Hepatitis B
Jennifer T. Wells, Robert Perrillo
An estimated 400 million persons in the world today are chronically infected with HBV. The majority of these individuals will not experience complications, but 15% to 40% will have serious sequelae such as cirrhosis or hepatocellular carcinoma (HCC), and many will die prematurely.1,2 In the United States, the rate of acute liver failure attributable to hepatitis B has been declining, as has the number of cases listed for transplantation for chronic liver failure.3,4 This decline is most likely due to broader vaccination and use of antiviral therapy. Unfortunately, these favorable trends are counterbalanced by a continuing increase in new cases of chronic hepatitis B and HCC.4,5
Effective vaccines against HBV have been available since the early 1980s, but perinatal and early life exposures continue to be major sources of infection in much of the developing world because of limited resources that preclude a policy of universal vaccination for newborns. From a global perspective, widespread implementation of early-life vaccination programs in high- and intermediate-risk countries will ultimately have the greatest impact on liver disease–related mortality in future generations. Promiscuous heterosexual contact and injection drug use account for most new cases of hepatitis B in adults in low-prevalence areas such as the United States and Western Europe. Even in these areas, however, a further reduction in the incidence of acute infections will remain a challenge in the future because people who inject drugs and promiscuous heterosexuals greatly underutilize vaccination.
Epidemiology
Geographic Distribution and Sources of Infection
The prevalence of HBV infection varies markedly around the world. In highly endemic regions, such as Southeast Asia (excluding Japan), China, and much of Africa, 8% or more of the population are chronic HBV carriers, and the lifetime risk of infection ranges from 60% to 80%.6 In these high-risk areas, perinatal transmission and horizontal spread among children are the major means of transmission. Approximately 60% of the world's population reside where HBV is highly endemic.7 Regions of intermediate risk include parts of southern and Eastern Europe, the Middle East, Japan, the Indian subcontinent, much of the former Soviet Union, and northern Africa. In these areas, the lifetime risk of infection is between 20% and 60%. Horizontal transmission occurs among a broad age range, but neonatal exposure is also presumed to be common. Areas of low prevalence include North America, Western Europe, certain parts of South America, and Australia, where the lifetime risk of HBV infection is less than 20% and transmission is primarily horizontal between young adults. Sexual transmission is the main mode of transmission in Europe and North America, but injecting drug use continues to be a major contributor to new cases as well.8
Perinatal transmission accounts for the majority of new infections in the world and is believed to account for as many as half of all hepatitis B surface antigen (HBsAg)-positive carriers. Sixty percent to 90% of hepatitis B e antigen (HBeAg)–positive mothers transmit the infection to their offspring, whereas mothers who are positive for antibody to HBeAg (anti-HBe) transmit the disease less frequently (5% to 15%) (see later). Fortunately, the incidence of new infections and childhood HCC has diminished greatly in countries such as Taiwan where universal vaccination has been in place for decades.9
Infectivity
HBV is transmitted efficiently by percutaneous and mucous membrane exposure to infectious body fluids. The virus is 50 to 100 times as infectious as HIV and 10 times as infectious as HCV. HBeAg seropositivity indicates a higher risk not only of transmission from mother to child, but also after needlestick exposure and in the setting of household contact. HBV DNA has been detected by sensitive techniques such as PCR methodology in most body fluids, except for stool that has not been contaminated with blood. Although HBV replicates primarily in hepatocytes, the presence of replicative intermediates and virally encoded proteins in other sites, such as the adrenal gland, testis, colon, nerve ganglia, and skin, suggests that a vast extrahepatic reservoir for infectious virus exists.10 Small amounts of HBV DNA have been demonstrated in peripheral mononuclear cells and liver tissue years after apparent resolution of chronic infection.11,12 Extrahepatic localization of low levels of replicating virus explains the relatively high rate of HBV transmission from organ donors positive for antibody to hepatitis B core antigen (anti-HBc) (see later).13
Prevalence
Estimates of chronic HBV infection report a prevalence of 2.2 million persons in the United States.14 This number is approximately 1 million higher than formerly believed. The older figure of 1.25 million persons was an underestimate because of changing immigration patterns since the 1980s and an underrepresentation of certain minority groups in the National Health and Nutrition Examination Survey (NHANES) study. Nearly 15 million Asians live in the United States, and even a conservative estimate of the prevalence of HBV infection such as 5% would raise the overall number of HBV carriers in the United States by more than 750,000. Of all immigrants to the United States between 1974 and 1998, an estimated 60% were born in regions of intermediate or high HBV prevalence. This finding explains why more than 90% of chronic HBV infections in the United States are imported14 (Fig. 79-1).
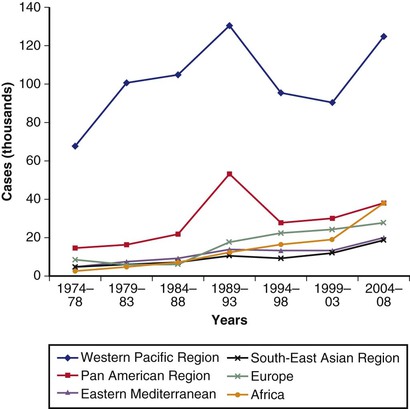
In contrast to the growth of new cases of chronic hepatitis B, a decline in acute cases of hepatitis B since the 1990s has been the result of universal vaccination of newborns, adult vaccination programs for high-risk persons, changes in sexual lifestyle, refinements in blood screening procedures, and the availability of virus-inactivated blood components.15 Health care workers have experienced a striking decline in HBV infection owing to high rates of vaccination.16
In the United States and much of the developed world, however, the highest incidence of acute cases continues to be in sexually active young adults.17 Since 1995, approximately 40% of cases of acute hepatitis B reported to the CDC were caused by intimate contact among heterosexuals. Fifteen percent to 20% were due to injecting drug use, and 12% occurred in men who have sex with men. No identifiable source of exposure was demonstrated in approximately 15% of cases.18 Cases of hepatitis B continue to result from hemodialysis, acupuncture, artificial insemination, and rarely, blood transfusion, but these cases account for a small contribution to the overall number of newly established acute infections.
Data from the CDC indicate that more than 95% of pregnant women in the United States are tested for HBsAg, and infant vaccine coverage levels are up to 93%. Despite these encouraging figures, however, the CDC also estimates that approximately 1000 new cases of hepatitis B in newborns each year are due to a missed birth dose or failure to complete the vaccine schedule.19
Several distinct trends in prevalence are noted when the results of the CDC-sponsored NHANES survey between 1988 and 1994 are compared with those obtained from 1999 to 2006. Although the prevalence of past and present infection was not different in the 2 groups as a whole, a significant decrease in the prevalence of infection was found in persons 6 to 19 and 20 to 49 years of age for the more recent time interval. Additionally, although the prevalence among foreign-born children continued to be higher than that among children born in the United States, it decreased by approximately 85%, most likely because of universal vaccination programs in the native countries of foreign-born children.20 Little difference in prevalence was found in foreign-born and native adults 50 years of age and older.
Clinical Outcomes
Acute Hepatitis B
The age at which a person becomes infected with HBV is a principal determinant of the clinical outcome. Perinatal exposure leads to the chronic HBV carrier state in as many as 95% of persons because of immunologic tolerance to the virus (see later). By contrast, children exposed during the first 5 years of life have a 30% chance of developing chronic HBV. Only 2% to 5% of adults with an intact immune system become chronically infected.21
Two thirds of patients with acute hepatitis B have an asymptomatic or subclinical illness that goes unrecognized. In the other one third, acute hepatitis, ranging from mild to moderate in severity, develops, with acute liver failure occurring in 1%. Although uncommon, hepatitis B accounts for 7% of all cases of acute liver failure and approximately 400 deaths annually in the United States.3,22 Rapid viral elimination may result in clearance of HBsAg from serum by the time of initial presentation. In these cases, the accurate diagnosis of acute hepatitis B may require testing with immunoglobulin (Ig) M anti-HBc (see later).
The rate of spontaneous survival in acute liver failure caused by HBV is only approximately 20%. Liver transplantation has resulted in survival rates of 50% to 60%. Recurrent disease in the allograft is uncommon because of administration of hepatitis B immune globulin (HBIG) and orally administered antiviral agents (see later and Chapter 97).
Chronic Hepatitis B
Progressive liver disease (including cirrhosis and HCC) can be expected to develop in one quarter to one third of people who acquire infection in the first few years of life. An estimated 15% to 25% of predominately middle-aged or older male patients with acquisition of infection early in life ultimately die of liver-related causes. Outcomes are related to host (age, male gender, genetic background, immune status) and viral (serum HBV DNA level, HBV genotype, mutation patterns) factors. HCC is 4 times as likely to develop in males as in females.
The presence of active viral replication and long-standing necroinflammatory liver disease caused by HBV strongly influences the rate of progression to cirrhosis. The major determinant of survival is the severity of the liver disease when the patient first comes to medical attention.23 Cirrhosis is associated with decreased survival and an increased frequency of HCC. Prior to the advent of antiviral therapy, 5- and 20-year survival rates of 55% and 25%, respectively, were reported in patients with HBV-related cirrhosis, compared with 97% and 63%, respectively, for those with mild (noncirrhotic) disease.24 In one study, an 84% 5-year survival rate was reported for patients with compensated HBV-related cirrhosis, compared with 14% for patients with cirrhosis complicated by ascites, jaundice, encephalopathy, or a history of variceal bleeding (see Chapters 74 and 92).25 Multivariate analyses in several large cohort studies have identified age, ascites, hyperbilirubinemia, and renal dysfunction as correlating independently with survival in patients with HBV-related cirrhosis. Therefore, early hepatic decompensation is an indication for antiviral therapy as well as assessment for liver transplantation (see later).
Clearance of HBsAg from serum in patients with HBV-related cirrhosis has been associated with an excellent prognosis, including improvement in liver histology and function, a decreased chance of viral reactivation, and improved long-term survival.23 HBsAg clearance, however, is not an absolute safeguard against the future development of HCC in persons who have preexisting cirrhosis.26
Virology
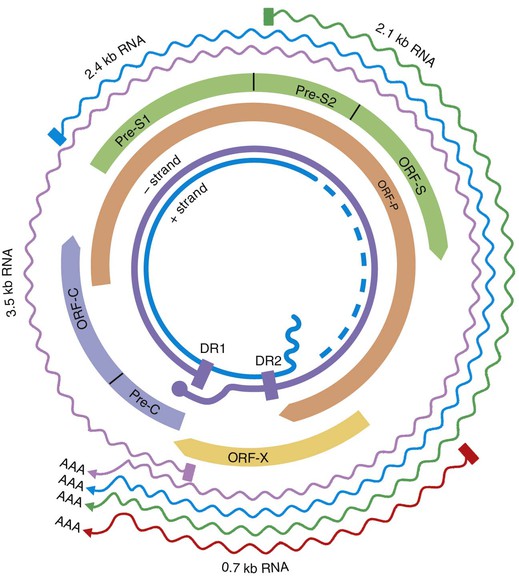
HBV is a small DNA virus that belongs to the Hepadnaviridae family. Other members of this virus family are human HBV-like agents that infect the woodchuck, ground and tree squirrels, woolly monkey, crane, heron, Ross goose, and duck. HBV is a small (3.2-kilobase [kb]) virus with a DNA genome that has a relaxed, circular, partially double-stranded configuration (Fig. 79-2). The genome is composed of 4 open reading frames (ORFs) and has a compact design in which several genes overlap and use the same DNA to encode different viral proteins. The 4 viral genes components include the core, surface, X, and polymerase genes. The core gene encodes the core nucleocapsid protein, which is important in viral packaging and production of HBeAg. The surface gene encodes the pre-S1, pre-S2, and S proteins (comprising the large [L], middle [M], and small [S] surface proteins). The X gene encodes the X protein, which has transactivating properties and may be important in hepatic carcinogenesis. The polymerase gene has a large ORF (≈800 amino acids) and overlaps the entire length of the surface ORF. It encodes a large protein with functions that are critical for packaging and DNA replication (including priming, RNA- and DNA-dependent DNA polymerase, and RNase H activities).
Viral Replication
Although HBV is a DNA virus, replication occurs through an RNA intermediate and requires an active viral reverse transcriptase/polymerase enzyme (Fig. 79-3). The mutation rate is higher for HBV than for other DNA viruses (an estimated 1013 to 1015 point mutations per day).27 Complete HBV genomic sequencing has identified a large number of mutations within the HBV genome, many of which are silent or do not alter the amino acid sequence of encoded proteins. Because of genomic overlap, however, some of the silent mutations in 1 ORF (e.g., the polymerase gene) may result in an amino acid substitution in an overlapping ORF (surface gene), although with uncertain clinical implications.
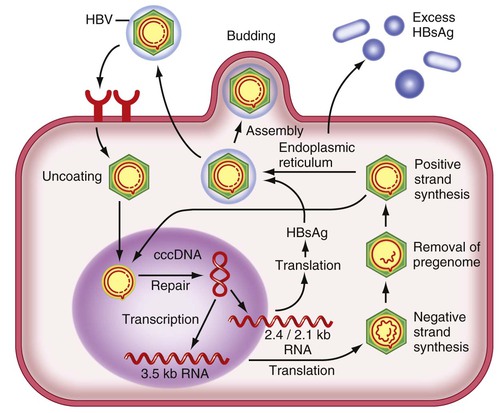
HBV replication begins with encapsidation of the pregenomic RNA through complex interactions between host and viral proteins. HBV DNA polymerase reverse transcribes the pregenomic RNA into a negative-strand HBV DNA, which in turn serves as the template for positive-strand synthesis to form a partially double-stranded genome. Concurrent with HBV DNA synthesis, the nucleocapsid undergoes maturation and, through an incompletely understood mechanism, interacts with the S protein to initiate viral assembly in the endoplasmic reticulum. S protein is synthesized in the endoplasmic reticulum, where monomer aggregates that exclude host membrane proteins subsequently bud into the lumen as subviral particles. When formed, HBsAg undergoes glycosylation in the endoplasmic reticulum and the Golgi apparatus. Noninfectious subviral particles (spherical and filamentous forms of HBsAg) are secreted in great abundance when compared with mature virions. These subviral HBsAg particles exceed virions in number by a variable factor of 102 to 105 and can accumulate up to concentrations of several hundred micrograms per milliliter of serum.28
Genotypes
A genetic classification based on comparisons of complete genomes has demonstrated 10 genotypes (designated A through J) and numerous subtypes of HBV (Box 79-1).29 These classifications are defined as a divergence in the entire HBV genomic sequence of 8% or more. Genotype A is the predominant genotype in northern Europe and the United States. Genotypes B and C are confined to populations in eastern Asia and the Far East, but changes in immigration patterns have resulted in an influx of Asian HBV carriers with these genotypes into the United States.30 Genotype D is found worldwide but is especially prevalent in the Mediterranean area, Middle East, and south Asia. Genotype E is indigenous to western sub-Saharan areas, and genotype F prevails in Central America. Cases of genotype G have been reported in the United States and France. Genotype H has been described in Mexico. Genotypes I and J are the most recently discovered and have been observed in Vietnam and the Ryukyu Islands in Japan, respectively.29
Clinical associations appear to exist with the various genotypes (see Box 79-1). The strongest clinical associations appear to be that (1) HBeAg seroconversion occurs earlier in patients with HBV genotype B than in those with genotype C, and (2) the response to therapy with interferon (IFN) is better with genotypes A and B than with C and D (see later).31 The viral genotype also has implications for the frequency of precore and core mutations (see later) and may have an effect on the frequency of HCC. There is no compelling evidence that genotypes affect the HBV DNA response to nucleoside analogs (see later).
The clinical associations with the various genotypes have not led to specific recommendations on routine testing because genotype classification does not generally lead to a difference in management. One exception to this rule, however, occurs when a patient is being considered for IFN therapy. In patients who are suitable candidates based on age and other factors (see later), genotype testing may have clinical value because genotypes A and B are associated with higher rates of a sustained virologic response and HBsAg clearance.32
Mutations
The vast majority of mutations in the HBV genome that are identified by comparing nucleotide sequences with those of wild-type HBV are silent or do not alter the amino acid sequence in a particular ORF. Some mutations have potentially important disease associations, however, and are described below.
Hepatitis B Surface Antigen Gene
HBsAg gene mutants result from a primary mutation in the HBsAg gene or a mutation in the overlapping DNA polymerase gene during nucleoside antiviral therapy (see later). Once the mutation appears, mutated virions can become selected immunologically as the dominant form of the virus.
Mutations in the HBsAg gene between amino acid positions 124 to 147 are potentially important because this region of the HBsAg gene includes the major “a” epitope that binds to neutralizing antibody to HBsAg (anti-HBs). The mutation can lead to failure to detect HBsAg by commercial assays, which depend on binding to anti-HBs, and to failure of neutralization by HBIG or of vaccination.
Infection with HBsAg gene-mutant HBV is accompanied by detection of anti-HBc. Serum HBV DNA levels can be as varied as they are in HBsAg carriers (see later).These mutants need to be distinguished from cases of “occult” hepatitis B, which has been linked to cryptogenic cirrhosis and an increased risk of HCC.33,34 In occult HBV infection, HBsAg-negative persons have detectable HBV DNA in serum.33 Some of these persons may lack evidence of other serologic markers of infection (e.g., anti-HBc). Occult HBV infection is considered to be due to active suppression of viral replication by the host immune system; as a result, when HBV DNA is detectable in serum, it is present in low levels (<200 IU/mL).
Large-scale vaccination programs in regions endemic for HBV have revealed a 2% to 3% frequency of vaccine-escape HBsAg mutants. It remains controversial whether or not these mutants will be further selected in the future, thereby leading to widespread vaccine failure, but most available evidence does not support this scenario. The importance of HBsAg gene mutants for HBIG failure is less controversial. Such escape mutants are detected in as many as 50% of HBIG-treated patients in whom recurrent HBV infection develops after liver transplantation, and the frequency of their occurrence appears to correlate with the length of time over which HBIG is repeatedly administered.35
Precore, Basal Core Promoter, and Core Genes
Mutations in the precore and basal core promoter regions of the HBV genome can influence the production of HBeAg. A precore mutation results in a stop codon at nucleotide 1896 that abolishes the synthesis of HBeAg,36 whereas mutations in the basal core promoter at nucleotides 1762 and 1764 decrease HBeAg synthesis by approximately 70% while maintaining pregenomic RNA levels.37 Both types of mutations have been observed in cases of severe hepatitis, which has been attributed to the loss of the immune-tolerizing effects of HBeAg antigen (see later). The presence of core promoter mutations has been linked to an increased risk of HCC, and a higher prevalence has been found in patients infected with HBV genotype C.29 Precore and basal core promoter mutants have been described in the same patients and are particularly common in Asian and European patients with chronic hepatitis B.38 A large serosurvey of HBV carriers residing in the United States has found that precore and core promoter mutations are common (frequencies of 27% and 44%, respectively). Both mutant forms of HBV were observed to occur far more commonly in HBeAg-negative patients (precore mutation in 38% of HBeAg-negative vs. 9% of HBeAg-positive patients; core promoter mutation in 51% vs. 36%).39 In addition to these mutations, upstream mutations in the core gene can influence immunologic responses to HBV. Core gene mutations have been shown to block recognition of HBV by cytotoxic T lymphocytes (CTLs), a key mode of viral clearance. Therefore, the mutations contribute to HBV immune escape and possibly influence the response to IFN.40 Core gene mutations within the immunodominant epitopes of the HBV nucleocapsid also can affect CD4+ T-cell reactivity.41
In patients with perinatally acquired chronic hepatitis B, a prolonged immune tolerant phase with minimal to absent hepatic necroinflammatory activity is typically seen for the first 20 to 30 years of HBV infection. Sequencing studies have shown stable core gene sequences during this phase. Precore mutations are also uncommon during this phase. Core gene mutations become more common as patients pass from the immune active phase and undergo HBeAg seroconversion, at which time a growing number of mutations are observed in the region of the core gene that includes many B- and T-cell epitopes. Both precore stop codon mutants and core gene mutants have been associated with a poor response to IFN therapy.
HBV DNA Polymerase
The polymerase gene encodes a DNA polymerase enzyme needed for encapsidation of viral RNA into core particles, conversion of the pregenomic viral RNA into a negative strand of viral DNA (reverse transcription), and conversion of this first HBV DNA strand into a second DNA strand of positive polarity. In general, the HBV reverse transcriptase function of the polymerase gene is highly conserved because major mutations that impair the efficiency of viral replication lead to selection pressure against such variants. As indicated earlier, HBV has low replication fidelity, however, meaning that it has a propensity to mispair nucleotide bases when it reverse transcribes viral RNA to DNA. HBV DNA polymerase also lacks any proofreading activity, so it cannot repair its mistakes. Therefore, when a nucleotide base is misplaced, it remains in the growing viral DNA strand as a base mutation, and the new HBV DNA genome has a different sequence from the original (wild-type) genome. The overall error rate of HBV DNA polymerase is estimated to be 1 per 10,000 nucleotides copied, which translates to the potential for 10 million base-pair errors per day in an infected person. All possible single-base mutations can be produced in a 24-hour period, although many such mutations will yield nonviable viruses.42
Single or double nucleotide substitutions alter the amino acid sequence in the reverse transcriptase domain of the HBV DNA polymerase enzyme, thereby decreasing binding of drugs to its active site on the enzyme. Mutations in the HBV polymerase gene can lead to clinically apparent resistance to nucleoside analog therapy whenever there is both decreased susceptibility to the antiviral drug and sufficient replication fitness to allow continued propagation in the expanding viral population (or “quasispecies”).
High levels of viral replication and high mutability of the virus allow the preexistence of single and even double polymerase mutants as a minor component of the viral quasispecies even before antiviral therapy is begun. Because of the limitations in sensitivity of currently available molecular assays (e.g., the line probe assay), these mutants are not detectable until they constitute at least 5% to 10% of the entire viral population. Ultradeep pyrosequencing is a research technique with the ability to detect HBV mutants that constitute less than 1% of the total population.43
Persistent infection with drug-resistant HBV has been associated with progression of disease and blunting of hepatic histologic improvement with antiviral therapy.44 Severe flares of hepatitis have also been reported after the emergence of drug-resistant mutants,45 and acquisition of these mutants may lead to rapidly progressive liver disease after liver transplantation.46 Horizontal transmission of these mutants also has been described.
Pathogenesis
HBV is generally not a cytopathic virus, and the severity of HBV-associated liver disease is considered to be related to the intensity of the host immunologic response to the virus. Whereas both cellular and humoral immune responses are needed for effective clearance and long-term protection against reinfection, the cellular immune response appears to be the arm principally involved in the pathogenesis of disease. The immunologic response to HBV encompasses both an innate, or nonspecific, response (e.g., natural killer cells and IFNs) and an adaptive immune response, including antibodies to viral antigens, HLA class II–restricted CD4+ T cells, and HLA class I–restricted CD8+ cytotoxic T cells (CTLs).47 Induction of the antigen-specific T-cell response is thought to occur in lymphoid organs, where the host T cells encounter viral peptide antigens (or epitopes) that are presented by antigen-presenting cells such as dendritic cells, B cells, and macrophages. This process results in the maturation and expansion of T cells that are specific for these viral epitopes and is followed by their migration to the liver, where they perform their effector function.
During acute HBV infection, most HBV DNA molecules are cleared rapidly from the liver via noncytopathic mechanisms mediated by cytokines that are released initially by cells of the innate immune system48 and later by liver-infiltrating HBV-specific CD8+ cells. Cell-mediated immune responses are efficient in self-limited infection because the responses are vigorous, multispecific, and oriented toward type 1 helper T-cell functions. Persons with chronic HBV infection, by contrast, exhibit infrequent, narrowly focused, and weak HBV-specific T-cell responses.49 In chronic hepatitis B, the majority of mononuclear cells in liver infiltrates of patients with chronic hepatitis B at any given time are non–antigen specific.50
CD8+ CTLs are thought to contribute to the disease process in the liver and result in apoptosis of infected hepatocytes. To be recognized by the CD8+ CTLs, targeted hepatocytes must present viral epitopes as short peptides that have been endogenously processed and fit within the peptide-binding groove of the class I major histocompatibility complex (MHC) molecules.51 The binding of the CTL T-cell receptor to the peptide-MHC complex on the hepatocyte surface can then result in the direct killing of the infected cell and release of potent antiviral cytokines by the activated CTL. Recognition by MHC class II–restricted CD4+ helper T cells requires the appropriate presentation of viral peptides in the context of class II MHC molecules. The CD4+ cells produce antiviral cytokines and provide help in neutralizing antibody production. Antibody neutralization limits intrahepatic spread of virus during primary infection and serves an important role in preventing reinfection.
Natural History
Four phases of HBV infection have been described: immune tolerance, immune clearance, the inactive carrier state, and reactivation (Fig. 79-4). These consecutive phases are much more likely to be apparent in patients with acquisition of chronic hepatitis B early in life.
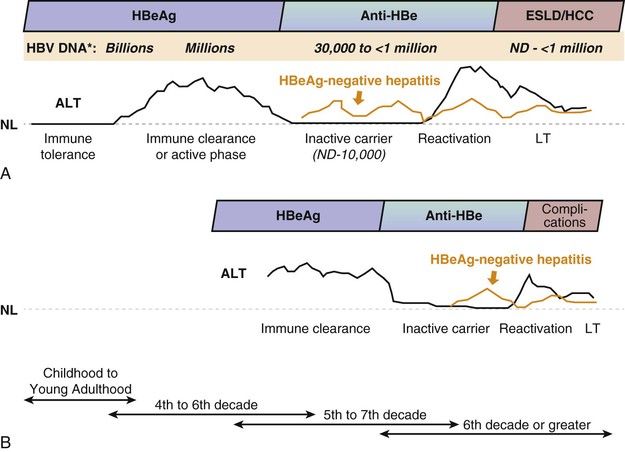
The immune tolerance phase is often the earliest phase to be recognized when there is a history of infection at birth or the first few years of life. It is characterized by HBeAg positivity, high levels of HBV DNA (≥107 IU/mL), low or normal levels of serum aminotransferases, and minimal or no necroinflammation or fibrosis in the liver. During this phase, the rates of HBeAg loss are low. Experiments in transgenic mice suggest that HBeAg induces a state of immunologic tolerance to HBV in neonates.52 Perinatal transmission of HBeAg is considered to be one of several potential mechanisms behind the immune-tolerance phase.53
The immune active phase often begins after several decades of HBV infection and is characterized by elevated serum aminotransferase levels, lower HBV DNA levels than in the immune tolerance phase, and histologic evidence of chronic hepatitis. The trigger mechanisms for this apparent immunologic activation against HBV are poorly understood, but CD8+ CTL-mediated lysis of infected hepatocytes has been shown to occur. The duration of the immune active phase varies and frequently is many years. Continued efforts by the host immune system against the virus may result in HBeAg seroconversion (loss of HBeAg with the development of anti-HBe in serum). The mean annual rate of spontaneous HBeAg seroconversion generally ranges from 8% to 15% in older children or adults with elevated serum ALT levels; however, the rate is considerably lower among Asian children and immunocompromised persons.23
HBeAg seroconversion does not always indicate quiescent disease, however. As many as 30% of persons who undergo HBeAg seroconversion enter into a subsequent phase of active disease that is caused by the selection of HBeAg-negative mutants (precore mutation, core promoter mutantion, or a combination of both).54 At least 50% of these persons demonstrate multiple fluctuations in HBV DNA and aminotransferase levels each year, and recognition of active disease and exclusion of the inactive HBsAg carrier state (see later) may require serial assessments of both serum HBV DNA and aminotransferase levels.
The majority of patients who undergo HBeAg seroconversion, however, enter into a third phase (inactive HBV carrier stage) that is characterized by normalization of serum ALT and low (<2000 IU/mL) or nondetectable serum HBV DNA levels (see Fig. 79-4). Over time, hepatic necroinflammation and fibrosis subside.55 The inactive HBsAg carrier phase may last a lifetime, but some patients ultimately develop reactivation, which may occur spontaneously because of a loss of immunologic control over viral replication or may be due to immunosuppressive drug therapy (see later). Reactivation is defined by the reappearance of high levels of HBV DNA in serum, with or without HBeAg seroreversion, and often a noticeable rise in serum ALT levels.
If the immune active phase of hepatitis B remains untreated, cirrhosis can be anticipated to develop in at least 20% of cases. Various factors have been determined to increase the risk of cirrhosis, and, of these, older age, male gender, the stage of fibrosis at presentation, and ongoing HBV replication are perhaps the most important clinically. Combined infection with HDV (see Chapter 81), HCV (see Chapter 80), or HIV, as well as concomitant alcohol abuse, has also been linked to a higher rate of development of cirrhosis and HCC.
When cirrhosis develops, 2 major complications may occur: hepatic decompensation and HCC. The estimated annual frequency of developing hepatic decompensation in HBV-associated cirrhosis is 5% to 8%, whereas that of HCC is 2% to 4%.56 Factors associated with an increased risk of HCC include male gender, age 45 years or greater, having a first-degree relative with HCC, the presence of cirrhosis, HBeAg positivity, reversion from anti-HBe to HBeAg positivity, and increased HBV DNA levels regardless of the HBeAg state.57 Some studies suggest that core promoter mutations (which are generally associated with higher levels of viral replication) and genotype C are additive risk factors for a high rate of eventual transformation to HCC.58 HCC can still develop in HBsAg-positive persons with none of the identified risk factors, but less frequently. In addition, HCC can occur in cirrhotic patients who have undergone HBsAg seroconversion, and all patients with cirrhosis need continued surveillance (Box 79-2 [see Chapter 96]).59
Serum ALT as a Surrogate Marker for Disease Activity
The serum ALT level has been used conventionally as a measure of disease activity in patients with chronic hepatitis B. A serum ALT level within the normal laboratory reference range, however, has been shown to be an imperfect surrogate marker for lack of disease activity. Clinical laboratories base their range of normal values on donors without known liver disease, but this population may include obese persons, alcohol consumers, and diabetics, each of whom tends to expand the supposedly upper limit of normal. In fact, all-cause and liver-related mortality is increased when the serum ALT level exceeds 20 U/L in women and 30 U/L in men, and these values should be accepted as the upper limits of the normal range (see Chapter 73).60 Studies in Asia and the United States have shown that as many as 20% to 30% of Asian HBV carriers with persistently normal serum ALT levels and serum HBV DNA levels over 2000 IU/mL (roughly equivalent to 10,000 copies/mL) have grade 2 or greater inflammation and stage 2 or greater fibrosis (on scales of 0 to 4) on a liver biopsy specimen.61 HBeAg-negative Asian HBV carriers with high-normal serum ALT levels by standard reference ranges tend to be older, have a greater frequency of serum HBV DNA levels in excess of 2000 IU/mL, and have a higher frequency of basal core promoter HBV mutations—all features that can be associated with adverse long-term outcomes.62 Therefore, liver biopsy or a noninvasive determination of hepatic fibrosis can be a useful tool to ensure that the severity of underlying liver disease is not underestimated in such persons (see Chapter 73).
HBV DNA Level and Long-Term Complications
Population-based Asian cohort studies have established that the serum HBV DNA level is the single best predictor of future progression to cirrhosis and HCC in HBV-infected persons.63,64 In the prospective REVEAL-HBV natural history cohort study, more than 3600 untreated HBsAg carriers from Taiwan were followed for more than 11 years. Of these, 60% were male, 40% were older than age 50, 85% were HBeAg negative, and 95% had normal serum ALT levels using standard reference ranges. The calculated relative risks for cirrhosis and HCC were shown to correlate with the level of HBV DNA on entry into the study when compared with a reference population of HBsAg carriers with undetectable serum HBV DNA by PCR assay.64 Even serum HBV DNA levels as low as 10,000 copies/mL (equivalent to 2000 IU/mL) were associated with a higher relative risk of cirrhosis and HCC. The relative risk was highest (hazard ratio of 10) in persons with a serum HBV DNA level that was greater than 100,000 copies/mL and intermediate (hazard ratio of 3.8) in persons in whom the serum HBV DNA level decreased spontaneously from greater than 100,000 copies/mL at the time of enrollment to less than 10,000 copies/mL at the last point of follow-up. These data can be interpreted to mean that both the duration and level of viremia are important risk factors for the development of HCC. The data also suggest that suppression of serum HBV DNA levels, whether spontaneously or as a result of antiviral therapy, lowers the risk of HCC.
Some authorities recommend that Asian men 50 years of age or older with serum HBV DNA levels 100,000 copies/mL or greater receive long-term therapy with a nucleoside (or nucleotide) analog to prevent HCC, even if serum ALT values are normal.65 Additional support for this recommendation can be found in a landmark study in which more than 600 Asian patients with advanced fibrosis and a serum HBV DNA level greater than 100,000 copies/mL were randomized in a ratio of 2 : 1 to active treatment with the nucleoside analog lamivudine or placebo.66 Disease progression and HCC occurred significantly more frequently in the group of patients randomized to placebo.
Clinical and Pathologic Features
Acute Hepatitis B
The incubation period of acute hepatitis B varies from a few weeks to 6 months (average, 60 to 90 days), depending on the amount of replicating virus in the inoculum. The disease may be more severe in patients coinfected with other hepatitis viruses and in those with established underlying liver disease.67 Acute infections are heralded by malaise, nausea, vomiting, and a serum sickness-like prodrome of fever, arthralgias or arthritis, and rash, which is most commonly maculopapular or urticarial, in 10% to 20% of patients. This prodrome results from circulating HBsAg–anti-HBs complexes that activate complement and are deposited in the synovium and walls of cutaneous blood vessels. These features generally abate before the manifestations of liver disease and peak serum aminotransferase elevations are observed. Jaundice develops in only about 30% of patients.
Clinical symptoms and jaundice generally disappear after 1 to 3 months. In general, elevated serum ALT levels and serum HBsAg titers decline and disappear together, and in approximately 80% of cases, HBsAg disappears by 12 weeks after the onset of illness. Persistence of HBsAg after 6 months implies development of a carrier state, with only a small likelihood of recovery during the next 6 to 12 months.
Serum aminotransferase levels of 1000 to 2000 U/L are typical during acute hepatitis B, with the ALT higher than the AST level. In patients with icteric hepatitis, the rise in serum bilirubin levels often lags behind the rise in ALT levels. The peak ALT level does not correlate with prognosis, and the prothrombin time (INR) is the best indicator of prognosis. If acute liver failure develops, patients usually present within 4 weeks of the onset of symptoms and have associated multiorgan dysfunction, coagulopathy, encephalopathy, and high mortality rates if they are not treated by prompt antiviral therapy and liver transplantation. Patients older than 40 years appear to be more susceptible than younger persons to “late-onset” liver failure, which occurs several months after the onset of acute symptoms and is associated with encephalopathy and renal dysfunction. The pathogenic mechanisms of this severe form of HBV-related hepatitis are poorly understood but are presumed to involve massive immune-mediated lysis of infected hepatocytes and possibly impaired regeneration of new hepatocytes (see Chapter 95).
Chronic Hepatitis B
A history of acute or symptomatic hepatitis is often lacking in patients with chronic HBV infection. When symptoms are present, fatigue tends to predominate over other constitutional symptoms, such as poor appetite and malaise. Patients may remain asymptomatic even during periods of reactivated hepatitis. In other instances, particularly when superimposed on cirrhosis, reactivation of HBV infection may be associated with frank jaundice and signs of liver failure.
Physical examination may be normal, or hepatosplenomegaly may be found. In decompensated cirrhosis, spider telangiectasias, jaundice, ascites, and peripheral edema are common. During exacerbations of disease, serum ALT levels may be as high as 1000 U/L or more, and the clinical and laboratory picture is indistinguishable from that of acute hepatitis B, including the presence in serum of IgM anti-HBc in some cases. Progression to cirrhosis should be suspected whenever hypersplenism, hypoalbuminemia (in the absence of nephropathy), or prolongation of the prothrombin time is found. The serum AST level is typically higher than the serum ALT level in patients with advanced cirrhosis.
Extrahepatic Manifestations
Although uncommon, extrahepatic syndromes can occur with acute or, more commonly, chronic hepatitis B and are important to recognize because they may occur without clinically apparent liver disease and can be mistaken for independent disease processes in other organ systems. The pathogenesis is not completely understood but likely involves an aberrant immunologic response to extrahepatic viral proteins.68 Many of the extrahepatic manifestations are observed in association with circulating immune complexes that activate serum complement. Serum complement levels are generally low, and antiviral therapy may be beneficial in reducing the amount of immunologically activating viral antigens.
Arthritis-Dermatitis
The dermatitis-arthritis prodromal manifestations of acute hepatitis B must be distinguished from inflammatory forms of arthritis, because glucocorticoid therapy, if mistakenly given to these patients, can lead to enhanced HBV replication, and abrupt withdrawal of these agents may be associated with a flare in disease activity.
Polyarteritis Nodosa
As many as 30% of patients with polyarteritis nodosa are infected with HBV, but the disorder develops in less than 1% of patients with chronic HBV infection. This association has been reported predominantly in North America and Europe and not observed in Asia, where HBV is acquired perinatally. Typical features include arthralgias, fever, rash, abdominal pain, renal disease, hypertension, mononeuritis multiplex, and central nervous system abnormalities. Plasmapheresis may be useful, and therapeutic responses have also been observed with antiviral agents, given alone or in combination with plasmapheresis.
Glomerulonephritis
Several types of glomerular lesions have been described in patients with chronic HBV infection; membranous glomerulonephritis and membranoproliferative glomerulonephritis are the most common.69 Renal biopsy specimens have demonstrated immune-complex deposition and cytoplasmic inclusions in the glomerular basement membrane. Nephrotic syndrome is the most common presentation of HBV-associated glomerulonephritis. The diagnosis requires the presence of immune-complex glomerulonephritis in a renal biopsy specimen and the demonstration of glomerular deposits of 1 or more HBV antigens, such as HBsAg, HBcAg, or HBeAg, by immunohistochemistry. The renal disease typically resolves in months to several years in children. Resolution may occur after HBeAg seroconversion. The natural history of HBV-related glomerulonephritis in adults has not been well defined, but several reports suggest that glomerular disease is often slowly and relentlessly progressive.70 Successful treatment has been accomplished with IFN-α and has been linked to long-term control of HBV replication.71 Therapy with nucleos(t)ide analogs has also resulted in improved renal function and diminished proteinuria.72
Cryoglobulinemia
Type II cryoglobulins consist of a polyclonal IgG and monoclonal IgM, whereas type III cryoglobulins contain polyclonal IgG and rheumatoid factor. Type II and type III cryoglobulinemia have been associated with hepatitis B, but the association is uncommon. Cryoglobulinemia may be associated with systemic vasculitis (purpura, arthralgias, peripheral neuropathy, and glomerulonephritis) but is often paucisymptomatic or asymptomatic. Nucleos(t)ide analog therapy has been used successfully to treat symptomatic cryoglobulinemia.73
Histopathologic Features
Chronic HBV infection is characterized by mononuclear cell infiltration in the portal tracts. Periportal inflammation often leads to the disruption of the limiting plate of hepatocytes (interface hepatitis), and inflammatory cells often can be seen at the interface between collagenous extensions from the portal tracts and liver parenchyma (referred to as active septa). During reactivated hepatitis B, lobular inflammation is more intense and reminiscent of that seen in acute viral hepatitis. Steatosis is not a feature of chronic hepatitis B, as it is of chronic hepatitis C.
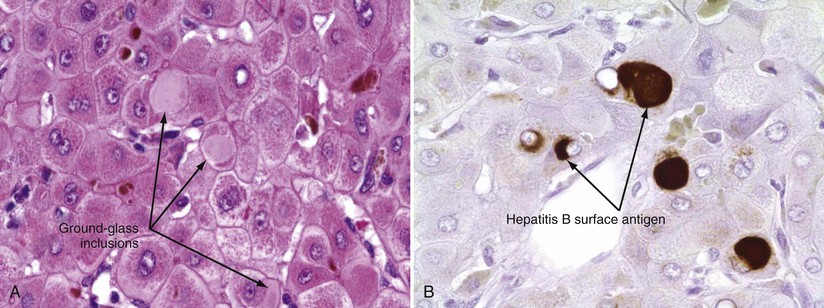
The only histologic feature noted on routine light microscopy that is specific for chronic hepatitis B is the presence of ground-glass hepatocytes (Fig. 79-5). This morphologic finding results from accumulation of HBsAg particles (20 to 30 nm in diameter) in the dilated endoplasmic reticulum. Because of high levels of cysteine in HBsAg, ground-glass cells have a high affinity for certain dyes, such as orcein, Victoria blue, and aldehyde fuchsin. Ground-glass hepatocytes may also be seen in HBV carriers, in whom they may be detected in up to 5% of cells. When present in abundance, ground-glass hepatocytes often indicate active viral replication.74 Immunofluorescence and electron microscopic studies have shown HBcAg inside the hepatocyte nuclei of affected cells. During periods of intense hepatitis activity, cytoplasmic core antigen staining is generally observed. After successful treatment of HBV infection with a nucleos(t)ide analog, the cytoplasmic core antigen staining often disappears, but nuclear core antigen staining due to persistence of the HBV cccDNA trancriptional template may remain.
Acute Flares
Chronic hepatitis B is often punctuated by sudden flares of disease activity that are characterized by a precipitous increase in serum aminotransferase levels. Although a uniform biochemical definition of a flare is lacking, it has frequently been described as an increase in serum ALT levels to at least 2 to 3 times the baseline value and at least 100 IU/mL. Flares are an important part of the natural history of hepatitis B because they can lead to histologic progression when they occur repeatedly and are moderate or severe. Acute flares in chronic hepatitis B occur in association with a number of circumstances and clinical situations (Table 79-1). Most flares are preceded by an increase in viral replication, which stimulates an enhanced cellular immune response that targets virus-infected hepatocytes. The mechanisms behind the increase in viral replication are unknown in many instances and are presumed to be due to weakening of immune control over viral replication or replication-fit viral mutants such as core promoter mutants or drug-resistant mutant HBV (see earlier). Irrespective of the cause of the increased viral replication, however, the biochemical abnormalities usually occur coincident to or immediately after an increase in serum HBV DNA levels.
TABLE 79-1
Causes of Hepatitis Flares in Patients with Chronic Hepatitis B
| Cause of Flare | Comment |
| Spontaneous | Factors that precipitate viral replication are unclear |
| Immunosuppressive therapy | Flares are often observed during withdrawal of the agent; preemptive antiviral therapy is required |
| Antiviral therapy for HBV | |
| Interferon | Flares are often observed during the second to third month of therapy in 30% of patients; may herald virologic response |
| Nucleoside analog | |
| During treatment | Flares are no more common than with placebo |
| Drug-resistant HBV | Severe consequences can occur in patients with advanced liver disease |
| On withdrawal | Flares are caused by the rapid re-emergence of wild-type HBV; severe consequences can occur in patients with advanced liver disease |
| HIV treatment | Flares can occur as a result of the direct toxicity of HAART or with immune reconstitution; HBV increases the risk of antiretroviral drug hepatotoxicity |
| Genotypic variation | |
| Precore and core promoter mutants | Fluctuations in serum ALT levels are common with precore mutants |
| Superinfection with other hepatitis viruses | May be associated with suppression of HBV replication |

Spontaneous Flares
Spontaneous flares have been observed in patients with HBeAg-positive chronic hepatitis B, in whom they occur in 5% to 10% of patients annually, and in those with HBeAg-negative chronic hepatitis B, in whom fluctuations of both serum HBV DNA and ALT levels are common. It is not clear if severe physical or emotional stress can weaken the immune system and lead to a secondary increase in viral replication.
In persons who acquire HBV infection early in life, flares become more common during adulthood, presumably because of a breakdown in immune tolerance to HBV.75 In this situation, the flares are almost certainly host derived rather than virally mediated, and although poorly understood, they are most likely the result of a change in the regulation of viral antigen-specific T cells.53
Immunosuppressive Therapy–Induced Flares
Reactivation of hepatitis B with flares of serum aminotransferase levels is a well-recognized complication of cytotoxic or immunosuppressive therapy, including conventional cancer chemotherapy and potent biologic response modifiers that are used to treat rheumatic, GI, and skin disorders.76 Although many drugs have been reported to induce HBV reactivation, they tend to fall into one of several classes of agents (Table 79-2). Suppression of the normal immunologic responses to HBV during therapy leads to enhanced viral replication and is thought to result in widespread infection of hepatocytes. On discontinuation of immunosuppressive medications, as occurs with cancer chemotherapy, immune competence is restored and infected hepatocytes are rapidly destroyed. In general, the more potent the immunosuppression, the higher the level of viral replication and, thus, the greater the potential for serious clinical consequences. Postmortem studies of liver tissue from patients with severe liver injury have documented sparse staining of viral antigens, suggesting that the patients were in an active state of immune clearance.77
TABLE 79-2
Drugs Known to Be Associated with Hepatitis B Reactivation*
| Category | Agent | Drug Class | Common Indications |
| Antineoplastic | |||
| Endocrinologic | |||
| Glucocorticoid | |||
| Immunomodulatory | |||
| Anti-rejection | |||
| Transplantation | |||

The literature provides ample evidence for HBV reactivation leading to severe hepatitis, death from acute liver failure, and delay or inability to continue treatment for the underlying disease. When reactivation occurs in the setting of cancer chemotherapy or systemic treatment for a severe autoimmune disorder, the patient may not be eligible for salvage liver transplantation. A growing body of evidence shows benefit to screening all patients in need of immunosuppressive drug therapy for HBsAg and anti-HBc and prophylactically treating HBsAg-positive patients with antiviral therapy.78 Whenever indicated, antiviral therapy should be started at least 1 week before the immunosuppressive drug therapy is initiated. Clinical outcomes are much better when prophylaxis is provided as compared with on-demand antiviral therapy after reactivation has become clinically apparent.78 In a prospective, randomized controlled study of lamivudine treatment of HBsAg-positive lymphoma patients undergoing chemotherapy, reactivated HBV infection developed in 53% of the “watchful-waiting” group but 0% of the prophylactically treated group.79 Nucleos(t)ide analog therapy should be given for 6 to 12 months after completion of therapy. Lamivudine can be used successfully when immunosuppressive therapy of finite duration is given.78 When immunosuppressive therapy is given indefinitely for conditions other than cancer or when the patient has a baseline serum HBV DNA level of 2000 IU/mL or more, a nucleos(t)ide analog with high antiviral potency and a high genetic barrier to resistance (e.g., entecavir, tenofovir) is recommended.
The vast majority of patients reported to have immunosuppressive therapy–induced HBV reactivation have been positive for HBsAg in serum before treatment, but some studies have described the reappearance of HBsAg in patients who were initially positive only for anti-HBs, anti-HBc, or both. This event is called reverse HBsAg seroconversion and is particularly apt to happen with bone marrow or hematopoietic stem cell transplantation or the use of rituximab, a B-cell depleting drug (see Chapter 35).80 Therefore, antiviral therapy is generally recommended for anti-HBc-positive patients in these clinical situations. A management dilemma exists when an HBsAg-negative/anti-HBc-positive patient is treated with immunosuppressive drug therapy in other circumstances, because adequate studies are lacking in these situations. Whether these patients are best managed by prophylactic antiviral treatment or serial HBV DNA monitoring with early treatment if there is an increase in the serum HBV DNA level of at least 1 log10 IU/mL is controversial.
There have been numerous reports of HBV reactivation after the use of TNF-α inhibitor therapy. TNF-α inhibits HBV replication in vitro, and all drugs that block this pro-inflammatory cytokine have been associated with HBV reactivation.81 In one large retrospective series from Spain, HBV reactivation occurred in 39% of HBsAg-positive patients and 5% of anti-HBc-positive patients given TNF-α inhibitors.81 Reactivation occurs considerably more frequently with infliximab than with adalimumab or etanercept, possibly owing to the high peak levels associated with infliximab, as well as its ability to induce complete elimination of TNF-α.82 The importance given to these drugs as a potential cause of reactivation cannot be overemphasized, because they are usually administered for prolonged periods, millions of patients are being treated with these agents in the United States alone, and they are used for a diverse number of medical conditions, including RA, ankylosing spondylitis, IBD, psoriasis, and several other autoimmune disorders.
Reactivated hepatitis B also occurs in patients who are given immunosuppressive medications to prevent organ transplant rejection. All HBsAg-positive and anti-HBc-positive transplant recipients should be considered to be at risk for HBV reactivation, with bone marrow and hematopoietic stem cell recipients having the highest level of risk (see Chapter 35). Post-transplantation treatment of organ rejection and graft-versus-host disease further contributes to this risk. In the setting of liver transplantation, indefinite nucleos(t)ide analog therapy is generally administered when grafts from anti-HBc-positive donors are given to previously nonexposed or anti-HBc-positive recipients.83 A few cases have been reported in which the use of anti-HBc-positive livers given to lamivudine-treated recipients resulted in de novo hepatitis due to lamivudine-resistant HBV mutants.84 Some experts routinely recommend the use of higher genetic barrier nucleos(t)ide analogs in the setting of anti-HBc-positive liver donation.
Antiviral Therapy–Induced Flares
Antiviral treatment of chronic hepatitis B can be associated with ALT increases and flares of hepatitis in several circumstances. Flares may occur during IFN or nucleos(t)ide analog therapy, after withdrawal of nucleos(t)ide analogs or glucocorticoid therapy, and in association with lamivudine-, adefovir-, entecavir-, or telbivudine-resistant mutants.
During Interferon Therapy.
IFN-induced flares of chronic hepatitis B occur in approximately one third of treated patients and result from the immunostimulatory properties of the drug. Flares occur with conventional and pegylated formulations of IFN (see later and Chapter 80) and have been reported to occur more frequently in patients infected with HBV genotype A than with other genotypes. This finding may explain the higher rate of sustained virologic response and HBsAg clearance in IFN-treated patients with genotype A infection. Serum ALT flares have been shown to be a predictor of sustained virologic response and may be especially important in achieving a sustained virologic remission in patients with a high level of viremia.85 Flares tend to be particularly common in patients who have decompensated liver disease, with rates as high as 50% reported in one series.86 Flares that occur in patients with advanced liver fibrosis have frequently been associated with clinical deterioration, and, as a result, IFN is generally considered to be contraindicated in patients with cirrhosis.
During Nucleos(t)ide Analog Therapy.
The registration studies for all nucleos(t)ide analogs have detected ALT flares during treatment in approximately 10% of patients, and these flares were no more common or severe than those occurring in nontreated patients. Whether a reduction in viral burden results in a transient restoration of immune competence is controversial, but the interaction does not appear to be clinically important. Aminotransferase increases are generally brief, even with continuation of therapy.
After Withdrawal of Nucleos(t)ide Analog.
Serum ALT flares occur in approximately 20% of patients after withdrawal of nucleos(t)ide analog therapy. These flares are thought to be caused by rapid resurgence of wild-type HBV, and although generally well tolerated, they too may be associated with serious clinical exacerbations in patients with advanced liver disease. Reinstitution of the original therapy is usually associated with a decline in HBV DNA levels.
During Antiretroviral Therapy.
Serum ALT flares occur in patients coinfected with HIV and HBV who receive HAART.87 The cause of these flares can be multifactorial. One of the most common causes is immunologic reconstitution due to the effectiveness of antiretroviral therapy.88 Patients with low CD4 counts before HAART therapy and high HBV DNA levels are often at greatest risk for this syndrome, and acute liver failure may occasionally result.89
Lamivudine resistance is less important as a cause of flares in HBV/HIV coinfected patients, because most patients in developed nations are now taking tenofovir or combination therapy with tenofovir and emtrictabine instead (see later). HBV infection increases the risk of hepatotoxicity from antiretroviral therapy, usually within 6 months after the initiation of treatment, and hepatotoxicity should be suspected if aminotransferase elevations occur despite an appropriate decline in HBV DNA levels. Affected HIV-infected patients may also be particularly susceptible to ALT flares because of a higher risk of infection with other hepatitis viruses.
Flares Associated with Genotypic Variation
Chronic infection with precore mutant HBV is often associated with periodic flares of liver cell necrosis interspersed with periods of normal serum ALT and low serum HBV DNA levels.54 These flares have been attributed to rises in the concentration of precore mutants in the liver and changes in the ratio of concentrations of precore to wild-type HBV.
Mutations at the basal core promoter region of the HBV genome are associated with increased histologic evidence of liver inflammation and viral replication.90 Multiple exacerbations of hepatitis resulting from reactivated HBV infection have been described in patients with basal core promoter mutations, either alone or in association with precore mutations.
Flares Caused by Infection with Other Viruses
Patients with chronic HBV infection may exhibit severe flares in serum aminotransferase levels and even frank liver failure when superinfected with another hepatotropic virus, such as HAV, HCV, or HDV. Increased mortality has been reported when HDV superinfection is superimposed on chronic hepatitis B, and chronic HDV infection is often associated with multiple fluctuations in serum aminotransferase levels (see Chapter 81).
Acute hepatitis C superimposed on chronic hepatitis B has been reported to be as clinically severe as HDV superinfection and has been associated with high rates of liver failure (34%) and death (10%).91 In a study involving 240 Chinese HBV carriers, those who became superinfected with HEV had a significantly higher rate of complications, including liver failure and death (33% vs. 2%), when compared to those who became superinfected with HAV.92 Because a vaccine for HEV is not commercially available, these data underscore the importance of clinical vigilance for acute hepatitis E in the right setting and consideration for early treatment with IFN if there is serologic confirmation of infection in an established HBV carrier (see Chapter 82).
Diagnosis
HBsAg appears in serum 2 to 10 weeks after exposure to HBV and before the onset of symptoms or elevation of serum aminotransferase levels. In self-limited acute hepatitis, HBsAg usually becomes undetectable after 4 to 6 months. Persistence of HBsAg for more than 6 months implies evolution to chronic HBV infection.
The disappearance of HBsAg is followed several weeks later by the appearance of anti-HBs. In most patients, anti-HBs persists for life and provides long-term immunity. In some patients, anti-HBs may not become detectable after disappearance of HBsAg, but these patients do not appear to be susceptible to recurrent HBV infection.93 Anti-HBs may not be detectable during a window period of several weeks to months after the disappearance of HBsAg. During this period, the diagnosis of acute HBV infection is made by the detection of IgM anti-HBc in serum.
Coexistence of HBsAg and anti-HBs in serum has been reported in approximately 10% to 20% of HBV carriers. The mechanisms of this finding are not clear but most likely relate to antibody formed against minor variants of the HBsAg protein. The presence of these heterotypic antibodies is not associated with specific risk factors or changes in clinical course and may occur in patients with or without active liver disease and viral replication.
Anti-HBc is detectable in acute and chronic HBV infection. During acute infection, anti-HBc is predominantly of the IgM class and is usually detectable for 4 to 6 months after an acute episode of hepatitis and rarely for up to 2 years. IgM anti-HBc may become detectable during exacerbations of chronic hepatitis B and is often used as a surrogate for active viral replication. Anti-HBc of the IgG class is found in persons who recover from acute hepatitis B and also is the form found in those who progress to chronic infection.
In low endemic areas of the world such as the United States, isolated anti-HBc in serum has been detected in 1% to 4% of the general population. Less than 5% of these patients can be anticipated to have HBV DNA detectable in serum and therefore occult viremia.94 By contrast, isolated anti-HBc may be found in more than 50% of patients in highly endemic regions of the world, and 10% to 30% of patients with this finding may have HBV DNA detectable in serum.95,96 Isolated reactivity for anti-HBc may occur in a number of other clinical situations also (Table 79-3). Perhaps the most clinically important to recognize is a false-positive test result, which is usually very weakly reactive and may not be reproducible. Failure to appreciate this possibility in patients who have no apparent risk of exposure to HBV may result in needless consultation, inappropriate exclusion from vaccination, and, unfortunately, rejection of the person from blood or organ donation. Such individuals often have a primary rather than anamnestic response to HBV vaccination.
TABLE 79-3
Possible Interpretations of an Isolated Positive Test Result for Antibody to Hepatitis B Core Antigen
| Interpretation | Comments |
| Remote infection | Most common cause; particularly common in endemic areas of the world where acquisition of infection early in life is frequent. Patients may have HBV DNA detectable in serum |
| False-positive result | Weakly positive. May be nonreproducible in a different clinical laboratory |
| Window period of acute hepatitis B | Seen relatively infrequently due to increased sensitivity of current anti-HBs assays |
| Occult infection | HBV DNA detectable in serum and liver tissue |
HBeAg is a viral protein that is found in serum early during acute HBV infection. HBeAg reactivity usually disappears at the time of or soon after the peak in serum aminotransferase levels, and persistence of HBeAg 3 or more months after the onset of illness indicates a high likelihood of transition to chronic HBV infection. The finding of HBeAg in the serum of an HBsAg-positive carrier indicates a high level of viral replication and greater infectivity for intimate contacts. With a commercially available PCR assay, nearly 90% of patients with HBeAg-positive chronic hepatitis B have been found to have serum HBV DNA levels persistently above 105 copies/mL, with a mean value of 8.37 log10 (>108 copies/mL).97 Serum HBV DNA values can be as high as 1012-13 during the immune tolerance phase. By contrast, anti–HBe-positive patients have much lower serum HBV DNA levels (105 to 108 copies/mL), with the highest values being found in those with persistently or intermittently elevated serum ALT levels.
HBV DNA can be measured in serum with qualitative or quantitative assays. Most clinical laboratories in the United States use a quantitative PCR method that is capable of detecting less than 400 copies/mL. The Cobas TaqMan (Roche Molecular Diagnostics, Pleasanton, Calif.) is commonly used and can reliably determine HBV DNA levels over a linear range from 25 IU/mL (≈100 copies/mL) to 108 IU/mL. A number of non–PCR-based assays are available, but they are less useful clinically.
The quantification of serum HBV DNA is commonly used to evaluate a patient's candidacy for antiviral therapy and to monitor response during treatment. Patients with a high serum HBV DNA level (>109 copies/mL) at baseline respond less commonly to therapy with IFN than do those with lower levels.98 By contrast, baseline serum HBV DNA levels have not been shown to correlate with response to nucleos(t)ide analog therapy because of the more potent inhibition of viral replication by these agents. Monitoring of HBV DNA levels at key intervals such as 12 and 24 weeks of therapy allows one to predict the likelihood of HBeAg clearance with both pegylated IFN and nucleos(t)ide analog therapy.99,100 In the past, reappearance of HBV DNA in serum during treatment predominantly suggested that drug resistance had occurred.101 Such is not the case, however, with high-genetic-barrier nucleos(t)ide analog therapy during which the reemergence of HBV DNA often indicates poor adherence to therapy.102
Qualitative PCR has been reported to be an even more sensitive method than quantitative PCR for detecting HBV DNA. Use of qualitative PCR has altered traditional concepts about the clearance of HBV DNA from serum in acute and chronic HBV infection. For example, small amounts of HBV DNA can be detected in serum and peripheral mononuclear cells years after recovery from acute or chronic hepatitis B.103 Qualitative PCR for HBV DNA has a relatively limited role in clinical decision making, however, and suffers from poor standardization of assay techniques.
Assays for quantification of HBsAg have become commercially available and are licensed in Europe and Asia (Architect Assay, Abbott Diagnostics, Chicago, Ill.; Elecys II assay, Roche Molecular Diagnostics). These assays are available for research purposes in the United States and are likely to become commercially available. Clinical trials in both HBeAg-positive and HBeAg-negative patients have demonstrated a rapid decline in HBsAg concentration during IFN therapy and a much slower decline during the first few years of nucleos(t)ide analog therapy. In HBeAg-negative hepatitis B, a decrease in HBsAg concentration of 0.5 log at week 12 or 1 log at week 24 of IFN therapy has been found to provide better prediction of a sustained virologic response than does the decline in serum HBV DNA levels.104 High negative predictive values at week 12 (>90%) have led to proposal of a stopping rule that would avoid needless extension of IFN therapy and lead to initiation of a different treatment regimen. The data are less clear for patients with HBeAg-positive chronic hepatitis B. Although the HBsAg kinetic data provide promising insights, there is no consensus on how serial measurement of HBsAg should be used. The rate of decline of HBsAg concentration has been found to vary according to HBV genotype, and the proposed stopping rule is unlikely to be applicable to all genotypes.105
Treatment
Seven drugs are approved for the treatment of chronic hepatitis B. Five of these agents are nucleos(t)ide analogs that suppress HBV replication through an inhibitory effect on the viral DNA polymerase. Nucleos(t)ide analogs have excellent oral bioavailability and a good safety record and are far more potent inhibitors of viral replication than IFN-α. They have proved to be particularly useful in the management of patients with decompensated cirrhosis, because even small doses of IFN can lead to worsening liver failure and severe infections. IFN therapy is used much less frequently because of its adverse effects, but continued interest in this agent stems from the fact that it is both immunostimulatory as well as antiviral. Moreover, IFN induces a more rapid decline in HBsAg concentration and a higher rate of HBsAg clearance.
Goals
The primary treatment goals for patients with chronic hepatitis B are to forestall progression of liver disease, prevent late complications (cirrhosis, liver failure, and HCC), and increase survival. All of these objectives are achievable with long-term suppression of viral replication with either IFN-α or nucleos(t)ide analogs. Unfortunately, less than 10% of patients with potentially treatable chronic hepatitis B are estimated to be given antiviral therapy. Missed opportunities for HBV screening and poor appreciation of the indications for treatment are the major impediments.106,107
Cultural Barriers
More than two thirds of immigrant populations with chronic hepatitis B were born in areas of the world where hepatitis B is highly endemic. Many of these persons do not seek health care, and the possibility of hepatitis B is often not explored when they do.
Significant cultural barriers to the effective management of these patients exist. Appreciation of these barriers is critical because the potential impact on future health and financial resources needed to care for late complications of hepatitis B are immense. One of the greatest barriers to acceptance of antiviral therapy is the limited proficiency in English language skills that leads to isolation and may negatively influence government support to an individual or community. Asian immigrants, for example, often own small businesses or have jobs in which insurance is not provided, thus preventing them from having access to care. If they are able to obtain medical care, the health care provider often is not able to communicate with first-generation immigrants in their native tongues, further leading to distrust, confusion, and embarrassment. Cultural, religious, and stigmatizing beliefs may impede care. These barriers may be overcome, however, with sensitivity on the part of the care provider. Whenever possible, the provider should arrange for a translator or seek the assistance of someone who speaks the patient's native language.108
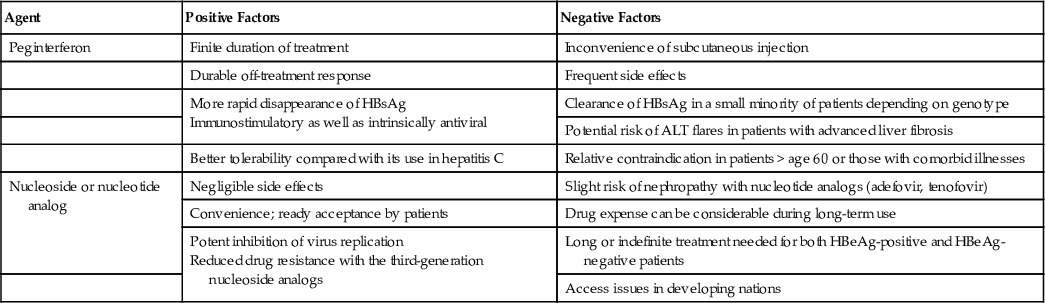
Choice of Agent
In deciding on the appropriate type of therapy for a patient with chronic hepatitis B, the physician should consider the serum ALT level, serum HBV DNA level, and liver histology, if available, as well as the expense of treatment, potential for adverse events, age of the patient, and presence of other comorbid conditions (Table 79-4). Moreover, before starting antiviral therapy, the patient should be committed to having serial blood samples and assessments.
TABLE 79-4
Positive and Negative Factors to Consider in the Decision to Treat Hepatitis B with Peginterferon or a Nucleoside or Nucleotide Analog
| Agent | Positive Factors | Negative Factors |
| Peginterferon | Finite duration of treatment | Inconvenience of subcutaneous injection |
| Durable off-treatment response | Frequent side effects | |
More rapid disappearance of HBsAg Immunostimulatory as well as intrinsically antiviral | Clearance of HBsAg in a small minority of patients depending on genotype | |
| Potential risk of ALT flares in patients with advanced liver fibrosis | ||
| Better tolerability compared with its use in hepatitis C | Relative contraindication in patients > age 60 or those with comorbid illnesses | |
| Nucleoside or nucleotide analog | Negligible side effects | Slight risk of nephropathy with nucleotide analogs (adefovir, tenofovir) |
| Convenience; ready acceptance by patients | Drug expense can be considerable during long-term use | |
Potent inhibition of virus replication Reduced drug resistance with the third-generation nucleoside analogs | Long or indefinite treatment needed for both HBeAg-positive and HBeAg-negative patients | |
| Access issues in developing nations |
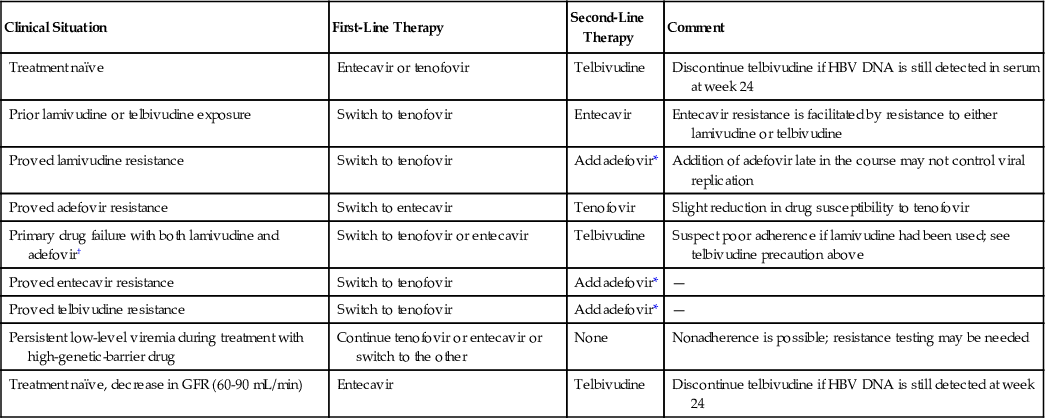
The latest generation of nucleos(t)ide analogs such as tenofovir and entecavir have a high genetic barrier to resistance and, therefore, can be used as monotherapy. Accordingly, these agents are generally preferred as first-line treatment when available (Table 79-5). In emerging nations, lamivudine and adefovir are often used as first-line therapy owing to financial constraints and limited availability of the newer oral agents.
TABLE 79-5
Choice of Nucleoside or Nucleotide Analog for HBV Infection
| Clinical Situation | First-Line Therapy | Second-Line Therapy | Comment |
| Treatment naïve | Entecavir or tenofovir | Telbivudine | Discontinue telbivudine if HBV DNA is still detected in serum at week 24 |
| Prior lamivudine or telbivudine exposure | Switch to tenofovir | Entecavir | Entecavir resistance is facilitated by resistance to either lamivudine or telbivudine |
| Proved lamivudine resistance | Switch to tenofovir | Add adefovir* | Addition of adefovir late in the course may not control viral replication |
| Proved adefovir resistance | Switch to entecavir | Tenofovir | Slight reduction in drug susceptibility to tenofovir |
| Primary drug failure with both lamivudine and adefovir† | Switch to tenofovir or entecavir | Telbivudine | Suspect poor adherence if lamivudine had been used; see telbivudine precaution above |
| Proved entecavir resistance | Switch to tenofovir | Add adefovir* | — |
| Proved telbivudine resistance | Switch to tenofovir | Add adefovir* | — |
| Persistent low-level viremia during treatment with high-genetic-barrier drug | Continue tenofovir or entecavir or switch to the other | None | Nonadherence is possible; resistance testing may be needed |
| Treatment naïve, decrease in GFR (60-90 mL/min) | Entecavir | Telbivudine | Discontinue telbivudine if HBV DNA is still detected at week 24 |
IFN and nucleos(t)ide analogs each have advantages and disadvantages that should be considered when making a treatment decision (see Table 79-4). One major advantage of IFN is that treatment is limited to 1 year or less, and virologic responses tend to be quite durable, especially in patients with HBeAg-positive hepatitis B. The shorter time required for treatment may be an important factor for some patients.
Definitions of Response
Phase 3 drug registration trials for nucleos(t)ide analogs utilized predefined biochemical, virologic, and histologic end points to evaluate the response to treatment. Biochemical response required normalization of serum ALT levels, virologic response required a sustained disappearance of HBV DNA from serum for at least 6 months after treatment, and histologic response required a 2-point or greater improvement in the necroflammatory score without worsening fibrosis. HBeAg seroconversion to anti-HBe, rather than HBeAg disappearance alone, was used as an additional virologic end point to minimize the chance of virologic relapse.
Some of the large clinical trials of IFN used a virologic end point of HBeAg loss rather than HBeAg seroconversion, and the persistent detection of low levels of serum HBV DNA (<2000 IU/mL) was allowed in the definition of a sustained virologic response, because nondetectable HBV DNA occurs less frequently with IFN-based therapy than with nucleos(t)ide therapy. These less stringent requirements did not preclude a durable response to IFN in approximately 80% of HBeAg-positive patients when evaluated years later.109 This durability has been hypothesized to be due to a continued immunoregulatory effect of IFN after treatment discontinuation. A sustained decline of serum HBV DNA levels to less than 2000 IU/mL was also included as a primary end point in many of the IFN trials in patients with HBeAg-negative hepatitis B. Long-term follow-up of these patients has indicated a higher rate of relapse when compared with HBeAg-positive patients, but sustained responses have been reported in 30% of the HBeAg-negative patients.
Irrespective of the type of antiviral therapy used, the best assurance that late relapses will not occur is provided by the disappearance of HBsAg, with or without seroconversion to anti-HBs; this occurrence comes closest to a clinical cure of hepatitis B. Unfortunately, this more stringent end point occurs uncommonly with IFN and is even more rarely achieved with nucleos(t)ide analog therapy despite the greater antiviral potency of the latter.
Nucleoside and Nucleotide Analogs
The vast majority of previously untreated (“treatment-naïve”) patients are treated with 1 or more nucleos(t)ide analogs rather than IFN. Between 70% and 85% of HBeAg-positive patients will become HBV DNA negative during the first year of treatment.110 The rate is as high as 85% to 95% after 2 years of therapy when drugs associated with a high genetic barrier to resistance are used. A small group of patients may still have HBV DNA detectable in serum after several years of therapy with high-genetic-barrier nucleos(t)ide analogs even though the clinical and biochemical response persists. The reason for the persistence of serum HBV DNA in these patients is not well understood, but the HBV DNA level is almost always below 2000 IU/mL, and drug-resistant mutants have not been demonstrated in this situation; most experts recommend continuing treatment with the same drug with a high genetic barrier to resistance.
The lack of side effects and excellent resistance profiles of the orally available antiviral agents are especially important properties because HBeAg seroconversion occurs slowly and often requires treatment for years or for an indefinite duration. Despite a rapid decline in serum HBV DNA levels on nucleos(t)ide analog therapy, only 20% to 25% of treated patients achieve HBeAg seroconversion after 1 year of treatment. The rate rises to 40% after 5 years of continuous treatment provided that the patient remains adherent and viral resistance does not occur.111 Long-term or maintenance therapy is particularly likely to be necessary in patients with HBeAg-negative hepatitis B because relatively high rates of relapse have been demonstrated after drug withdrawal despite years of treatment during which HBV DNA has remained nondetectable.
From 15% to 20% of patients with HBeAg-positive hepatitis B have a virologic relapse, including reappearance of HBeAg as well as HBV DNA, within 1 year of treatment discontinuation. Continuation of nucleos(t)ide analog therapy for several months beyond the period when HBeAg seroconversion is first detected diminishes the chance of relapse.112 Treatment for a minimum of 6 months after HBeAg seroconversion has been recommended by all the international liver societies. Even when this practice has been followed, however, some studies have reported relapse rates as high as 30% to 50% in Asian patients with HBeAg-positive chronic hepatitis B.113 The reasons for differences in relapse rates in various studies are not clear, but interstudy differences in HBV genotype and patient-related factors, such as the duration of infection, may be potential explanations.
Lamivudine
Owing to a high rate of resistance, lamivudine is no longer recommended as first-line therapy except in persons who require only short-term therapy, such as patients undergoing cancer chemotherapy. Prolonged lamivudine resistance has been associated with a blunted histologic response and more frequent hepatitis flares. The drug is still used widely as first-line therapy in developing countries. Before starting antiviral therapy in patients born in endemic areas for hepatitis B, care should be taken to inquire if antiviral therapy had ever been taken previously. In such patients, it may be best not to use entecavir because of the high likelihood that the patient had been exposed to lamivudine, prior exposure to which may predispose the patient to entecavir resistance (see later).
Adefovir Dipivoxil
Adefovir was licensed by the FDA a few years after lamivudine became available and was used frequently because it was effective against both wild-type and lamivudine-resistant HBV. Because of its limited potency, however, primary treatment failure (<1 log decline in the serum HBV DNA level at week 12) was observed in 30% of patients. Resistance to this drug due to 1 or 2 nucleotide substitutions in HBV DNA was documented in nearly 30% of patients by the end of 5 years of continuous therapy.114
Although adefovir use in the United States has largely been supplanted by tenofovir (see later), adefovir is still used widely outside of this country as primary therapy or whenever lamivudine resistance has emerged. In such cases, it is often used in combination with lamivudine in patients with advanced disease, because lamivudine is capable of suppressing adefovir-resistant mutants. Adefovir has the disadvantage of potential nephrotoxicity and should not be used in patients with preexisting compromised renal function.
Entecavir
Entecavir is a nucleoside analog that is more potent than lamivudine or adefovir and has a much higher genetic barrier to resistance, requiring an additional DNA polymerase mutation superimposed on the backbone of preexisting lamivudine-resistant mutations. This situation is rare in treatment-naïve patients, thus explaining the fact that resistance has been found in only 2% of treatment-naïve patients during 5 years of continuous treatment. In phase 3 clinical trials, an entecavir dose of 0.5 mg was used in treatment-naïve HBeAg-positive and HBeAg-negative patients, whereas 1 mg was used in patients who were resistant to lamivudine. The latter strategy is not recommended any longer because of a shortened time to acquisition of entecavir resistance in lamivudine-resistant patients115; tenofovir should be used instead (see later). The benefits of long-term use of entecavir include progressive regression of fibrosis, reversal of the features of cirrhosis, and a decreased incidence of HCC.116,117
Telbivudine
Telbivudine is a nucleoside analog that has been shown to be more potent than lamivudine in both HBeAg-positive and HBeAg-negative patients. Unfortunately, after 2 years of treatment, genotypic resistance was found in 25% of HBeAg-positive patients and 11% of HBeAg-negative patients, respectively. The highest rates of virologic response, and, conversely, the lowest rates of resistance, were found in patients who achieved nondetectable HBV DNA levels at 24 weeks of treatment. This drug shares cross resistance with lamivudine and should never be used as replacement therapy in lamivudine-resistant patients.
Telbivudine has fallen out of favor as a first-line treatment for HBV infection in the United States because of an unacceptably high rate of resistance when used as monotherapy. Resistance can be avoided in most instances, however, by careful attention to patient selection. The package insert states that this drug is only indicated in HBeAg-positive patients with a baseline serum HBV DNA level less than 109 copies/mL and a baseline serum ALT level greater than 2-fold elevated, and patients who are HBeAg-negative should have a serum HBV DNA level less than 107 copies/mL. Moreover, the drug should be discontinued if HBV DNA is still detected at week 24 of treatment. These recommendations on patient selection and early discontinuation of treatment are based on observations from the pivotal registration trial in which meeting these thresholds was predictive of low resistance and high virologic response. For example, if HBV DNA is not detectable at week 24 of treatment, telbivudine can be continued because drug resistance is much less common (3% to 4%) after 2 years of treatment.
Data collected on more than 500 HBeAg-positive and HBeAg-negative patients with chronic hepatitis B have revealed an increase in glomerular filtration rate (GFR) of 14.9 mL/min from baseline to 4 years of treatment. Improvement in renal function was observed in the first year of treatment and was sustained during subsequent years. Most of the improvement was confined to patients with minimal renal impairment (GFR of 60 to 90 mL/min) at baseline, and the renal functional improvement was not related to the degree of viral suppression.118 Therefore, the drug may be a safer alternative to adefovir or tenofovir in patients with preexisting renal impairment or those who are predisposed to this risk (e.g., obese patients with diabetes mellitus and hypertension).
Tenofovir Disoproxil Fumarate
Tenofovir is similar chemically to adefovir, but it has significantly more antiviral potency. Although head-to-head comparisons are not available, tenofovir may have slightly greater antiviral potency than entecavir. It is associated with a few safety issues, however, that are not found with entecavir. Tenofovir therapy has rarely been associated with decreased bone density in HIV-infected patients. As with adefovir, renal tubular damage and Fanconi syndrome have been observed in rare instances, and the elderly or persons with preexisting mild renal disease may be at particular risk.
Remarkably, resistance to tenofovir has not occurred after 5 years of treatment in either HBeAg-positive or HBeAg-negative patients. In a study that included more than 300 patients, 240 weeks of tenofovir-based therapy was associated with significant histologic regression, including reversal of cirrhosis in 74% of patients with pretreatment cirrhosis.111 High serum HBV DNA levels (>109 copies/mL), previously considered a treatment challenge when resistance-prone antiviral agents are used, are suppressed to undetectable levels in more than 90% of patients during long-term treatment.111,119 Because of its high genetic barrier to resistance and strong antiviral potency, tenofovir is used frequently as first-line therapy, particularly in cases of heavy prior exposure to lamivudine, known lamivudine resistance, or suboptimal response to adefovir.
Emtricitabine
Structurally similar to lamivudine, emtricitabine also inhibits HBV DNA polymerase and HIV reverse transcriptase. It is not FDA approved for use in hepatitis B, either alone or in a combined tablet with tenofovir that is commonly used for HIV infection. Because of its structural similarity to lamivudine, it shares the same resistance profile.
HBV DNA Monitoring
Patients who undergo treatment with a nucleos(t)ide analog should be followed with serial ALT and HBV DNA assessments at 4-month intervals during the first year of treatment.100 Once the HBV DNA level is nondetectable or less than 2000 IU/mL, follow-up intervals can be extended to every 6 months provided that the patient is taking a drug with a high genetic barrier to resistance and has demonstrated adherence to therapy.
As mentioned earlier, not all patients have nondetectable HBV DNA within the first several years of treatment even with drugs that have a high genetic barrier to resistance. In such cases, continuing the antiviral agent is reasonable, with the expectation of an eventual response or maintenance of virologic remission. In cases in which low-level viremia persists during maintenance treatment with a low-genetic-barrier drug such as lamivudine, adefovir, or telbivudine, switching to tenofovir is most appropriate. Entecavir is an equally suitable alternative if the patient fails to clear HBV DNA on adefovir (see Table 79-5).
Drug Failure
Primary drug failure at 12 weeks of treatment or a 1-log or greater increase in HBV DNA level (virologic breakthrough) is rarely seen with entecavir and tenofovir and usually indicates that the patient is not taking the drug appropriately. Based on a database of prescription utilization, at least 10% to 15% of patients fail to take their medication appropriately and miss 1 or more doses each month.120 Patients who experience early treatment failure or virologic breakthrough at any time should be questioned carefully in a culturally sensitive manner about drug adherence, and treatment goals should be reinforced.
Virologic breakthrough can also be due to drug resistance with low-genetic-barrier nucleos(t)ide analogs. Genotypic resistance is the term that describes the finding of a nucleotide substitution in the HBV DNA polymerase gene that has been associated with clinical evidence of drug resistance. Mutations that have commonly been associated with antiviral drug resistance can be detected by a commercially available reverse hybridization assay (InnoLipa, Innogenetics, Belgium). This method can only detect HBV mutants that have been proved to be associated with drug resistance. Furthermore, to be detectable, the drug-resistant mutant HBV has to constitute at least 10% of the viral population. Ultradeep pyrosequencing is a research method for detecting minor HBV variants that constitute less than 1% of the total HBV quasispecies.121 As a result of its greatly enhanced sensitivity, this method may ultimately provide clues as to why some adherent patients fail to become negative for serum HBV DNA after several years of treatment.
If a patient is unknowingly continued on the same therapy after genotypic resistance is detected, virologic rebound (defined as an increase in serum HBV DNA levels to >100,000 copies, or 20,000 IU/mL) will follow, and subsequently the serum ALT level will become elevated. Rescue therapy can modify this sequence of events if the patient is switched to a second agent that lacks cross resistance to the original drug (see Table 79-5). Preferably, the second agent should be a high-genetic-barrier drug. In clinical practice, tenofovir monotherapy has been used successfully in cases of lamivudine, adefovir, or entecavir resistance.122 Entecavir could be used for adefovir resistance. When neither tenofovir nor entecavir is available, another alternative would be to add a second low-genetic-barrier nucleos(t)ide analog that is effective against the original drug-resistant mutant. The disadvantage of this approach is that combined therapy is more cumbersome and expensive than switching to a high-genetic-barrier drug.
Interferon
Both standard and pegylated forms of IFN-α are available. They have similar immunologic effects, but pegylated IFN has greater antiviral potency and is better tolerated.123 Pegylated IFN is used more frequently worldwide. The FDA-approved duration of therapy is 48 weeks at a dose of 180 µg for both HBeAg-positive and HBeAg-negative chronic hepatitis B.
Predictors of response are the same for both types of IFN. Patients with baseline ALT values of at least 2 to 3 times the upper limit of normal, an HBV DNA level less than 109 IU/mL, and genotype A or B respond more frequently than patients without these features. Because ALT flares may occur during therapy, IFN is contraindicated in patients with even mildly decompensated cirrhosis and must be used with caution in patients with clinically stable cirrhosis.
Loss of HBsAg occurs in approximately 5% of HBeAg-positive patients within the first 6 months after completion of treatment.31 This rate compares with rates of disappearance of HBsAg in 5% of patients treated for 96 weeks with entecavir and 3% of those on tenofovir for 48 weeks.124,125 Although these differences may seem minimal, sustained responses to IFN are often associated with incremental HBsAg loss during long-term follow up, a finding that has not been demonstrated for nucleos(t)ide analogs.109,126 A 3-year follow-up study of patients who lost HBeAg and had sustained virologic response after a year of pegylated IFN-α demonstrated that initial responders with HBV genotypes A or B had the highest rates of durable virologic response (96% and 86%, respectively) and the highest rate of HBsAg clearance (58% and 14%, respectively).109 By contrast, the same end points were achieved, respectively, in only 76% and 6% of initial responders with genotype D. Unlike nucleos(t)ide analogs, IFN use is not associated with drug resistance.
The more rapid decline in HBsAg concentration during IFN treatment of both HBeAg-positive and HBeAg-negative hepatitis B (see earlier) is thought to reflect immune-mediated elimination of HBV cccDNA (the genomic template for viral transcription). These differences in viral kinetics emphasize that the mechanisms of action of the 2 major classes of antiviral agents differ and provides a rationale for the use of combined IFN and nucleoside analog therapy.
Combination Interferon and Nucleos(t)ide Analog Therapy
From a conceptual standpoint, treatment with the combination of IFN and a nucleos(t)ide analog may prove to be more effective than either drug alone and may permit a shorter course of nucleos(t)ide analog therapy, thereby reducing the risk of viral resistance to low-genetic-barrier drugs. Three large multicenter studies evaluated responses to a combination of pegylated IFN and lamivudine compared with IFN alone. Although suppression of HBV DNA was greater in the combined-therapy arms, the response was not sustained after discontinuation of treatment.31,127,128 Trials of pegylated IFN combined with entecavir or tenofovir are in progress, as are studies in which sequential therapy, starting with a nucleos(t)ide analog, is given.129 It is hoped that IFN combined with a high-genetic-barrier nucleos(t)ide analog given for a longer interval than previously reported may be a more effective approach than prior treatment regimens.
Guidelines
Consensus guidelines for the treatment of hepatitis B have been published by the AASLD, Asian-Pacific Association for the Study of the Liver, and European Association for the Study of the Liver.102,130,131 The recommendations made in the 3 sets of published guidelines have many similarities. In general, they recommend treatment of persons who have both biochemical evidence of liver injury and serum HBV DNA levels in excess of 20,000 IU/mL in HBeAg-positive patients and greater than 2000 IU/mL in HBeAg-negative patients.132 Nucleos(t)ide analog therapy is recommended specifically as first-line treatment in patients with decompensated cirrhosis. Emphasis also is given to the treatment of patients with serum ALT levels that are at least double the upper limit of normal, although some experts disagree with setting arbitrary serum ALT and HBV DNA thresholds.132 The guidelines indicate that treatment decisions ideally should be made in the context of moderately severe disease (stage 2 or greater fibrosis). Liver biopsy is no longer mandated in the AASLD guidelines but is encouraged whenever doubt about the extent of disease exists. Noninvasive assessment of fibrosis, such as US (e.g., transient) elastography and MR elastography, can be helpful in situations in which a liver biopsy is either not possible or contraindicated (see Chapters 73, 74, and 80).133
The guidelines recommend continuing treatment for 6 to 12 months after HBeAg seroconversion to minimize the chance of virologic relapse. Defining the duration of treatment remains an even greater challenge in patients with HBeAg-negative hepatitis because of the absence of secure virologic end points. It is worth noting that although the published treatment guidelines are helpful in assessing the need for treatment, their reliance on grade A evidence (randomized controlled clinical trials) has led to the absence of definitive recommendations in special patient populations for which the data have been less stringently acquired.
Special Populations
Pregnant Women
HBV infection may still develop in 5% to 10% of appropriately immunized newborns of HBV-infected mothers and may occur when vaccination is delayed or incomplete (see later). Preliminary studies have shown that infected spermatozoa or maternal oocytes are associated with HBsAg-expressing embryos.134 Threatened preterm labor, premature rupture of maternal membranes, and detection of HBV in the placenta have also been linked to intrapartum transmission.135
There are only 2 reasons to treat HBsAg-positive pregnant women: to forestall disease progression in the mother or to prevent breakthrough infection in newborns of highly viremic mothers. Epidemiologic studies have shown that HBeAg-positive mothers with a serum HBV DNA level greater than 106 or 108 copies/mL present a significantly higher risk of maternal transmission to the neonate even when properly immunized at birth.136,137 Several studies, most of which were neither prospective nor randomized and controlled in design, have shown that treatment of such highly viremic mothers in the third trimester reduces the rate of neonatal breakthrough infection.137 Many experts have recommended administering antiviral therapy during the last trimester and even continuing it for a brief period after delivery to avoid postpregnancy flares of disease caused by recovery of immunologic function.138
None of the current antiviral agents is licensed for use in pregnancy. Telbivudine and tenofovir are considered category B drugs by the FDA (defined as a lack of animal embryologic toxicity without studies in humans). Lamivudine is a category B drug in HIV-infected pregnant women but a category C drug in HBV-infected women (defined as embryotoxic or teratogenic in animals, without study in humans). A large amount of safety data exists for lamivudine in HIV-infected mothers, and there is increasing data on tenofovir (category B) in the U.S. Antiretroviral Pregnancy Registry.139 Of the more than 10,000 reported pregnancies in which the mother received an oral nucleos(t)ide analog, 95% had HIV infection, and less than 1% had HBV monoinfection. Nevertheless, the overall frequency of birth defects in the infants of these women has not been shown to be significantly different from that reported in the general U.S. population.
Women who are of child-bearing age should be warned not to become pregnant while undergoing treatment with a nucleos(t)ide analog. If pregnancy occurs during treatment, however, the risk of drug withdrawal and subsequent ALT flares must be balanced against the slight uncertainty of harmful effects to the fetus. Figure 79-6 shows a proposed algorithm for the treatment of HBsAg-pregnant mothers with a high level of viremia. Because lamivudine has an excellent safety record and the most extensive use during pregnancy, its use can been recommended for highly viremic mothers. The risk of developing drug-resistant HBV can be anticipated to be small because of the need for short-term treatment. Telbivudine has also been used successfully during pregnancy to prevent neonatal infection from highly viremic HBeAg-positive mothers and has an FDA status B classification.137 Tenofovir is another reasonable choice not only because of its category B status, but also because it is more potent than lamivudine or telbivudine. IFN is contraindicated during pregnancy because of its antiproliferative effects. Breast-feeding is not contraindicated in infants of HBsAg-positive women. Mothers who remain on a nucleos(t)ide analog after delivery, however, are advised not to breast-feed because of a small amount of maternal transfer of drug to the newborn.140
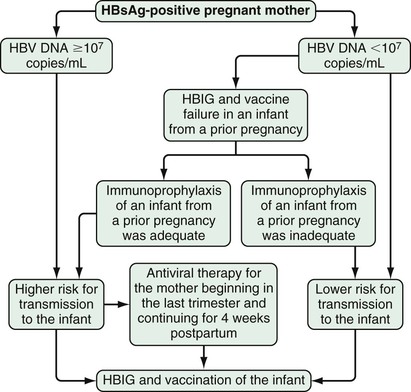
Severe Acute Hepatitis
Because of the high rate (>95%) of complete immunologic recovery from acute hepatitis B, definitive recommendations about drug treatment cannot be made. Some experts recommend nucleos(t)ide analog therapy when HBeAg remains detectable in serum for more than 10 to 12 weeks because of the high likelihood of evolution to chronic HBV infection without treatment. It has been suggested that persons with acute viral hepatitis complicated by an increase in the INR above 1.5 and deep jaundice persisting for more than 4 weeks should receive antiviral therapy.141 Antiviral treatment of patients with hepatitis B–induced acute liver failure is recommended in the practice guidelines of the AASLD because of the safety of nucleos(t)ide analog therapy and the need for liver transplantation in many of these patients.130
The duration of antiviral therapy is not established for severe or life-threatening acute hepatitis. It has been recommended that therapy continue for 3 months after HBsAg seroconversion or at least 12 months after HBeAg seroconversion if HBsAg loss does not occur.102
Cirrhosis
Nucleos(t)ide analog therapy has been shown to be safe in patients with cirrhosis and has made a major difference in the care of patients with advanced liver disease. Antiviral therapy with a high-genetic-barrier drug is preferred in all cases of HBV-related cirrhosis. Marked improvement in hepatic function and regression of fibrosis, including reversal of the histologic features of cirrhosis, has been shown after prolonged viral suppression with entecavir and tenofovir.111 Nucleos(t)ide analog therapy has been associated with a remarkable decline in the need for liver transplantation in patients with HBV-associated chronic liver failure.142 HCC may still occur in cirrhotic patients who have lost HBsAg, and regular US surveillance should be continued in these persons (see Chapter 96).143 The mechanisms behind such late development of HCC have not been defined, but continued hepatocyte regeneration is likely to have a promotional effect on tumorigenesis, and cumulative chromosomal damage from integrated viral sequences may also play a role.144
HBV-HIV Coinfection
With improved control of HIV disease with HAART, liver disease has emerged as one of the leading causes of death in patients with HIV infection (see Chapter 34).145 Antiviral therapy for moderate to severe hepatitis B should be considered for all HIV-HBV coinfected patients, irrespective of the CD4 count. Guidelines enacted by the Department of Health and Human Services suggest that early antiretroviral therapy for all patients who are to undergo treatment for hepatitis B is not required.146 Entecavir treatment is associated with a decline in HIV RNA levels and is not recommended for use in patients who are not receiving concomitant HIV treatment.147 In patients with a CD4 count less than 350/mm3, the use of agents with activity against both HIV and HBV, such as lamivudine and tenofovir, is recommended. Combination therapy with either emtricitabine and tenofovir or lamivudine and tenofovir should ideally be used to avoid or delay the development of HIV resistance to tenofovir.
HBV-HCV Coinfection
When compared with monoinfected patients, HBV-HCV–coinfected patients tend to have more severe liver injury and a higher probability of cirrhosis.148 Limited data are available, however, to define the best approach to treatment in this group of patients. The typical patient is positive for HCV RNA but negative for HBV DNA in serum. Close monitoring has been recommended before and after treatment is initiated, because some patients exhibit alternating viremia, and in some instances resurgence of HBV replication has been documented after successful treatment of hepatitis C.149 The optimal therapy for patients who are positive in serum for both HBV DNA and HCV RNA is also unclear. In such instances, the authors have had success in treating both infections simultaneously with a combination of a nucleos(t)ide analog, pegylated IFN-α, and ribavirin. What effect direct-acting antiviral agents for hepatitis C will have on the treatment of combined HBV and HCV infection remains to be explored (see Chapter 80).
Urgent Treatment
Hepatitis B seldom needs to be treated immediately. There are clinical situations, however, for which prompt or even urgent therapy is required to forestall disease progression, decrease morbidity, or clinically stabilize the patient. Antiviral therapy for hepatitis B should be started as soon as possible whenever a patient with active disease has advanced liver fibrosis or potentially life-threatening disease (Table 79-6). The strongest evidence of benefit for antiviral treatment is for advanced fibrosis, cirrhosis, and decompensated chronic hepatitis B. The data are less certain for reactivation of chronic hepatitis B and even less so for acute severe hepatitis, because available studies have been small and mostly retrospective in nature. The benefit of antiviral treatment for severe acute hepatitis B associated with liver failure remains particularly problematic because disparate rates of efficacy have been reported and HBV DNA may be present in low levels or even nondetectable when the patient is first seen. Based on the expectation that antiviral therapy may be beneficial and will not be harmful, however, a controlled clinical trial is no longer ethically justified.
TABLE 79-6
Indications for Prompt or Urgent Treatment of Hepatitis B
| Indications | Preferred Agent | Principal Supportive Data | |
| Cirrhosis* | |||
| Decompensated | Clinical stabilization; minimizing risk for recurrence post transplant | Entecavir (0.5 mg) or tenofovir (300 mg)† | Open label; multiple large case series |
| Borderline compensated | Forestalling disease progression; avoidance of transplantation | As above | Undefined |
| Well compensated | As above | As above | Randomized controlled trials |
| Acute Liver Failure | |||
| HBV reactivation | Minimizing further liver injury; reducing risk of recurrence after liver transplantation, if needed | Entecavir (0.5 mg) or tenofovir (300 mg)† | Open label with comparison with historical controls |
| Severe acute hepatitis | Minimizing further liver injury and enhancing full recovery | Consider lamivudine or telbivudine‡ | Small case series |
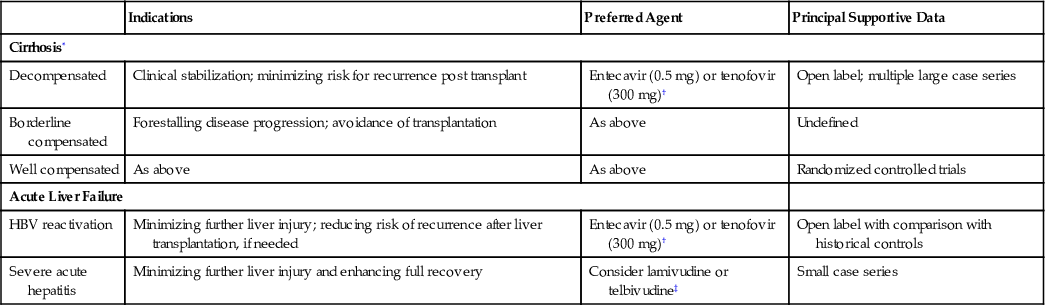
Prevention
Immunoprophylaxis against HBV is of 2 types: passive immunization using HBIG and active immunization using inactive HBsAg. Active immunization gives long-term immunity, whereas passive immunization confers only immediate and short-lived protection.
Hepatitis B Immune Globulin
HBIG is prepared from plasma that is known to contain high titers of anti-HBs. Numerous clinical trials have established the efficacy of HBIG in preventing HBV infection in high-risk persons, such as hemodialysis patients, sexual partners of patients with hepatitis B, and newborn infants of HBsAg-positive mothers within 12 hours of birth along with simultaneous vaccination. HBIG licensed in the United States has an anti-HBs titer of 1 : 100,000. In Europe, several preparations of HBIG with different concentrations and pharmacokinetic properties are available. HBIG is safe, although rare anaphylactic reactions can occur. Myalgias, rash, and arthralgias have also been reported and are believed to result from formation of antigen-antibody complexes.
Hepatitis B Vaccine
Currently marketed HBV vaccines make use of recombinant DNA technology by introducing the gene for HBsAg (S gene) into the genome of yeast. The 2 vaccines available in the United States are Recombivax HB (Merck, licensed in 1986) and Engerix-B (GlaxoSmithKline, licensed by SmithKline Beecham in 1989). No serious side effects of the HBV vaccine have been reported. Aluminum hydroxide is used as an adjuvant in both vaccines. Because thimerosal, a preservative used in the vaccines, contains mercury, thimerosal-free vaccines have become available, especially for use in infants. The HBV vaccine is administered intramuscularly in the deltoid muscle of adults and the anterolateral thigh of infants and neonates. Because the vaccines contain HBsAg only, anti-HBs is the sole antibody produced. Consequently, vaccinees who test positive for anti-HBc after vaccination should be considered to have either resolved infection or active infection depending on the results of reflex testing for HBsAg.
The vaccines typically achieve an anti-HBs titer greater than 100 mIU/mL. Antibody titers greater than 100 mIU/mL confer 100% protection against HBV infection, and a lower antibody titer (up to 10 mIU/mL) is seroprotective in most instances. Peak antibody titers and persistence of antibody levels vary among different persons. The titers drop steadily over the first 2 years after vaccination, sometimes to levels less than 10 mIU/mL. Two studies in different populations have demonstrated that anti-HBs titers decrease to nonprotective levels in at least 25% to 50% of recipients over a period of 5 to 10 years.150
Although protective anti-HBs response rates after HBV vaccination occur in approximately 90% of patients, a number of factors can impede an adequate antibody response. Smoking; obesity; injection into the buttock; chronic liver disease; presence of HLA-DR3, DR7, and DQ2 alleles; absence of the HLA-A2 allele; and extremes of age may be associated with reduced immunogenicity. Such “hyporesponders” may benefit from a higher dose of vaccine. Response rates are also lower in immunocompromised patients, such as transplant recipients, patients receiving chemotherapy, and those with end-stage liver disease. Only 50% to 60% of patients on hemodialysis respond adequately to vaccination. Therefore, patients with chronic kidney disease should be vaccinated early in the course of their disease, before renal disease progresses, to ensure optimal response to vaccination. Five percent to 8% of HBV vaccine recipients do not achieve detectable anti-HBs levels (“nonresponders”).
Because HBV vaccination results in strong immunologic memory capable of preventing infection even in patients with low or undetectable antibody titers, recommendations do not call for a booster vaccine dose in immunocompetent adults and children. Booster doses are, however, recommended for patients undergoing hemodialysis, in whom anti-HBs titers should be tested annually and a booster dose given if the titer is lower than 10 mIU/mL.151 Data from a study in Taiwan demonstrated a surprisingly high rate of HBsAg positivity in adolescents who were vaccinated at birth, especially those born to HBeAg-positive mothers who did not receive HBIG or had less than 4 doses of vaccine. The suggestion was made that a booster dose may be needed at age 15 or older in such highly exposed individuals.152 Confirmation of this finding would have important implications.
Vaccination Schedule
The typical vaccination schedule is 0, 1, and 6 months. The first 2 doses have no effect on the final anti-HBs titer. The third dose acts as a booster to achieve a high anti-HBs titer. In immunocompromised patients and those undergoing hemodialysis, 4 vaccine doses are recommended, with the fourth dose given to ensure the highest possible anti-HBs titer. If vaccination is interrupted, the second dose should be administered as soon as possible after the first.153 If the third dose is not given on schedule, it should be given at least 2 months after the second dose.
In the United States and elsewhere, the HBV vaccine is administered to all children and infants as a part of the universal immunization program. Unfortunately, although more than 160 countries have proposed and approved universal vaccination, not all have the financial resources to sponsor a national health program that incorporates affordable or free vaccination.154
Combination HBV vaccines with diphtheria-pertussis-tetanus (DPT) and Haemophilus influenzae type B (Hib) (DTPw-HB/Hib) are in use for immunization of infants. The other component antigens do not reduce the immunogenicity against HBV.155 Adolescents who have not been vaccinated in infancy or childhood should also be vaccinated.
Postexposure and Perinatal Prophylaxis
Table 79-7 lists recommendations for prophylaxis after exposure to a known HBsAg-positive source. Postexposure vaccination should be considered for any percutaneous, ocular, or mucous membrane exposure. The type of immunoprophylaxis is determined by the HBsAg status of the source and the vaccination-response status of the exposed person. If a patient is a known responder to previous vaccination, then nothing need be done.
TABLE 79-7
Recommended Postexposure Prophylaxis for HBV According to the Vaccination Status of the Exposed Person
| Vaccination Status of Exposed Person | Recommended Prophylaxis |
| Unvaccinated | HBIG (0.06 mL/kg) and initiate the hepatitis B vaccine series |
| Previously vaccinated: | |
| Known responder* | None |
| Known nonresponder | HBIG × 2 doses (1 month apart) |
| OR | |
| HBIG × 1 dose and initiate revaccination | |
| Antibody response unknown | Test for anti-HBs If adequate*: No treatment If inadequate†: HBIG × 1 dose and give vaccine booster dose |
Bivalent Vaccine
A combined HAV and HBV vaccine has been licensed commercially (TWINRIX, GlaxoSmithKline, Research Triangle Park, N.C.) and has been shown to be highly immunogenic and protective against both infections. This vaccine offers ease of administration for persons at increased risk of both HAV and HBV infection (e.g., world travelers, men who have sex with men) and in patients with underlying chronic liver disease.156
Recommendations
In 2008, the CDC extended its original recommendations for routine HBsAg screening to include persons born in countries in which the prevalence of hepatitis B exceeds 2%.153 In 2011, the CDC added a recommendation for HBV vaccination in persons 19 to 59 years of age with diabetes mellitus type 1 or 2. Health care professionals may vaccinate diabetics 60 years of age or older after assessing their risk of exposure.157
The CDC has revised its recommendations for health care providers and students chronically infected with hepatitis B.158 In general, the CDC suggests that such persons can maintain their current form of practice. They do not recommend notification of patients of the caregivers' HBV status, nor do they broadly recommend antiviral therapy. Another basic recommendation is that all health care providers and students receive the HBV vaccine followed by testing for anti-HBs to verify immunity. If anti-HBs is not detectable, reflex testing with anti-HBc and HBsAg is the suggested next step and will allow identification of those persons with active HBV infection.
In the past, the CDC did not recommend prevaccination screening for health care providers and students who perform exposure-prone tasks (i.e., surgical procedures in which access is difficult and a needlestick injury may occur). In the 2012 guidelines, the CDC recommended that persons who perform exposure-prone procedures and those at high risk for HBV infection (e.g., those born in endemic regions of the world) undergo prevaccination testing for hepatitis B.158 This recommendation is not considered necessary for providers and students who do not perform exposure-prone procedures. It is further recommended that for surgeons who are found to be HBV carriers, including oral surgeons, obstetricians/gynecologists, surgical residents, and others who perform exposure-prone procedures, their procedures should be reviewed by a duly constituted expert review panel with balanced perspective that will oversee the procedures and practice of the individual. A serum HBV level of 1000 IU/mL (equivalent to 5000 copies/mL) is a suggested appropriate threshold for review by the expert panel. Regular activities can be maintained if a lower or undetectable HBV DNA level is documented by regular testing at least every 6 months. Spontaneous fluctuations above the threshold may require that the health care provider or student abstain from performing exposure-prone procedures until retested and, if positive at this level again, “reasonable” steps are taken; the term reasonable is meant to include antiviral therapy.
The 2012 CDC guidelines strongly emphasize the need for strict confidentiality but admit that confidentiality may not be possible. It may be difficult to implement all of these guidelines at institutions that lack the necessary infrastructure or for health care providers who perform exposure-prone procedures and choose not to receive antiviral therapy.