The Peripheral Blood Smear
Barbara J. Bain
With the development of sophisticated automated instruments to count and characterize blood cells, Romanowsky (Wright-Giemsa or May-Grünwald-Giemsa)−stained peripheral blood smears are now performed on only a minority of blood specimens received in a hematology laboratory. Nevertheless, the blood smear remains important for a number of reasons: it can (1) verify the result of an automated instrument, (2) provide an immediate specific diagnosis, or (3) indicate a narrow range of diagnostic possibilities, permitting a focused rather than indiscriminate investigation.1-3 A blood film can provide a rapid diagnosis in cases in which speed is crucial, such as in acute promyelocytic leukemia, thrombotic thrombocytopenic purpura, Burkitt lymphoma, and certain infections.4,5 Sometimes a smear provides unexpected information that is of value in patient management.
Usually, blood smears are initially interpreted by a laboratory scientist. In some countries, it is customary for clinicians to examine the blood smears of their own patients because the clinician has the final responsibility for integrating all information and making a diagnosis. However, the interpretation of a blood smear can be difficult, and a trained laboratory hematologist or hematopathologist has a major role in interpreting smears that may have been initially examined by a laboratory scientist. It is crucial that, when requesting a blood count, the clinician provides all the essential information needed to interpret the count and any associated smear. Regardless of whether the clinician examines the blood film, he or she must be able to interpret the written report issued by the laboratory. To do so, the clinician must be familiar with the terms generally used by laboratory staff and the possible significance of the abnormalities described. The most important of these terms are illustrated in Figures 157-1 through 157-20.
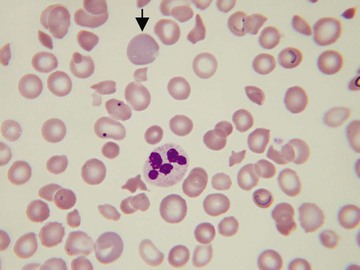
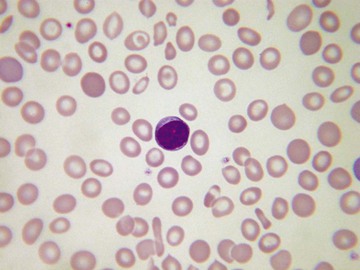
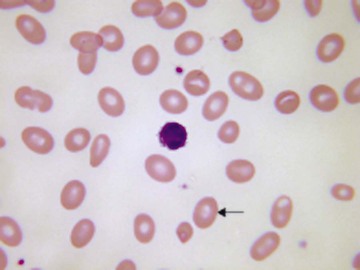
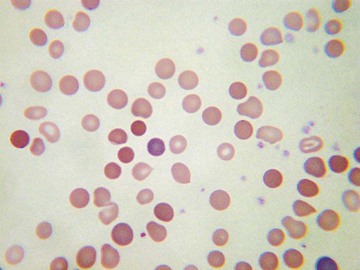
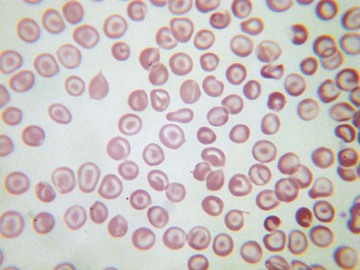
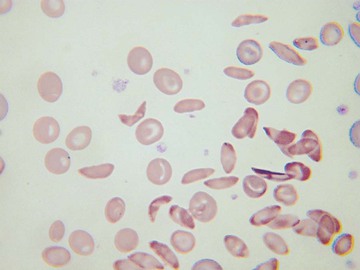
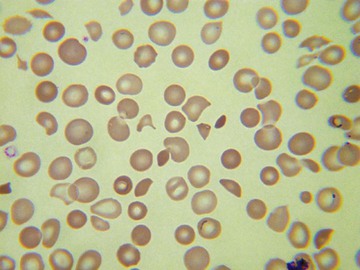
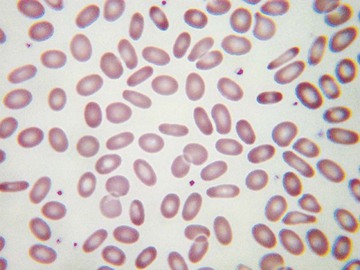
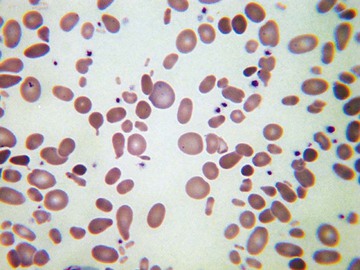
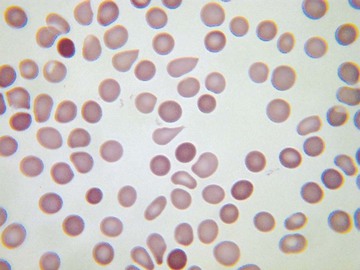
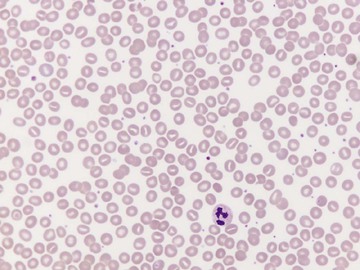
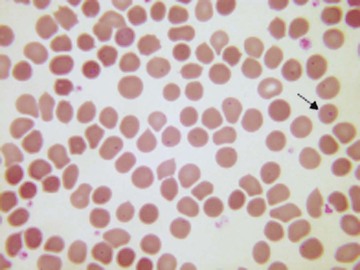
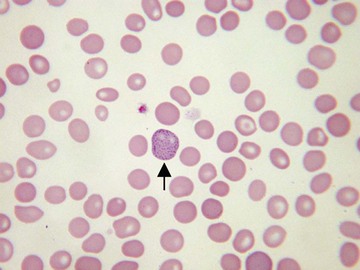
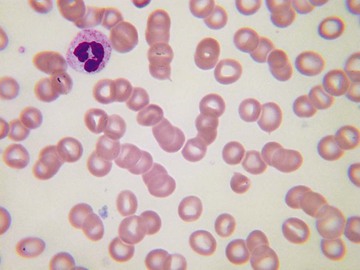
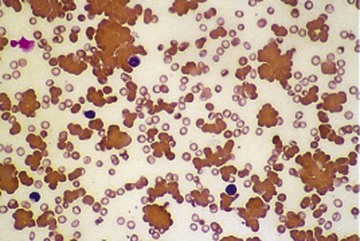
 Reasons for Performing a Blood Smear
Reasons for Performing a Blood Smear
A blood smear may be requested by a clinician or initiated by a laboratory scientist or a laboratory hematologist. Clinical findings that should lead a clinician to request a blood smear are summarized in Table 157-1.
TABLE 157-1
CLINICAL FEATURES SUGGESTING THE NEED FOR A BLOOD SMEAR
| CLINICAL FEATURE | REASON TO PERFORM A BLOOD SMEAR |
| Lymphadenopathy or splenomegaly | May be indicative of infectious mononucleosis or another reactive condition, or of leukemia or lymphoma |
| Clinically evident anemia | Helps in the differential diagnosis |
| Bruising or bleeding tendency, including unexplained retinal hemorrhages | May confirm thrombocytopenia or show morphologically abnormal platelets (which may have defective function); sometimes shows acute leukemia or other condition causing bone marrow failure |
| Acute renal failure | Hemolytic-uremic syndrome and thrombotic thrombocytopenic purpura should be confirmed or excluded |
| Jaundice and hypertension in a pregnant woman | May show schistocytes, supporting a diagnosis of HELLP syndrome |
| Bone pain | May indicate multiple myeloma, bone marrow infiltration, or sickle cell disease |
| Unexplained chest or abdominal pain or acute splenic enlargement in a child | Possible sickle cell disease |
| Unexplained hyperbilirubinemia | Assessment of possible hemolysis |
Laboratory scientists and physicians may initiate a blood smear that has not been requested by the clinician if the clinical details indicate the possibility of a significant hematologic abnormality. However, they are also likely to evaluate a blood smear in response to abnormalities revealed by an automated instrument. These abnormalities may be quantitative or qualitative. Quantitative abnormalities that require evaluation include anemia, polycythemia, macrocytosis, microcytosis, neutrophilia, lymphocytosis, eosinophilia, thrombocytopenia, and thrombocytosis.
Modern automated instruments are able to “flag” the presence of qualitative abnormalities that require a blood film to be examined for confirmation of the abnormality or for further elucidation. “Flags” are generated in response to the electrical impedance or the light scattering characteristics of individual cells. Some instruments are dependent on cytochemical reactions of cells or on the cells' ability to polarize light. Most instruments can indicate the possibility of the presence of blast cells, reactive or other atypical lymphocytes, granulocyte precursors, or nucleated red blood cells. Some instruments can enumerate nucleated red blood cells. Instruments using a cytochemical reaction for peroxidase to help identify neutrophils, eosinophils, and monocytes may flag the appearance of large, unstained (i.e., peroxidase-negative) cells; such cells may indicate a harmless inherited peroxidase deficiency, but sometimes such cells are lymphoma cells, reactive lymphocytes, or leukemic blast cells. Instruments often flag the possibility of an erroneous platelet count, such as when there is an overlap in size between platelets and red cells or when light-scattering characteristics suggest the presence of platelet aggregates (a potential cause of pseudothrombocytopenia). A reported increase in basophil count should generally also be regarded as a flag because it often represents a pseudobasophilia, resulting from the presence of leukemia or lymphoma cells. Some instruments alert the instrument operator to the possible presence of malaria parasites.
International consensus guidelines indicate which automated instrument results require blood smear review. Whether a review is needed is determined in part by whether that specimen is the first one obtained from that patient and whether there has been a significant change from a previously validated result (referred to as a delta check). Laboratory computers can be programmed to indicate when a result meets the criteria for smear review.
Automated red cell measurements can, to some extent, replace examination of the blood film.6 An increased red cell distribution width indicates the presence of anisocytosis. A decreased mean cell volume is usually a reliable indicator of microcytosis. A reduction in the mean cell hemoglobin concentration indicates hypochromia (for most instruments, however, this measurement is less sensitive than the human eye in the detection of hypochromia). An increased mean cell volume usually indicates macrocytosis, but examination of a smear is necessary both to confirm that the result is not artifactual and to elucidate the cause. Some instruments can measure the hemoglobin concentration in individual cells and thus flag the presence of hyperdense cells; however, a blood film is still necessary to distinguish spherocytes from irregularly contracted cells, sickle cells, and other cells that have an increased hemoglobin concentration. Most instruments produce a histogram of the size distribution of red cells, and some do the same for the distribution of hemoglobin concentration; either of these graphic representations may show dimorphic red cells (i.e., two populations of cells).
The Blood Smear in the Differential Diagnosis of Anemia
 Microcytic Anemias
Microcytic Anemias
In microcytic anemias (Chapter 159), the automated count is of considerable importance and may permit a distinction between iron deficiency and thalassemia heterozygosity. In iron deficiency, there is initially a normocytic normochromic anemia; only when the deficiency becomes more severe is there microcytosis. Conversely, in β-thalassemia heterozygosity, there is usually a normal or near-normal hemoglobin concentration, but the red blood cell count is increased, and there is marked microcytosis (low mean cell volume) together with a parallel reduction in mean cell hemoglobin. The blood film provides supplementary information that can favor one diagnosis or the other. Iron deficiency is more likely to be associated with hypochromia and elliptocytes (“pencil cells”), whereas in β-thalassemia heterozygosity, there is microcytosis, hypochromia is less marked, and there are more likely to be target cells and basophilic stippling. Individuals with α-thalassemia involving the deletion of two α genes (–α/–α) or (– –/α α) have red cell indices similar to those of β-thalassemia heterozygosity; in this case, the blood film usually does not provide any additional diagnostically useful information, although individuals with nondeletional α-thalassemia due to hemoglobin Constant Spring have prominent basophilic stippling. When there is deletion of a single α gene, the blood count is either less abnormal or normal, and the blood smear does not provide any extra diagnostically useful information. The blood smear is, however, a useful supplement to the blood count in suggesting a diagnosis of hemoglobin H disease (Chapter 162). The count shows anemia, marked microcytosis (low mean cell volume and mean cell hemoglobin), and usually a reduction in the mean cell hemoglobin concentration. The smear usually shows marked poikilocytosis in addition to microcytosis, and there may be polychromasia, correlating with an elevated reticulocyte count. Iron deficiency anemia also needs to be distinguished from anemia of chronic disease. The blood counts may be quite similar, but in anemia of chronic disease, the smear often shows features of inflammation, such as increased rouleaux formation, background staining (as a result of increased plasma proteins), and sometimes neutrophilia. Other rare microcytic anemias that must be distinguished from iron deficiency include congenital sideroblastic anemia (Chapter 159) and lead poisoning. In congenital sideroblastic anemia, the film is dimorphic, with one population of hypochromic microcytes and another of normochromic normocytic cells. In lead poisoning, the presence of basophilic stippling and polychromasia in a patient with microcytosis can suggest the diagnosis.
Macrocytic Anemias
A blood film can be important in distinguishing true macrocytosis from factitious macrocytosis as a result of the presence of red cell agglutinates (see Fig. 157-16). Diagnostic features that can suggest the cause of macrocytosis are shown in Table 157-2.
TABLE 157-2
USEFUL FEATURES FOR DETERMINING THE CAUSE OF MACROCYTIC ANEMIA
| CAUSE | SMEAR FEATURES |
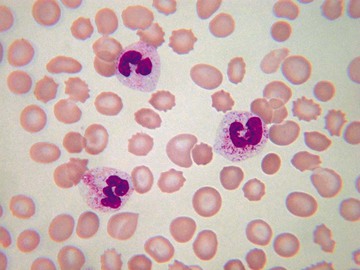
| Megaloblastic anemia (vitamin B12 or folic acid deficiency) | Oval macrocytes, teardrop poikilocytes, hypersegmented neutrophils; when severe, marked anisocytosis and poikilocytosis, which may include red cell fragments |
| Ethanol excess | Target cells and stomatocytes; anisocytosis and poikilocytosis less than in megaloblastic anemia |
| Liver disease | Target cells, stomatocytes |
| Myelodysplastic syndromes, including sideroblastic anemias | Other dysplastic features such as hypogranular and hypolobulated neutrophils; if erythropoiesis is sideroblastic, a population of hypochromic microcytic cells and Pappenheimer bodies |
| Chronic hemolytic anemia | Polychromasia; characteristic poikilocytes sometimes present (e.g., irregularly contracted cells if there is an unstable hemoglobin) |
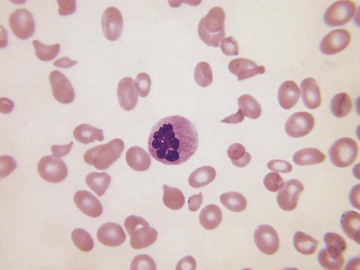
A smear is particularly important in evaluating the possibility of a megaloblastic anemia (Chapter 164) (Figs. 157-4 and 157-21). Sometimes assays of vitamin B12 and folate are normal despite a deficiency, and only the blood film features suggest the true diagnosis and indicate the need for further investigation.
Normocytic Normochromic Anemia
A blood smear is only occasionally useful in determining the cause of a normocytic normochromic anemia. Signs of inflammation may be present in anemia of chronic disease. Increased rouleaux formation and background staining can also indicate multiple myeloma. Polychromasia suggests the possibility of young red cells as a result of recent blood loss or hemolysis. Small numbers of acanthocytes may indicate hypothyroidism or anorexia nervosa. Dysplastic features, such as hypogranular or pseudo-Pelger neutrophils, suggest a myelodysplastic syndrome.
Hemolytic Anemias
The possibility of hemolysis is suggested by the presence of polychromasia and macrocytosis. Specific causes of hemolysis are suggested by the presence of various poikilocytes, as shown in Table 157-3.
TABLE 157-3
BLOOD SMEAR FEATURES SUGGESTING A SPECIFIC CAUSE OF INHERITED OR ACQUIRED HEMOLYTIC ANEMIA
| BLOOD SMEAR FEATURES | CONDITIONS SUGGESTED |
| Spherocytes | Hereditary spherocytosis, autoimmune hemolytic anemia, alloimmune hemolytic anemia (e.g., hemolytic disease of the newborn, delayed hemolytic transfusion reaction), drug-induced immune hemolytic anemia, Clostridium perfringens sepsis |
| Elliptocytes | Hereditary elliptocytosis |
| Oval macrocytes plus stomatocytes | South-East Asian ovalocytosis |
| Irregularly contracted cells | Glucose-6-phosphate dehydrogenase deficiency, oxidant damage from chemicals or drugs in individuals with normal red cell enzymes (e.g., dapsone administration), liver failure due to Wilson disease (release of copper from liver), unstable hemoglobin, hemoglobin C homozygosity |
| Sickle cells and boat-shaped cells | Sickle cell disease (e.g., sickle cell anemia or compound heterozygous states such as S/C, S/D-Punjab, S/O-Arab, S/β-thalassemia) |
| Target cells | Hemoglobin C homozygosity, other hemoglobinopathies, hereditary xerocytosis |
| Stomatocytes | Hereditary stomatocytosis |
| Acanthocytes | Liver failure (spur cell hemolytic anemia) |
| Basophilic stippling | Lead poisoning, pyrimidine 5′-nucleotidase deficiency |
| Red cell fragments (schistocytes) | Microangiopathic hemolytic anemia (including hemolytic-uremic syndrome, thrombotic thrombocytopenic purpura, HELLP syndrome, and sometimes hemolysis associated with disseminated intravascular coagulation), mechanical hemolytic anemia (e.g., defective cardiac prosthetic valve, march hemoglobinuria) |
The distinction between spherocytes and irregularly contracted cells is important; both are dense cells with absent central pallor, but the differential diagnosis is quite different. Recognition of the features of oxidant damage is important in diagnosing glucose-6-phosphate dehydrogenase (G6PD) deficiency because sometimes an assay for G6PD performed during an acute hemolytic episode is normal (Chapter 161). In addition to irregularly contracted cells, there may be ghost cells, hemi-ghost cells (“blister cells”), and even Heinz bodies protruding from the red cells and confirmed on a Heinz body preparation. The observation of these features is an indication to repeat the assay when the acute hemolytic episode is over.
Assessment of Thrombocytopenia, Thrombocytosis, and Platelet Morphology
A blood smear is essential to validate the cell count whenever an automated count shows thrombocytopenia (e.g., a count <60 × 109/L) (Chapter 172). This should be done quickly before patient management is altered, such as by postponing surgery or initiating further diagnostic workup. A platelet transfusion should never be given for an unexpected thrombocytopenia without microscopic confirmation of the count. A factitiously low platelet count is often the result of in vitro platelet aggregation (Chapter 171) and is occasionally the result of platelet satellitism, possibly also with platelet phagocytosis. To detect aggregates reliably, the edges and the tail of the smear should be examined. The presence of fibrin strands also suggests the activation of coagulation and an erroneous platelet count.
If a low platelet count is confirmed, the film may give clues to the cause (Chapter 172). Giant platelets (Figs. 157-22 and 157-23) occur in a number of inherited thrombocytopenias, including Bernard-Soulier syndrome and MYH9-related disorders (the May-Hegglin anomaly and related conditions). Small platelets are less common but are a feature of Wiskott-Aldrich syndrome and familial platelet disorder with propensity to myeloid malignancy. Agranular platelets occur in the gray platelet syndrome7 and platelets with a reduced number of larger than normal granules are seen in Jacobsen/Paris-Trousseau syndrome. The presence of May-Hegglin inclusions (Döhle-like bodies; see Fig. 157-19) in neutrophils indicates that the cause of the thrombocytopenia is a mutation in MYH9. In acquired thrombocytopenias, increased platelet turnover is often accompanied by the presence of large platelets, whereas bone marrow failure is associated with platelets of normal size. It is important to look for red cell fragments to confirm or exclude a diagnosis of thrombotic thrombocytopenic purpura and atypical hemolytic-uremic syndrome in any patient with the apparent recent onset of thrombocytopenia; because platelet transfusions are usually contraindicated in these conditions, the smear should be examined before platelet transfusion is contemplated. The smear of any patient with the apparent recent onset of severe thrombocytopenia should be examined carefully for evidence of acute promyelocytic leukemia; the leukemic cells may be infrequent in the circulating blood. Hemorrhagic manifestations and a low platelet count can also be indicative of meningococcal septicemia; in some patients, organisms are seen in the blood smear and the diagnosis is confirmed; in other patients, only marked toxic changes in neutrophils are detected.
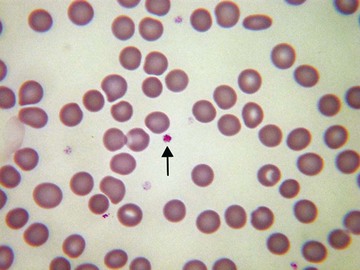
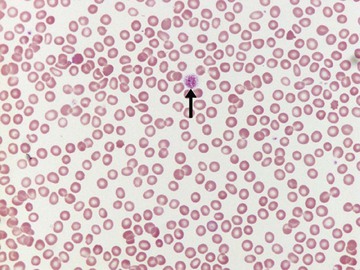
Thrombocytosis should also be confirmed on a smear. Factitiously elevated counts may be the result of the presence of red cell fragments (in microangiopathic or mechanical hemolytic anemia, burns, or accidental in vitro heating of the blood sample), white cell fragments (in acute leukemia and, less often, in lymphoma), cryoglobulin precipitates, or microorganisms (particularly Candida species). If the count is confirmed, the blood smear may be useful to indicate a likely cause (e.g., features of hyposplenism or the presence of basophilia in a myeloproliferative disorder).
It is sometimes necessary to examine a smear to confirm that an apparently normal platelet count is valid. This should always be done in patients with acute leukemia and an elevated white cell count; the presence of white cell fragments of a similar size to platelets can suggest that the platelet count is at a safe level when it is in fact dangerously low. Counting the ratio of platelets to other particles of similar size permits the count to be corrected. Any unexpectedly normal count should be confirmed; for example, the sudden rise of the automated platelet count in a patient being treated for a hematologic neoplasm may be the result of fungi that have colonized an indwelling intravenous line and are being shed into the blood stream.
Leukocytosis and Leukopenia
The finding of leukocytosis is not necessarily an indication for a blood smear (Chapter 170). For example, this finding would be expected in a patient with infection or following surgery or trauma, in which case smear confirmation is not required. However, unexpected leukocytosis requires a smear. Artifactual elevation is unusual but can occur as a result of cryoglobulinemia, hyperlipidemia, or the presence of Candida species. Distinguishing reactive changes from leukemia on the basis of morphology is usually straightforward for myelocytosis but more difficult for lymphocytosis.8 In reactive leukocytosis, there is usually toxic granulation, and Döhle bodies may be present (see Figs. 157-18 and 157-19). Vacuolation is particularly characteristic of bacterial infection, and there may be some degranulation of neutrophils. The hematologist should be aware of the changes induced by granulocyte colony-stimulating factor so that they are not confused with a response to infection; this cytokine can cause toxic granulation, Döhle bodies, vacuolation, and the presence of macropolycytes (giant neutrophils) and circulating neutrophil precursors. The changes typical of leukemia are discussed later.
Leukopenia (Chapter 167) usually requires a film for confirmation and elucidation. The exception is when it is expected in a given clinical context, such as when a patient has had recent chemotherapy. Rarely, an apparent leukopenia is artifactual, owing to the aggregation of neutrophils mediated by an autoantibody or resulting from infection-induced changes in adhesion molecules of the leukocyte surface membrane.
With some automated instruments, it is necessary to confirm that neutropenia is real. If the automated count is based on peroxidase cytochemistry, the presence of an inherited deficiency will lead to an apparent neutropenia associated with an increase of large, unstained (i.e., peroxidase-negative) cells. The scatter plot is characteristic, but because the same features could be due to acute leukemia with neutropenia and circulating blast cells, a smear is needed for confirmation.
Leukemias and Lymphomas
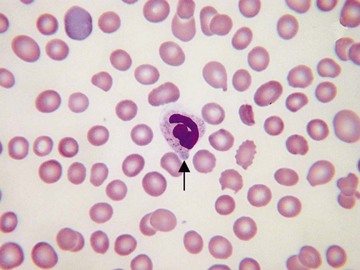
The blood smear is critical in the diagnosis of leukemias and lymphomas. The lymphoblasts of acute lymphoblastic leukemia are usually medium-sized agranular cells with relatively scanty cytoplasm (Fig. 157-24), whereas in acute myeloid leukemia, blast cells are generally larger, with more plentiful cytoplasm that may contain granules or Auer rods (Fig. 157-25). Myeloid blast cells vary in appearance according to whether they are myeloblasts or monoblasts. Myeloblasts are usually medium sized and may have plentiful granules, scanty granules, or no visible granules; they may contain Auer rods. Monoblasts are much larger cells with plentiful cytoplasm containing few granules and, very rarely, Auer rods. Megakaryoblasts are present in some patients and are sometimes cytologically distinctive because of their tendency to form cytoplasmic blebs or develop platelet-type granules.
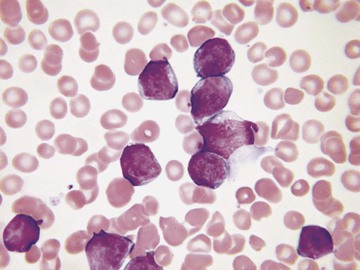
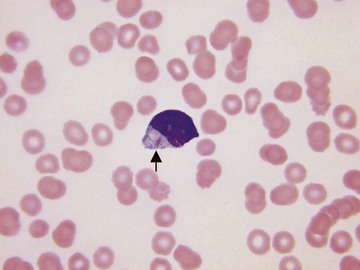
Chronic myelogenous leukemia has a very characteristic blood smear (Fig. 157-26) in which the most numerous cells are myelocytes and mature neutrophils. Eosinophils and basophils are also present. Dysplastic features are generally absent. In atypical Philadelphia chromosome–negative chronic myeloid leukemia, monocytosis is more frequent and dysplastic features are present. Chronic myelomonocytic leukemia is characterized by increased monocytes, some of them immature, with inconspicuous dysplastic features and infrequent granulocyte precursors.
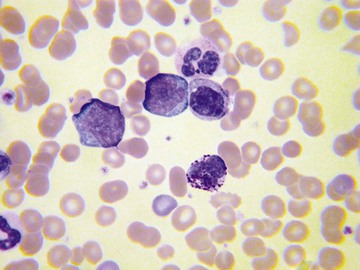
Chronic lymphocytic leukemia also has a very characteristic blood film, with an increase of small, mature lymphocytes of rather uniform appearance. The chromatin is often irregularly clumped, creating a mosaic effect (Fig. 157-27). Smear cells are almost always increased in number but are not pathognomonic.
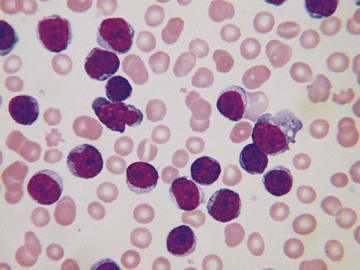
Lymphoma in leukemic phase often has cytologic features that aid in the diagnosis. Follicular lymphoma, Burkitt lymphoma, and splenic marginal zone lymphoma can all be distinctive.
The Incidental Detection of Clinically Significant Abnormalities
Sometimes the examination of a blood film reveals unexpected but clinically significant information. Examples are given in Table 157-4. Detection of microorganisms is particularly likely in patients with acquired immunodeficiency syndrome (AIDS), hyposplenic patients,9 and patients with overwhelming sepsis, but sometimes they are detected in immunologically normal patients with only trivial symptoms. It should be noted that microorganisms in blood smears sometimes represent contaminants, particularly if specimens have been obtained by skin prick or from the umbilical cord and if there has been a delay in making the film.
TABLE 157-4
INCIDENTAL BUT CLINICALLY RELEVANT BLOOD SMEAR OBSERVATIONS
| OBSERVATION | POSSIBLE SIGNIFICANCE |
| Acanthocytes | Abetalipoproteinemia, neuroacanthocytosis (includes choreoacanthocytosis, McLeod phenotype, Huntington-like disease 2 and pantothenate-kinase associated neurodegeneration) |
| Howell-Jolly bodies, target cells, and acanthocytes | Hyposplenism (congenital, previous splenectomy, celiac disease, amyloidosis) |
| Cryoglobulin | Hepatitis C, multiple myeloma, Waldenström macroglobulinemia |
| Vacuolated lymphocytes | Inherited metabolic disorders |
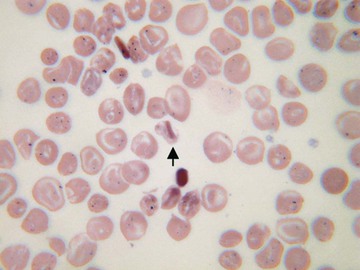
| Parasites (e.g., malaria parasites, Babesia, microfilaria, trypanosomes, Leishmania) | Parasitic infection |
| Fungi (Candida spp, Histoplasma capsulatum, Penicillium marneffei, Cryptococcus neoformans, Malassezia furfur) | Disseminated fungal infection or, in the case of Candida, colonization of an indwelling intravenous line |
| Bacteria (e.g., pneumococcus, meningococcus, Capnocytophaga canimorsus, Borrelia, Ehrlichia, Anaplasma, Yersinia pestis) | Bacterial infection |
| Leukoerythroblastic blood film | Bone marrow infiltration (e.g., metastatic malignancy) |
Conclusion
Despite major advances in other diagnostic methods, the blood smear remains very useful in hematologic diagnosis. Sometimes it is critical either because it yields a diagnosis very quickly or because it provides information that is not available in any other way.
Review Questions
1. A 72-year-old woman presents with gradual onset of digital paresthesia and difficulty with fine movement of the hands. More recently, she has noticed an electrical sensation running down her trunk and into her limbs when she bends her head forward. She is found to have a hemoglobin concentration of 9.8 g/dL and a mean cell volume of 107 fL. A blood film shows macrocytes, oval macrocytes, and hypersegmented neutrophils. Which of the following is the most likely diagnosis?
Answer: B The patient has described the features of peripheral neuropathy and Lhermitte syndrome. These neurologic features in a patient with macrocytic anemia and hypersegmented neutrophils are indicative of vitamin B12 deficiency causing megaloblastic anemia, peripheral neuropathy, and subacute combined degeneration of the spinal cord (Chapter 164). (Larkman N, Hulson O, Gilhooly M. Picture quiz: a man with tingling fingers. BMJ. 2013;346:37-38.)
2. A 67-year-old man presents with right heart failure and extensive bruising. The cardiac silhouette is normal on chest radiography. Electrocardiography shows reduced voltage of QRS complexes. Coagulation screen shows prolongation of the prothrombin time and activated partial thromboplastin time. A full blood count is normal apart from a slight increase of the platelet count to 487 × 109/L. A blood film shows Howell-Jolly bodies, target cells, occasional acanthocytes, and platelet anisocytosis. There is no relevant previous medical history. Which of the following is the most likely diagnosis?
C. AL (light-chain associated) amyloidosis
E. Disseminated intravascular coagulation
Answer: C The blood film features are those of hyposplenism, which also provides an explanation for the slight increase in the platelet count. The hyposplenism is the result of extensive amyloid deposition in the spleen. AL amyloidosis causes a variety of coagulation abnormalities, including acquired factor X deficiency, which could explain the prolonged prothrombin and activated partial thromboplastin times (Chapter 188). Acquired hemophilia would not explain the hyposplenism. (Gatt ME, Palladini G. Light chain amyloidosis 2012: a new era. Br J Haematol. 2013;160:582-598; and Gillmore JD, Wechalekar A, Bird J, et al. Guidelines on the diagnosis and investigation of AL amyloidosis. Br J Haematol. 2015;168:207-218.)
3. A young Greek woman presents to the emergency department because of the sudden onset of jaundice and fatigue. She has just returned from a brief trip to Greece for a family wedding. She is taking no medications. She is noted to be pale and has tachycardia. Her hemoglobin concentration is 8.7 g/dL and mean corpuscular volume is 109 fL. Reticulocyte count is increased to 280 × 109/L. Bilirubin is elevated and is found to be mainly unconjugated. Her blood film confirms marked anemia and shows polychromatic macrocytes, irregularly contracted cells, hemighosts (blister cells), and a few ghost cells. An assay for glucose-6-phosphate dehydrogenase is normal. Which of the following is the most likely diagnosis?
B. Autoimmune hemolytic anemia
C. Glucose-6-phosphate dehydrogenase deficiency
Answer: C Unconjugated hyperbilirubinemia does not suggest acute liver failure. The blood film features are those of oxidant-induced hemolysis, and a diagnosis of glucose-6-phosphate dehydrogenase (G6PD) deficiency must therefore be considered. The blood film features are not consistent with the other types of hemolytic anemia listed. Most patients with acute hemolysis due to G6PD deficiency are men because this is an X-linked condition. However, hemolysis can occur in women because a proportion of red cells will be deficient. If there is marked lyonization, hemolysis may be severe. The G6PD assay is likely to be normal because the remaining red cells are not deficient and the G6PD concentration will be increased by the presence of reticulocytes. In this circumstance, the blood film is very important in suggesting the right diagnosis. A dietary history should be taken. It is possible that the patient has recently eaten broad beans (Chapter 161). (Bain BJ. Sudden onset of jaundice in a Sardinian man. Am J Hematol. 2008;83:810; Bain BJ. A ghostly presence. Am J Hematol. 2010;85:271.)
4. A 35-year-old pregnant woman complains of severe fatigue and swollen ankles. Her blood count shows the following: red blood cells, 4.22 × 1012/L; hemoglobin concentration, 7 g/dL; hematocrit, 0.29 L/L; mean corpuscular volume, 67 fL; mean corpuscular hemoglobin, 16.6 pg; mean corpuscular hemoglobin concentration, 24.5 g/dL. Her blood film shows hypochromia, microcytosis, and poikilocytosis including elliptocytes (pencil cells), target cells, and acanthocytes. Some Howell-Jolly bodies are present. Which of the following is the most likely diagnosis?
A. β-Thalassemia heterozygosity
D. Congenital sideroblastic anemia
Answer: C The blood count is suggestive of iron deficiency, not β-thalassemia heterozygosity (in which moderately severe anemia would not be expected). The blood film shows the features of iron deficiency (hypochromia, microcytosis, and pencil cells) and of hyposplenism (acanthocytes, target cells, and Howell-Jolly bodies). The combination of hyposplenism and iron deficiency suggests a diagnosis of celiac disease with splenic atrophy (Chapter 159). (Croese J, Harris O, Bain BJ. Coeliac disease. Haematological features and delay in diagnosis. Med J Aust. 1979;ii:335-338.)
5. A 24-year-old woman who was hypertensive during pregnancy has an epileptiform convulsion 24 hours postpartum. She has jaundice, edema, and proteinuria. An urgent blood count shows leukocytosis, neutrophilia, anemia, and thrombocytopenia. Her blood film confirms the thrombocytopenia and in addition shows left shift, toxic granulation, and red cell fragments (schistocytes). Which of the following is the most appropriate diagnosis?
A. Disseminated intravascular coagulation
B. Hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome
E. Thrombotic thrombocytopenic purpura
Answer: B This syndrome is a severe form of preeclampsia/eclampsia, occurring during pregnancy or within 48 hours of delivery. The hemolytic anemia is microangiopathic in nature, and the detection of schistocytes is diagnostically useful. (Bain BJ, Riches J. Help with HELLP. Am J Hematol. 2010;85:70; Zini G, d'Onofrio G, Briggs C, et al; International Council for Standardization in Haematology [ICSH]. ICSH recommendations for identification, diagnostic value, and quantitation of schistocytes. Int J Lab Hematol. 2012;34:107-116.)