Recombinant DNA and Biotechnology
 The recombinant DNA speaks :
The recombinant DNA speaks :The term biotechnology represents a fusion or an alliance between biology and technology. Frankly speaking, biotechnology is a newly discovered discipline for age-old practices e.g. preparation of wine, beer, curd, bread. These natural processes are regarded as old or traditional biotechnology.
The new or modern biotechnology embraces all the genetic manipulations, cell fusion techniques, and improvements made in the old biotechnological processes. The biotechnology with particular reference to recombinant DNA in human health and disease is briefly described in this chapter.
Genetic engineering primarily involves the manipulation of genetic material (DNA) to achieve the desired goal in a pre-determined way. Some other terms are also in common use to describe genetic engineering.
Brief history of recombinant DNA technology
The present day DNA technology has its roots in the experiments performed by Boyer and Cohen in 1973. In their experiments, they successfully recombined two plasmids (pSC 101 and pSC 102) and cloned the new plasmid in E.coli. In the later experiments the genes of a frog could be successfully transplanted, and expressed in E.coli. This made the real beginning of modern rDNA technology and laid foundations for the present day molecular biotechnology.
Some biotechnologists who admire Boyer-Cohen experiments divide the subject into two chronological categories.
Recombinant DNA technology is a vast field. The basic principles and techniques of rDNA technology along with the most important applications are briefly described in this chapter.
Basic principles of rDNA technology
There are many diverse and complex techniques involved in gene manipulation. However, the basic principles of recombinant DNA technology are reasonably simple, and broadly involve the following stages (Fig.27.1).

1. Generation of DNA fragments and selection of the desired piece of DNA (e.g. a human gene).
2. Insertion of the selected DNA into a cloning vector (e.g. a plasmid) to create a recombinant DNA or chimeric DNA (Chimera is a monster in Greek mythology that has a lion's head, a goat's body and a serpent's tail. This may be comparable to Narasimha in Indian mythology).
3. Introduction of the recombinant vectors into host cells (e.g. bacteria).
4. Multiplication and selection of clones containing the recombinant molecules.
Recombinant DNA technology with special reference to the following aspects is described
Molecular tools of genetic engineering
The term genetic engineer may be appropriate for an individual who is involved in genetic manipulations. The genetic engineer's toolkit or molecular tools namely the enzymes most commonly used in recombinant DNA experiments are briefly described.
Restriction endonucleases—DNA cutting enzymes
Restriction endonucleases are one of the most important groups of enzymes for the manipulation of DNA. These are the bacterial enzymes that can cut/split DNA (from any source) at specific sites. They were first discovered in E.coli restricting the replication of bacteriophages, by cutting the viral DNA (The host E.coli DNA is protected from cleavage by addition of methyl groups). Thus, the enzymes that restrict the viral replication are known as restriction enzymes or restriction endonucleases.
Nomenclature
Restriction endonucleases are named by a standard procedure, with particular reference to the bacteria from which they are isolated. The first letter (in italics) of the enzymes indicates the genus name, followed by the first two letters (also in italics) of the species, then comes the strain of the organism and finally a Roman numeral indicating the order of discovery. A couple of examples are given below.
EcoRI is from Escherichia (E) coli (co), strain Ry13 (R), and first endonuclease (I) to be discovered. HindIII is from Haemophilus (H) influenzae (in), strain Rd (d) and, the third endonucleases (III) to be discovered.
Cleavage patterns
Majority of restriction endonucleases (particularly type II) cut DNA at defined sites within recognition sequence. A selected list of enzymes, recognition sequences, and their products formed is given in Table 27.1.
Table 27.1
Some restriction enzymes with sources, recognition sequences and the products formed
| Enzyme (source) | Recognition sequence | Products |
| EcoRI |  |
A–A–T–T–C…… |
| (Escherichia coli) | G…… | |
| ……G | ||
| C–T–T–A–A | ||
| BamHI |  |
G–A–T–C–C…… |
| (Bacillus amyloliquefaciens) | G…… | |
| ……G | ||
| ……C–C–T–A–G | ||
| HaeIII |  |
*C–C…… |
| (Haemophilus aegyptius) | G–G…… | |
| ……*G–G | ||
| ……C–C | ||
| HindIII |  |
A–G–C–T–T…… |
| (Haemophilus influenzae) | A…… | |
| ……A | ||
| ……T–T–C–G–A | ||
| NotI |  |
G–G–C–C–G–C…… |
| (Nocardia otitidis) | C–G…… | |
| ……G–C | ||
| ……C–G–C–C–G–G…… |
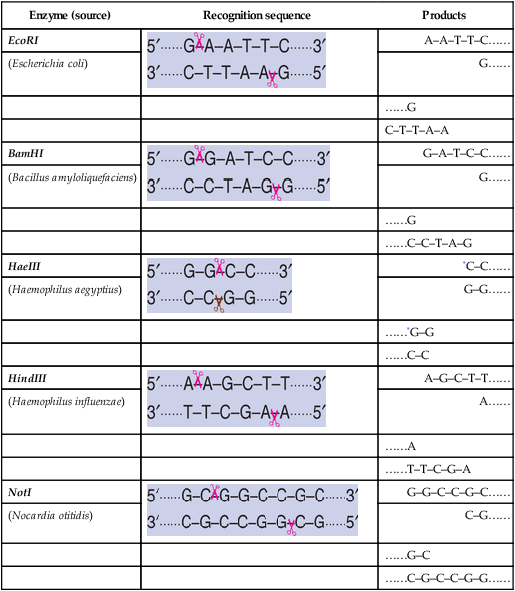
Note : Scissors indicate the sites of cleavage
*The products are with blunt ends while for the rest, the products are with sticky ends
The cut DNA fragments by restriction endonucleases may have mostly sticky ends (cohesive ends) or blunt ends, as given in Table 27.1. DNA fragments with sticky ends are particularly useful for recombinant DNA experiments. This is because the single-stranded sticky DNA ends can easily pair with any other DNA fragment having complementary sticky ends.
DNA ligases—DNA joining enzymes
The cut DNA fragments are covalently joined together by DNA ligases. These enzymes were originally isolated from viruses. They also occur in E.coli and eukaryotic cells. DNA ligases actively participate in cellular DNA repair process.
The action of DNA ligases is absolutely required to permanently hold DNA pieces. This is so since the hydrogen bonds formed between the complementary bases (of DNA strands) are not strong enough to hold the strands together. DNA ligase joins (seals) the DNA fragments by forming a phosphodiester bond between the phosphate group of 5’-carbon of one deoxyribose with the hydroxyl group of 3’-carbon of another deoxyribose (Fig.27.2).

Many enzymes are used in the recombinant DNA technology/genetic engineering. A selected list of these enzymes and the reactions catalysed by them is given in Table 27.2.
Table 27.2
The most commonly used enzymes in recombinant DNA technology/genetic engineering
| Enzyme | Use/reaction |
| Alkaline phosphatase | Removes phosphate groups from 5′-ends of double/single-stranded DNA (or RNA). |
| Bal 31 nuclease | For the progressive shortening of DNA. |
| DNA ligase | Joins DNA molecules by forming phosphodiester linkages between DNA segments. |
| DNA polymerase I | Synthesizes DNA complementary to a DNA template. |
| DNase I | Produces single-stranded nicks in DNA. |
| Exonuclease III | Removes nucleotides from 3′-end of DNA. |
| λ exonuclease | Removes nucleotides from 5′-end of DNA. |
| Polynucleotide kinase | Transfers phosphate from ATP to 5′-OH ends of DNA or RNA. |
| Restriction enzymes | Cut double-stranded DNA with a specific recognition site. |
| Reverse transcriptase | Synthesizes DNA from RNA. |
| RNase A | Cleaves and digests RNA (and not DNA). |
| RNase H | Cleaves and digests the RNA strand of RNA-DNA heteroduplex. |
| Taq DNA polymerase | Used in polymerase chain reaction |
| SI nuclease | Degrades single-stranded DNA and RNA. |
| Terminal transferase | Adds nucleotides to the 3′-ends of DNA or RNA. Useful in homopolymer tailing. |
Host cells—the factories of cloning
The hosts are the living systems or cells in which the carrier of recombinant DNA molecule or vector can be propagated. There are different types of host cells-prokaryotic (bacteria) and eukaryotic (fungi, animals and plants). Some examples of host cells used in genetic engineering are given in Table 27.3.
Table 27.3
Some examples of host cells used in genetic engineering
| Group | Examples |
| Prokaryotic | |
| Bacteria | Escherichia coli |
| Bacillus subtilis | |
| Streptomyces sp | |
| Eukaryotic | |
| Fungi | Saccharomyces cerevisiae |
| Aspergillus nidulans | |
| Animals | Insect cells |
| Oocytes | |
| Mammalian cells | |
| Whole organisms | |
| Plants | Protoplasts |
| Intact cells | |
| Whole plants | |

Host cells, besides effectively incorporating the vector's genetic material, must be conveniently cultivated in the laboratory to collect the products. In general, microorganisms are preferred as host cells, since they multiply faster compared to cells of higher organisms (plants or animals).
Eukaryotic hosts
Eukaryotic organisms are preferred to produce human proteins since these hosts with complex structure (with distinct organelles) are more suitable to synthesize complex proteins. The most commonly used eukaryotic organism is the yeast, Saccharomyces cerevisiae.
Mammalian cells
Despite the practical difficulties to work with and high cost factor, mammalian cells (such as mouse cells) are also employed as hosts. The advantage is that certain complex proteins which cannot be synthesized by bacteria can be produced by mammalian cells e.g. tissue plasminogen activator. This is mainly because the mammalian cells possess the machinery to modify the protein to the active form (post-translational modifications).
Plasmid
Plasmids are extrachromosomal, doublestranded, circular, self-replicating DNA molecules. Almost all the bacteria have plasmids containing a low copy number (1–4 per cell) or a high copy number (10–100 per cell). The size of the plasmids varies from 1 to 500 kb. Usually, plasmids contribute to about 0.5 to 5.0% of the total DNA of bacteria (Note : A few bacteria contain linear plasmids e.g. Streptomyces sp, Borella burgdorferi).
Nomenclature of plasmids
It is a common practice to designate plasmid by a lower case p, followed by the first letter(s) of researcher(s) names and the numerical number given by the workers. Thus, pBR322 is a plasmid discovered by Bolivar and Rodriguez who designated it as 322. Some plasmids are given names of the places where they are discovered e.g. pUC is plasmid from University of California.
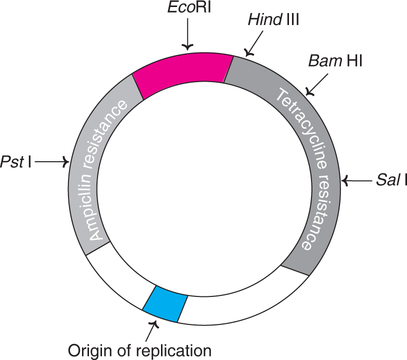
pBR322 – the most common plasmid vector
pBR322 of E.coli is the most popular and widely used plasmid vector, and is appropriately regarded as the parent or grand parent of several other vectors.
pBR322 has a DNA sequence of 4,361 bp. It carries genes resistance for ampicillin (Ampr) and tetracycline (Tetr) that serve as markers for the identification of clones carrying plasmids. The plasmid has unique recognition sites for the action of restriction endonucleases such as EcoRI, HindIII, BamHI, SalI and PstlI (Fig.27.3).
Bacteriophages
Bacteriophages or simply phages are the viruses that replicate within the bacteria. In case of certain phages, their DNA gets incorporated into the bacterial chromosome and remains there permanently. Phage vectors can accept short fragments of foreign DNA into their genomes. The advantage with phages is that they can take up larger DNA segments than plasmids. Hence phage vectors are preferred for working with genomes of human cells. The most commonly used phages are bacteriophage λ (phage λ) and bacteriophage (phage M13).
Cosmids
Cosmids are the vectors possessing the characteristics of both plasmid and bacteriophage λ. Cosmids can be constructed by adding a fragment of phage λ DNA including cos site, to plasmids. A foreign DNA (about 40 kb) can be inserted into cosmid DNA. The recombinant DNA so formed can be packed as phages and injected into E.coli. Once inside the host cell, cosmids behave just like plasmids and replicate. The advantage with cosmids is that they can carry larger fragments of foreign DNA compared to plasmids.
Artificial chromosome vectors
Human artificial chromosome (HAC)
Developed in 1997 (by H. Willard), human artificial chromosome is a synthetically produced vector DNA, possessing the characteristics of human chromosome. HAC may be considered as a self-replicating microchromosome with a size ranging from 1/10th to ⅕th of a human chromosome. The advantage with HAC is that it can carry human genes that are too long. Further, HAC can carry genes to be introduced into the cells in gene therapy.
Choice of vector
Among the several factors, the size of the foreign DNA is very important in the choice of vectors. The efficiency of this process is often crucial for determining the success of cloning. The sizes of DNA insert that can be accepted by different vectors is shown in Table 27.4.
Table 27.4
The different cloning vectors with the corresponding hosts and the sizes of foreign insert DNAs
| Vector | Host | Foreign insert DNA size |
| Phage λ | E. coli | 5–25 kb |
| Cosmid λ | E. coli | 35–45 kb |
| Plasmid artifical chromosome (PAC) | E. coli | 100–300 kb |
| Bacterial artificial chromosome (BAC) | E. coli | 100–300 kb |
| Yeast chromosome | S. cerevisiae | 200–2000 kb |
Methods of gene transfer
Introducing a foreign DNA (i.e. the gene) into the cells is an important task in biotechnology. The efficiency of this process is often crucial for determining the success of cloning. The most commonly employed gene transfer methods, namely transformation, conjugation, electroporation and lipofection, and direct transfer of DNA are briefly described.
Transformation
Transformation is the method of introducing foreign DNA into bacterial cells (e.g. E.coli). The uptake of plasmid DNA by E.coli is carried out in ice-cold CaCl2 (0-5°C), and a subsequent heat shock (37–45°C for about 90 sec). By this technique, the transformation frequency, which refers to the fraction of cell population that can be transferred, is reasonably good e.g. approximately one cell per 1000 (10–3) cells.
Conjugation
Conjugation is a natural microbial recombination process. During conjugation, two live bacteria (a donor and a recipient) come together, join by cytoplasmic bridges and transfer singlestranded DNA (from donor to recipient). Inside the recipient cell, the new DNA may integrate with the chromosome (rather rare) or may remain free (as is the case with plasmids).
The natural phenomenon of conjugation is exploited for gene transfer. This is achieved by transferring plasmid-insert DNA from one cell to another. In general, the plasmids lack conjugative functions and therefore, they are not as such capable of transferring DNA to the recipient cells. However, some plasmids with conjugative properties can be prepared and used.
Electroporation
Electroporation is based on the principle that high voltage electric pulses can induce cell plasma membranes to fuse. Thus, electroporation is a technique involving electric field-mediated membrane permeabilization. Electric shocks can also induce cellular uptake of exogenous DNA (believed to be via the pores formed by electric pulses) from the suspending solution. Electroporation is a simple and rapid technique for introducing genes into the cells from various organisms (microorganisms, plants and animals).
Liposome-mediated gene transfer
Liposomes are circular lipid molecules, which have an aqueous interior that can carry nucleic acids. Several techniques have been developed to encapsulate DNA in liposomes. The liposomemediated gene transfer is referred to as lipofection.
On treatment of DNA fragment with liposomes, the DNA pieces get encapsulated inside liposomes. These liposomes can adhere to cell membranes and fuse with them to transfer DNA fragments. Thus, the DNA enters the cell and then to the nucleus. The positively charged liposomes very efficiently complex with DNA, bind to cells and transfer DNA rapidly.
Gene cloning strategies
A clone refers to a group of organisms, cells, molecules or other objects, arising from a single individual. Clone and colony are almost synonymous.
Gene cloning strategies in relation to recombinant DNA technology broadly involve the following aspects (Fig.27.4).

Cloning from genomic DNA or mRNA?
DNA represents the complete genetic material of an organism which is referred to as genome. Theoretically speaking, cloning from genomic DNA is supposed to be ideal. But the DNA contains non-coding sequences (introns), control regions and repetitive sequences. This complicates the cloning strategies, hence DNA as a source material is not preferred, by many workers. However, if the objective of cloning is to elucidate the control of gene expression, then genomic DNA has to be invariably used in cloning.
The use of mRNA in cloning is preferred for the following reasons.
• mRNA represents the actual genetic information being expressed.
• Selection and isolation mRNA are easy.
• As introns are removed during processing, mRNA reflects the coding sequence of the gene.
• The synthesis of recombinant protein is much easier with mRNA cloning.
Besides the direct use of genomic DNA or mRNA, it is possible to synthesize DNA in the laboratory (by polymerase chain reaction), and use it in cloning experiments. This approach is useful if the gene sequence is short and the complete sequence of amino acids is known.
Basic techniques in genetic engineering
There are several techniques used in recombinant DNA technology or gene manipulation. The most frequently used methods are listed.
• Isolation and purification of nucleic acids.
• Nucleic acid blotting techniques.
• Methods of gene transfer (described already).
• Production of monoclonal antibodies (![]() Chapter 41).
Chapter 41).
Isolation and purification of nucleic acids
Almost all the experiments dealing with gene manipulations require pure forms of either DNA or RNA, or sometimes even both. Hence there is a need for the reliable isolation of nucleic acids from the cells. The purification of nucleic acids broadly involves three stages.
1. Breaking or opening of the cells to expose nucleic acids.
2. Separation of nucleic acids from other cellular components.
The basic principles and procedures for nucleic acid purification are briefly described.
Purification of cellular DNA
The first step for DNA purification is to open the cells and release DNA. The method should be gentle to preserve the native DNA. Due to variability in cell structure, the approaches to break the cells are also different.
Lysis of cells
Bacterial cells
The bacterial cells (e.g. E. coli) can be lysed by a combination of enzymatic and chemical treatments. The enzyme lysozyme and the chemical ethylenediamine tetraacetate (EDTA) are used for this purpose. This is followed by the addition of detergents such as sodium dodecyl sulfate (SDS).
Methods to purify DNA
There are two different approaches to purify DNA from the cellular extracts.
1. Purification of DNA by removing cellular components : This involves the degradation or complete removal of all the cellular components other than DNA. This approach is suitable if the cells do not contain large quantities of lipids and carbohydrates.
The cellular extract is centrifuged at a low speed to remove the debris (e.g. pieces of cell wall) that forms a pellet at the bottom of the tube. The supernatant is collected and treated with phenol to precipitate proteins at the interface between the organic and aqueous layers. The aqueous layer, containing the dissolved nucleic acids, is collected and treated with the enzyme ribonuclease (RNase). The RNA is degraded while the DNA remains intact. This DNA can be precipitated by adding ethanol and isolated after centrifugation, and suspended in an appropriate buffer.
2. Direct purification of DNA : In this approach, the DNA itself is selectively removed from the cellular extract and isolated. There are two ways for direct purification of DNA.
In one method, the addition of a detergent cetyltrimethyl ammonium (CTAB) results in the formation of an insoluble complex with nucleic acids. This complex, in the form of a precipitate is collected after centrifugation and suspended in a high-salt solution to release nucleic acids. By treatment with RNase, RNA is degraded. Pure DNA can be isolated by ethanol precipitation.
The second technique is based on the principle of tight binding between DNA and silica particles in the presence of a denaturing agent such as guanidinium thiocyanate. The isolation of DNA can be achieved by the direct addition of silica particles and guanidinium thiocyanate to the cellular extract, followed by centrifugation. Alternately, a column chromatography containing silica can be used, and through this the extract and guanidinium thiocyanate are passed. The DNA binds to the silica particles in the column which can be recovered.
Purification of mRNA
Among the RNAs, mRNA is frequently required in a pure form for genetic experiments.
After the cells are disrupted on lysis by different techniques (described above), the cellular extract is deproteinised by treatment with phenol or phenol/chloroform mixtures. On centrifugation, the nucleic acids get concentrated in the upper aqueous phase which may then be precipitated by using isopropanol or ethanol.
The purification of mRNA can be achieved by affinity chromatography using oligo (dT)- cellulose (Fig.27.5). This is based on the principle that oligo (dT)-cellulose can specifically bind to the poly (A) tails of eukaryotic mRNA. Thus, by this approach, it is possible to isolate mRNA from DNA, rRNA and tRNA.

As the nucleic acid solution is passed through an affinity chromotographic column, the oligo(dT) binds to poly(A) tails of mRNA. By washing the column with high-salt buffer, DNA, rRNA and tRNA can be eluted, while the mRNA is tightly bound. This mRNA can be then eluted by washing with low-salt buffer. The mRNA is precipitated with ethanol and collected by centrifugation (Fig.27.5).
Nucleic acid blotting techniques
Blotting techniques are very widely used analytical tools for the specific identification of desired DNA or RNA fragments from thousands of molecules. Blotting refers to the process of immobilization of sample nucleic acids on solid support (nitrocellulose or nylon membranes). The blotted nucleic acids are then used as targets in the hybridization experiments for their specific detection. An outline of the nucleic acid blotting technique is depicted in Fig.27.6.

Types of blotting techniques
The most comonly used blotting techniques are listed below
The Southern blotting is named after the scientist Ed Southern (1975) who developed it. The other names Northern blotting and Western blotting are laboratory jargons which are now accepted. Western blotting involves the transfer of protein blots and their identification by using specific antibodies.
A diagrammatic representation of a typical blotting apparatus is depicted in Fig.27.7.

Southern blotting
Southern blotting technique is the first nucleic acid blotting procedure developed in 1975 by Southern. It is depicted in Fig.27.8, and briefly described.

The genomic DNA isolated from cells/tissues is digested with one or more restriction enzymes. This mixture is loaded into a well in an agarose or polyacrylamide gel and then subjected to electrophoresis. DNA, being negatively charged migrates towards the anode (positively charged electrode); smaller DNA fragments move faster.
The separated DNA molecules are denatured by exposure to a mild alkali and transferred to nitrocellulose or nylon paper. This results in an exact replica of the pattern of DNA fragments on the gel. The DNA can be annealed to the paper on exposure to heat (80°C). The nitrocellulose or nylon paper is then exposed to labeled cDNA probes. These probes hybridize with complementary DNA molecules on the paper.
The paper after thorough washing is exposed to X-ray film to develop autoradiograph. This reveals specific bands corresponding to the DNA fragments recognized by cDNA probe.
Zoo blot
This is a specialized Southern blot technique used to compare DNA sequences (genomes) between humans and other organisms. e.g. hemoglobin gene sequences in humans compared to that of chimpanzee, horse and pig. Zoo blot technique is also useful to distinguish between coding and non-coding regions and their evolution in different organisms.
Applications of Southern blotting
• It is an invaluable method in gene analysis.
• Important for confirmation of DNA cloning.
• Forensically applied to detect minute quantities of DNA (to identify parenthood, thieves, rapists etc.).
• Highly useful for the determination of restriction fragment length polymorphism (RFLP) associated with pathological conditions.
Northern blotting
Northern blotting is the technique for the specific identification of RNA molecules. The procedure adopted is almost similar to that described for Southern blotting and is depicted in Fig.27.9. RNA molecules are subjected to electrophoresis, followed by blot transfer, hybridization and autoradiography.

RNA molecules do not easily bind to nitrocellulose paper or nylon membranes. Blottransfer of RNA molecules is carried out by using a chemically reactive paper prepared by diazotization of aminobenzyloxymethyl to create diazobenzyloxymethyl (DBM) paper. The RNA can covalently bind to DBM paper.
Northern blotting is theoretically, a good technique for determining the number of genes (through mRNA) present on a given DNA. But this is not really practicable since each gene may give rise to two or more RNA transcripts. Another drawback is the presence of exons and introns.
Dot-blotting
Dot-blotting is a modification of Southern and Northern blotting techniques described above. In this approach, the nucleic acids (DNA or RNA) are directly spotted onto the filters, and not subjected to electrophoresis. The hybridization procedure is the same as in original blotting techniques.
Dot-blotting technique is particularly useful in obtaining quantitative data for the evaluation of gene expression.
Western blotting
Western blotting involves the identification of proteins. It is very useful to understand the nucleic acid functions, particularly during the course of gene manipulations.
The technique of Western blotting involves the transfer of electrophoresed protein bands from polyacrylamide gel to nylon or nitrocellulose membrane. These proteins can be detected by specific protein-ligand interactions. Antibodies or lectins are commonly used for this purpose.
Autoradiography
Autoradiography is the process of localization and recording of a radiolabel within a solid specimen, with the production of an image in a photographic emulsion. These emulsions are composed of silver halide crystals suspended in gelatin.
When a β-particle or a γ-ray from a radiolabel passes through the emulsions, silver ions are converted to metallic silver atoms. This results in the development of a visible image which can be easily detected.
DNA Sequencing
Determination of nucleotide sequence in a DNA molecule is the basic and fundamental requirement in biotechnology. DNA sequencing is important to understand the functions of genes, and basis of inherited disorders. Further, DNA cloning and gene manipulation invariably require knowledge of accurate nucleotide sequence.
Maxam and gilbert technique
The first DNA sequencing technique, using chemical reagents, was developed by Maxam and Gilbert (1977). This method is briefly described below (Fig.27.10).

A strand of source DNA is labeled at one end with 32P. The two strands of DNA are then separated. The labeled DNA is distributed into four samples (in separate tubes). Each sample is subjected to treatment with a chemical that specifically destroys one (G, C) or two bases (A + G, T + C) in the DNA. Thus, the DNA strands are partially digested in four samples at sites G, A + G, T + C and C. This results in the formation of a series of labeled fragments of varying lengths. The actual length of the fragment depends on the site at which the base is destroyed from the labeled end. Thus for instance, if there are C residues at positions 4, 7, and 10 away from the labeled end, then the treatment of DNA that specifically destroys C will give labeled pieces of length 3, 6 and 9 bases. The labeled DNA fragments obtained in the four tubes are subjected to electrophoresis side by side and they are detected by autoradiograph. The sequence of the bases in the DNA can be constructed from the bands on the electrophoresis.
Dideoxynucleotide method
Currently, the preferred technique for determining nucleotide sequence in DNA is the one developed by Sanger (1980). This is an enzymatic procedure commonly referred to as the dideoxynucleotide method or chain termination method (Note : Fredrick Sanger won Nobel prize twice, once for determining the structure of protein, insulin; the second time for sequencing the nucleotides in an RNA virus).
A dideoxynucleotide is a laboratory-made chemical molecule that lacks a hydroxyl group at both the 2′ and 3′carbons of the sugar (Fig.27.11). This is in contrast to the natural deoxyribonucleotide that possesses at 3′ hydroxyl group on the sugar.
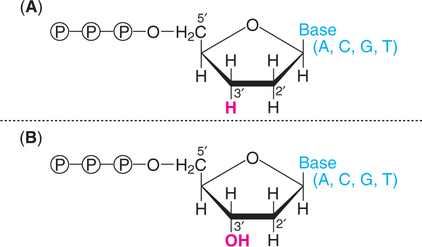
Termination role of dideoxynucleotide
In the normal process of DNA replication, an incoming nucleoside triphosphate is attached by its 5′-phosphate group to the 3′-hydroxyl group of the last nucleotide of the growing chain (Refer ![]() Chapter 24) when a dideoxynucleotide is incorporated to the growing chain, no further replication occurs. This is because dideoxynucleotide, lacking a 3′-hydroxyl group, cannot form a phosphodiester bond and thus the DNA synthesis terminates.
Chapter 24) when a dideoxynucleotide is incorporated to the growing chain, no further replication occurs. This is because dideoxynucleotide, lacking a 3′-hydroxyl group, cannot form a phosphodiester bond and thus the DNA synthesis terminates.
Sequencing method
The process of sequencing DNA by dideoxynucleotide method is briefly described. A single-stranded DNA to be sequenced is chosen as a template. It is attached to a primer (a short length of DNA oligonucleotide) complementary to a small section of the template. The 3′-hydroxyl group of the primer initiates the new DNA synthesis.
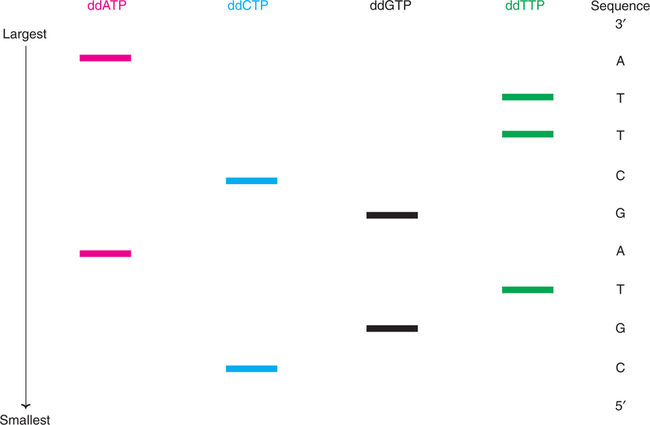
DNA synthesis is carried out in four reaction tubes. Each tube contains the primed DNA, Klenow subunit (the larger fragment of DNA polymerase of E. coli), four dideoxyribonucleotides (ddATP, ddCTP, ddGTP or ddTTP). It is necessary to radiolabel (with 32P) the primer or one of the deoxyribonucleotides.
As the new DNA synthesis is completed, each one of the tubes contains fragments of DNA of varying length bound to primer. Let us consider the first reaction tube with dideoxyadenosine (ddATP). In this tube, DNA synthesis terminates whenever the growing chain incorporates ddA (complementary to dT on the template strand). Therefore, this tube will contain a series of different length DNA fragments, each ending with ddA. In a similar fashion, for the other 3 reaction tubes, DNA synthesis stops as the respective dideoxynucleotides are incorporated.
The synthesis of new DNA fragments in the four tubes is depicted in Fig.27.12.
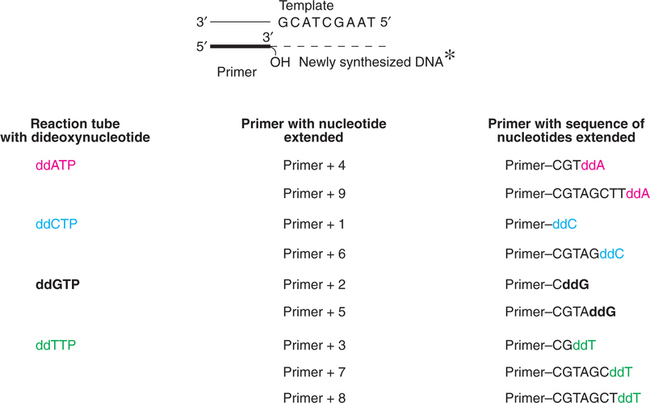
The DNA pieces are denatured to yield free strands with radiolabel. The samples from each tube are separated by polyacrylamide gel electrophoresis. This separation technique resolves DNA pieces, different in size even by a single nucleotide. The shortest DNA will be the fastest moving on the electrophoresis.
The sequence of bases in a DNA fragment is determined by identifying the electrophoretic (radiolabeled) bands by autoradiography. In the Fig.27.13, the sequence of the newly synthesized DNA fragment that is complementary to the original DNA piece is shown. It is conventional to read the bands from bottom to top in 5′to 3′ direction. By noting the order of the bands first C, second G, third T and so on, the sequence of the DNA can be determined accurately. As many as 350 base sequences of a DNA fragment can be clearly identified by using autoradiographs.
Modifications of dideoxynucleotide method
Replacement of 32P-radiolabel by 33P or 35S improves the sharpness of autoradiographic images. DNA polymerase of the thermophilic bacterium, Thermus aquaticus (in place of Klenow fragment of E. coli DNA polymerase I) or a modified form of phage T7 DNA polymerase (sequenase) improves the technique.
Automated DNA sequencing
DNA sequencing in the recent years is carried out by an automated DNA sequencer. In this technique, flourescent tags are attached to chainterminating nucleotides (dideoxynucleotides). This tag gets incorporated into the DNA molecules, while terminating new strand synthesis. Four different fluorescent dyes are used to identify chain-terminating reactions in a sequencing gel. The DNA bands are separated by electrophoresis and detected by their fluorescence. Recently, four dyes that exhibit strong absorption in laser are in use for automated sequencing.
DNA chips (microarrays)
DNA chips or DNA microarrays are recent developments for DNA sequencing as result of advances made in automation and miniarization. A large number of DNA probes, each one with different sequence, are immobilized at defined positions on the solid surface, made up of either nylon or glass. The probes can be short DNA molecules such as cDNAs or synthetic oligonucleotides.
For the preparation of high density arrays, oligonucleotides are synthesized in situ on the surface of glass or silicon. This results in an oligonucleotide chip rather than a DNA chip.
Technique of DNA sequencing
A DNA chip carrying an array of different oligonucleotides can be used for DNA sequencing. For this purpose, a fluorescently labeled DNA test molecule, whose sequence is to be determined, is applied to the chip. Hybridization occurs between the complementary sequences of the test DNA molecule and oligonucleotides of the chip. The positions of these hybridizing oligonucleotides can be determined by confocal microscopy. Each hybridizing oligonucleotide represents an 8- nucleotide sequence that is present in the DNA probe. The sequence of the test DNA molecule can be deduced from the overlaps between the sequences of the hybridizing oligonucleotides (Fig.27.14).

Applications of DNA chips
There have been many successes with this relatively new technology of DNA chips. Some of them are listed.
• Identification of genes responsible for the development of nervous systems.
• Detection of genes responsible for inflammatory diseases.
• Construction of microarrays for every gene in the genome of E. coli, and almost all the genes of the yeast Saccharomyces cerevisiae.
• Expression of several genes in prokaryotes has been identified.
• Detection and screening of single nucleotide polymorphisms (SNPs).
• Rapid detection of microorganisms for environmental monitoring.
The future of DNA chips
The major limitation of DNA chips at present is the unavailability of complete genome arrays for higher eukaryotes, including humans. It is expected that within the next few years such DNA chips will be available. This will help the biotechnologists to capture the functional snapshots of the genome in action for higher organisms.
Polymerase chain reaction (DNA amplification)
The polymerase chain reaction (PCR) is a laboratory (in vitro) technique for generating large quantities of a specified DNA. Obviously, PCR is a cell-free amplification technique for synthesizing multiple identical copies (billions) of any DNA of interest. Developed in 1984 by Karry Mullis (Nobel Prize, 1993), PCR is now considered as a basic tool for the molecular biologist. As is a photocopier a basic requirement in an office, so is the PCR machine in a molecular biology laboratory!
Principle of PCR
The double-stranded DNA of interest is denatured to separate into two individual strands. Each strand is then allowed to hybridize with a primer (renaturation). The primer-template duplex is used for DNA synthesis (the enzyme-DNA polymerase). These three steps— denaturation, renaturation and synthesis are repeated again and again to generate multiple forms of target DNA.
Technique of PCR
The essential requirements for PCR are listed below
1. A target DNA (100–35,000 bp in length).
2. Two primers (synthetic oligonucleotides of 17–30 nucleotides length) that are complementary to regions flanking the target DNA.
3. Four deoxyribonucleotides (dATP, dCTP, dGTP, dTTP).
4. A DNA polymerase that can withstand at a temperature up to 95°C (i.e., thermostable).
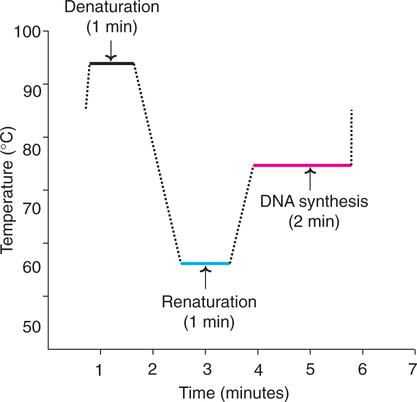
The actual technique of PCR involves repeated cycles for amplification of target DNA. Each cycle has three stages.
1. Denaturation : On raising the temperature to about 95°C for about one minute, the DNA gets denatured and the two strands separate.
2. Renaturation or annealing : As the temperature of the mixutre is slowly cooled to about 55°C, the primers base pair with the complementary regions flanking target DNA strands. This process is called renaturation or annealing. High concentration of primer ensures annealing between each DNA strand and the primer rather than the two strands of DNA.
3. Synthesis : The initiation of DNA synthesis occurs at 3′-hydroxyl end of each primer. The primers are extended by joining the bases complementary to DNA strands. The synthetic process in PCR is quite comparable to the DNA replication of the leading strand (Refer ![]() Chapter 24). However, the temperature has to be kept optimal as required by the enzyme DNA polymerase. For Taq DNA polymerase, the optimum temperature is around 75°C (for E. coli DNA polymerase, it is around 37°C). The reaction can be stopped by raising the temperature (to about 95°C).
Chapter 24). However, the temperature has to be kept optimal as required by the enzyme DNA polymerase. For Taq DNA polymerase, the optimum temperature is around 75°C (for E. coli DNA polymerase, it is around 37°C). The reaction can be stopped by raising the temperature (to about 95°C).
The 3 stages of PCR in relation to temperature and time are depicted in Fig.27.15. Each cycle of PCR takes about 3–5 minutes. In the normal practice, the PCR is carried out in an automated machine.
As is evident from the Fig.27.16 (cycle I), the new DNA strand joined to each primer is beyond the sequence that is complementary to the second primer. These new strands are referred to as long templates, and they will be used in the second cycle.

 indicate primers).
indicate primers).For the second cycle of PCR, the DNA strands (original + newly synthesized long template) are denatured, annealed with primers and subjected to DNA synthesis. At the end of second round, long templates, and short templates (DNA strands with primer sequence at one end, and sequence complementary to the other end primer) are formed.
In the third cycle of PCR, the original DNA strands along with long and short templates are the starting materials. The technique of denaturation, renaturation and synthesis are repeated. This procedure is repeated again and again for each cycle. It is estimated that at the end of 32nd cycle of PCR, about a million-fold target DNA is synthesized. The short templates possessing precisely the target DNA as doublestranded molecules accumulate.
Sources of DNA polymerase
In the original technique of PCR, Klenow fragment of E. coli DNA polymerase was used. This enzyme, gets denatured at higher temperature, therefore, fresh enzyme had to be added for each cycle. A breakthrough occurred (Lawyer 1989) with the introduction of Taq DNA polymerase from thermophilic bacterium, Thermus aquaticus. The Taq DNA polymerase is heat resistant, hence it is not necessary to freshly add this enzyme for each cycle of PCR.
Applications of PCR
The advent of PCR had, and continues to have tremendous impact on molecular biology. The applications of PCR are too many to be listed here. Some of them are selectively and very briefly described. Other applications of PCR are discussed at appropriate places.
PCR in comparative studies of genomes
The differences in the genomes of two organisms can be measured by PCR with random primers. The products are separated by electrophoresis for comparative identification. Two genomes from closely related organisms are expected to yield more similar bands.
PCR is very important in the study of evolutionary biology, more specifically referred to as phylogenetics. As a technique which can amplify even minute quantities of DNA from any source (hair, mummified tissues, bone, or any fossilized material), PCR has revolutionized the studies in palaentology and archaelogy. The movie ‘Jurassic Park’, has created public awareness of the potential applications of PCR!
Gene libraries
The collection of DNA fragments (specifically genes) from a particular species represents gene libraries. The creation or construction of gene libraries (broadly genomic libraries) is accomplished by isolating the complete genome (entire DNA from a cell) which is cut into fragments, and cloned in suitable vectors. Then the specific clone carrying the desired (target) DNA can be identified, isolated and characterized. In this manner, a library of genes or clones (appropriately considered as gene bank) for the entire genome of a species can be constructed.
Establishing a gene library for humans
The human cellular DNA (the entire genome) may be subjected to digestion by restriction endonucleases (e.g., EcoRI). The fragments formed on an average are of about 4 kb size. (i.e., 4000 nitrogenes bases). Each human chromosome, containing approximately 100,000 kb can be cut into about 25,000 DNA fragments. As the humans have 23 different chromosomes (24 in man), there are a total of 575,000 fragments of 4 kb length formed. Among these 575,000 DNA fragments is the DNA or gene of interest (say insulin gene).
Now is the selection of a vector and cloning process. E.coli, a harmless bacterium to humans is most commonly used. The plasmids from E. coli are isolated. They are digested by the same restriction enzyme as was used for cutting human genome to form open plasmids. The human chromosomal DNA fragments and open plasmids are joined to produce recombined plasmids. These plasmids contain different DNA fragments of humans. The recombined plasmids are inserted into E. coli and the cells multiply (Fig.27.17). The E. coli cells possess all the human DNA in fragments. It must, however be remembered that each E. coli cell contains different DNA fragments. All the E. coli cells put together collectively represent genomic library (containing about 575,000 DNA fragments).

Screening strategies
Once a DNA library is created, the clones (i.e., the cell lines) must be screened for identification of specific clones. The screening techniques are mostly based on the sequence of the clone or the structure/function of its product.
Screening by DNA hybridization
The target sequence in a DNA can be determined with a DNA probe (Fig.27.18). To start with, the double-stranded DNA of interest is converted into single strands by heat or alkali (denaturation). The two DNA strands are kept apart by binding to solid matrix such as nitrocellulose or nylon membrane. Now, the single strands of DNA probe (100–1,000 bp) labeled with radioisotope are added. Hybridization (i.e., base pairing) occurs between the complementary nucleotide sequences of the target DNA and the probe. For a stable base pairing, at least 80% of the bases in the two strands (target DNA and the probe) should be matching. The hybridized DNA can be detected by autoradiography.

 indicates radioisotope label in the DNA probe)
indicates radioisotope label in the DNA probe)(Note : DNA probe or gene probe represents a segment of DNA that is tagged with a label (i.e. isotope) so as to detect a complementary base sequence with sample DNA after hybridization)
Site-directed mutagenesis and protein engineering
Modifications in the DNA sequence of a gene are ideal to create a protein with desired properties. Site-directed mutagenesis is the technique for generating amino acid coding changes in the DNA (gene). By this approach specific (site-directed) change (mutagenesis) can be made in the base (or bases) of the gene to produce a desired enzyme. The net result in site-directed mutagenesis is incorporation of a desired amino acid (of one's choice) in place of a specific amino acid in a protein or a polypeptide. By employing this technique, enzymes that are more efficient and more suitable than the naturally occurring counterparts can be created for industrial applications. But it must be remembered that site-directed mutagenesis is a trial and error method that may or may not result in a better protein.
A couple of proteins developed by sitedirected mutagenesis and protein engineering are given next.
Tissue plasminogen activator (tPA)
Tissue plasminogen activator is therapeutically used to lyse the blood clots that cause myocardial infarction. Due to its shorter half-life (around 5 minutes), tPA has to be repeatedly administered. By replacing asparagine residue (at position 120) with glutamine, the half-life of tPA can be substantially increased. This is due to the fact that glutamine is less glycosylated than asparagine and this makes a difference in the half-life of tPA.
DNA in disease diagnosis and medical forensics
DNA, being the genetic material of the living organisms, contains the information that contributes to various characteristic features of the specific organism. Thus, the presence of a disease-causing pathogen can be detected by identifing a gene or a set of genes of the organism. Likewise an inherited genetic defect can be diagnosed by identifying the alterations in the gene. In the modern laboratory diagnostics, DNA analysis is a very useful and a sensitive tool.
The basic principles underlying the DNA diagnostic systems, and their use in the diagnosis of certain pathogenic and genetic diseases are described. Besides these, the various approaches for DNA fingerprinting (or DNA profiling) are also discussed.
Methods of DNA assay
The specific identification of the DNA sequence is absolutely essential in the laboratory diagnostics. This can be achieved by employing the following principles/tools.
Nucleic acid hybridization
Hybridization of nucleic acids (particularly DNA) is the basis for reliable DNA analysis. Hybridization is based on the principle that a single-stranded DNA molecule recognizes and specifically binds to a complementary DNA strand amid a mixture of other DNA strands. This is comparable to a specific key and lock relationship. The general procedure adopted for nucleic acid hybridization has been described (See p. 597 and Fig.27.18). Some more information is given below (Fig.27.19).

The single-stranded target DNA is bound to a membrane support. Now the DNA probe (single-stranded and labeled with a detector substance) is added. Under appropriate conditions (temperature, ionic strength), the DNA probe pairs with the complementary target DNA. The unbound DNA probe is removed. Sequence of nucleotides in the target DNA can be identified from the known sequence of DNA probe.
There are two types of DNA hybridizationradioactive and non-radioactive respectively using DNA probes labeled with isotopes and non-isotopes as detectors.
The DNA chip-microarray of gene probes
The DNA chip or Genechip contains thousands of DNA probes (4000,000 or even more) arranged on a small glass slide of the size of a postage stamp. By this recent and advanced approach, thousands of target DNA molecules can be scanned simultaneously.
Technique for use of DNA chip
The unknown DNA molecules are cut into fragments by restriction endonucleases. Fluorescent markers are attached to these DNA fragments. They are allowed to react with the probes of the DNA chip. Target DNA fragments with complementary sequences bind to DNA probes. The remaining DNA fragments are washed away. The target DNA pieces can be identified by their fluorescence emission by passing a laser beam. A computer is used to record the pattern of fluoresence emission and DNA identification.
The technique of employing DNA chips is very rapid, besides being sensitive and specific for the identification of several DNA fragments simultaneously. Scientists are trying to develop Genechips for the entire genome of an organism.
Applications of DNA chip
The presence of mutations in a DNA sequence can be conveniently identified. In fact, Genechip probe array has been successfully used for the detection of mutations in the p53 and BRCA I genes. Both these genes are involved in cancer (See p. 593 also).
DNA in the diagnosis of infectious diseases
The use of DNA analysis (by employing DNA probes) is a novel and revolutionary approach for specifically identifying the disease-causing pathogenic organisms. This is in contrast to the traditional methods of disease diagnosis by detection of enzymes, antibodies etc., besides the microscopic examination of pathogens. Although at present not in widespread use, DNA analysis may soon take over the traditional diagnostic tests in the years to come. Diagnosis of selected diseases by genetically engineered techniques or DNA probes or direct DNA analysis is briefly described.
Tuberculosis
Tuberculosis is caused by the bacterium Mycobacterium tuberculosis. The commonly used diagnostic tests for this disease are very slow, and sometimes may take several weeks. This is because M. tuberculosis multiplies very slowly (takes about 24 hrs. to double; E. coli takes just 20 minutes to double).
A novel diagnostic test for tuberculosis was developed by genetic engineering, and is illustrated in Fig.27.20. A gene from firefly, encoding the enzyme luciferase is introduced into the bacteriophage specific for M. tuberculosis. The bacteriophage is a bacterial virus, frequently referred to as luciferase reporter phage or mycophage. The genetically engineered phage is added to the culture of M. tuberculosis. The phage attaches to the bacterial cell wall, penetrates inside, and inserts its gene (along with luciferase gene) into the M. tuberculosis chromosome. The enzyme luciferase is produced by the bacterium. When luciferin and ATP are added to the culture medium, luciferase cleaves luciferin. This reaction is accompanied by a flash of light which can be detected by a luminometer. This diagnostic test is quite sensitive for the confirmation of tuberculosis.

The flash of light is specific for the identification of M. tuberculosis in the culture. For other bacteria, the genetically engineered phage cannot attach and enter in, hence no flash of light would be detected.
Malaria
Malaria, mainly caused by Plasmodium falciparum, and P. vivax, affects about one-third of the world's population. The commonly used laboratory tests for the diagnosis of malaria include microscopic examination of blood smears, and detection of antibodies in the circulation. While the former is time consuming and frequently gives false-negative tests, the latter cannot distinguish between the past and present infections.
A specific DNA diagnostic test for identification of the current infection of P. falciparum has been developed. This is carried out by using a DNA probe that can bind and hybridize with a DNA fragment of P. falciparum genome, and not with other species of Plasmodium. It is reported that this DNA probe can detect as little as 1 ng of P. falciparum in blood or 10 pg of its purified DNA.
DNA in the diagnosis of genetic diseases
Traditional laboratory tests for the diagnosis of genetic diseases are mostly based on the estimation of metabolites and/or enzymes. This is usually done after the onset of symptoms.
The laboratory tests based on DNA analysis can specifically diagnose the inherited diseases at the genetic level. DNA-based tests are useful to discover, well in advance, whether the individuals or their offsprings are at risk for any genetic disease. Further, such tests can also be employed for the prenatal diagnosis of hereditary disorders, besides identifying the carriers of genetic diseases.
Although not in routine use in the laboratory service, methods have been developed or being developed for the analysis of DNA in the diagnosis of several genetic diseases. These include sickle-cell anemia, cystic fibrosis, Duchenne's muscular dystrophy, Huntington's disease, fragile X syndrome, Alzheimer's disease, certain cancers (e.g. breast cancer, colon cancer), type II diabetes, obesity, Parkinson's disease and baldness.
Sickle-cell anemia
Sickle-cell anemia is a genetic disease characterized by the irregular sickle (crescent like) shape of the erythrocytes. Biochemically, this disease results in severe anemia and progressive damage to major organs in the body (heart, brain, lungs, joints).
Sickle-cell anemia occurs due to a single amino acid change in the β-chain of hemoglobin. Specifically, the amino acid glutamate at the 6th position of β-chain is replaced by valine. At the molecular level, sickle-cell anemia is due to a single-nucleotide change (A → T) in the β-globin gene of coding (or antisense) strand. In the normal β-globin gene the DNA sequence is CCTGAGGAG, while in sickle-cell anemia, the sequence is CCTGTGGAG. This single-base mutation can be detected by using restriction enzyme MstII to cut DNA fragments in and around β-globin gene, followed by the electrophoretic pattern of the DNA fragments formed. The change in the base from A to T in the β-globin gene destroys the recognition site (CCTGAGG) for MstII (Fig.27.21). Consequently, the DNA fragments formed from a sickle-cell anemia patient for β-globin gene differ from that of a normal person. Thus, sickle-cell anemia can be detected by digesting mutant and normal β-globin genes by restriction enzyme and performing a hybridization with a cloned β-globin DNA probe.

Gene banks —a novel concept
As the search continues by scientists for the identification of more and more genes responsible for various diseases, the enlightened public (particularly in the developed countries), is very keen to enjoy the fruits of this research outcome. As of now, DNA probes are available for the detection a limited number of diseases. Researchers continue to develop DNA probes for a large number of genetically predisposed disorders.
Gene banks are the centres for the storage of individual's DNAs for future use to diagnose diseases. For this purpose, the DNA isolated from a person's cells (usually white blood cells) is stored. As and when a DNA probe for the detection of a specific disease is available, the stored DNA can be used for the diagnosis or risk assessment of the said genetic disease.
In fact, some institutions have established gene banks. They store the DNA samples of the interested customers at a fee (one firm was charging $200) for a specified period (say around 20–25 years). For the risk assessment of any disease, it is advisable to have the DNAs from close relatives of at least 2–3 generations.
DNA fingerprinting or DNA profiling
DNA fingerprinting is the present day genetic detective in the practice of modern medical forensics. The underlying principles of DNA fingerprinting are briefly described.
The structure of each person's genome is unique. The only exception being monozygotic identical twins (twins developed from a single fertilized ovum). The unique nature of genome structure provides a good opportunity for the specific identification of an individual.
It may be remembered here that in the traditional fingerprint technique, the individual is identified by preparing an ink impression of the skin folds at the tip of the person's finger. This is based on the fact that the nature of these skin folds is genetically determined, and thus the fingerprint is unique for an individual. In contrast, the DNA fingerprint is an analysis of the nitrogenous base sequence in the DNA of an individual.
History and terminology
The original DNA fingerprinting technique was developed by Alec Jaffreys in 1985. Although the DNA fingerprinting is commonly used, a more general term DNA profiling is preferred. This is due to the fact that a wide range of tests can be carried out by DNA sequencing with improved technology.
Applications of DNA fingerprinting
The amount of DNA required for DNA fingerprint is remarkably small. The minute quantities of DNA from blood strains, body fluids, hair fiber or skin fragments are enough. Polymerase chain reaction is used to amplify this DNA for use in fingerprinting. DNA profiling has wide range of applications—most of them related to medical forensics. Some important ones are listed below.
In general, the fingerprinting technique is carried out by collecting the DNA from a suspect (or a person in a paternity or immigration dispute) and matching it with that of a reference sample (from the victim of a crime, or a close relative in a civil case).
DNA markers in disease diagnosis and fingerprinting
The DNA markers are highly useful for genetic mapping of genomes. There are four types of DNA sequences which can be used as markers.
1. Restriction fragment length polymorphisms (RFLPs, pronounced as rif-lips).
2. Minisatellites or variable number tandem repeats (VNTRs, pronounced as vinters).
3. Microsatellites or simple tandem repeats (STRs).
4. Single nucleotide polymorphisms (SNPs, pronounced as snips).
The general aspects of the above DNA markers are described along with their utility in disease diagnosis and DNA fingerprinting.
Restriction fragment length polymorphisms (RFLPs)
A RFLP represents a stretch of DNA that serves as a marker for mapping a specified gene. RFLPs are located randomly throughout a person's chromosomes and have no apparent function.
A DNA molecule can be cut into different fragments by a group of enzymes called restriction endonucleases (See Table 27.1). These fragments are called polymorphisms (literally means many forms).
An outline of RFLP is depicted in Fig.27.22. The DNA molecule 1 has three restriction sites (R1, R2, R3), and when cleaved by restriction endonucleases forms 4 fragments. Let us now consider DNA 2 with an inherited mutation (or a genetic change) that has altered some base pairs. As a result, the site (R2) for the recognition by restriction endonuclease is lost. This DNA molecule 2 when cut by restriction endonuclease forms only 3 fragments (instead of 4 in DNA 1).
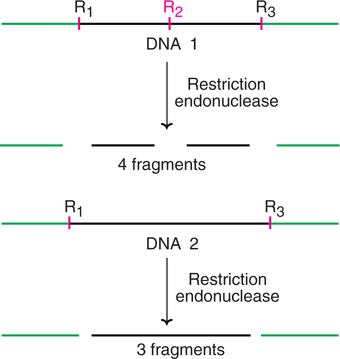
As is evident from the above description, a stretch of DNA exists in fragments of various lengths (polymorphisms), derived by the action of restriction enzymes, hence the name restriction fragment length polymorphisms.
RFLPs in the diagnosis of diseases
If the RFLP lies within or even close to the locus of a gene that causes a particular disease, it is possible to trace the defective gene by the analysis of RFLP in DNA. The person's cellular DNA is isolated and treated with restriction enzymes. The DNA fragments so obtained are separated by electrophoresis. The RFLP patterns of the disease suspected individuals can be compared with that of normal people (preferably with the relatives in the same family). By this approach, it is possible to determine whether the individual has the marker RFLP and the disease gene. With 95% certainity, RFLPs can detect single gene-based diseases.
Methods of RFLP scoring
Two methods are in common use for the detection of RFLPs (Fig.27.23).
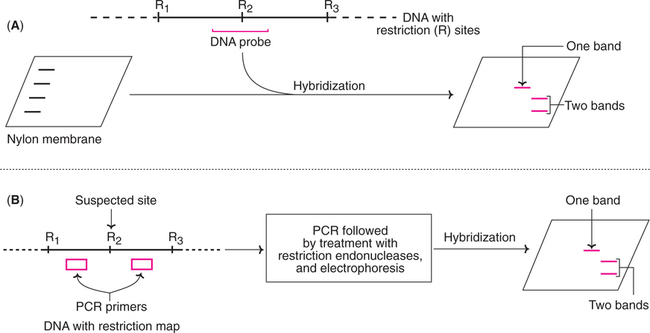
1. Southern hybridization : The DNA is digested with appropriate restriction enzyme, and separated by agarose gel electrophoresis. The so obtained DNA fragments are transferred to a nylon membrane. A DNA probe that spans the suspected restriction site is now added, and the hybridized bands are detected by autoradiograph. If the restriction site is absent, then only a single restriction fragment is detected. If the site is present, then two fragments are detected (Fig.27.23A).
2. Polymerase chain reaction : RFLPs can also be scored by PCR. For this purpose, PCR primers that can anneal on either side of the suspected restriction site are used. After amplification by PCR, the DNA molecules are treated with restriction enzyme and then analysed by agarose gel electrophoresis. If the restriction site is absent only one band is seen, while two bands are found if the site is found (Fig.27.23B).
Variable Number Tandem Repeats (VNTRs)
VNTRs, also known as minisatellites, like RFLPs, are DNA fragments of different length. The main difference is that RFLPs develop from random mutations at the site of restriction enzyme activity while VNTRs are formed due to different number of base sequences between two points of a DNA molecule. In general, VNTRs are made up of tandem repeats of short base sequences (10–100 base pairs). The number of elements in a given region may vary, hence they are known as variable number tandem repeats.
An individual's genome has many different VNTRs and RFLPs which are unique to the individual. The pattern of VNTRs and RFLPs forms the basis of DNA fingerprinting or DNA profiling.
In the Fig.27.24, two different DNA molecules with different number of copies (bands) of VNTRs are shown. When these molecules are subjected to restriction endonuclease action (at two sites R1 and R2), the VNTR sequences are released, and they can be detected due to variability in repeat sequence copies. These can be used in mapping of genomes, besides their utility in DNA fingerprinting.
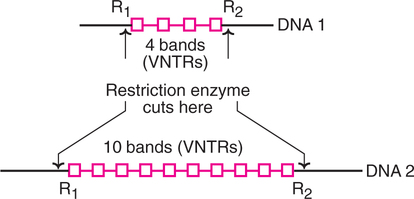
VNTRs are useful for the detection of certain genetic diseases associated with alterations in the degree of repetition of microsatellites e.g. Huntington's chorea is a disorder which is found when the VNTRs exceed 40 repeat units.
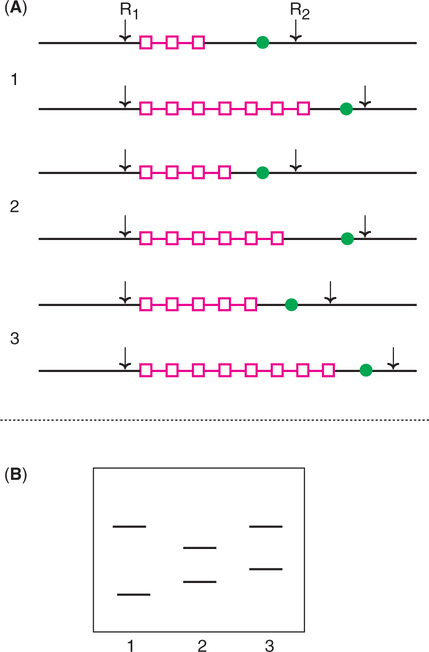
Use of RFLPs and VNTRs in genetic fingerprinting
RFLPs caused by variations in the number of VNTRs between two restriction sites can be detected (Fig.27.25). The DNAs from three individuals with different VNTRs are cut by the specific restriction endonuclease. The DNA fragments are separated by electrophoresis, and identified after hybridization with a probe complementary to a specific sequence on the fragments.
Microsatellites (Simple tandem repeats)
Microsatellites are short repeat units (10–30 copies) usually composed of dinucleotide or tetranucleotide units. These simple tandem repeats (STRs) are more popular than minisatellites (VNTRs) as DNA markers for two reasons.
1. Microsatellites are evenly distributed throughout the genome.
2. PCR can be effectively and conveniently used to identify the length of polymorphism.
Two variants (alleles) of DNA molecules with 5 and 10 repeating units of a dimer nucleotides (GA) are depicted in Fig.27.26.

By use of PCR, the region surrounding the microsatellites is amplified, separated by agarose gel electrophoresis and identified.
Single Nucleotide Polymorphisms (SNPs)
SNPs represent the positions in the genome where some individuals have one nucleotide (e.g. G) while others have a different nucleotide (e.g. C). There are large numbers of SNPs in genomes. It is estimated that the human genome contains at least 3 million SNPs. Some of these SNPs may give rise to RFLPs.
SNPs are highly useful as DNA markers since there is no need for gel electrophoresis and this saves a lot of time and labour. The detection of SNPs is based on the oligonucleotide hybridization analysis (Fig.27.27).
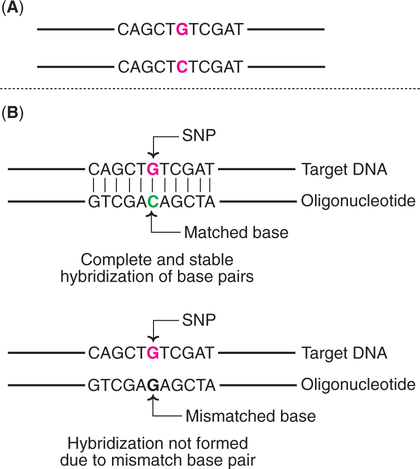
An oligonucleotide is a short single-stranded DNA molecule, synthesized in the laboratory with a length not usually exceeding 50 nucleotides. Under appropriate conditions, this nucleotide sequence will hybridize with a target DNA strand if both have completely base paired structure. Even a single mismatch in base pair will not allow the hybridization to occur.
DNA chip technology is most commonly used to screen SNPs hybridization with oligonucleotide (See p. 593).
Current technology of DNA Fingerprinting
In the forensic analysis of DNA, the original techniques based on RFLPs and VNTRs are now largely replaced by microsatellites (short tandem repeats). The basic principle involves the amplification of microsatellites by polymerase chain reaction followed by their detection.
It is now possible to generate a DNA profile by automated DNA detection system (comparable to the DNA sequencing equipment).
Pharmaceutical products of DNA technology
The advent of recombinant DNA technology heralded a new chapter for the production of a wide range of therapeutic agents in sufficient quantities for human use. The commercial exploitation of recombinant DNA (rDNA) technology began in late 1970s by a few biotechnological companies to produce proteins. There are at least 400 different proteins being produced (by DNA technology) which may serve as therapeutic agents for humans. A selected list of some important human proteins produced by recombinant DNA technology potential for the treatment of human disorders is given in Table 27.5. As of now, only a selected few of them (around 30) have been approved for human use, and the most important among these are given in Table 27.6.
Table 27.5
A selected list of human proteins produced by recombinant DNA technology for treatment of human disorders
| Disorder | Recombinant protein(s) |
| Anemia | Hemoglobin, erythropoietin |
| Asthma | Interleukin-I receptor |
| Atherosclerosis | Platelet-derived growth factor |
| Delivery | Relaxin |
| Blood clots | Tissue plasminogen activator, urokinase |
| Burns | Epidermal growth factor |
| Cancer | Interferons, tumor necrosis factor, colony stimulating factors, interleukins, lymphotoxin, macrophageactivating factor |
| Diabetes | Insulin, insulin-like growth factor |
| Emphysema | α1-Antitrypsin |
| Female infertility | Chorionic gonadotropin |
| Free radical damage (minimizing) | Superoxide dismutase |
| Growth defects | Growth hormone, growth hormone-releasing factor, somatomedin-C |
| Heart attacks | Prourokinase |
| Hemophilia A | Factor VIII |
| Hemophilia B | Factor IX |
| Hepatitis B | Hepatitis B vaccine |
| Hypoalbuminemia | Serum albumin |
| Immune disorders | Interleukins, β-cell growth factors |
| Kidney disorders | Erythropoietin |
| Lou Gehrig's disease (amytrophic lateral sclerosis) | Brain-derived neurotropic factor |
| Multiple sclerosis | Interferons (α, β, γ) |
| Nerve damage | Nerve growth factor |
| Osteomalacia | Calcitonin |
| Pain | Endorphins and enkephalins |
| Rheumatic disease | Adrenocorticotropic hormone |
| Ulcers | Urogastrone |
| Viral infections | Interferons (α, β, γ) |
Table 27.6
A selected list of rDNA-derived therapeutic agents (approved by FDA) with trade names and their applications in humans
| rDNA product | Trade name(s) | Applications/uses |
| Insulin | Humulin | Diabetes |
| Growth hormone | Protropin/Humatrope | Pituitary dwarfism |
| α-Interferon | Intron A | Hairy cell leukemia |
| Hepatitis B vaccine | Recombinax HB/Engerix B | Hepatitis B |
| Tissue plasminogen activator | Activase | Myocardial infarction |
| Factor VIII | Kogenate/Recombinate | Hemophilia |
| DNase | Pulmozyme | Cystic fibrosis |
| Erythropoietin | Epogen/Procrit | Severe anemia with kidney damage |
Insulin and diabetes
Diabetes mellitus is characterized by increased blood glucose concentration (hyperglycemia) which occurs due to insufficient or inefficient insulin. In the early years, insulin isolated and purified from the pancreases of pigs and cows was used for the treatment of severe diabetics. This often resulted in allergies. Recombinant DNA technology has become a boon to diabetic patients.
Production of recombinant insulin
Attemps to produce insulin by recombinant DNA technology started in late 1970s. The basic technique consisted of inserting human insulin gene and the promoter gene of lac operon on to the plasmids of E. coli. By this method human insulin was produced. It was in July 1980, seventeen human volunteers were, for the first time, administered recombinant insulin for treatment of diabetes at Guy's Hospital, London. And in fact, insulin was the first ever pharmaceutical product of recombinant DNA technology administered to humans. Recombinant insulin worked well, and this gave hope to scientists that DNA technology could be successfully employed to produce substances of medical and commercial importance. An approval, by the concerned authorities, for using recombinant insulin for the treatment of diabetes mellitus was given in 1982. And in 1986, Eli Lilly company received approval to market human insulin under the trade name Humulin.
Technique for production of recombinant insulin
The orginal technique (described briefly above) of insulin synthesis in E. coli has undergone several changes, for improving the yield. e.g. addition of signal peptide, synthesis of A and B chains separately etc.
The procedure employed for the synthesis of two insulin chains A and B is illustrated in Fig.27.28. The genes for insulin A chain and B chain are separately inserted to the plasmids of two different E. coli cultures. The lac operon system (consisting of inducer gene, promoter gene, operator gene and structural gene Z for β-galactosidase) is used to express both the genes. The presence of lactose in the culture medium induces the synthesis of insulin A and B chains in separate cultures. The so formed insulin chains can be isolated, purified and joined together to give a full-fledged human insulin.

Recombinant vaccines
Recombinant DNA technology in recent years, has become a boon to produce new generation vaccines. By this approach, some of the limitations (low yield, high cost, side effects) of traditional vaccine production could be overcome.
The list of diseases for which recombinant vaccines are developed or being developed is given in Table 22.7. It may be stated here that due to very stringent regulatory requirements to use in humans, the new generation vaccines are first tried in animals, and it may take some more years before most of them are approved for use in humans.
Table 27.7
A selected list of diseases along with the pathogenic organisms for which recombinant vaccines are developed or being developed
| Disease | Pathogenic organism |
| Viral diseases | |
| Acute infantile gastroenteritis | Rotavirus |
| Acute respiratory diseases | Influenza A and B viruses |
| AIDS | Human immunodeficiency virus |
| Chicken pox | Varicella–zoster virus |
| Encephalitis | Japanese encephalitis virus |
| Genital ulcers | Herpes simplex virus type–2 |
| Hemorrhagic fever | Dengue virus |
| Liver damage | Hepatitis A virus |
| Liver damage | Hepatitis B virus |
| Upper and lower respiratory tract lesions | Yellow fever virus |
| Bacterial diseases | |
| Cholera | Vibrio cholerae |
| Diarrhea | E. coli |
| Dysentery | Shigella strain |
| Gonorrhea | Niesseria gonorroheae |
| Leprosy | Mycobacterium leprae |
| Meningitis | Neisseria meningitidis |
| Pneumonia | Streptococcus pneumoniae |
| Rheumatic fever | Streptococcus group A |
| Tetanus | Clostridium tetani |
| Tuberculosis | Mycobacterium tuberculosis |
| Typhoid | Salmonella typhi |
| Urogenital tract infection | Streptococcus group B |
| Parasitic diseases | |
| Filariasis | Wuchereria bancrofti |
| Malaria | Plasmodium sp |
| River blindness | Onchocerca volvulus |
| Schistosomiasis | Schistosoma mansoni |
| Sleeping sickness | Trypanosoma sp |
Hepatitis B vaccine—the first synthetic vaccine
In 1987, the recombinant vaccine for hepatitis B (i.e. HBsAg) became the first synthetic vaccine for public use. It was marketed by trade names Recombivax and Engerix-B. Hepatitis B vaccine is safe to use, very effective and produces no allergic reactions. For these reasons, this recombinant vaccine has been in use since 1987. The individuals must be administered three doses over a period of six months. Immunization against hepatitis B is strongly recommended to anyone coming in contact with blood or body secretions. All the health professionals—physicians, surgeons, medical laboratory technicians, nurses, dentists, besides police officers, firefighters etc., must get vaccinated against hepatitis B.
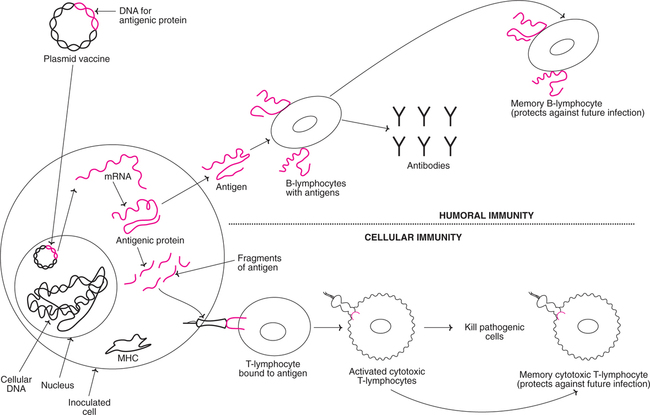
DNA Vaccines (genetic immunization)
Genetic immunization by using DNA vaccines is a novel approach that came into being in 1990. The immune response of the body is stimulated by a DNA molecule sequence of pathogen's genome. This DNA is basically a bacterial plasmid engineered to include the sequence of an antigenic protein from the pathogen. After its entry into different cell types, this DNA can be expressed there using cellular transcription and translation machinary. Thus, DNA vaccines behave like viruses. They cannot, however become infectious due to limited amout of genetic information they contain.
DNA vaccine—plasmids can be administered to the animals by one of the following delivery methods.
Dna vaccine and immunity
An illustration of a DNA vaccine and the mechanism of its action in developing immunity is given in Fig.27.29. The plasmid vaccine carrying the DNA (gene) for antigenic protein enters the nucleus of the inoculated target cell of the host. This DNA produces RNA, and in turn the specific antigenic protein. The antigen can act directly for developing humoral immunity or as fragments in association with major histocompatability class (MHC) molecules for developing cellular immunity. DNA vaccines may hold some promise for vaccination against cancer, HIV, malaria, tuberculosis etc.

 Biotechnology is a newly discovered discipline for age-old practices (e.g. preparation of curd, wine, beer), with special emphasis on genetic manipulations.
Biotechnology is a newly discovered discipline for age-old practices (e.g. preparation of curd, wine, beer), with special emphasis on genetic manipulations.
Human artificial chromosome (HAC) is a synthetic vector, possessing the characteristics of human chromosome. HAC is capable of carrying large-sized human genes that may be useful in gene therapy.
Southern blotting technique (that specifically detects DNA) is employed for the identification of thieves, rapists, and settlement of parenthood.
Polymerase chain reaction is useful for the diagnosis of inherited diseases, in DNA sequencing, and in forensic medicine.
By employing site-directed mutagenesis, it is possible to produce more efficient and more suitable enzymes for therapeutic and industrial purposes.
The analysis of genetic material DNA (gene/genes) is employed for the diagnosis of certain diseases, and in medical forensics e.g. AIDS, sickle-cell anemia, certain cancers, DNA fingerprinting.
The pharmaceutical products of rDNA technology have revolutionized the treatment of certain diseases e.g. diabetes, asthma, atherosclerosis, heart attacks, hemophilia.
Recombinant vaccine for hepatitis B is the first synthetic vaccine. It is effective, safe and produces no allergic reactions.
Genetic immunization by using DNA vaccines is a novel concept. It has been shown that the immune response (humoral and cellular) of the body can be stimulated by a DNA molecule.
Transgenic mice that serve as animal models for human diseases have been developed. These include human mouse (model for immune system), Alzheimer's mouse, oncomouse (model for cancer), prostate mouse, knockout mice (for allergy, transplantation etc.).
Transgenic animals serve as bioreactors for the production of therapeutically important proteins e.g. interferon, lactoferrin, urokinase.
Certain pet animals (cats, dogs) are being cloned by some companies.
Transgenic animals
With the advent of modern biotechnology, it is now possible to carry out manipulations at the genetic level to get the desired characteristics in animals. Transgenesis refers to the phenomenon of introduction of exogeneous DNA into the genome to create and maintain a stable heritable character. The foreign DNA that is introduced is called transgene. And the animal whose genome is altered by adding one or more transgenes is said to be transgenic. The transgenes behave like other genes present in the animals’ genome, and are passed on to the offsprings. Thus, transgenic animals are genetically engineered or genetically modified organisms (GMOs) with a new heritable character.
Importance of transgenic animals—general
Transgenesis has now become a powerful tool for studying the gene expression and developmental processes in higher organisms, besides the improvement in their genetic characteristics. Transgenic animals serve as good models for understanding the human diseases. Further, several proteins produced by transgenic animals are important for medical and pharmaceutical applications. Thus, the transgenic farm animals are a part of the lucrative world-wide biotechnology industry, with great benefits to mankind.
Transgenic mice and their applications
Mouse, although not close to humans in its biology, has been and continues to be the most exploited animal model in transgenesis experiments. The common feature between man and mouse is that both are mammals. Transgenic mice are extensively used as animal models for understanding human diseases, and for the production of therapeutic agents. Adequate care, however, must be exercised before extrapolating data of transgenic mice to humans.
Mouse models for several human diseases (cancers, muscular dystrophy, arthritis, Alzheimer's disease, hypertension, allergy, coronary heart disease, endocrine diseases, neurodegenerative disorders etc.) have been developed. A selected few of them are listed.
• The human mouse, the transgenic mouse that displays human immune system.
• The Alzheimer's mouse to understand the pathological basis of Alzheimer's disease.
• The oncomouse, the animal model for cancer.
• The prostate mouse, the transgenic mouse to understand prostate cancer.
• The knockout mice, (developed by eliminating specific genes) for certain diseases e.g. SCID mouse, knockout mouse for transplantation.
Animal bioreactors
Transgenesis is wonderfully utilized for production of proteins for pharmaceutical and medical use. In fact, any protein synthesized in the human body can be made in the transgenic animals, provided that the genes are correctly programmed. The advantage with transgenic animals is to produce scarce human proteins in huge quantities. Thus, the animals serving as factories for production of biologically important products are referred to as animal bioreactors or sometimes pharm animals. Some transgenic animals that serve as bioreactors are listed
Dolly – the transgenic clone
Dolly, the first ever mammal clone was developed by Wilmut and Campbell in 1997. It is a sheep (female lamb) with a mother and no father.
The technique primarily involves nuclear transfer and the phenomenon of totipotency. The character of a cell to develop into different cells, tissues, organs, and finally an organism is referred to as totipotency or pluripotency. Totipotency is the basic character of embryonic cells. As the embryo develops, the cells specialize to finally give the whole organism. As such, the cells of an adult lack totipotency. Totipotency was induced into the adult cells for developing Dolly.
The cloning of sheep for producing Dolly, illustrated in Fig.27.30, is briefly described here. The mammary gland cells from a donor ewe were isolated. They were subjected to total nutrient deprivation (starvation) for five days. By this process, the mammary cells abandon their normal growth cycle, enter a dormant stage and regain totipotency character. An ovum (egg cell) was taken from another ewe, and its nucleus was removed to form an enucleated ovum. The dormant mammary gland cell and the enucleated ovum were fused by pulse electricity. The mammary cell outer membrane was broken, allowing the ovum to envelope the nucleus. The fused cell, as it had gained totipotency, can multiply and develop into an embryo. This embryo was then implanted into another ewe which served as a surrogate/foster mother. Five months later, Dolly was born.

As reported by Wilmut and Campbell, they fused 277 ovum cells, achieved 13 pregnancies, and of these only one pregnancy resulted in live birth of the offspring-Dolly.
Cloning of pet animals
Some of the companies involved in transgenic experiments have started cloning pet animals like cats and dogs. Little Nicky was the first pet cat that was cloned at a cost of $50,00 by an American company (in Dec. 2004). More cloned cats and dogs will be made available to interested parties (who can afford) in due course.
Some people who own pet animals are interested to continue the same pets which is possible through cloning. There is some opposition to this approach as the cloned animals are less healthy, and have shorter life span, besides the high cost factor.
Biotechnology and society
Advances in biotechnology, and their applications are most frequently associated with controversies. Based on their perception to biotechnology, the people may be grouped into three broad categories.
1. Strong opponents who oppose the new technology, as it will give rise to problems, issues and concerns humans have never faced before. They consider biotechnology as an unnatural manipulative technology.
2. Strong proponents who consider that the biotechnology will provide untold benefits to society. They argue that for centuries the society has safely used the products and processes of biotechnology.
3. A neutral group of people who have a balanced approach to biotechnology. This group believes that research on biotechnology (with regulatory systems), and extending its fruits to the society should be pursued with a cautious approach.
Benefits of biotechnology
The fruits of biotechnology are beneficial to the fields of healthcare, agriculture, food production, manufacture of industrial enzymes and appropriate environmental management.
It is a fact that modern technology in various forms is woven tightly into the fabric of our lives. Our day-to-day life is inseparable from technology. Imagine life about 1–2 centuries ago where there was no electricity, no running water, sewage in the streets, unpredictable food supply and an expected life span of less than 40 years. Undoubtedly, technology has largely contributed to the present day world we live in. Many people consider biotechnology as a technology that will improve the quality of life in every country, besides maintaining living standards at a reasonably higher level.
ELSI of biotechnology
Why so much uproar and negativity to biotechnology? This is mainly because the major part of the modern biotechnology deals with genetic manipulations. These unnatural genetic manipulations, as many people fear, may lead to unknown consequences.
ELSI is the short form to represent the ethical, legal and social implications of biotechnology. ELSI broadly covers the relationship between biotechnology and society with particular reference to ethical and legal aspects.
Risks and ethics of biotechnology
The modern biotechnology deals with genetic manipulations of viruses, bacteria, plants, animals, fish and birds. Introduction of foreign genes into various organisms raises concerns about the safety, ethics and unforeseen consequences. Some of the popular phrases used in the media while referring to experiments on recombinant DNA technology are listed.
The major apprehension of genetic engineering is that through recombinant DNA experiments, unique microorganisms or viruses (either inadvertently, or sometimes deliberately for the purpose of war) may be developed that would cause epidemics and environmental catastrophes. Due to these fears, the regulatory guidelines for research dealing with DNA manipulation were very stringent in the earlier years.
So far, risk assessment studies have failed to demonstrate any hazardous properties acquired by host cells/organisms due to transfer of DNA. Thus, the fears of genetic manipulations may be unfounded to a large extent. Consequently, there has been some relaxation in the regulatory guidelines for recombinant DNA research.
It is now widely accepted that biotechnology is certainly beneficial to humans. But it should not cause problems of safety to people and environment, and create unacceptable social, moral and ethical issues.
Summary
1. Recombinant DNA (rDNA) technology is primarily concerned with the manipulation of genetic material (DNA) to achieve the desired goal in a pre-determined way.
2. The procedure for rDNA technology involves molecular tools (enzymes e.g. restriction endonucleases), host cells (E. coli, S. cerevisiae), vectors (plasmids, bacteriophages), gene transfer (transformation, electroporation) and the strategies of gene cloning.
3. Blotting techniques are employed for the identification of desired DNA (Southern blot), RNA (Northern blot), and protein (Western blot).
4. Polymerase chain reaction is an in vitro technique for generating large quantities of a specified DNA i.e. cell-free amplification.
5. Gene libraries or genomic libraries represents the collection of DNA fragments (i.e. genes) from a genome of a particular species.
6. Site-directed mutagenesis is the technique for generating amino acid coding changes in the DNA (gene) to produce a desired protein/enzyme.
7. Analysis of DNA (i.e. detection of gene/genes) can be used as a diagnostic system for the detection of many pathogenic and genetic diseases e.g. tuberculosis, malaria, AIDS, sickle-cell anemia, certain cancers.
8. DNA fingerprinting or DNA profiling is the present day genetic detective in the practice of modern medical forensics. Four types of DNA markers are used in DNA fingerprinting–RFLFs, VNTRs, STRs, and SNPs.
9. Many pharmaceutical compounds of health importance (for disease treatment) are being produced by rDNA technology e.g. insulin, growth hormone, interferons, erythropoietin, hepatitis B vaccine.
10. Transgenic animals can be developed by introducing a foreign DNA (transgene). These animals are genetically modified or engineered with new heritable characters e.g. oncomouse, knockout mouse, prostate mouse.