pH greater than 7.15. Because the study was begun before the publication of the ARDS Network trial on tidal volume limitation, the conventional ventilation arm of this study targeted tidal volumes of 6 to 10 mL/kg. The trial was not powered to detect a mortality difference, and there was no significant difference in 30- or 90-day mortality, or in any other outcome. The authors performed a retrospective analysis of predictors of mortality, and discovered that being on conventional ventilation for more than 5 days correlated with a poor outcome.
More recently, two large randomized trials of HFO were published and showed no benefit in terms of survival or the duration of mechanical ventilation247,248 compared with conventional lung-protective ventilation. Indeed, one of the studies suggested that HFO might be harmful due to the amount of sedation required or due to impairment of hemodynamics. Because of these findings, the routine use of HFO for ARDS cannot be recommended, although its role as “rescue” therapy is not clear.
Liquid Ventilation
Liquid ventilation relies on the oxygen and carbon dioxide carrying capacity of organic liquids such as perfluorocarbons. Perfluorocarbons are modified hydrocarbons in which hydrogen atoms are replaced with fluorine, generating inert liquids that are nontoxic and minimally absorbed through the respiratory epithelium. The most widely studied perfluorocarbon, perflubron (perfluorooctyl bromide, eFig. 100-2), can dissolve about 17 times more O2 than saline solution and almost 4 times more CO2.249 Total liquid ventilation is a technique in which the lungs are completely filled with liquid and an extracorporeal exchanger is used to add O2 and remove CO2 from the liquid. Partial liquid ventilation, which is much easier to use clinically, involves partially filling the lung with liquid and then using a traditional ventilator to deliver gas tidal volumes.250
The theoretical benefits of liquid ventilation stem largely from improved lung recruitment, due to the lower surface tension of the perfluorocarbons and because the liquid tends to distribute to the dependent regions of the lung. Deposition of liquid with low surface tension in these areas may enhance alveolar recruitment; in addition, the weight of the liquid is thought to cause diversion of pulmonary blood flow to the nondependent (better ventilated) areas, improving  matching. Clearance of secretions (due to their displacement by the liquid) is also improved. Anti-inflammatory effects of perflubron have been described, although the clinical relevance of these effects is not well understood. To date, two randomized controlled trials of partial liquid ventilation in adults with ARDS have been published.251 The first enrolled 90 patients with ARDS to partial liquid ventilation with perflubron or to conventional mechanical ventilation. Other than very general guidelines (e.g., target arterial So2 > 90%), neither of the two ventilation strategies was protocolized. In addition, the inclusion and exclusion criteria were modified slightly during the course of the study. There was no significant difference in the number of ventilator-free days (the primary outcome measure) or in any other predefined outcome. More patients in the liquid ventilation arm experienced hypoxia, bradycardia, and respiratory acidosis, although the increase in the incidence of these adverse events was not statistically significant.
matching. Clearance of secretions (due to their displacement by the liquid) is also improved. Anti-inflammatory effects of perflubron have been described, although the clinical relevance of these effects is not well understood. To date, two randomized controlled trials of partial liquid ventilation in adults with ARDS have been published.251 The first enrolled 90 patients with ARDS to partial liquid ventilation with perflubron or to conventional mechanical ventilation. Other than very general guidelines (e.g., target arterial So2 > 90%), neither of the two ventilation strategies was protocolized. In addition, the inclusion and exclusion criteria were modified slightly during the course of the study. There was no significant difference in the number of ventilator-free days (the primary outcome measure) or in any other predefined outcome. More patients in the liquid ventilation arm experienced hypoxia, bradycardia, and respiratory acidosis, although the increase in the incidence of these adverse events was not statistically significant.
In 2006 a multicenter randomized trial of partial liquid ventilation with perfluorocarbon compared with conventional mechanical ventilation (Vt 10 mL/kg) enrolled more than 300 patients with ARDS. The study found a trend toward higher mortality in the groups receiving perfluorocarbon in addition to a higher incidence of barotrauma, hypoxia, and hypotension.252 Based on these data, partial liquid ventilation cannot be recommended for patients with ARDS.
Extracorporeal Membrane Oxygenation (see Chapter 103)
Extracorporeal membrane oxygenation (ECMO), also referred to as extracorporeal life support or extracorporeal lung assist, refers to the process by which the patient's blood is circulated to an external machine that provides oxygenation and/or carbon dioxide removal.253 In theory, ECMO could be used to oxygenate patients with ARDS while minimizing VILI and oxygen toxicity and allowing the lungs time to heal. There are multiple case reports of ECMO being used in ARDS, and it is used routinely in neonates with severe respiratory failure. A randomized controlled trial of ECMO in ARDS was completed almost 30 years ago and did not show any benefit.254 It is worth pointing out that the mortality rate in that study was approximately 90% in both the ECMO and the control arm. Subsequently, a variant of ECMO dedicated to carbon dioxide removal (extracorporeal CO2 removal, or ECco2R) was developed and showed promise in a case series when compared with a historical control group.255 This was followed by a randomized controlled trial using pressure-controlled inverse-ratio ventilation (IRV) and ECco2R in ARDS that failed to find any improvement in survival.256 This study illustrates the importance of using concurrent controls when assessing a novel therapy.
One of the complications of ECMO is bleeding; in an early randomized trial, patients randomized to the device were transfused an average of 1.7 L of blood per day. More recent data suggest that, because of many technological improvements, ECMO can be relatively safely performed by specialized centers. In 2009, a randomized controlled trial of conventional ventilation versus ECMO for acute respiratory failure was reported.257 The vast majority of patients in each arm had ARDS; referral to an ECMO center conferred a statistically significant survival benefit (relative risk of death at 6 months, 0.69; confidence interval, 0.05–0.97). However, patients in the conventional ventilation arm were cared for in multiple hospitals, did not have a standardized ventilation protocol, and experienced a mortality rate of 53%. In contrast, of the patients referred to the ECMO center, 75% actually received ECMO and a significantly higher percentage of patients in this group received lung-protective ventilation. Thus, it is unclear whether the improvement in survival was attributable to ECMO or to better care in a specialized hospital. A cohort study by the same authors examining ECMO for H1N1 influenza-induced ALI also suggested lower mortality from the treatment.258
There is also growing interest in using extracorporeal life support as an adjunctive therapy to maximize the benefit of lung-protective ventilation. Even at the recommended tidal volume of 6 mg/kg, the lungs of patients with ARDS may be damaged by overdistention and VILI. Thus, the promise of ECMO is that tidal volumes of less than 6 mL/kg predicted body weight can be used and the resultant respiratory acidosis can be managed by extracorporeal removal of CO2.259 Because of ongoing technological improvements that have reduced the requirement for anticoagulation and the invasiveness of the devices,260,261 this is likely to be a fertile area of research in coming years.
Summary
The modern approach to mechanical ventilation of patients with ARDS is based on two facts: first, that the diffuse lung injury of ARDS affects the lung heterogeneously; second, that mechanical ventilation itself can cause ALI. Thus, until new data emerge, the lung-protective ventilatory strategy adopted by the ARDS Network study109 is the standard of care and is associated with both short-term and long-term improvement262 in survival. Plateau pressure should be maintained at less than 30 cm H2O, and tidal volume should be limited to 6 mL/kg predicted body weight as much as possible. The prone position should be seriously considered in patients with P/F less than approximately 150 mm Hg in centers with medical staff experienced in proning patients. Given the dangers inherent in this approach, proning must be performed with caution and following published protocols.
The optimal level of PEEP in ARDS remains unclear, although randomized trial data indicate that using higher PEEP levels than the original ARDS Network study is safe and may improve oxygenation. We favor high levels of PEEP in order to prevent atelectrauma, reduce Fio2 and prevent hyperoxic lung injury in patients with greater hypoxemia (i.e., P/F < approximately 150 mm Hg), as long as the patient's hemodynamic status remains stable. We use the lowest possible Fio2 that maintains oxygen saturation above 90%, and we empirically adopt a target Fio2 of less than 0.6.
The use of neuromuscular blocking agents may also improve outcome by reducing ventilator-induced lung injury when used early in the clinical course of patients with relatively severe ARDS. Finally, despite the theoretical appeal of HFO ventilation, randomized controlled trials have shown that it does not improve survival over conventional lung-protective ventilation.
Long-Term Outcomes
Despite the profound derangement in oxygenation and respiratory system compliance that is characteristic of ARDS, it is remarkable that patients who survive often have near-normal pulmonary function tests 6 to 12 months later. Lung volumes and flows are generally slightly reduced or normal at 6 to 12 months, while the diffusion capacity may remain slightly reduced.263,264 Follow-up chest radiographs are usually normal, with a minority showing subtle abnormalities including pleural thickening or small cysts.264
Despite this impressive physiologic and radiologic recovery, patients who survive ARDS continue to have important functional limitations and a decreased health-related quality of life for at least 5 years after their illness.265,266 This reduction in quality of life seems attributable to ARDS or to its management or complications because, in one parallel cohort study in which patients with ARDS were matched with patients with sepsis or trauma with an equivalent severity of illness, the ARDS survivors reported significant decrements in health-related quality of life, particularly as related to physical function and pulmonary symptoms.267 This reduced quality of life (in areas reflecting physical function) has been correlated with the presence of persistent abnormalities in pulmonary function tests.263 In other patients, functional capacity (as measured by a 6-minute walk test) was persistently reduced and was ascribed to persistent muscle weakness and wasting.264 In a multivariate analysis in that study, better functional capacity was associated with a lack of systemic corticosteroid administration, absence of illness acquired in the intensive care unit, and rapid resolution of lung injury and multiorgan dysfunction. Patients who ultimately survive an episode of ARDS do not appear, however, to have increased mortality compared with other, similarly ill intensive care unit survivors.268
Psychological problems, including depressive symptoms,269 have been described in survivors of ARDS. In a retrospective case-control study, survivors of ARDS were found to have significantly more signs and symptoms of posttraumatic stress disorder than patients who had undergone maxillofacial (and presumably elective) surgery; they also had more signs and symptoms of posttraumatic stress disorder than soldiers who had served for prolonged periods of time in Bosnia.270 Persistent cognitive impairment has also been observed in survivors of ARDS for as long as 1 year after hospital discharge.271 Most patients in this study had impairments in at least one of memory, concentration, attention, or mental processing speed. Interestingly, these abnormalities were correlated to the degree and duration of hypoxemia of the patients.
ARDS is, by definition, a syndrome caused by a heterogeneous group of insults and is not a specific diagnosis. In particular, the clinician must search for an underlying cause to begin appropriate therapy. What has changed in the past 20 years, however, is our realization that in ARDS, regardless of the precipitant, inappropriate mechanical ventilation can do more harm than good. Amidst all the heterogeneity that is ARDS, it is perhaps ironic that this potential for iatrogenic injury may turn out to be one of the few common elements.
eFigure Image Gallery