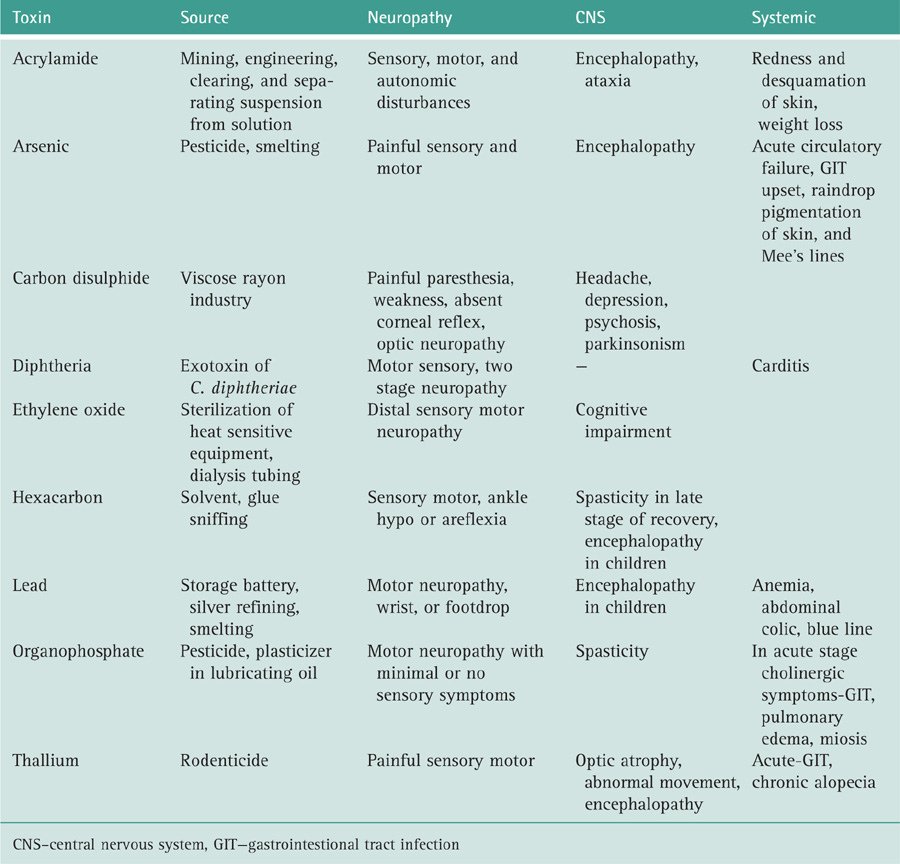
AAN. Consensus Report. Report and recommendations of the San Antonio Conference on diabetic neuropathy. American diabetes Association. American Academy of Neurology. 1988;11:592-597. Diabetes Care
Albers JW. AAEM Case report #4. Guillain Barre Syndrome. Muscle Nerve. 1989;12:705.
Albers JW, Donofrio PD, McGonagle TK. Sequential electrodiagnostic abnormalities in acute inflammatory demyelinating polyradiculoneuropathy.
Muscle Nerve. 1985;8:5128.
Albers JW, Kelly JJ. Acquired inflammatory demyelinating polyneuropathies: clinical and electrodiagnostic features. Muscle Nerve. 1989;12:435.
Albers JW, Robertson WC, Daube JR. Electrodiagnostic findings in acute porphyric neuropathy. Muscle Nerve. 1978;1:292.
Ad Hoc Subcommittee of the American Academy of Neurology AIDS Task Force. Criteria for diagnosis of chronic inflammatory demyelinating polyneuropathy (CIDP). Neurology. 1991;41:617.
Albert DA, Rimon D, Silverstein MD. The diagnosis of polyarteritis nodosa: a literature based decision analysis approach. Arthritis Rheumatoid. 1988;31:1117.
Arezzo JC. The use of electrophysiology for the assessment of diabetic neuropathy. Neurosci Res Comm. 1997;21:13.
Arnason BGW. Acute inflammatory demyelinating polyradiculoneuropathy. In: Dyck PJ, Thomas PK, Lambert EH, Bunge R, eds. Peripheral Neuropathy. Philadelphia: WB Saunders; 1984:2050. Vol. II
Asbury AK. Proximal diabetic neuropathy. Ann Neurol. 1977;2:179-180.
Asbury AK, Amason BG, Karp KR, et al. Criteria for diagnosis of Guillain Barre Syndrome. Ann Neurol. 1978;3:565.
Badenoch J, Bedford PD, Evans JR. Massive diverticulosis of the small intestine with steatorrhea and megaloblastic anemia. Q J Med. 1955;24:321.
Bailey RO, Baltch AL, Venkatesh R, et al. Sensory motor neuropathy associated with AIDS. Neurology. 1988;38:886.
Balagtas-Balmaseda OM, Grabois M, Balmaseda PF, et al. Cubital tunnel syndrome in rheumatoid arthritis. Arch Phys Med Rehabil. 1983;64:163.
Bansal R, Kalita J, Misra UK. Pattern of sensory conduction in Guillain–Barre syndrome. Electromyogr Clin Neurophysiol. 2001;41:433.
Bedlack RS, Wu T, Hammons S, et al. MINGE neuropathy: five cases mimicking chronic inflammatory demyelinating polyneuropathy. Muscle Nerve. 2004;29:364.
Behse F, Buchthal F, Carlsen F. Nerve biopsy and conduction studies in diabetic neuropathy. J Neurol Neurosurg Psychiat. 1977;40:1072.
Ben Hamida C, Doerfinger N, Blel S, et al. Localisation of Friedrich’s ataxia phenotype with selective vitamin E deficiency to chromosome 8q by homozygosity mapping. Nature Genetics. 1993;5:195.
Benstead TJ, Kuntz NL, Miller RG, et al. The electro physiologic profile of Dejerine Sotta’s disease (HMSN III). Muscle Nerve. 1990;13:586.
Berciano J, Combarros O, Callija J, et al. The application of nerve conduction and clinical studies to genetic counselling in hereditary motor and sensory neuropathy type I. Muscle Nerve. 1984;12:302.
Berek K, Margreiter J, Willeit J, et al. Polyneuropathies in critically ill patients: a prospective evaluation. Intensive Care Med. 1996;22:849.
Bolton CF, Gilbert JJ, Hahn AF, et al. Polyneuropathy in critically ill patients. J Neurol Neurosurg Psychiat. 1984;87:1223.
Bolton CF. Clinical neurophysiology of the respiratory system. AAEM minimonograph #40. Muscle Nerve. 1993;16:809.
Bolton CF. Critical illness polyneuropathy. In: Asbury AK, Thomas PK, eds. Peripheral Nerve Disorders. Oxford: Butterworth Heinemann; 1995:262. Vol. 2
Bolton CF. Peripheral neuropathies associated with chronic renal failure. Can J Neurol Sci. 1980;7:89.
Bouche P, Leger JM, Travers MA. Peripheral neuropathy in systemic vasculitis: clinical and electrophysiologic study of 22 patients. Neurology. 1986;36:1598.
Bradley WG, Verma A. Painful vasculitic neuropathy in HIV-I infection: relief of pain by prednisone therapy. Neurology. 1996;47:1446.
Breuer AC. Critical care neuropathy an outdated concept. Muscle Nerve. 1999;22:419.
Briemberg H, Levin KH, Amato AA. Multifocal conduction block in peripheral nerve vasculitis. J Clin Neuromuscl Dis. 2002;3:1353-1358.
Britt RP, Harper C, Spray GH. Megaloblastic anemia among Indians in Britain. Q J Med. 1971;40:499.
Brittand ST, Young RJ, Sharma AK, et al. Acute and remitting diabetic polyneuropathy: a comparison of peripheral nerve fibre pathology. Pain. 1992;48:361.
Bromberg MB, Albers JW. Patterns of sensory nerve conduction abnormalities in demyelinating and axonal peripheral nerve disorders. Muscle Nerve. 1993;16:262.
Brown MJ, Greene DA. Diabetic neuropathy: pathophysiology and management. In: Asbury AK, Gilliatt RW, eds. Peripheral Nerve Disorders. London: Butterworth; 1984:126.
Brown MJ, Pleasure DE, Asbury AK. Painful diabetic neuropathy. a morphometric study. Arch Neurol. 1976;33:164.
Brown WF, Feasby TE. Conduction block and Guillain Barre polyneuropathy. Brain. 1984;107:219.
Brown WF, Snow R. Patterns and severity of conduction abnormalities in Guillain Barre syndrome. J Neurol Neurosurg Psychiat. 1991;54:768.
Bruggen JP, Vander Meche FGA, de Gager AEJ, et al. Ophthalmoplegic and lower cranial nerve variants merge into each other and into classical Guillain–Barre syndrome. Muscle Nerve. 1998;21:239.
Bruyn GW, Garland H. Neuropathies of endocrine origin. In: Vinken PJ, Bruyn GW, eds. Handbook of Clinical Neurology. Amsterdam: North Holland; 1970:29.
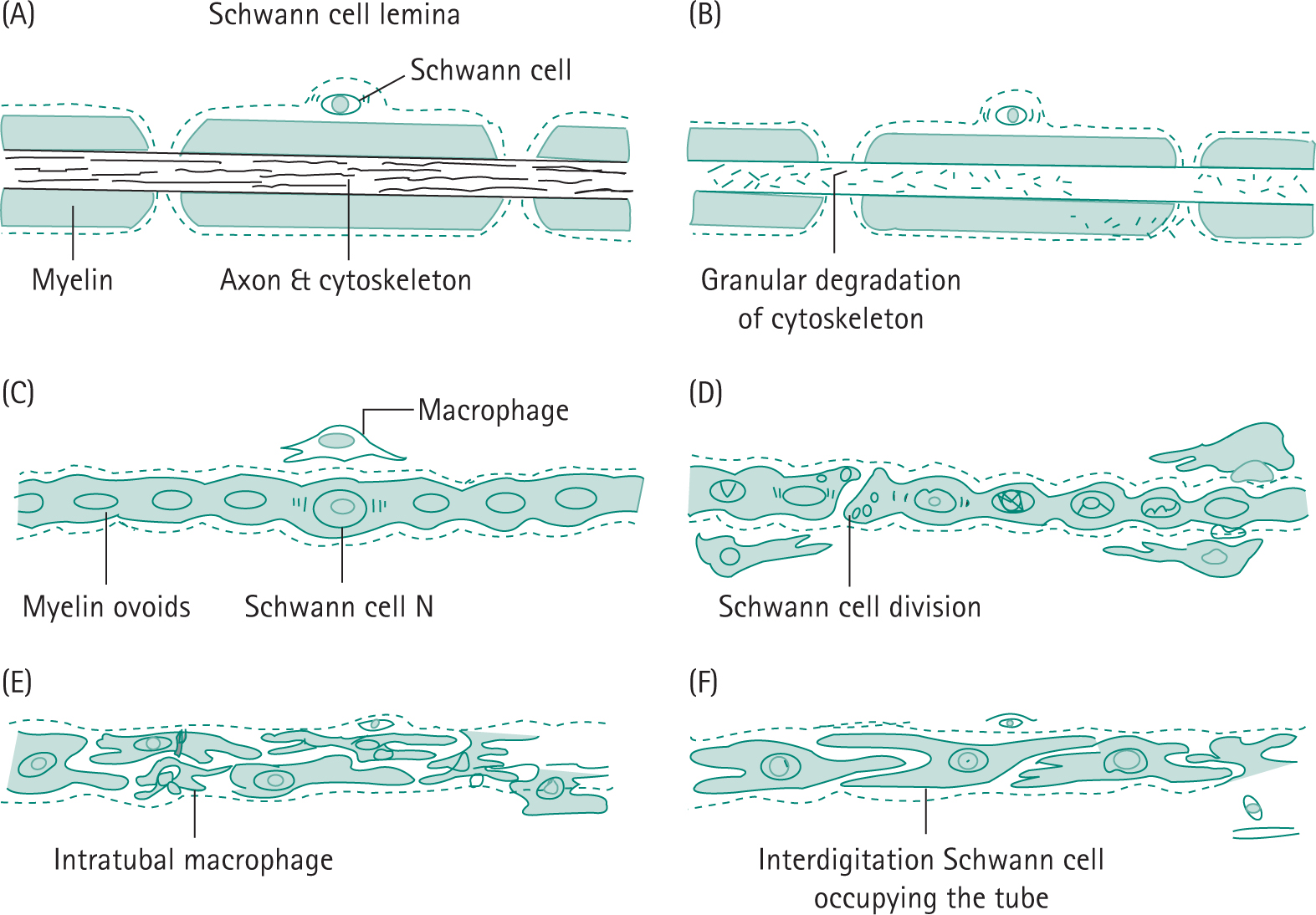
Bunge R, GritIin JW. The cell of Schwann. In: Asbury AK, Mckhann GM, McDonald WI, eds. Diseases of the Nervous System: Clinical Neurology. Philadelphia: WB Saunders; 1992:87.
Caselli RJ, Daube JR, Hunder GG, et al. Peripheral neuropathic syndrome in giant cell (temporal) arteritis. Neurology. 1988;38:685.
Casey EB, Jelliffe AM, Lequesne PM, et al. Vincristine neuropathy clinical and electrophysiological observation.
Brain. 1973;96:69.
CaValier SJ, Gambetti P. Dystrophic axons and spinal cord demyelination in cystic fibrosis. Neurology. 1998;31:714.
Chaco J, Magora A, Jauberman H, et al. An electromyographic study of lagophthalmos in leprosy. Int J Lepr. 1968;36:288.
Charosky CB, Gatti JC, Cardama JE. Neuropathies in Hansen’s disease. Int J Lepr. 1983;51:576.
Chassande B, Leger JM, Younes-Chennoufi AB, et al. Peripheral neuropathy associated with IgM monoclonal gammopathy correlations between clinical, electrophysiological features in 40 cases. Muscle Nerve. 1998;21:55.
Chimelli L, Scaravilli F. Morphological changes in autonomic nervous system of patients with AIDS (abstract). Proceedings of the Seventh International Conference on Neuroscience of HIV Infection. Italy: Pandova; 1991. 89
Chopra JS, Fannin T. Pathology of diabetic neuropathy. J Pathol. 1971;104:175.
Chopra S, Hurwitz LJ, Montogomery DAD. The pathogenesis of sural nerve in diabetes mellitus. Brain. 1969;92:391.
Cohen RD, Conn DL, Ilstrup DM. The clinical features and response to treatment in polyarteritis. Mayo Clin Proc. 1980;55:146.
Conn DL, Dyck PI. Angiopathic neuropathy in connective tissue diseases. In: Dyck PJ, Thomas PK, Lambert EH, eds, et al. Peripheral Neuropathy. Philadelphia: Saunders; 1984:2027. Vol. 2.
Conn DL, McDuffie FC, Dyck PJ. Immunopathological study of sural nerves in rheumatoid arthritis. Arthritis Rheum. 1972;15:135.
Cornblath DR. Treatment of neuromuscular complications of human immuno deficiency virus infection. Ann Neurol. 1988;23:(S)88.
Cornblath DR. Electrophysiology in Guillain Barre syndrome. Ann Neurol. 1990;27:S17.
Cornblath DR, McArthur JC. Predominantly sensory neuropathy in patients with AIDS and AIDS related complex. Neurology. 1988;38:794.
Cornblath DR, Mellitis ED, Griffin JW, et al. The Guillian Barre syndrome study group, motor conduction studies in Guillain Barre syndrome: description and prognostic value. Ann Neurol. 1988;23:354.
Croft PB, Eurich H, Wilkinson M. Peripheral neuropathy of sensorimotor type associated with malignant disease. Brain. 1967;90:31.
Cruickshank B. The arteritis of rheumatoid arthritis. Ann Rheum Dis. 1954;13:136.
Dalakas MC, Pezesckpour GH. Neuromuscular diseases associated with human immunodeficiency virus infection. Ann Neurol. 1988;23:(S)38.
Dastur DK, Anita NH, Divekar SC. The facial nerve in leprosy II: pathology, pathogenesis, electromyography and clinical correlation. Int J Lepr. 1968;34:118.
Dastur DK. Cutaneous nerve in leprosy: the relationship between histology and cutaneous sensibility. Brain. 1955;78(6):1-3.
Davies L, Spies JM, Pollard ID, et al. Vasculitis confined to peripheral nerves. Brain. 1996;II:1441.
de Gans J, Portegies P, Tiessens G, et al. Therapy for cytomegalo virus polyradiculopathy in patients with AIDS. Treatment with ganciclovir. AIDS. 1990;4:421.
De Jong JGY. Over families met hereditary disposite tot het optreten van neuritiden, gecorreleard met migraine. Psychiat Neurol BI. 1947;50:60.
De Silva KL, Pearce J. Neurology of metachromatic leucodystrophy. J Neurol Neurosurg Psychiat. 1973;36:30.
Divekar SC. Electrodiagnostic studies in leprosy. In: Antia NH, Dastur DK, eds. Symposium on Leprosy (University of Bombay). Bombay: JJ Group of Hospitals; 1965:72. February–March
Donofrio PD, Albers JW. AAEM mini monograph #34: Polyneuropathy: classification by nerve conduction studies and electromyography. Muscle Nerve. 1990;13:889.
DuBois DC, Almon RR. A possible role for glucocorticoid in denervation atrophy. Muscle Nerve. 1981;4:370.
Dulbourg O, Mouton P, Brice A, et al. Guidelines for diagnosis of hereditary neuropathy with liability to pressure palsies. Neuromuscul Disord. 2000;10:206.
Dutta AK, Mandal SB, Jopling WH. Surface temperature bald and hairy scalp in reference to leprosy affection. Ind J Dermatol. 1983;28:1.
Dyck PJ. Detection, characterization and staging of polyneuropathy: assessed in diabetes. Muscle Nerve. 1988;11:21.
Dyck PJ, Benstead TJ, Conn DL, et al. Nonsystemic vasculitic neuropathy. Brain. 1987;110:843.
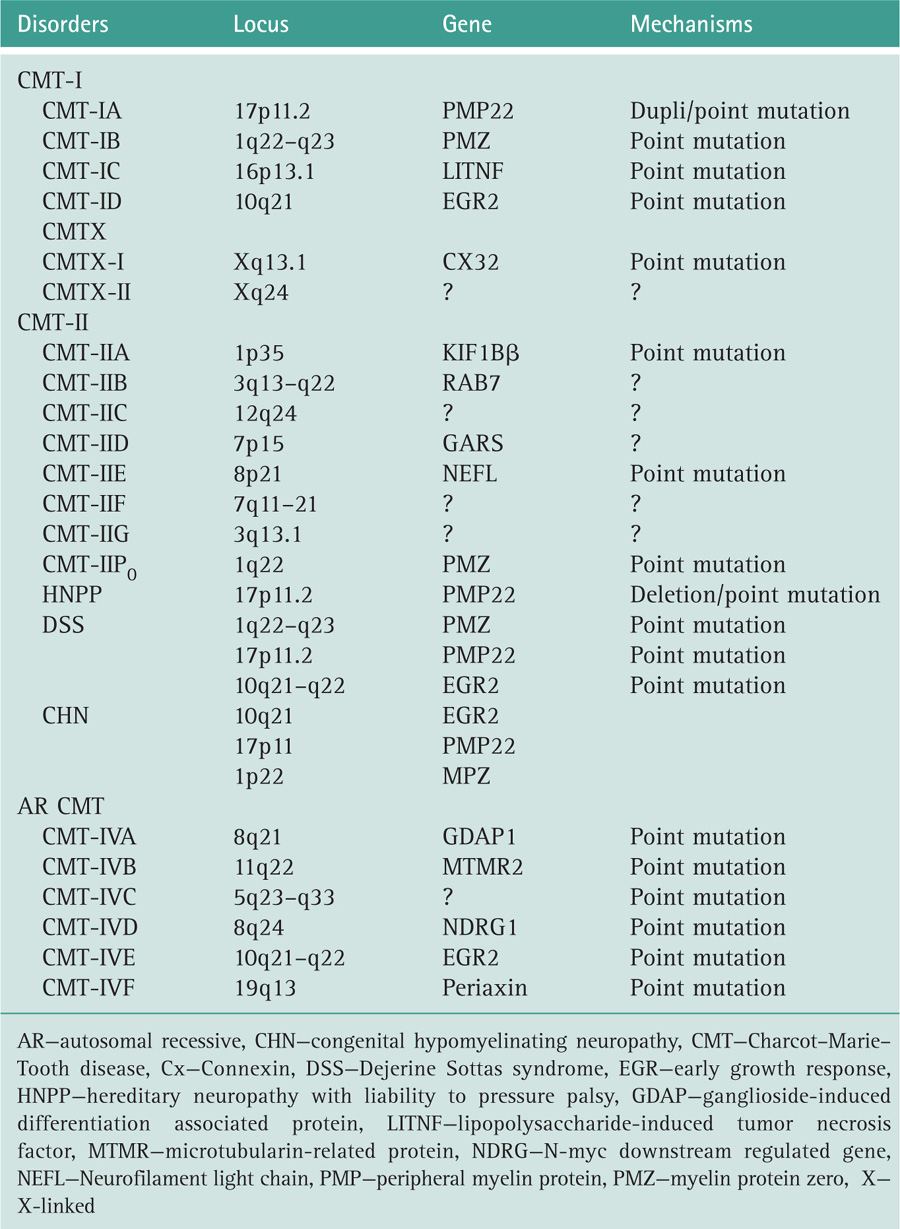
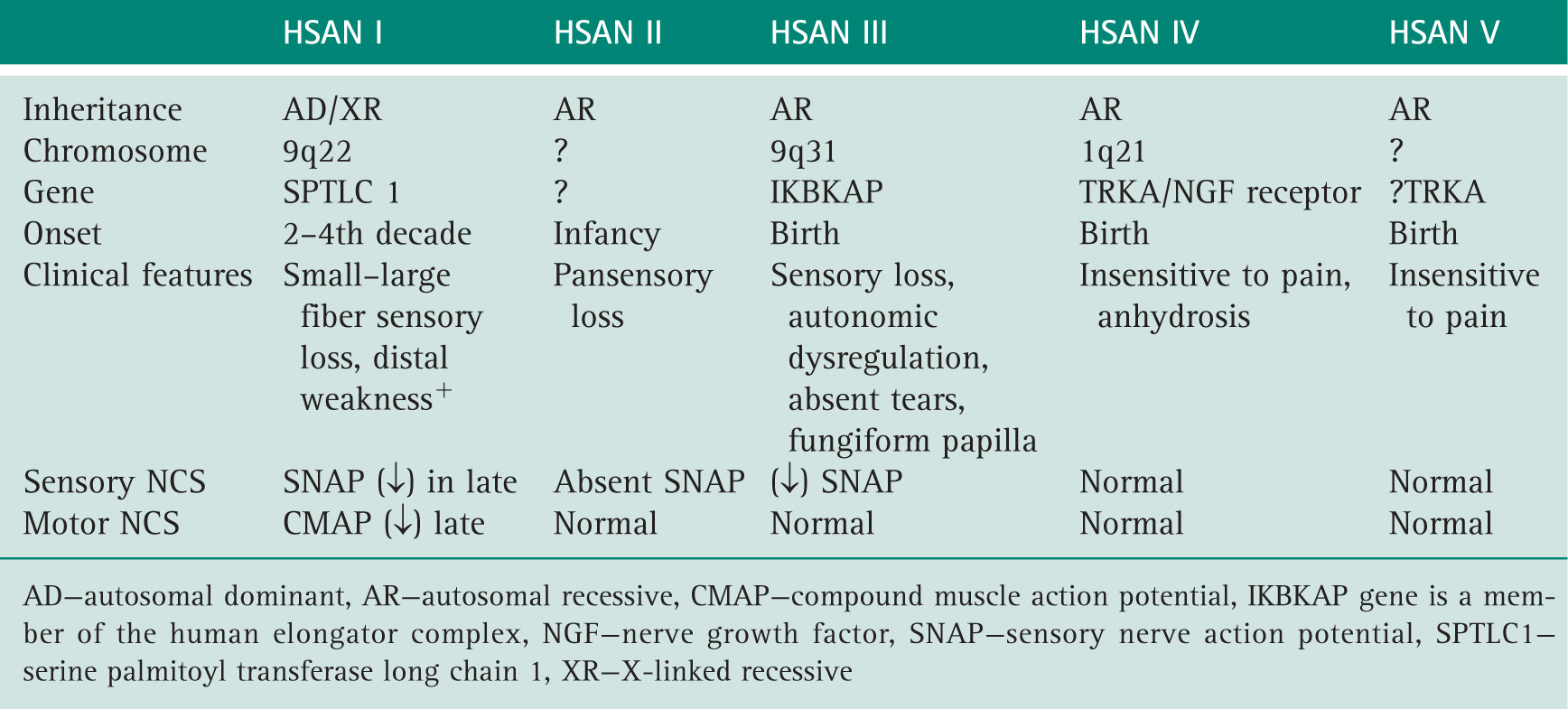
Dyck PJ, Chance PF, Lebo RV, et al. Hereditary motor and sensory neuropathies. In: Dyck PJ, Thomas PK, Griffin JW, eds, et al. Peripheral Neuropathy. Philadelphia: WB Saunders; 1993.
Dyck PJ, Conn DL, Okazaki H. Necrotizing angiopathic neuropathy. Mayo Clin Proc. 1972;47:461.
Dyck PJ, Johnson WJ, Lambert EH, et al. Segmental demyelination secondary to axonal degeneration in uremic neuropathy. Mayo Clin Proc. 1971;46:400.
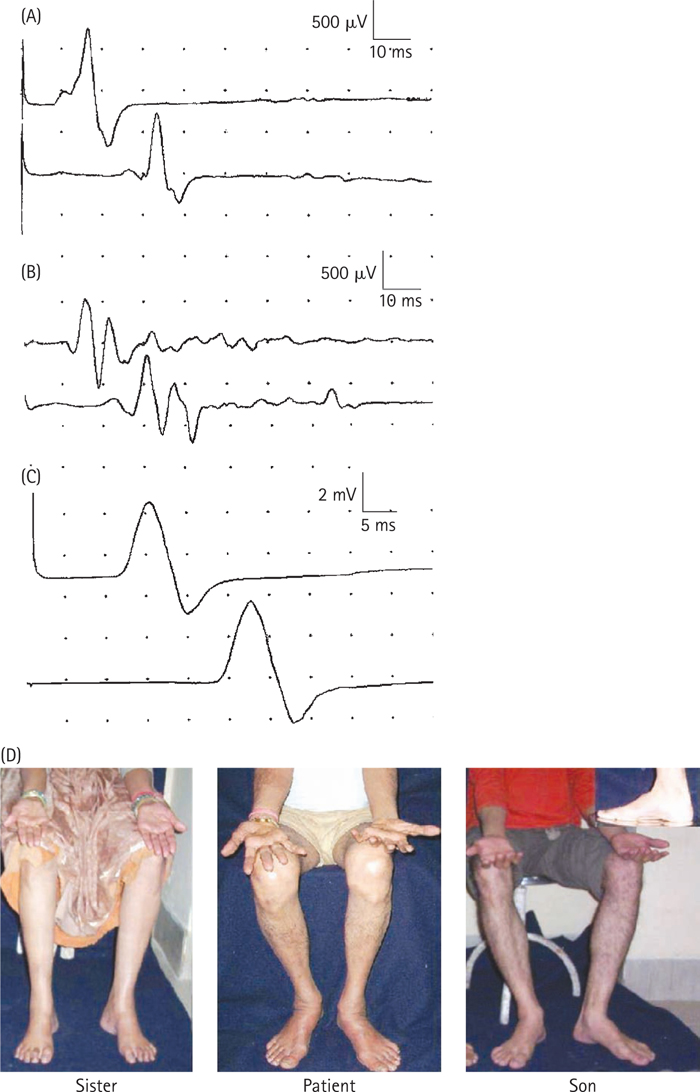
Dyck PJ, Karies JL, Lambert EH. Longitudinal study of neuropathic deficits and nerve conduction abnormalities in hereditary motor and sensory neuropathy type I. Neurology. 1989;39:1302.
Dyck PJ, Lambert EH. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy (i) neurologic, genetic and electrophysiologic findings in hereditary polyneuropathies (ii) neurologic, genetic and electrophysiologic findings in various neuronal degenerations. Arch Neurol. 1968;18:603.
Dyck PJB, Sinnreich M. Diabetic neuropathies. Continuum. 2003;9:19.
Dyck PJB, Winderbark AJ. Diabetic and non diabetic lumbosacral radiculoplexus neuropathies. New insights into pathophysiology and treatment. Muscle Nerve. 2002;25:477.
Ebright JR, Crane LR. Ganciclovir resistant cytomegalovirus.
AIDS. 1991;5:604.
Evenhouse M, Hans E, Snell E, et al. Hypotension in infection with the human immune deficiency virus. Ann Int Med. 1987;107:598.
Farrel DA, Medsger TA. Trigeminal neuropathy in progressive systemic sclerosis. Ann J Med. 1982;73:57.
Fisher M. An unusual variant of acute idiopathic polyneuritis (syndrome of ophthalmoplegia, ataxia and areflexia). N Engl J Med. 1956;255:57.
Friedman SA, Schulman RH, Weiss S. Hearing and diabetic neuropathy. Arch Int Med. 1975;135:573.
Frohnert PP, Sheps SG. Long term followup study of polyarteritis nodosa. Am J Med. 1967;43:8.
Fross RD, Daube JR. Neuropathy in the Miller Fisher syndrome: clinical and electrophysiologic findings. Neurology. 1987;37:1493.
Fuller GN, Jacoba JM, Guiloff RJ. Subclinical peripheral nerve involvement in AIDS. An electrophysiological and pathological study. J Neurol Neurosurg Psychiat. 1991;54:318.
Gheradi R, Lebargy F, Gaulard P, et al. Necrotising vasculitis and HIV replication in peripheral nerves. N Engl J Med. 1989;321:685.
Gherardi RK, Chretien F, Delfau-Larue MH, et al. Neuropathy in diffuse infiltrative lymphocytosis syndrome. An HIV neuropathy not a lymphoma. Neurology. 1998;50:1041.
Gilliatt RW, Goodman HV, Willison RG. The recording of lateral popliteal nerve action potential in man. J Neurol Neurosurg Psychiat. 1961;24:305.
Gilliatt RW. Nerve conduction in human and experimental neuropathies. Proc R Soc Med. 1966;58:989.
Gocke OJ, HSu K, Morgan C, et al. Association between polyarteritis and Australia antigen. Lancet. 1970.1149.
Gooch JL, Suchyta MR, Balbierz JM, et al. Prolonged paralysis after treatment with neuromuscular blocking drugs. Crit Care Med. 1991;19:1125.
Good AE, Christopher RP, Koepke GH, et al. Peripheral neuropathy associated with rheumatoid arthritis. Ann Intern Med. 1965;63:87.
Gordon PH, Wilbourn AJ. Early electrodiagnostic findings in Guillain-Barré syndrome. Arch Neurol. 2001;58:913.
Gorson KC, Ropper AH, Adelman LS, et al. Influence of diabetes mellitus on chronic inflammatory demyelinating polyneuropathy. Muscle Nerve. 2000;23:37.
Griffin JW, Cornblath DR, Alexander E, et al. Ataxic sensory neuropathy and dorsal root ganglionitis associated with Sjögren’s syndrome. Ann Neurol. 1990;23:304.
Griffin JW, Crawford TO, Tyor WR, et al. Predominantly sensory neuropathy in AIDS. Distal axonal degeneration and unmyelinated fibre loss. Neurology. 1991;41:(S)374.
Griffin JW, Ho TW. The Guillain–Barre syndrome at 75: the campylobacter connection. Ann Neurol. 1993;34:125.
Guillevin L, Dul LTH, Godeu P, et al. Clinical findings and prognosis of polyarteritis nodosa and Churg–Strauss angitis: a study of 165 patients. Brit J Rheumatoid. 1988;27:258.
Guiloff RJ. Peripheral nerve conduction in Miller Fisher syndrome. J Neurol Neurosurg Psychiat. 1977;40:801.
Gupta SK, Taly AB, Suresh TG, et al. Acute idiopathic axonal neuropathy (AIAN): a clinical and electrophysiological observation. Acta Neurol Scand. 1994;89:220.
Hackett ER, Shipley DE, Livengood E. Motor nerve conduction velocity studies in patients with leprosy. Int J Lepr. 1968;36:282.
Hanson P, Dive A, Brucher JM, et al. Acute corticosteroid myopathy in intensive care patients. Muscle Nerve. 1997;20:1371.
Haq RU, Pendlebury WW, Fries TJ, et al. Chronic inflammatory demyelinating polyradiculoneuropathy in diabetic patients. Muscle Nerve. 2003;27:465.
Harding AE. Spinocerebeller degeneration associated with a selective defect of vitamin E absorption. N Engl J Med. 1985;313:32.
Hastings RC, Brand PW, Mansfield ER, et al. Bacterial density in skin in lepromatous leprosy as related to temperature. Leprosy Rev. 1968;39:71.
Hellmann DB, Laing TJ, Petri M, et al. Mononeuritis multiplex: the yield of evaluations for rheumatic diseases. Medicine. 1988;67:145.
Herbison GL, Teng C, Martin JH, et al. Carpal tunnel syndrome in rheumatoid arthritis. Am J Phys Med. 1973;52:68.
Herman KH, Kennedy L. For the GOAL AIC study group. Physician perception of neuropathy in a large diabetes type 2 population (GOAL AIC study) confirms under diagnosis of neuropathy in everyday clinical practice. Diabetologia. 2003;46(S2):A71.
Hoogedijk JE, DeVisser M, Bolhuis PA, et al. Hereditary motor and sensory neuropathy Type 1: clinical and neurophysiological features of the 17p duplication subtype. Muscle nerve. 1994;17:85.
Horwich MS, Cho L, Porro RS, et al. Subacute sensory neuropathy. A remote effect of carcinoma. Ann Neurol. 1977;2:7.
Hughes RAC, Saunders E, Hall S, et al. Subacute idiopathic demyelinating polyradiculopathy. Arch Neurol. 1992;49:612.
Infante J, Garcia A, Combarros O, et al. Diagnostic strategy for familial and sporadic cases of neuropathy associated with 17p 11.1 deletion. Muscle Nerve. 2001;24:1149.
Jacobson J, Smith T, Gaub J, et al. Progressive neurological dysfunction during latent HIV infection. Brit Med J. 1989;299:225.
Jamal GA, Ballantyne JP. The localization of the lesion in patients with acute ophthalmoplegia ataxia and areflexia (Miller Fisher syndrome). Brain. 1988;111:95.
Jardim MR, Antunes SL, Santos AR, et al. Criteria for diagnosis of pure neural leprosy. J Neurol. 2003;250:806.
Judzewitsch RG, Jaspan JB, Polonsky KS, et al. Aldose reductase inhibition improves nerve conduction velocity in diabetic patients. N Engl J Med. 1983;308:119.
Kaji R, Oka N, Tsuji T, et al. Pathological findings at the site of conduction block in multifocal neuropathy. Ann Neurol. 1993;33:152.
Kaji R, Shibasaki H, Kimura J. Multifocal demyelinating motor neuropathy: cranial nerve involvement and immunoglobulin therapy.
Neurology. 1992;42:506.
Kaku DA, Parry GJ, Malamut A, et al. Nerve conduction studies in Charcot–Marie Tooth poly neuropathy associated with a segmental duplication of chromosome 17. Neurology. 1993;43:1806.
Kalita J, Misra UK, Sharma RK, et al. Femoral and radial neuropathy following vascular access cannulation for hemodialysis. Nephron. 1995;69:362.
Kaplan JG. Neurotoxicity of selected biological toxins. In: Spencer PS, Schaumburg HH, eds. Experimental and Clinical Neurotoxicology. Baltimore: Williams and Wilkins; 1980:631.
Katz J, Barohn R, Kojan S, et al. Axonal multifocal motor neuropathy without conduction block or other features of demyelination. Neurology. 2002;58:615.
Katz J, Wolfe G, Bryan W, et al. Electrophysiological findings in multifocal motor neuropathy. Neurology. 1997;48:700.
Kelly JJ, Kyle RA, Miles JM, et al. The spectrum of peripheral neuropathy in myeloma. Neurology (NY). 1981;31:24.
Kelly JJ, Kyle RA, O’Brien PC, et al. The prevalence of monoclonal gammopathy in peripheral neuropathy. Neurology (NY). 1981;31:1480.
Kennedy WR, Wendelshafer-Crabb G, Johnson T. Quantification of epidermal nerves in diabetic neuropathy. Neurology. 1996;47:1040.
Kissel JT, Slivka AP, Warmolts JR, et al. The clinical spectrum of necrotising angiopathy of the peripheral nervous system. Ann Neurol. 1985;18:251.
Klein CJ, Cunningham JM, Atkinson EJ, et al. The gene for hereditary motor and sensory neuropathy type 2C maps to 12q24. Ann Neurol. 2002;52:S1.
Kobayashi T, Yamanaka T, Jacobs JM, et al. Twitcher mouse an enzymatically athenic model of human globoid cell leucodystrophy (Krabbe disease). Brain Res. 1980;202:479.
Kohn J. Benign paraproteinemias. J Clin Pathol. 1976;8(S6):77.
Krarup C, Stuwart JD, Sumner AJ, et al. A syndrome of asymmetric limb weakness with motor conduction block. Neurology. 1990;40:118.
Krendel DA, Gilchrist JM, Johnson A, et al. Isolated deficiency of vitamin E with progressive neurological deterioration. Neurology. 1990;40:118.
Krendel DA, Lostigan DA, Hopkins LC. Successful treatment of neuropathy in patients with diabetes mellitus. Arch Neurol. 1995;52:1053.
Kumar A, Dalela D, Bhandari M, et al. Femoral neuropathy—an unusual complication of renal transplantation. Transplantation. 1991;51(6):1305.
Kurdi A, Abdul Kadier M. Clinical and electrophysiological studies of diphtheric neuritis in Jordan. J Neurol Sci. 1979;42:243.
Kuritsky A, Yerginer YM, Korezyn AD. Peripheral neuropathies in cerebrotendinous xanthomatosis. Neurology (Miniap). 1979;29:880.
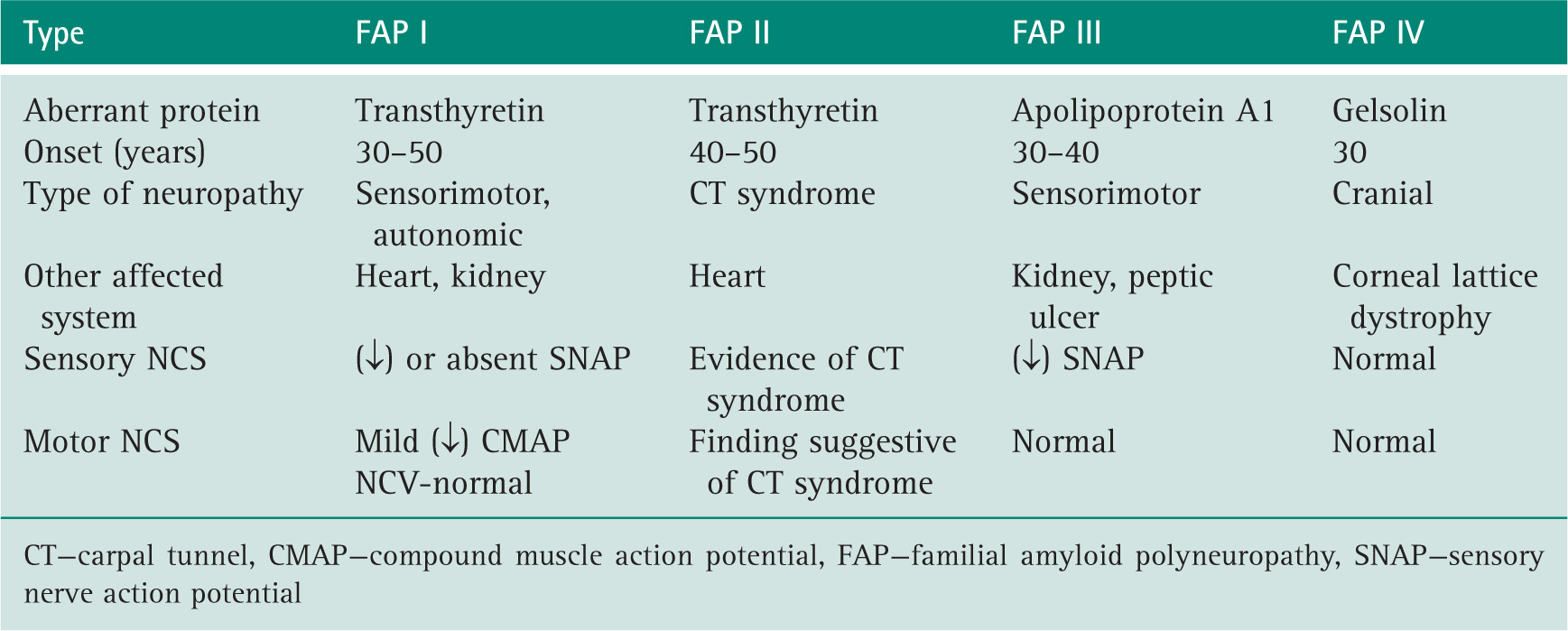
Kyle RA, Dyck PJ. Amyloidosis and neuropathy. In: Dyck PJ, Thomas PK, Griffin JW, eds, et al. Peripheral Neuropathy. 3rd ed Philadelphia: WB Saunders; 1993:1294. Vol. II
Kyle RA, Therneau TM, Rajkumar SV, et al. A longterm study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346:564.
Lamontagne A, Buchthal F. Electrophysiological studies in diabetic neuropathy. J Neurol Neurosurg Psychiat. 1970;33:442.
Lange DJ, Britton CB, Younger DS, et al. The neuromuscular manifestations of human immunodeficiency virus infection. Arch Neurol. 1988;45:1084.
Lecky BRF, Hughes RAC, Murray NMF. Trigeminal sensory neuropathy. Brain. 1987;110:1463.
Leger JM, Salachas F. Diagnosis of motor neuropathy. Eur J Neurol. 2001;8:201.
Levin KH. Neuropathy of ischemia. Muscle Nerve. 2002;26:435.
Lewis RA, Sumner AJ, Brown MJ, et al. Multifocal demyelinating neuropathy with persistent conduction block. Neurology. 1982;32:958.
Lipkin WI, Parry G, Kiprov D, et al. Inflammatory neuropathy in homosexual men with lymphadenopathy. Neurology. 1985;35:1479.
Logina I, Donaghy M. Diphtheritic polyneuropathy: a clinical study and comparison with Guillain Barre syndrome. J Neurol Neurosurg Psychiat. 1999;67:433.
Logothetis J, Kennedy WR, Ellington A, et al. Cryoglobulinemic neuropathy: incidence and clinical characteristics. Arch Neurol. 1968;19:389.
Logothetis J, Silverstein P, Coc J. Neurologic aspects of Waldenstrom’s macroglobulinemia. Arch Neurol (Chicago). 1960;5:564.
Low P, Dotson R. Symptom treatment of painful neuropathy. JAMA. 1998;280:1863-1864.
Low PA, Walsh JC, Huang CY, et al. The sympathetic nervous system in diabetic neuropathy—a clinical and pathological study. Brain. 1975;98:341.
Lubec D, Meillbacher WC, Fimsterer J, et al. Diagnostic workup in peripheral neuropathy: an analysis of 171 cases. Postgrad Med J. 1999;73:723.
Magora A, Sagher F, Chaco J, et al. An electrodiagnostic study of lower motor unit in leprosy. Int J Lepr. 1965;33:829.
Mah V, Yartavarian LM, Akers MA, et al. Abnormalities of peripheral nerve in patients with human immunodeficiency virus infection. Ann Neural. 1988;24:713.
Marsden CD, Obeso JS, Lang AE. Adrenoleukomyelo neuropathy presenting as spinocerebellar degeneration. Neurology (NY). 1982;32:1031.
Martin MM. Diabetic neuropathy: a clinical study of 150 cases. Brain. 1953;76:594.
Mayer RF. Peripheral nerve function in vitamin B 12 deficiency. Arch Neurol. 1965;B:355.
McCombe PA, Pollard JD, Mcleod JG. Chronic inflammatory demyelinating polyradiculopathy. Brain. 1987;110:1617.
McKahn GM, Cornblath DR, Ho TN, et al. Clinical and electrophysiological aspects of acute paralytic disease of children and young adults in Northern China.
Lancet. 1991;338:593.
Mcleod JG. Paraneoplastic neuropathies. In: Dyck PJ, Thomas PK, Griffin JW, eds, et al. Peripheral Neuropathy. Philadelphia: WB Saunders; 1993:1583.
McLeod JG. Investigations in peripheral neuropathy. J Neuro Neurosurg Psychiat. 1995;58:274.
McLeod JG, Hargrave JC, Walsh JC, et al. Nerve conduction studies in leprosy. Int J Lepr. 1975;43:21.
McLeod JG, Walsh JC. Neuropathies associated with paraproteinemias and dysproteinemias. In: Dyck PJ, Thomas PK, Lambert EH, eds. Peripheral Neuropathy. Philadelphia: WB Saunders; 1975:1012.
Meer J, Gilliatt RW. Proximal and distal conduction velocity in the motor nerves of patients with hereditary motor and sensory neuropathy (abstract). Muscle Nerve. 1989;12:762.
Mellgren SI, Conn DL, Stevens JC, et al. Peripheral neuropathy in primary Sjögren’s syndrome. Neurology. 1989;39:390.
Mendell JR, Kolkin S, Kissel JT, et al. Evidence for central nervous system demyelination of chronic inflammatory demyelinating polyradiculoneuropathy. Neurology. 1987;37:1291.
Meulstee J, Van der Meche FGA. The Dutch Guillain–Barre Study Group. Electrodiagostic criteria for polyneuropathy and demyelination: application in 135 patients with Guillain–Barre syndrome. J Neurol Neurosurg Psychiatry. 1995;59:482-486.
Meyer P. Anatomische untersuchungen uber diphtheritische Lahmung. Yirchows Arch Pathol Anat Physiol. 1881;85:181.
Miller RG, Parry GJ, Pfaeftl W, et al. The spectrum of peripheral neuropathy associated with ARC and AIDS. Muscle Nerve. 1988;11:857.
Miller RG, Peterson GW, Daube JR, et al. Prognostic value of electrodiagnosis in Guillian Barre syndrome. Muscle Nerve. 1988;11:769.
Miller RG, Storey JR, Grecoc M. Ganciclovir in the treatment of progressive AIDS related polyradiculopathy. Neurology. 1990;40:569.
Millers GD, O’Fallon JR, Tally JN. Plasma cell dyscrasia with polyneuropathy—the spectrum of POEM syndrome. N Engl J Med. 1992;327:1919.
Misra UK, Kalita J, Pandey R. Primary amyloid neuropathy—a case report. Neurol India. 1994;42:32.
Misra UK, Kalita J, Das A. Vitamin B12 deficiency syndrome. Electromyogr Clin Neurophysiol. 2003;43:57.
Molina R, Provost IT, Alexander EL. Peripheral inflammatory vascular disease in Sjögren’s syndrome. Arthritis Rheum. 1985;28:1341.
Monrad-Krohn BH. The neurological aspects of leprosy. Jacob Dybwad: Christiana; 1923.
Moody JF. Electrophysiological investigations in the neurological complications of carcinoma. Brain. 1965;88:1023.
Moore PM, Cupps TR. Neurological complications of vasculitis. Ann Neurol. 1983;14:155.
Morton DL, Itubashi HH, Grimes OF. Nonmetastatic neurological complications of bronchogenic carcinoma. The carcinomatous neuromyopathies. J Thorac Cardiovasc Surg. 1967;5:14.
Mossa A, Dubowitz V. Peripheral neuropathy in Cockayne’s syndrome. Arch Des Child. 1970;45:674.
Mouton R, Tardieu S, Gouider R, et al. Spectrum of clinical and electrophysiologic features in HNP patients with the 17p11.1 deletion. Neurology. 1993;52:1440.
Mulder DW, Bastron JA, Lambert EH. Hyperinsulin neuropathy. Neurology. 1956;6:627.
Mulder DW, Lambert EH, Dastron JA, et al. The neuropathies associated with diabetes. A clinical and electromyographic study of 103 unselected diabetic patients. Neurology. 1961;11:275.
Nakano KK. The entrapment neuropathies of rheumatoid arthritis. Orthop Clin North Ann. 1975;6:837.
Ndiaye-Naiang M, Diagne M, Ndiaye IP, et al. The value of electromyographic studies in leprosy. Acta Leprol (Geneva). 1986;100:51.
Nicolas G, Massonobe T, Le Forestier NN, et al. Proposed revised electrophysiological criteria for chronic inflammatory demyelinating polyradiculoneuropathy. Muscle Nerve. 2002;25:25.
Nielsen VK, Kardel T. Decremental conduction in normal human nerves subjected to ischaemia?. Acta Physiol Scand. 1974;92:249-262.
Noordeen SK. A look at world leprosy. Leprosy Rev. 1991;62:72.
Oh SJ, Chang CW. Conduction block and dispersion in hereditary motor and sensory neuropathy (abstract). Muscle Nerve. 1987;10:656.
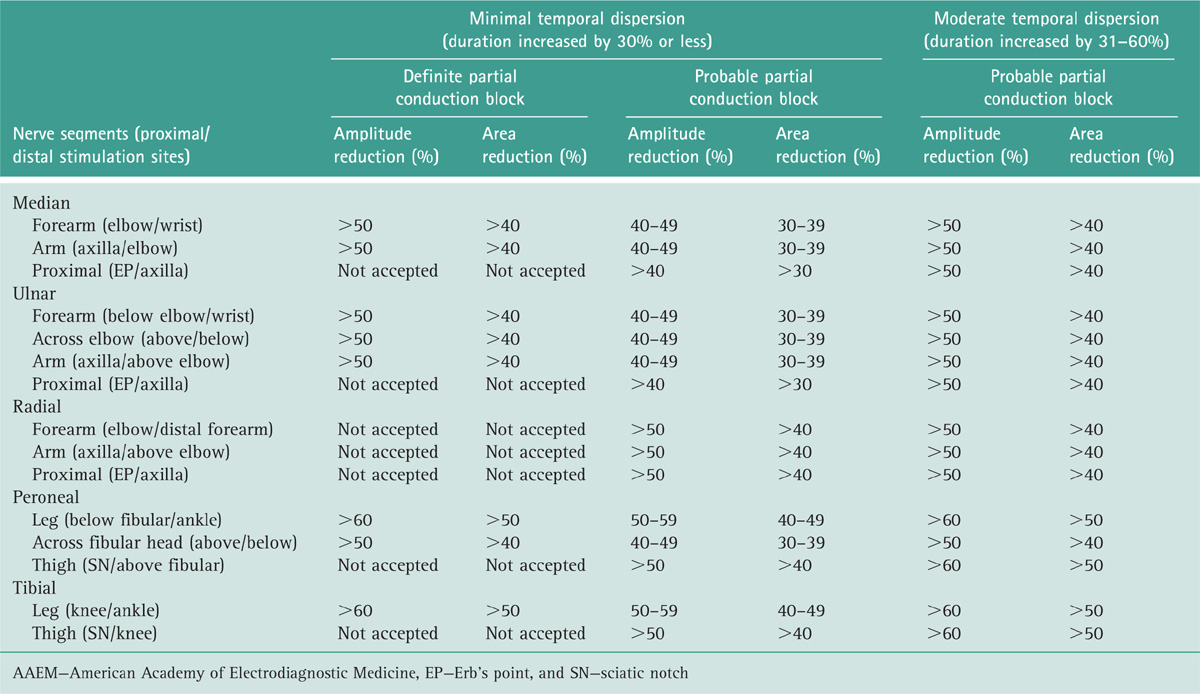
Oh SJ, Kim DE, Kuruoglu HR. What is the best diagnostic index of conduction block and temporal dispersion?. Muscle Nerve. 1994;17:489.
Oh SJ, Joy JL, Kuruoglu R. “Chronic sensory demyelinating neuropathy”: chronic inflammatory demyelinating polyneuropathy presenting as a pure sensory neuropathy. J Neurol Neurosurg Psychiat. 1992;53:677.
Olney RK. Neuropathies in connective tissue disease. AAEM mini monograph #38. Muscle Nerve. 1992;15:531.
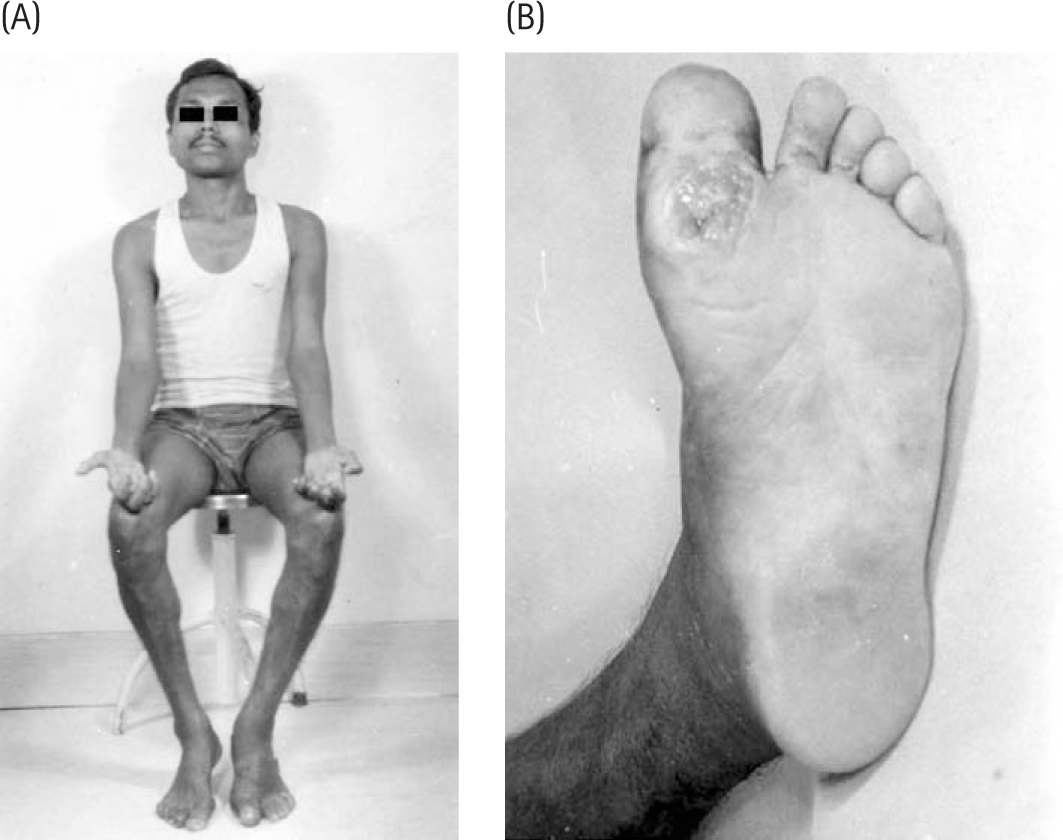
Ooi WW, Srinivasan J. Leprosy and the peripheral nervous system: basic and clinical aspects. Muscle Nerve. 2004;30:393.
Pakalins A, Drake ME, Barotin RJ, et al. Evoked potentials in chronic inflammatory demyelinating polyneuropathy. Arch Neurol. 1988;45:1014.
Partanen J, Niskanen L, Lehtinen J, et al. Natural history of peripheral neuropathy in patients with non-insulin dependent diabetes mellitus. N Engl J Med. 1995;333:89-94.
Pascoe MK, Low PA, Windebank AJ, et al. Subacute diabetic proximal neuropathy. Mayo Clin Proc. 1997;72:1123.
Pedley JC, Harman OJ, Waudby H, et al. Leprosy in peripheral nerves. Histopathological findings in 119 patients in Nepal. J Neurol Neurosurg Psychiatr. 1980;43:198.
Perry GJ, Brown MJ. Selective fibre vulnerability in acute ischemic neuropathy.
Ann Neurol. 1982;11:147.
Perry GJ, Clarke S. Pure motor neuropathy with multifocal conduction block masquerading as motor neuron disease. Muscle Nerve. 1985;8:617.
Perry GJ, Comblath DR, Brown MJ. Transient conduction block in acute peripheral nerve ischemia. Muscle Nerve. 1981;4:441.
Pestronk A, Chaudhry V, Feldman EL, et al. Lower motor neuron syndromes defined by patterns of weakness, nerve conduction abnormalities, and high titers of antiglycolipid antibodies. Ann Neurol. 1990;27:316.
Pestronk A, Cornblath DR, Illys AA, et al. Antibodies to GMI ganglioside. Ann Neurol. 1988;24:73.
Pestronk A, Cornblath DR, Ilyas AA, et al. A treatable multifocal motor neuropathy with antibodies to GM1 ganglioside. Ann Neurol. 1988;24:73.
Pfeifer MA, Cook D, Brodsky J, et al. Quantitative evaluation of cardiac parasympathetic activity in normal and diabetic man. Diabetes. 1982;31:339.
Pirat J. Diabetes mellitus and its degenerative complications, a prospective study of 4400 patients observed between 1947 and 1973. Diabetes Care. 1978;1:168. 252
Pourmand R. Evaluation of patients with suspected peripheral neuropathy: do the right thing and not everything. Muscle Nerve. 2002;26:286.
Quattrini C, Jeriorska M, Gawkrodger D, et al. Dermal nerve depletion and angiogenesis in diabetic neuropathy. Diabetologia. 2003;46(S):A72.
Reske Nielson E, Harmsen A, Vorre P. Ultrastructure of muscle biopsies in recent, short term and long term juvenile diabetes. Acta Neurol Scand. 1977;55:345.
Richard KO, Richard AL, Timothy DP, et al. Consensus criteria for the diagnosis of multifocal motor neuropathy. Muscle Nerve. 2003;27:117.
Ricoy JR, Cabello A, Rodriguez J, et al. Neuropathological studies related to the toxic syndrome related to adulterated rapeseed oil in Spain. Brain. 1983;106:817.
Ridley A. The neuropathy of acute intermittent porphyria. Q J Med. 1969;28:307.
Roelefs RI, Hrusheshy W, Rogin J. Peripheral sensory neuropathy and cisplatin chemotherapy. Neurology. 1984;34:934.
Ropert A, Metral S. Conduction block in neuropathies with necrotizing vasculitis. Muscle Nerve. 1990;13:102.
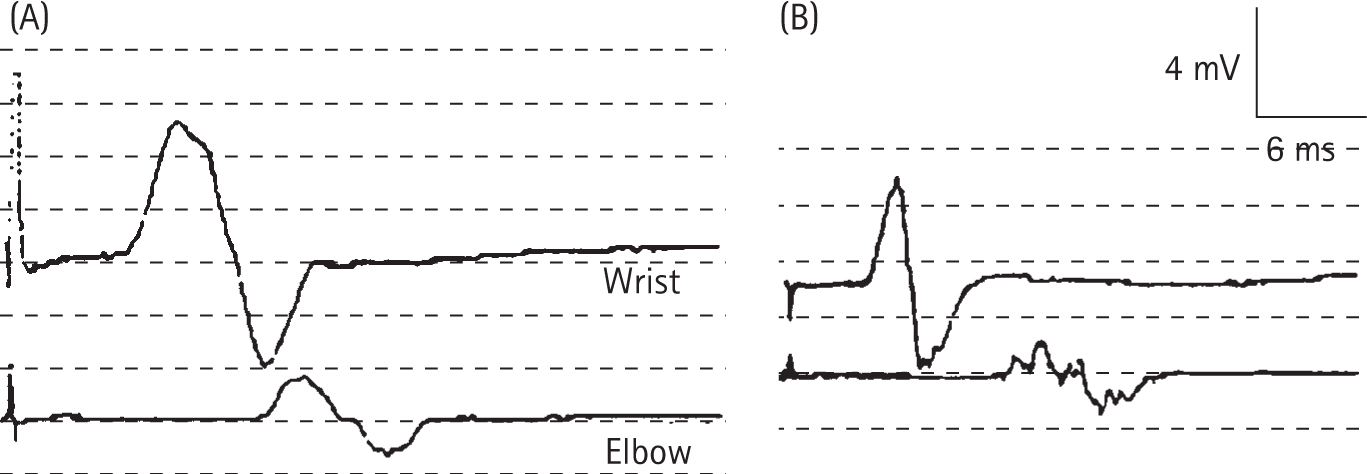
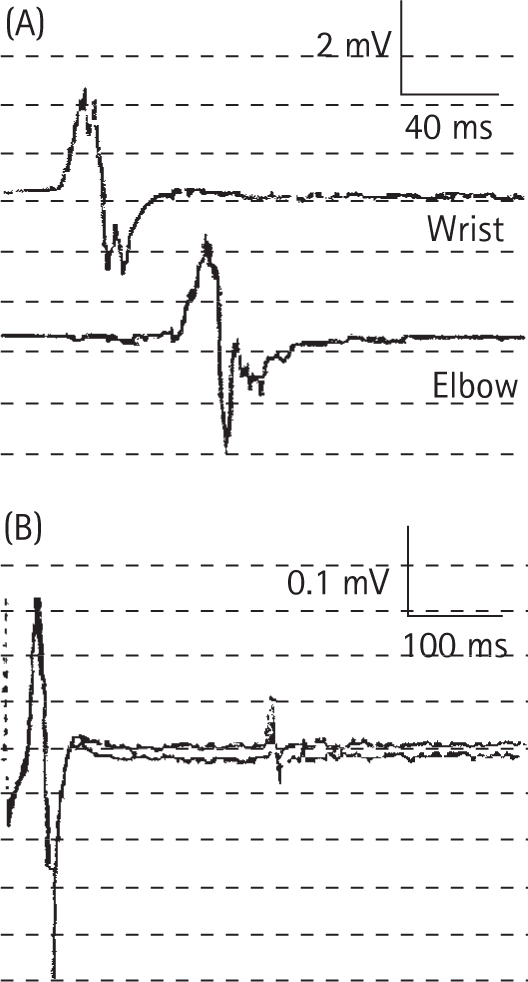
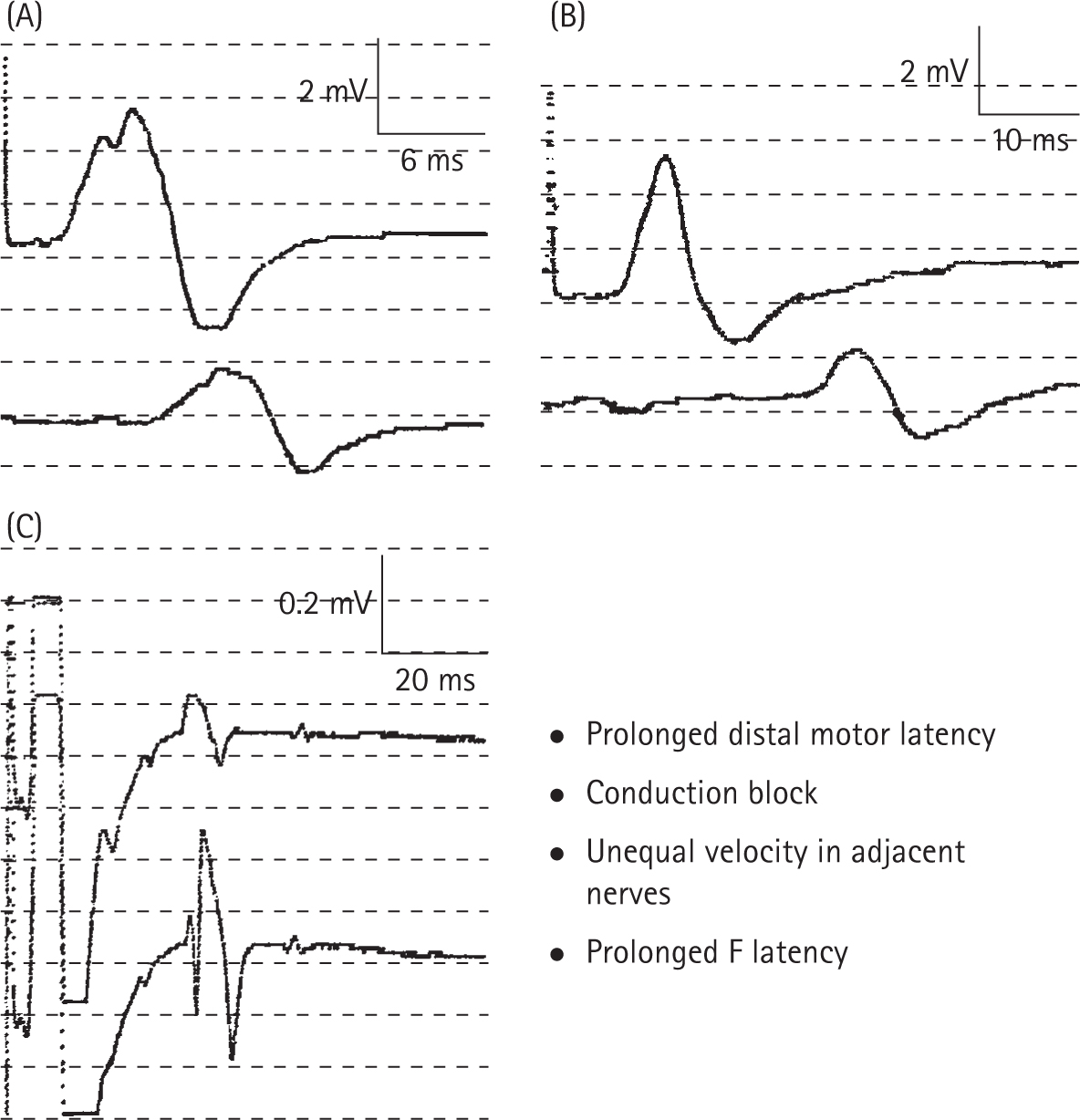
Ropper AH, Wijdicks EEM, Shahani BT. Electrodiagnostic abnormalities in 113 consecutive patients with Guillain–Barre syndrome. Arch Neurol. 1990;47:881-887.
Rosenberg RN, Lovelace RE. Mononeuvitis multiplex in lepromatous leprosy. Arch Neurol. 1969;20:257.
Rousseau LL, Frank G, Grisar T, et al. Osteosclerotic myeloma with polyneuropathy and ectopic secretion of calcitonin. Cancer. 1978;14:133.
Roy A, Kalita J, Gayathri N, et al. Reversible neuropathy in chronic renal failure. Nephron. 1998;80:63.
Roy EP, Gutmann L, Riggs JE. Longitudinal conduction studies in hereditary motor and sensory neuropathy type. Muscle Nerve. 1989;12:52.
Sack GH Jr. Acute intermittent porphyria. JAMA. 1990;264:1290.
Said G. A clinicopathologic study of acrodystrophic neuropathies. Muscle Nerve. 1980;3:491.
Said G, Lacroix-Ciaudo C, Fujimura H, et al. The peripheral neuropathy of necrotising arteritis: a clinicopathological study. Ann Neurol. 1988;23:46J.
Saito M, Hayashi Y, Suzuki T, et al. Linkage mapping of the gene for Charcot-Marie-Tooth disease type 2 to CMT2A. Neurology. 1997;49:1630.
Schaumburg HH, Spencer PS. Clinical and experimental studies of distal axonopathy: a frequent form of brain and nerve damage produced by environmental chemical hazards. Ann NY Acad Sci. 1979;329:14.
Schaumburg HH, Caplan J, Windebank A, et al. Sensory neuropathy from pyridoxine abuse: a new megavitamin syndrome. New Engl J Med. 1983;309:445.
Schaumburg HH, Kaplan JQ. Toxic neuropathies. In: Asbury AK, Thomas PK, eds. Peripheral Nerve Disorders. Oxford: Butterworth Heinnemann; 1995:238.
Schaumburg HH. Occupational environmental and biologic agents. In: Schaumburg HH, Berger AR, Thomas PK, eds. Disorder of Peripheral Nerves. Philadelphia: FA Davis; 1991:274.
Scott DGL, Bacon PA, Tribe CR. Systemic rheumatoid vasculitis: a clinical and laboratory study of 50 cases. Medicine. 1981;60:288.
Sebille A. Respective importance of different nerve conduction velocities in leprosy. J Neurol Sci. 1978;38:89.
Selikoff IJ, Robitzek EH, Ornstin GG. Treatment of pulmonary tuberculosis with hydrazine derivatives of isonicotinic acid. JAMA. 1952;150:251.
Sellman MS, Mayer RF. Conduction block in hereditary neuropathy with susceptibility to pressure palsies. Muscle Nerve. 1987;10:621.
Sharma K, Lross J, Farronay O, et al. Demyelinating neuropathy in diabetes mellitus. Arch Neurol. 2002;59:758.
Shy ME. Inherited peripheral neuropathies. Continum. 2003;9:117.
Sidiq M, Kirsner AB, Sheon RP. Carpal tunnel syndrome. First manifestation of the systemic lupus erythematosus. JAMA. 1972;222:1416.
Singleton JR, Smith AG, Bromberg MB. Painful sensory polyneuropathy associated with impaired glucose tolerance. Muscle Nerve. 2001;24:1225-1228.
Skillman TG, Johnson EW, Hamwi GJ, et al. Motor nerve conduction velocity in diabetes mellitus. Diabetes. 1961;10:46.
Snider WD, Simpson DM, Nielsen S, et al. Neurological complications of acquired immune deficiency syndrome: analysis of 50 patients. Ann Neurol. 1983;14:403.
So YT, Engstrom JW, Olney RK. The spectrum of electrodiagnostic abnormalities in patients with human immunodeficiency virus infection. Muscle Nerve. 1990;13:855.
So YT, Holtzman DM, Abrams DJ, et al. Peripheral neuropathy associated with acquired immune deficiency syndrome: prevalence and clinical features from a population based survey.
Arch Neurol. 1988;45:945.
Sokol RJ, Kaydent H, Bettis DB, et al. Isolated vitamin deficiency in the absence of fat malabsorption familial and sporadic cases: characterisation and investigation of causes. J Lab Clin Med. 1988;111:548.
Sokoloff L, Wilens SL, Bunim LL. Arteritis of striated muscle in rheumatoid arthritis. Am J Pathol. 1951;27:157.
Solders G, Nennesmo I, Persson A. Diptheritic neuropathy: an analysis based on muscle and nerve biopsy and repealed neurophysiological and autonomic function test. J Neurol Neurosurg Psychiat. 1989;52:876.
Sorensen AWS, With TK. Persistent paresis after prophyric attacks. S Afr Med J. 1971;45(special issue):101.
Stewart PM, Hersley WJ. An acute attack of variegate porphyria complicated by severe autonomic neuropathy. Aust NZ Med. 1981;11:82.
Sumner CJ, Sheth S, Griffin JW, et al. The spectrum of neuropathy in diabetes and impaired glucose tolerance. Neurology. 2003;60:108.
Swift TR, Hackett ER, Shipley DE, et al. The peroneal and tibial nerves in lepromatous leprosy. Clinical and lectrophysiologic observations. Int J Lepr Other Mycobact Dis. 1973;41:25.
Tesfaye S, Malik R, Harris N, et al. Arterio venous shunting and proliferating new vessels in acute painful neuropathy of rapid glycemic control (insulin neuritis). Diabetologia. 1996;39:329-335.
Thomas PK. Central and peripheral demyelination. In: Serratrice G, Pellisiel JF, Desnuelle D, eds, et al. Myelopathies. Paris expansion scientifique Francaise: Neuropathies et Myopathy; 1989:14. I
Thomas PK. Diabetic neuropathy. In: Kin H, Jarrett J, eds. Complications of Diabetes. London: Edward Arnold; 1982.
Thomas PK. Understanding neuropathies: the impact of ancillary investigations. Baitheres Clin Neurol. 1996;5:157.
Thomas PK, Eliasson SG. Diabetic neuropathy. In: Dyck PJ, Thomas PK, Lambert EH, eds, et al. Peripheral Neuropathy. Philadelphia: WB Saunders; 1984:1773.
Thomas PK, King RHM, Koeen RS, et al. Comparative ultrastructural observations on peripheral nerve abnormalities in the late infantile, juvenile and late onset forms of metachromatic. I. Eucodystrophies. Acta Neuropathol (Berl). 1977;39:237.
Thomas PK, Lascelles RG. The pathology of diabetic neuropathy. Q J Med. 1966;35:489.
Thorton CA, Latif AS, Emmanuel JC. Guillain Barre syndrome associated with human immunodeficiency virus infection in Zimbabwe. Neurology. 1991;41:812.
Travers RL, Allison DJ, Brettle RP, et al. Polyarteritis nodosa: a clinical and angiopathic analysis of 17 cases. Semin Arthritis Rheum. 1979;8:184.
Uncini A, De Angelis MV, Di Muzio A, et al. Chronic inflammatory demyelinating polyneuropathy in diabetics: motor conductions are important in the differential diagnosis with diabetic polyneuropathy. Clin Neurophysiol. 1999;110:705.
Uncini A, DiGuglielmo G, DiMuzio A, et al. Differential electrophysiological features of neuropathy associated with 17p11.2 deletion and duplication. Muscle Nerve. 1995;18:628.
Uplekar MW, Antia NH. Clinical and histopathological observations on pure neuritic leprosy. Ind J Lepr. 1986;58:513.
Van der Meche FGA, Meulstee J, Kleyweg RP. Current diagnostic criteria for Guillain–Barre syndrome. Ann Neurol. 1991;30:851-852.
Van der Meche FGA, Meulstee J. Guillain–Barre syndrome, a model for random conduction block. J Neurol Neurosurg Psychiat. 1988;55:1158.
Van Heel DA, Levitt NS, Winter TA. Diabetic neuropathic cachexia—the importance of positive recognition and early nutritional support. Int J Clin Pract. 1998;52:591.
Van Heel DA, Levitt NS, Winter TA. Diabetic neuropathic cachexia—the importance of positive recognition and early nutritional support. Int J Clin Pract. 1998;52:591-592.
Veith G. Untersuchnger uber die histologie der polyneuritis diptheria. Beitr Pathol Anat. 1949;110:567.
Verghese M, Ithiamani KV, Satyanarayan KR, et al. A study of conduction velocity of ulnar and median nerves in leprosy. Int J Lepr. 1970;38:271.
Vital C, Vallat JM. Ultrastructural Study of Human Diseased Peripheral Nerve. New York: Masson; 1980. 76
Vital A, Vital C. Polyarteritis nodosa and peripheral neuropathy. Acta Neuropathol (Berlin). 1985;71:136.
Waldinger TP, Siegle RJ, Weber W, Voorhees JJ. Dapsone induced peripheral neuropathy case report and review. Arch Dermatol. 1984;120:356.
Walsh JC. Neuropathy of multiple myeloma. Arch Neurol. 1971;25:404.
Ward JD, Barnes CG, Fisher DJ, et al. Improvement in nerve conduction following treatment in newly diagnosed diabetes. Lancet. 1971;1:428.
Weber F, Ziegler A. Axonal neuropathy in chronic peripheral arterial occlusive disease. Muscle Nerve. 2002;26:47.
Weiss JA, White JC. Correlation of a 1a afferent conduction with the ataxia of Fisher syndrome. Muscle Nerve. 1986;9:327.
Widenbank AJ. Metal Neuropathy. In: Dyck PJ, Thomas PK, Griffin JW, eds, et al. Peripheral Neuropathy. Philadelphia: WB Saunders; 1993:1549.
Wilbourn AJ. Serial conduction studies in human nerve degeneration. Electroencephalogr Clin Neurophysiol. 1977;43:616.
Wilbourn AJ, Furlan AJ, Hulley W, Ruschhaupt W. Ischemic monomelic neuropathy. Neurology. 1983;33:447.
Willison HJ, Veitch J, Patterson G, et al. Miller Fisher syndrome is associated with serum antibodies to GQ1b ganglioside. J Neurol Neurosurg Psychiat. 1993;56:204.
Wilson SAK.
Neurology. 2nd ed London: Butterworth; 1954. Vol. 2, 197
Wilt NJ, Zochodne DW, Bolton CF, et al. Peripheral nerve functions in sepsis and multiple organ failure. Chest. 1991;99:176.
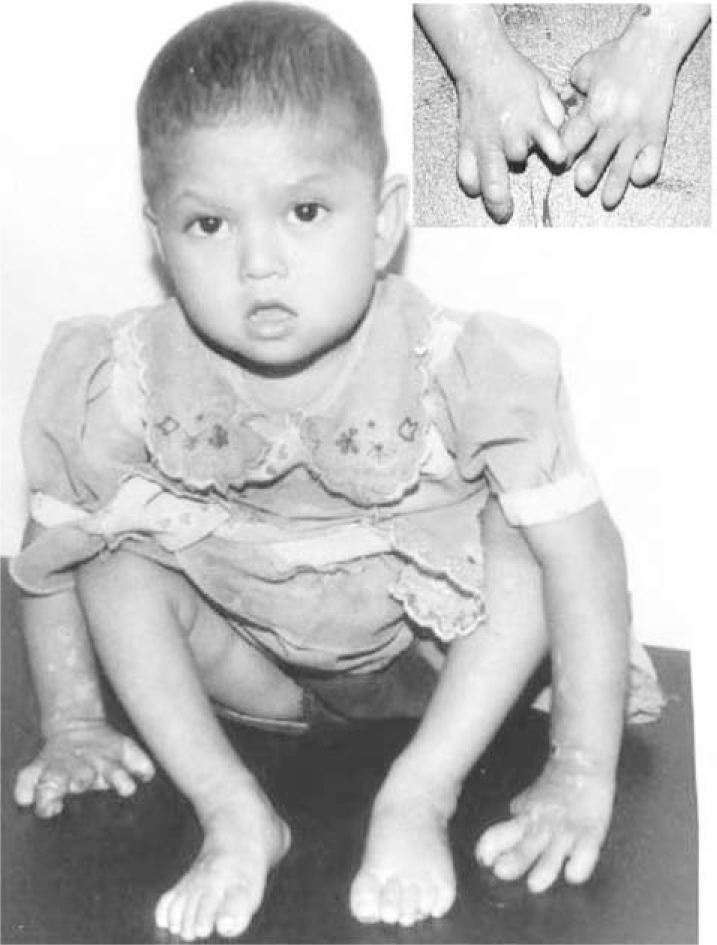
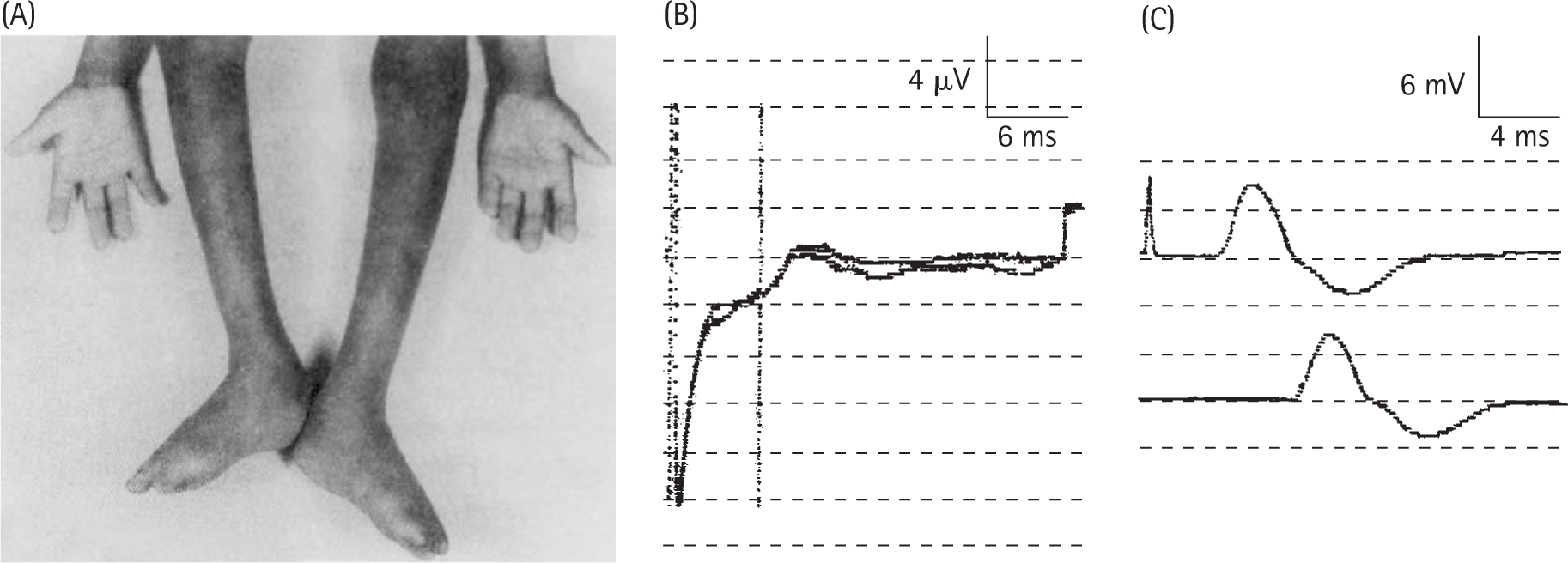
Winkelman RK, Lambert EH, Hayles AB. Congenital absence of pain report of a case and experimental studies. Arch Dermatol. 1962;85:325.
Yao JK, Herbert PN. Lipoprotein deficiency and neuromuscular manifestations. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Poduslo JF, eds. Peripheral Neuropathy. 3rd ed. Philadelphia: WB Saunders; 1993:1179. Vol. II
Yuki N. Infectious origins of and molecular mimicry in Guillain–Barre and Fisher syndromes. Lancet Infect Dis. 2001;1:29.
Zochodne DW, Bolton CF, Wells, et al. Critical illness polyneuropathy: a complication of sepsis and multiple organ failure. Brain. 1987;110:819.